

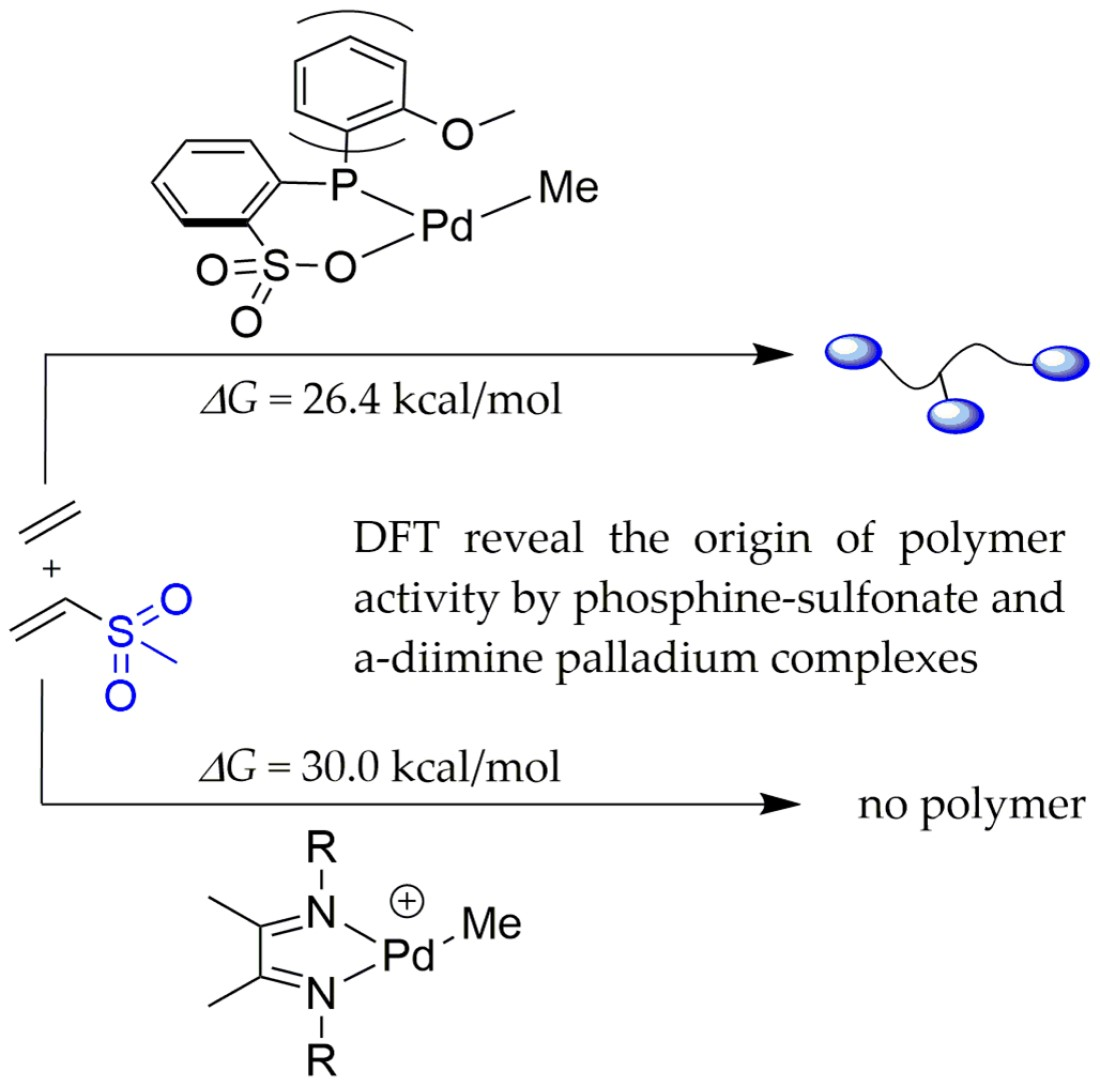
A DFT Study of the Copolymerization of Methyl Vinyl Sulfone and Ethylene Catalyzed by Phosphine–Sulfonate and α-Diimine Palladium Complexes

Abstract
:
1. Introduction
2. Results and Discussion
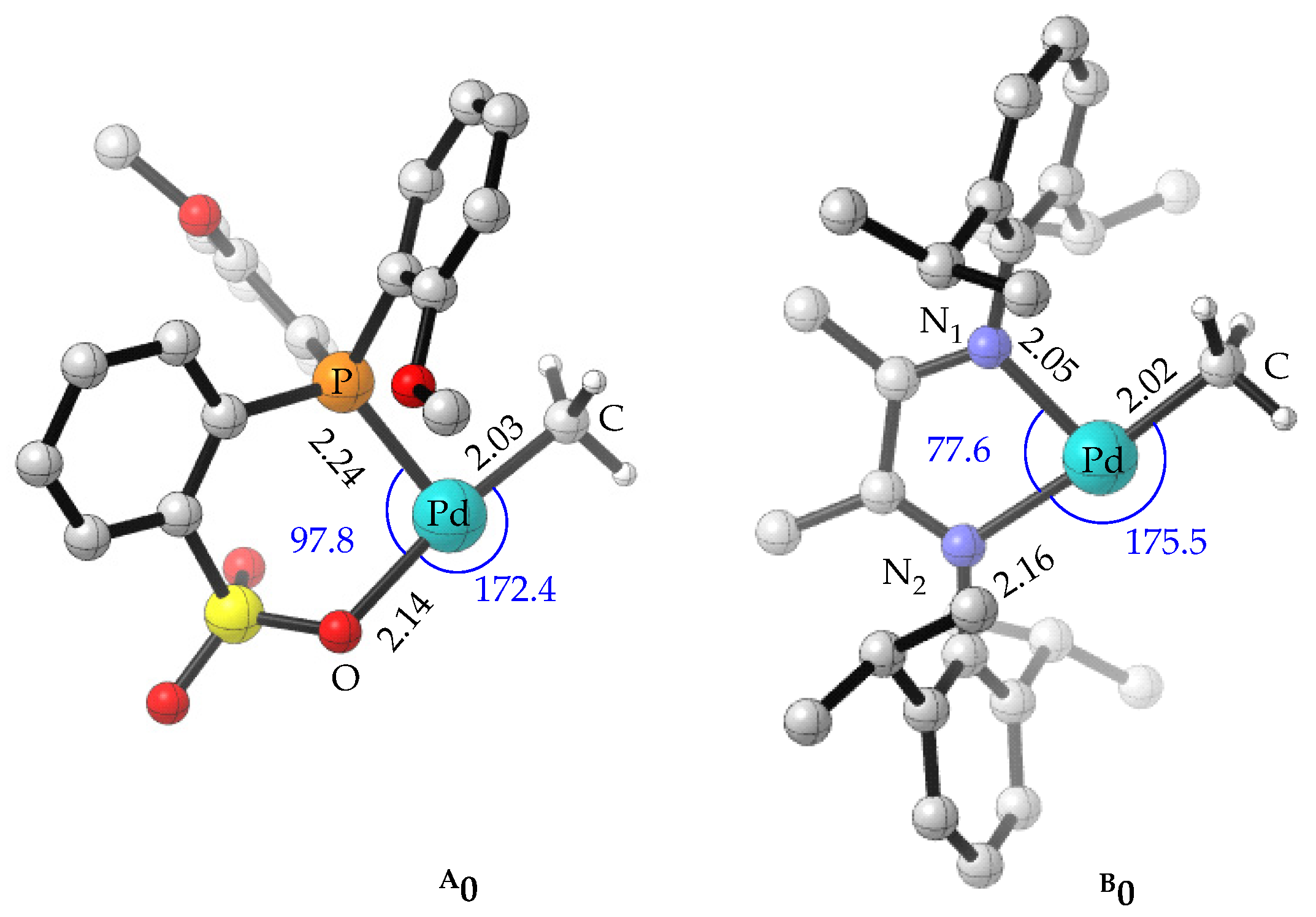
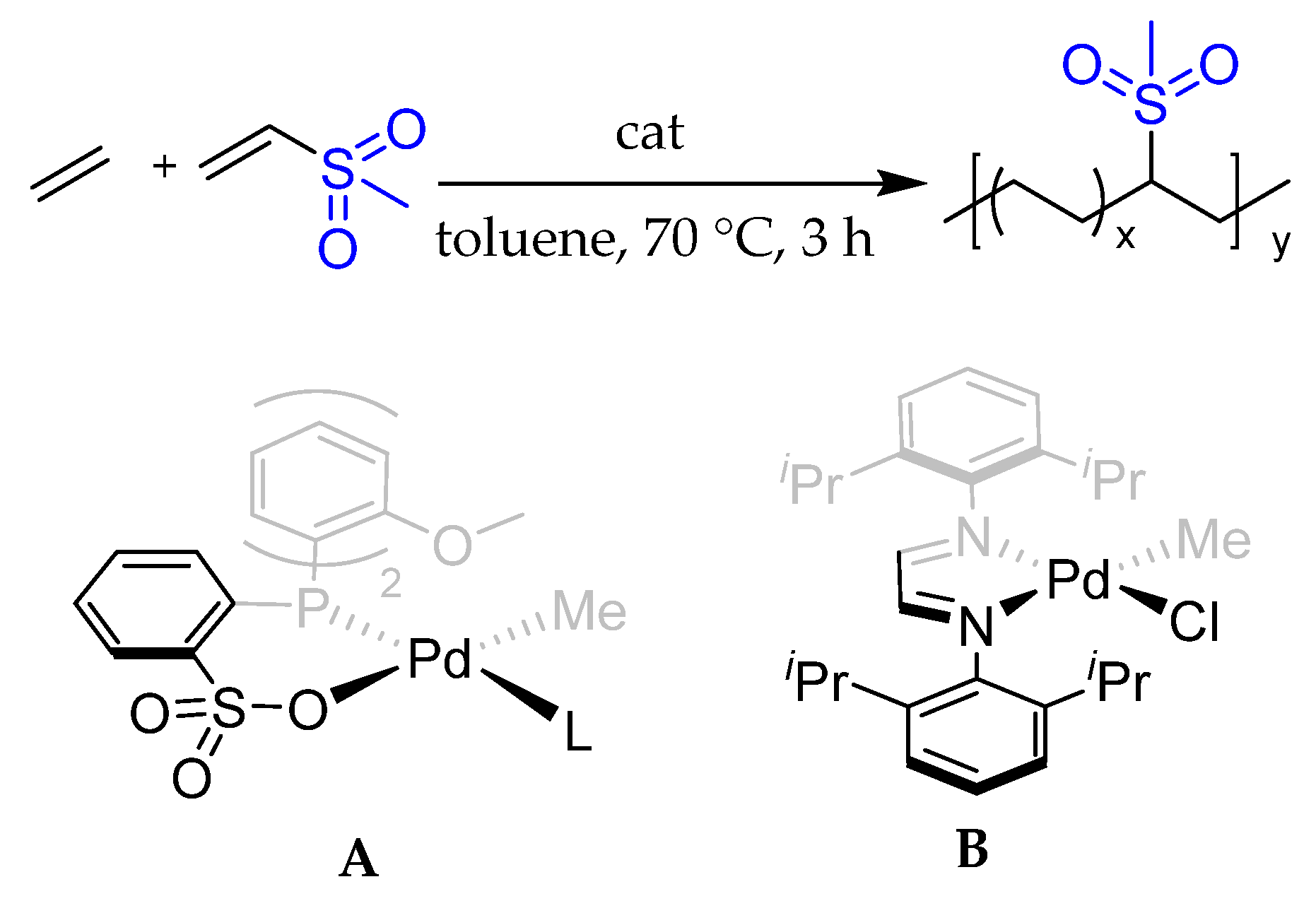
2.1. Structures of Active Species A0 and B0
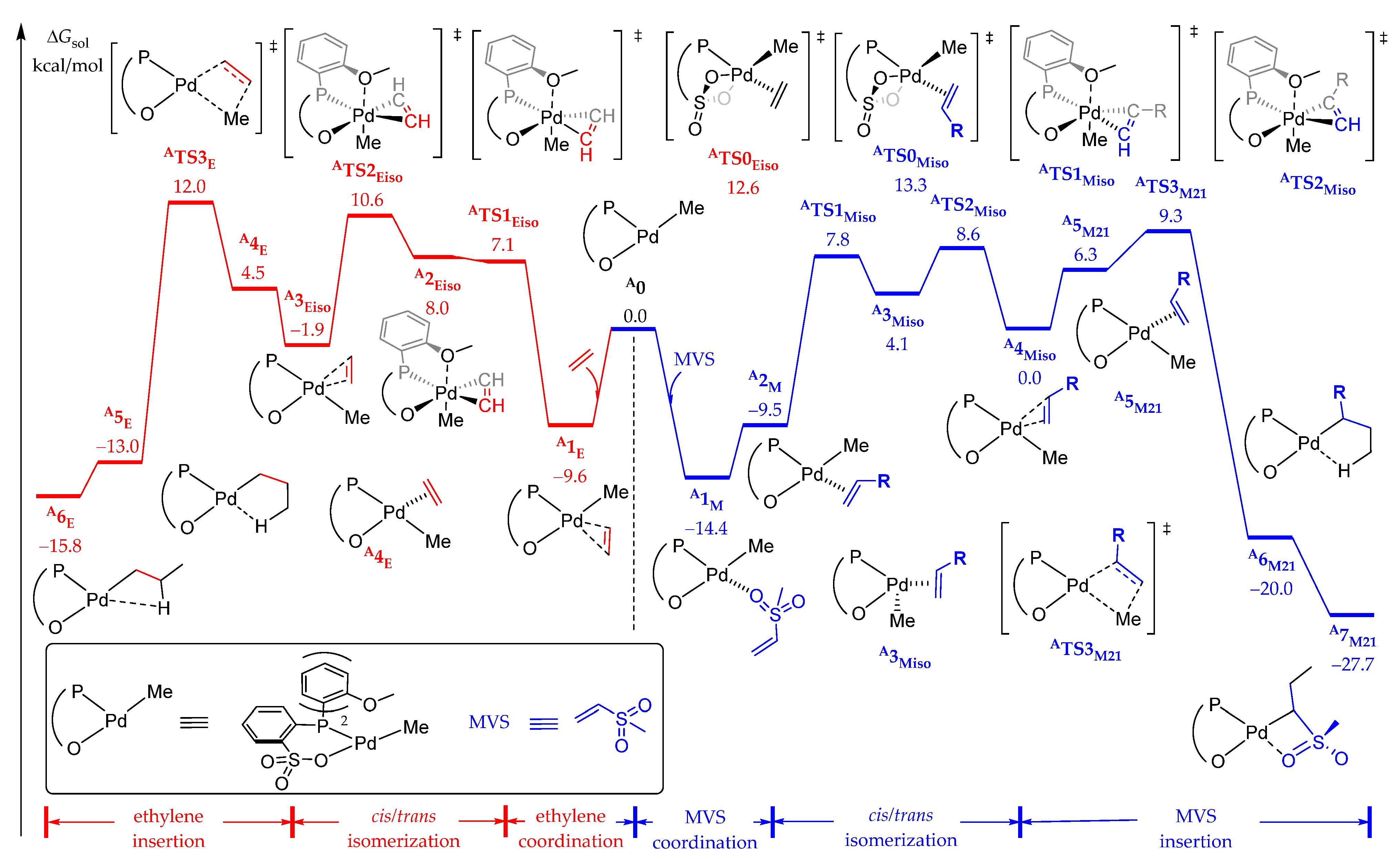
2.2. Copolymerization Mechanism of Ethylene and MVS by the Phosphine–Benzene Sulfonate Catalyst A
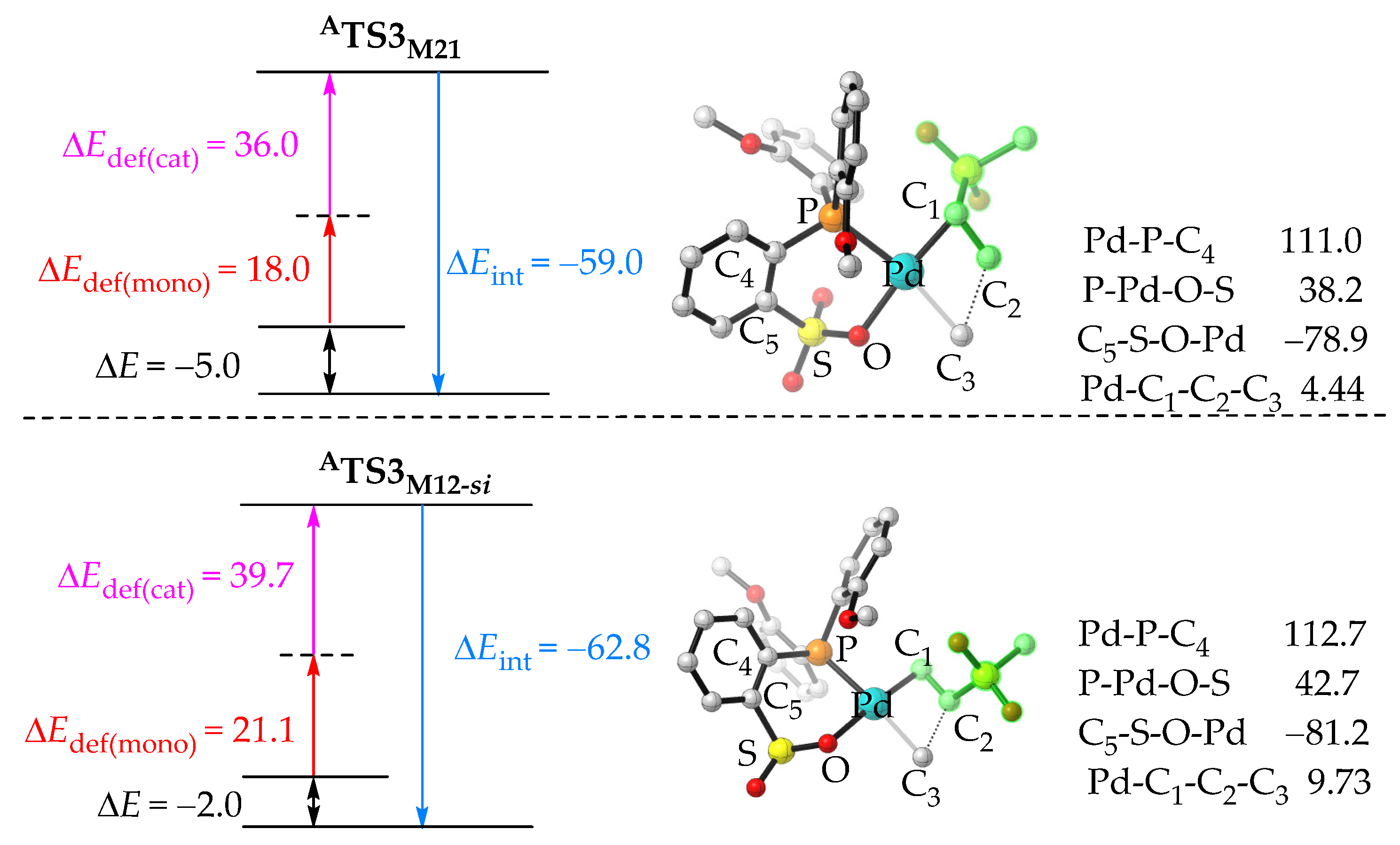
2.2.1. Chain Initiation
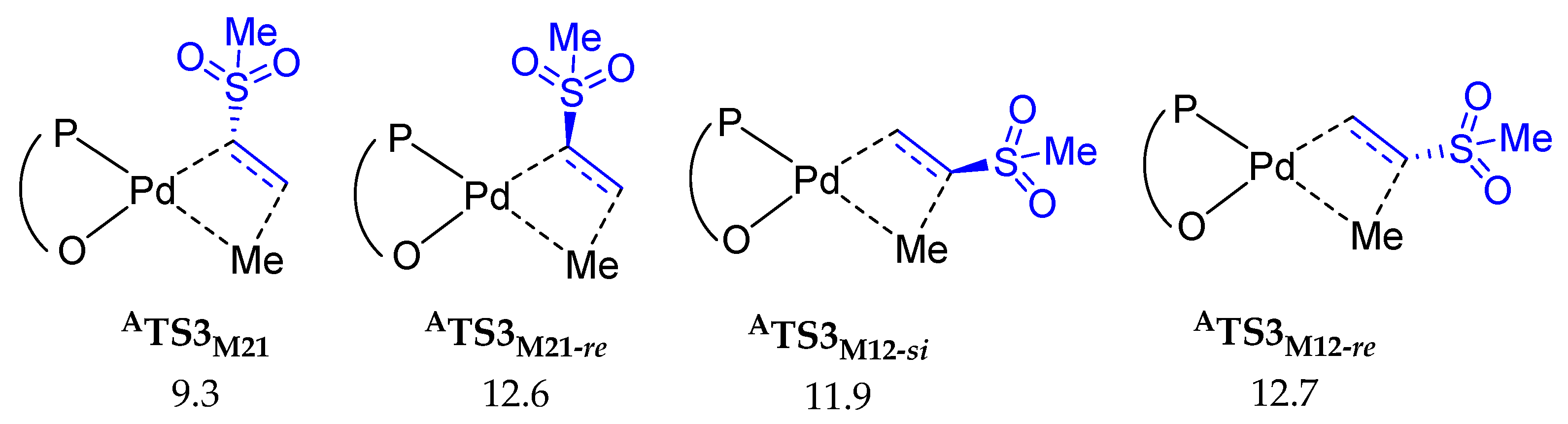
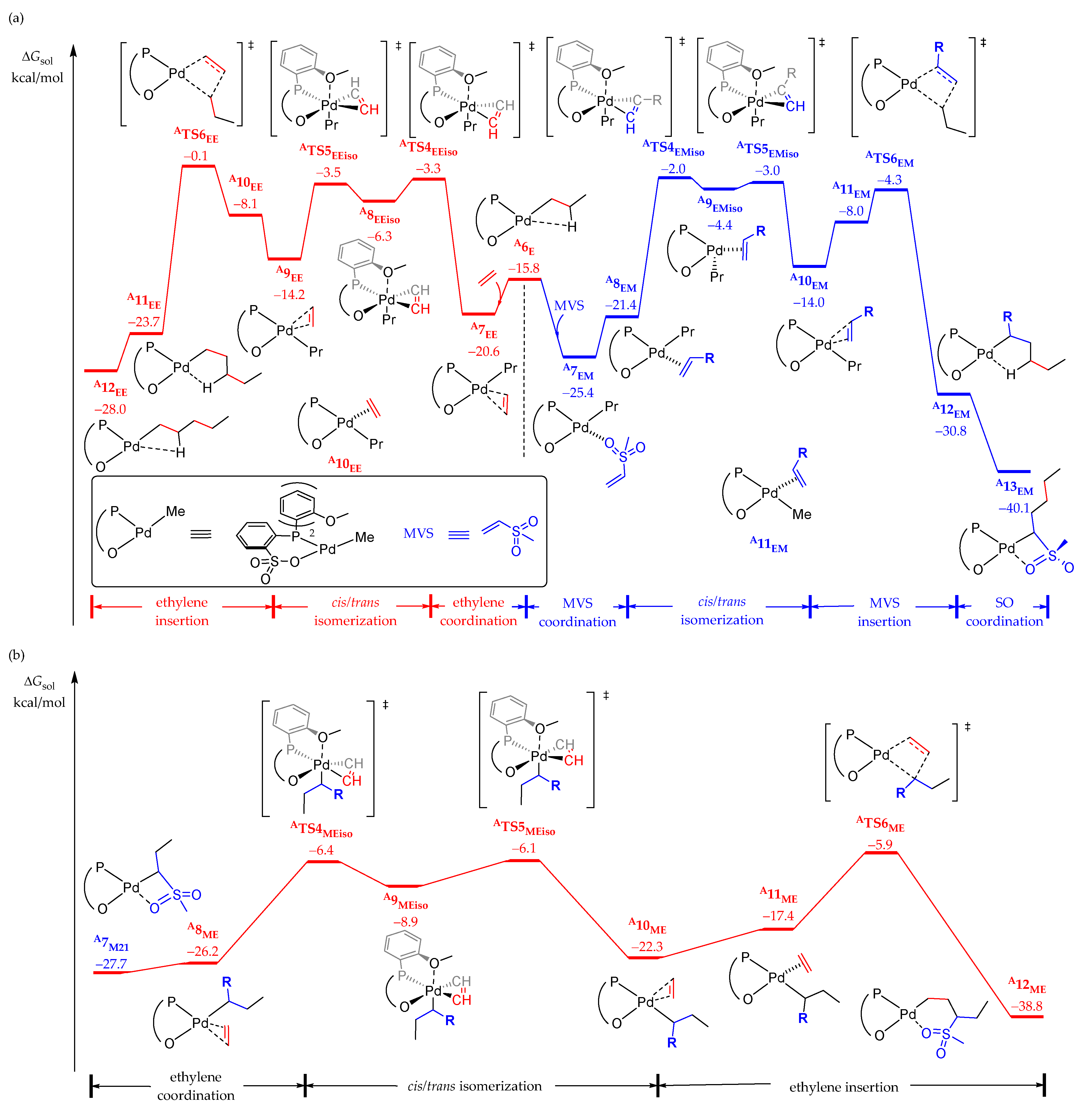
2.2.2. Chain Propagation
2.3. Copolymerization Mechanism of Ethylene and MVS by the α-Diimine Palladium Catalyst B
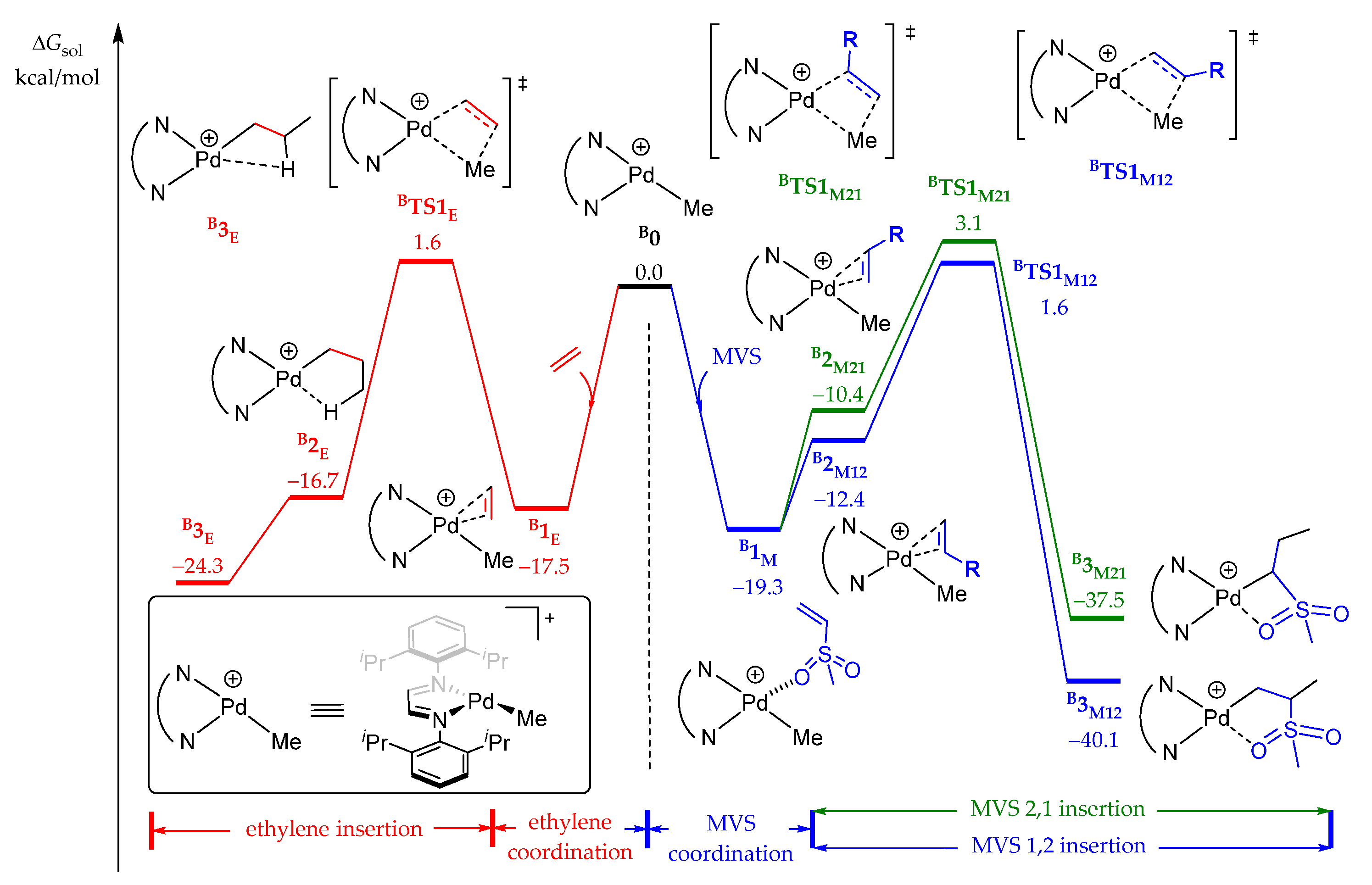
2.3.1. Chain Initiation
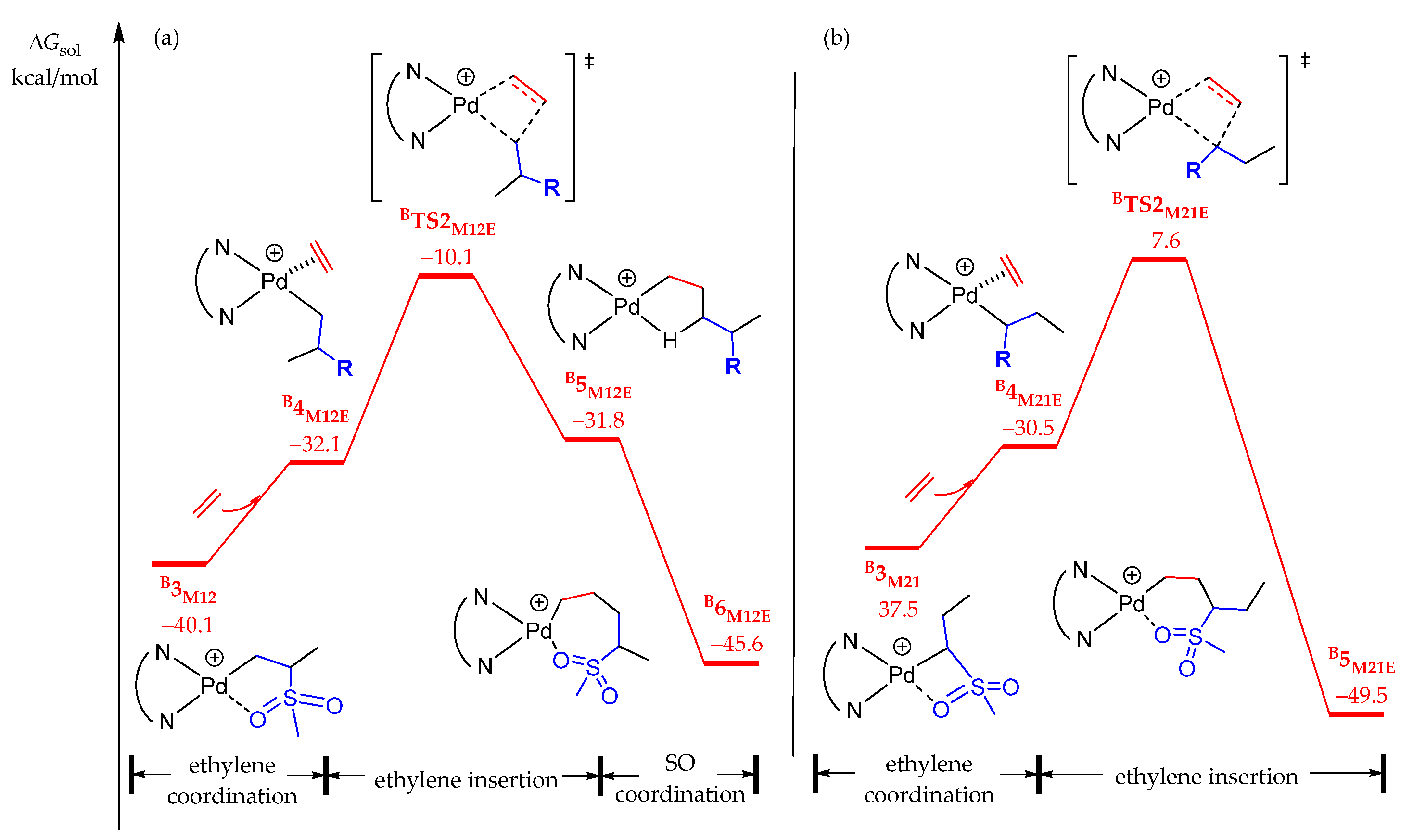
2.3.2. Chain Propagation
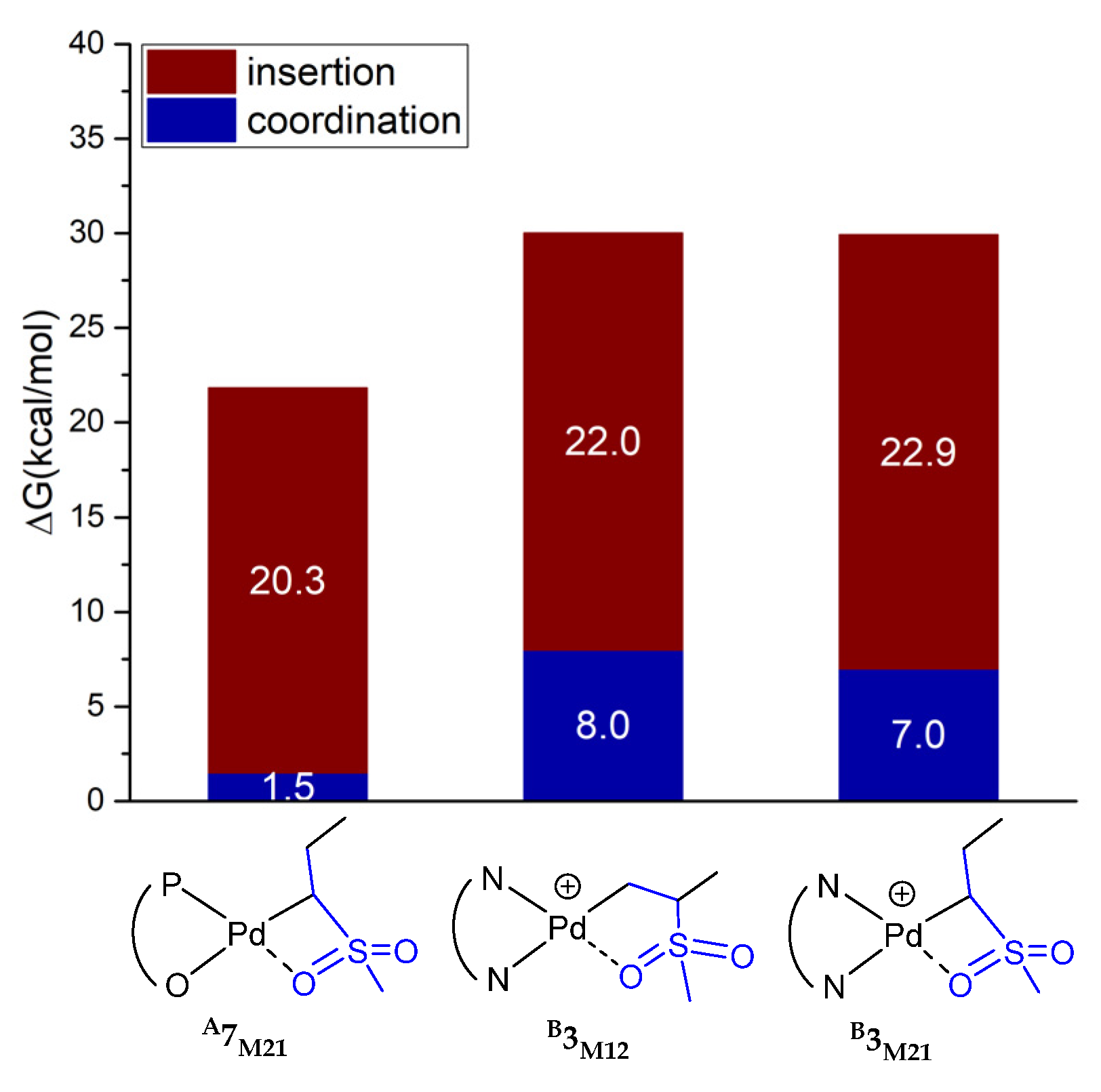
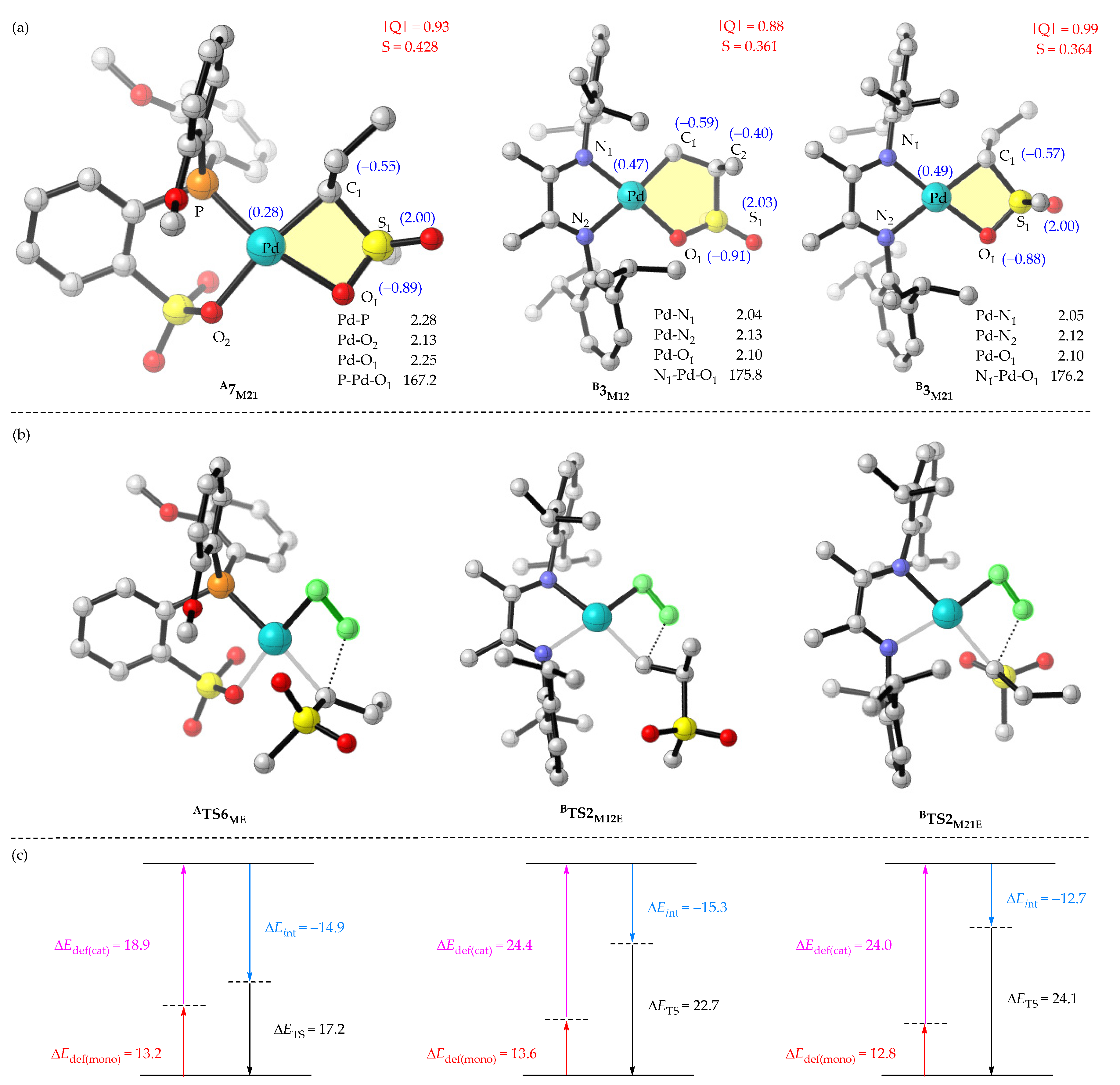
2.4. Comparisons of A- and B-Medicated Copolymerization of Ethylene and MVS
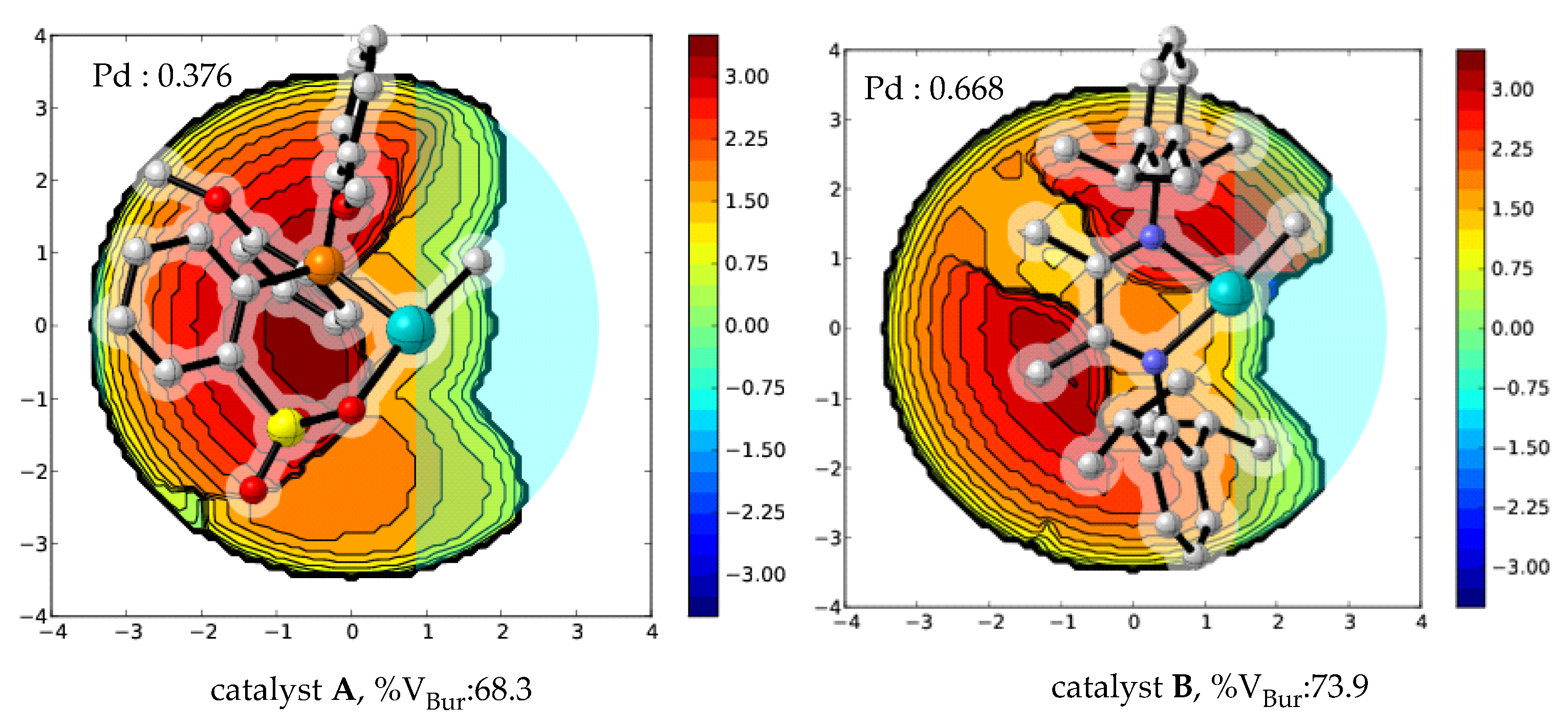
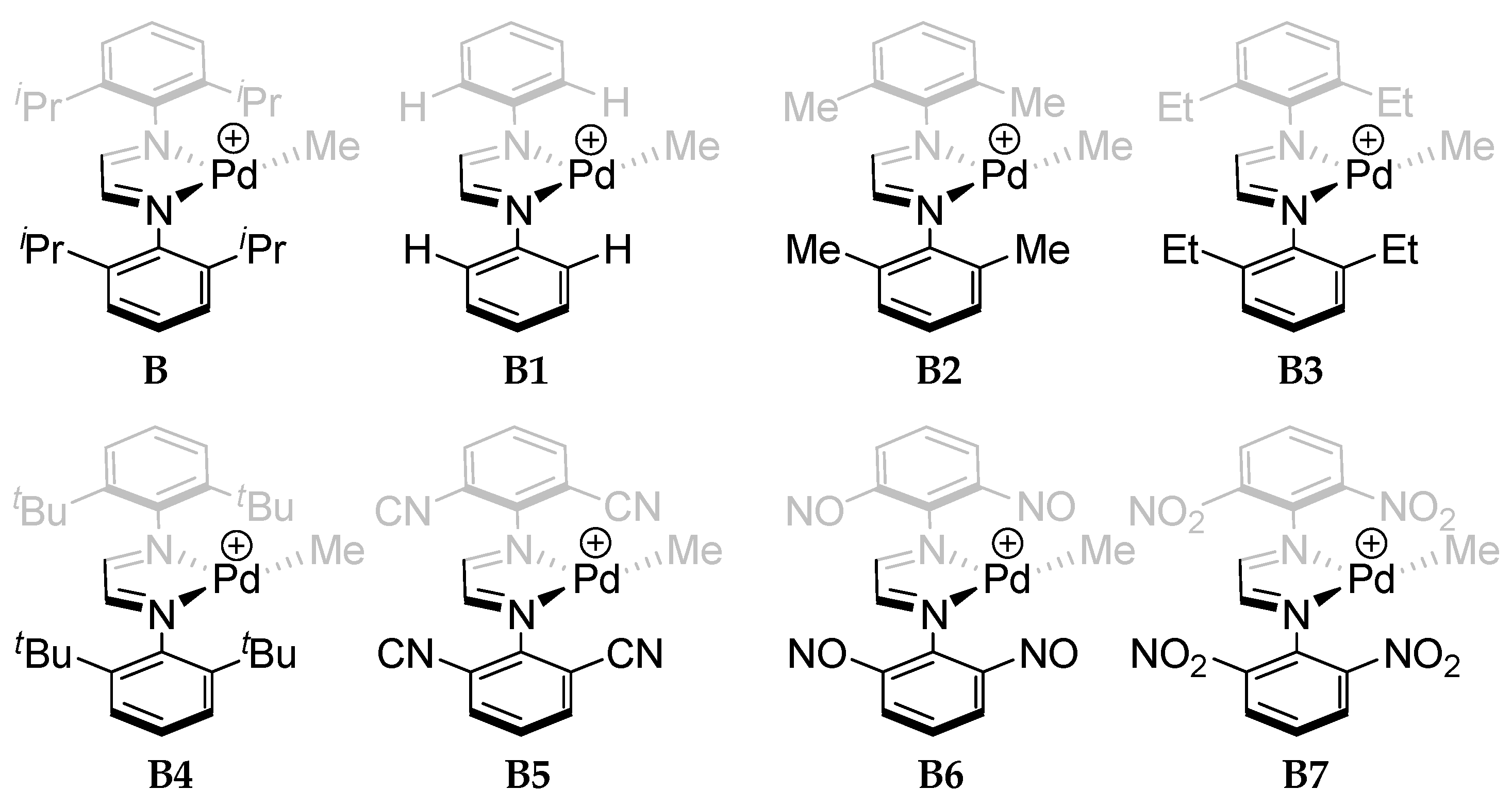
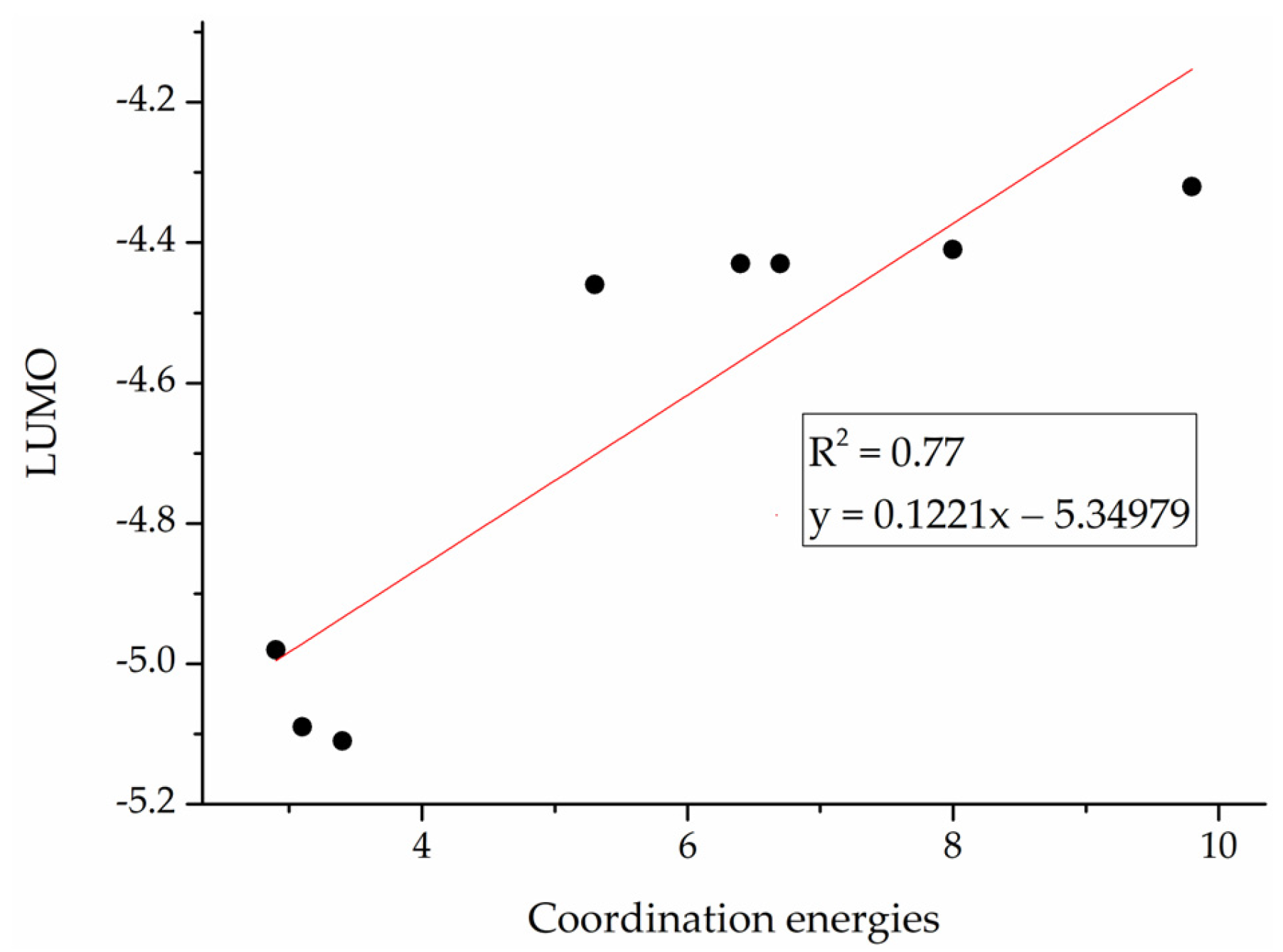
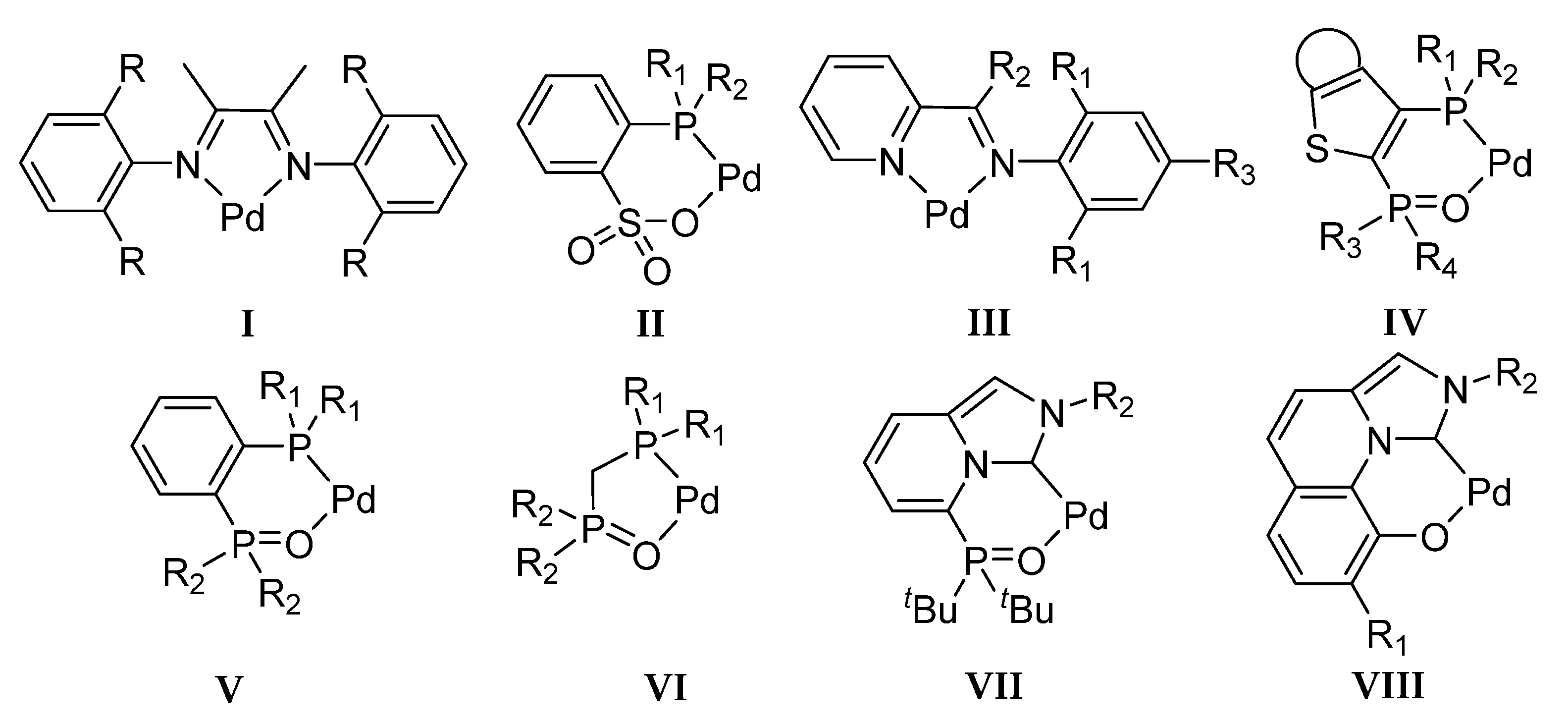
2.5. Catalyst Design
