
Machine Learning: A Suitable Method for Biocatalysis


1
Computação e Cognição Centrada nas Pessoas, Lusofona University, Campo Grande, 376, 1749-024 Lisbon, Portugal
2
BioRG (Biomedical Research Group) and Faculty of Engineering, Lusofona University (ULHT), Campo Grande, 376, 1749-024 Lisbon, Portugal
3
iBB—Institute for Bioengineering and Biosciences, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisbon, Portugal
4
Associate Laboratory i4HB—Institute for Health and Bioeconomy at Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisbon, Portugal
*
Authors to whom correspondence should be addressed.
Catalysts 2023, 13(6), 961; https://doi.org/10.3390/catal13060961
Submission received: 28 March 2023
/
Revised: 14 May 2023
/
Accepted: 31 May 2023
/
Published: 1 June 2023
(This article belongs to the Special Issue Editorial Board Member's Collection Series: Innovative Biocatalysts and Enzyme Immobilization)
Abstract
:Biocatalysis is currently a workhorse used to produce a wide array of compounds, from bulk to fine chemicals, in a green and sustainable manner. The success of biocatalysis is largely thanks to an enlargement of the feasible chemical reaction toolbox. This materialized due to major advances in enzyme screening tools and methods, together with high-throughput laboratory techniques for biocatalyst optimization through enzyme engineering. Therefore, enzyme-related knowledge has significantly increased. To handle the large number of data now available, computational approaches have been gaining relevance in biocatalysis, among them machine learning methods (MLMs). MLMs use data and algorithms to learn and improve from experience automatically. This review intends to briefly highlight the contribution of biocatalysis within biochemical engineering and bioprocesses and to present the key aspects of MLMs currently used within the scope of biocatalysis and related fields, mostly with readers non-skilled in MLMs in mind. Accordingly, a brief overview and the basic concepts underlying MLMs are presented. This is complemented with the basic steps to build a machine learning model and followed by insights into the types of algorithms used to intelligently analyse data, identify patterns and develop realistic applications in biochemical engineering and bioprocesses. Notwithstanding, and given the scope of this review, some recent illustrative examples of MLMs in protein engineering, enzyme production, biocatalyst formulation and enzyme screening are provided, and future developments are suggested. Overall, it is envisaged that the present review will provide insights into MLMs and how these are major assets for more efficient biocatalysis.
1. Bioprocesses in Biotechnology
Bioprocess engineering is an interdisciplinary science that combines biology and engineering to optimize the growth of organisms and/or the generation of target materials. Traditional approaches to develop and improve bioprocesses, e.g., the microbial production of antibiotics, have evolved based on different techniques, namely genetic engineering and molecular biology [1].
Biochemical engineering is a multidisciplinary research area that combines natural sciences (e.g., biology, chemistry) with engineering (e.g., chemical engineering, process engineering) to address global challenges and strategic national priorities, including energy, health and sustainability. It involves several topics related to the development of biological processes, from the preparation of raw materials to the synthesis of bioproducts and recovery of biowaste, fostering a circular economy approach, with several applications relevant mainly for industrial biotechnology and medical and health biotechnology [2]. Some relevant milestones in biotechnology achieved through bioprocesses are given in Table 1.
Plenty of mechanistic-chemistry- and physics-based phenomenological and empirical models have been presented so far to simulate the kinetics of microbial biomass growth and product synthesis [7,8,9]; to quantify metabolic flux analysis [10,11]; for bioprocess optimization and biomanufacturing [12,13,14,15,16]; for bioreactor design, modelling and scale-up [17,18,19,20]; for the design of bioseparation units [20,21,22]; for protein and enzyme design [23,24,25,26]; and to characterize the kinetics of enzyme catalysis [24,27,28,29,30]. Nevertheless, and despite advanced mathematical theories such as optimization and statistical analysis, both the research community and industry are still short of efficient and robust modelling models that can accurately translate the complex knowledge of the bioengineering domain into mathematical formulation [2]. However, in recent years, there has been a shift from physical modelling to data-driven modelling with the application of machine training techniques and a large number of data recorded and stored by the biochemical industry. Such an approach, which involves the combination of machine learning algorithms and information sets, shows significant potential for the identification of intricate relationships [31,32,33,34,35,36,37].