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zou, C.; Chen, C.L. Polar-functionalized, crosslinkable, self-healing and photoresponsive polyolefins. Angew. Chem. Int. Ed. 2020, 59, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Huang, Z. Recent advances in coordination-insertion copolymerization of ethylene with polar functionalized comonomers. Chin. J. Chem. 2020, 38, 1445–1448. [Google Scholar] [CrossRef]
- Karimi, M.; Arabi, H.; Sadjadi, S. New advances in olefin homo and copolymerization using neutral, single component palladium/nickel complexes ligated by a phosphine-sulfonate. J. Catal. 2022, 412, 59–70. [Google Scholar] [CrossRef]
- Mu, H.; Zhou, G.; Hu, X.; Jian, Z. Recent advances in nickel mediated copolymerization of olefin with polar monomers. Coord. Chem. Rev. 2021, 435, 213802. [Google Scholar] [CrossRef]
- Johnson, L.K.; Killian, C.M.; Brookhart, M. New Pd(II)- and Ni(II)-based catalysts for polymerization of ethylene and α- olefins. J. Am. Chem. Soc. 1995, 117, 6414–6415. [Google Scholar] [CrossRef]
- Johnson, L.K.; Mecking, S.; Brookhart, M. Copolymerization of ethylene and propylene with functionalized vinyl monomers by palladium(II) catalysts. J. Am. Chem. Soc. 1996, 118, 267–268. [Google Scholar] [CrossRef]
- Drent, E.; van Dijk, R.; van Ginkel, R.; van Oort, B.; Pugh, R.I. Palladium catalysed copolymerisation of ethene with alkylacrylates: Polar comonomer built into the linear polymer chain. Chem. Commun. 2002, 7, 744–745. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, H.; Li, S.; Dai, S. Direct synthesis of various polar functionalized polypropylene materials with tunable molecular weights and high incorporation ratios. Polym. Chem. 2021, 12, 5495–5504. [Google Scholar] [CrossRef]
- Li, S.; Dai, S. Highly efficient incorporation of polar comonomers in copolymerizations with ethylene using iminopyridyl palladium system. J. Catal. 2021, 393, 51–59. [Google Scholar] [CrossRef]
- Mu, H.L.; Ye, J.H.; Zhou, G.L.; Li, K.K.; Jian, Z.B. Ethylene polymerization and copolymerization with polar monomers by benzothiophene-bridged BPMO-Pd catalysts. Chin. J. Polym. Sci. 2020, 38, 579–586. [Google Scholar] [CrossRef]
- Ye, J.; Mu, H.; Wang, Z.; Jian, Z. Heteroaryl backbone strategy in bisphosphine monoxide palladium-catalyzed ethylene polymerization and copolymerization with polar monomers. Organometallics 2019, 38, 2990–2997. [Google Scholar] [CrossRef]
- Carrow, B.P.; Nozaki, K. Synthesis of functional polyolefins using cationic bisphosphine monoxide-palladium complexes. J. Am. Chem. Soc. 2012, 134, 8802–8805. [Google Scholar] [CrossRef]
- Mitsushige, Y.; Carrow, B.P.; Ito, S.; Nozaki, K. Ligand-controlled insertion regioselectivity accelerates copolymerisation of ethylene with methyl acrylate by cationic bisphosphine monoxide-palladium catalysts. Chem. Sci. 2016, 7, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsushige, Y.; Yasuda, H.; Carrow, B.P.; Ito, S.; Kobayashi, M.; Tayano, T.; Watanabe, Y.; Okuno, Y.; Hayashi, S.; Kuroda, J.; et al. Methylene-bridged bisphosphine monoxide ligands for palladium-catalyzed copolymerization of ethylene and polar monomers. ACS Macro Lett. 2018, 7, 305–311. [Google Scholar] [CrossRef]
- Tao, W.; Akita, S.; Nakano, R.; Ito, S.; Hoshimoto, Y.; Ogoshi, S.; Nozaki, K. Copolymerisation of ethylene with polar monomers by using palladium catalysts bearing an N-heterocyclic carbene phosphine oxide bidentate ligand. Chem. Commun. 2017, 53, 2630–2633. [Google Scholar] [CrossRef]
- Nakano, R.; Nozaki, K. Copolymerization of propylene and polar monomers using Pd/IzQO catalysts. J. Am. Chem. Soc. 2015, 137, 10934–10937. [Google Scholar] [CrossRef]
- Bouilhac, C.; Rünzi, T.; Mecking, S. Catalytic copolymerization of ethylene with vinyl sulfones. Macromolecules 2010, 43, 3589–3590. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, F.; Pan, L.; Wang, B.; Li, Y. Facile synthesis of high-molecular-weight vinyl sulfone (sulfoxide) modified polyethylenes via coordination-insertion copolymerization. Macromolecules 2020, 53, 5177–5187. [Google Scholar] [CrossRef]
- Liu, F.; Hu, H.; Xu, Y.; Guo, L.; Zai, S.; Song, K.; Gao, H.; Zhang, L.; Zhu, F.; Wu, Q. Thermostable α-Diimine Nickel(II) catalyst for ethylene polymerization: Effects of the substituted backbone structure on catalytic properties and branching structure of polyethylene. Macromolecules 2009, 42, 7789–7796. [Google Scholar] [CrossRef]
- Rosar, V.; Meduri, A.; Montini, T.; Fini, F.; Carfagna, C.; Fornasiero, P.; Balducci, G.; Zangrando, E.; Milani, B. Analogies and differences in palladium-catalyzed CO/ Styrene and ethylene/methyl acrylate copolymerization reactions. Chem. Cat. Chem. 2014, 6, 2403–2418. [Google Scholar] [CrossRef]
- Bahri-Laleh, N.; Hanifpour, A.; Mirmohammadi, S.; Poater, A.; Nekoomanesh-Haghighi, M.; Talarico, G.; Cavallo, L. Computational modeling of heterogeneous Ziegler-Natta catalysts for olefins polymerization. Prog. Polym. Sci. 2018, 84, 89–144. [Google Scholar] [CrossRef]
- Li, K.; Mu, H.; Kang, X.; Jian, Z. Suppression of chain transfer and promotion of chain propagation in neutral anilinotropone nickel polymerization catalysis. Macromolecules 2022, 55, 2533–2541. [Google Scholar] [CrossRef]
- Sun, J.; Chen, M.; Luo, G.; Chen, C.; Luo, Y. Diphosphazanemonoxide and phosphine-sulfonate palladium catalyzed ethylene copolymerization with polar monomers: A computational study. Organometallics 2019, 38, 638–646. [Google Scholar] [CrossRef]
- Guironnet, D.; Roesle, P.; Rünzi, T.; Göttker-Schnetmann, I.; Mecking, S. Insertion polymerization of acrylate. J. Am. Chem. Soc. 2009, 131, 422–423. [Google Scholar] [CrossRef] [Green Version]
- Noda, S.; Nakamura, A.; Kochi, T.; Chung, L.W.; Morokuma, K.; Nozaki, K. Mechanistic studies on the formation of linear polyethylene chain catalyzed by palladium phosphine sulfonate complexes: Experiment and theoretical studies. J. Am. Chem. Soc. 2009, 131, 14088–14100. [Google Scholar] [CrossRef]
- Rezabal, E.; Ugalde, J.; Frenking, G. The trans effect in palladium phosphine sulfonate complexes. J. Phys. Chem. A 2017, 121, 7709–7716. [Google Scholar] [CrossRef]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. A theoretical study of the ethylene-metal bond in complexes between copper(1+), silver(1+), gold(1+), platinum(0) or platinum (2+) and ethylene, based on the Hartree-Fock-slater transition-state method. Inorg. Chem. 1979, 18, 1558–1565. [Google Scholar] [CrossRef]
- Deng, L.; Woo, T.K.; Cavallo, L.; Margl, P.M.; Ziegler, T. The role of bulky substituents in Brookhart-type Ni(II) diimine catalyzed olefin polymerization: A combined density functional theory and molecular mechanics study. J. Am. Chem. Soc. 1997, 119, 6177–6186. [Google Scholar] [CrossRef]
- Hu, X.; Kang, X.; Zhang, Y.; Jian, Z. Facile access to polar-functionalized ultrahigh molecular weight polyethylene at ambient conditions. CCS Chem. 2022, 4, 1680–1694. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the ColleSalvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Ehlers, A.W.; Böhme, M.; Dapprich, S.; Gobbi, A.; Höllwarth, A.; Jonas, V.; Köhler, K.F.; Stegmann, R.; Veldkamp, A.; Frenking, G. A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc-Cu, Y-Ag and La-Au. Chem. Phys. Lett. 1993, 208, 111–114. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Andrae, D.; Haüβermann, U.; Dolg, M.; Stoll, H.; Preuβ, H. Energy-adjusted ab initio pseudopotentials for the 2nd and 3rd row transition-elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Legault, C.Y. CYLView, Version 1.0b; Université de Sherbrooke: Sherbrooke, QB, Canada, 2009; Available online: http://www.cylview.org (accessed on 11 April 2023).
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A web tool for analyzing catalytic pockets with topographic steric maps. Organometallics 2016, 35, 2286–2293. Available online: https://www.molnac.unisa.it/OMtools/sambvca2.1/index.html (accessed on 11 April 2023). [CrossRef] [Green Version]
- Liu, C.; Qin, Z.X.; Ji, C.L.; Hong, X.; Szostak, M. Highly-chemoselective step-down reduction of carboxylic acids to aromatic hydrocarbons via palladium catalysis. Chem. Sci. 2019, 10, 5736–5742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.Y.; Pu, M.; Cheng, H.G.; Sperger, T.; Schoenebeck, F. Arylation of axially chiral phosphorothioate salts by dinuclear PdI catalysis. Angew. Chem. Int. Ed. 2019, 58, 11395–11399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Coordination Energy | Relative Insertion Energy | Chain Propagation Energy Barrier | LUMO |
|---|---|---|---|---|
| B | 8.0 | 22.0 | 30.0 | −4.41 |
| B1 | 5.3 | 21.6 | 26.9 | −4.46 |
| B2 | 6.4 | 20.1 | 26.5 | −4.43 |
| B3 | 6.7 | 20.2 | 26.9 | −4.43 |
| B4 | 9.8 | 19.7 | 29.5 | −4.32 |
| B5 | 2.9 | 21.2 | 24.1 | −4.98 |
| B6 | 3.1 | 22.6 | 25.7 | −5.09 |
| B7 | 3.4 | 20.7 | 24.1 | −5.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Li, S.; Kang, X.; Zhang, W.; Luo, Y. A DFT Study of the Copolymerization of Methyl Vinyl Sulfone and Ethylene Catalyzed by Phosphine–Sulfonate and α-Diimine Palladium Complexes. Catalysts 2023, 13, 1026. https://doi.org/10.3390/catal13061026
Zhu L, Li S, Kang X, Zhang W, Luo Y. A DFT Study of the Copolymerization of Methyl Vinyl Sulfone and Ethylene Catalyzed by Phosphine–Sulfonate and α-Diimine Palladium Complexes. Catalysts. 2023; 13(6):1026. https://doi.org/10.3390/catal13061026
Chicago/Turabian StyleZhu, Ling, Shuang Li, Xiaohui Kang, Wenzhen Zhang, and Yi Luo. 2023. "A DFT Study of the Copolymerization of Methyl Vinyl Sulfone and Ethylene Catalyzed by Phosphine–Sulfonate and α-Diimine Palladium Complexes" Catalysts 13, no. 6: 1026. https://doi.org/10.3390/catal13061026