Natural products can have different chemical structures and, according to studies carried out on the biosynthesis of these natural products, a wide range of biosynthetic enzymes have been identified [38,39,40]. Biosynthetic enzymes of natural origin are therefore a fundamental source of several effects. From a biocatalytic point of view, the selection of a potential biosynthesis enzyme depends on substrate specificity, cofactors, turnover, stability, functional expression and the ability to perform its autonomous function outside its natural pathway in a specific cell. Still, the biosynthetic enzymes involved in secondary metabolism are often only moderately efficient catalysts, with turnover numbers (kcat) typically about 30-fold lower than those of primary metabolism, which is probably due to their evolutionary histories [41,42]. Moreover, it has been highlighted that the average enzyme is far from the kinetic perfection (kcat~106 to 107 s−1; kcat/KM~108 to 109 s−1M−1, KM: Michaelis constant) of textbook examples of enzymes [41,43]. Additionally, native enzymes also display several other notable limitations, among them poor catalytic efficiency over non-cognate substrates that are often of interest for practical applications; limited activity and stability under harsh industrial conditions (e.g., relatively extreme pH, temperature, ionic strength) and substrate and/or product inhibition [42,44,45]. These limitations and strategies to overcome them through enzyme engineering have been summarized recently [46]. Nonetheless, biosynthetic enzymes have been deemed starting points for defined evolution efforts to improve catalysis rates, substrate tolerance and scope, stability in given solvents, e.g., ionic liquids or organic solvents, and to minimize or eliminate cofactor requirements [47].
Several strategies based on machine learning methods (MLMs) have been presented to make enzymes more performant in real-life applications. As MLMs can be used to gain insight into relationships involving the sequence, structure and function of enzymes in a data-driven manner, they enable the identification of proper sequences from characterized enzymes that are likely to display the improved properties sought after [48,49], such as, e.g., structural stability [50]; to predict the effect of mutations on catalytic power and selectivity [51]; to screen potential substrates for a given enzyme [52]; to identify substrate promiscuity from sequence data [53]; and to predict new beneficial mutant combinations in engineered enzymes that broaden the substrate range of a given enzyme [54]. These last three foster enzyme promiscuity. Regarding this particular matter, novel algorithms to predict enzyme–substrate compatibility that rely on curated information from metagenomic enzyme family screens have been proposed. These are expected to contribute to establishing the basis and standards for reliable enzyme–substrate compatibility models and to enhance our ability to predict enzymes’ ability to act on non-cognate substrates. It has previously been hinted that there might be an optimal range in which the residues should position themselves; this is related to the active site, which may be pertinent for promiscuity [55]. Moreover, it has been highlighted that widening the catalytic site may diversify the binding orientations of different substrates, impair selectivity and ultimately lead to the formation of multiple isomers. Thus, care has to be taken when increasing the space in the catalytic site for enhanced substrate tolerance as not to compromise selectivity [56]. Research dedicated to gaining insights into enzyme promiscuity, in particular to better understand the structure–function relationship, has gained relevance in the last decade. Accordingly, a database dedicated to gathering information on promiscuous activities and helping to identify new catalytic activities and their underlying mechanisms has been recently presented [57]. Improving operational stability may be achieved through enzyme immobilization. By collecting and processing information from published works involving enzyme and carrier properties, a predictive model can be developed to assist in the design of robust immobilized biocatalysts [58]. A package to assist in the rational immobilization of enzymes was developed based on an algorithm that collects information on enzyme features, e.g., active site, surface and residue clustering, and retrieves the literature on immobilization to assist the user in identifying the proper immobilization approach for a given enzyme [59]. Models to identify peptides that bind to specific materials, more specifically polystyrene, have also been presented, which negate the need for expensive and time-consuming wet-lab experiments [60].
2. Artificial Intelligence and Machine Learning
2.1. An Overview and Basic Concepts
Artificial intelligence (AI) gives computers the ability to make decisions via analysing data independently, following predefined rules or pattern recognition models. In the field of biotechnology, AI is widely used for various research challenges, most notably for de novo protein design, where new proteins with envisaged functions are assembled using amino acid sequences not found naturally according to the physical principles underlying intra- and intermolecular interactions [61,62,63,64]; in protein engineering, where selected proteins are manipulated to tailor selected key properties, e.g., activity, selectivity and stability [61,65]. Here, AI has been particularly useful when used to assist directed evolution experiments, namely by enabling a reduction in the number of wet-lab iterations required to generate a protein with the intended features [61,66,67]. Additionally, AI has been used in the field of biopharmaceuticals (drugs and vaccines) to develop new drugs, redefine existing and marketed drugs, understand drugs’ mechanism and mode of operation, design and optimize clinical trials and identify biomarkers. AI is also used in the analysis of genomic interaction and the study of interaction pathways, protein-D, cancer diagnosis and analysis of genetics, among other applications [68,69,70]. Machine learning (ML) is a subfield of AI that allows the development of computer programs that learn and improve their performance automatically based on experience and without being explicitly programmed. In various studies, ML improvement strategies from large datasets generated by different techniques have been advantageously used for different purposes, such as the identification of weight-associated biomarkers [71], discovery of food identity markers [72], elucidation of animal metabolism [73] and investigation of many other areas of metabolomic development [74,75]. Many studies highlight the essential advantage of using ML and systems biology in pathway discovery and analysis, identifying enzymes, modelling metabolism and growth, genome annotation, the study of multiomics datasets, and 3D protein modelling [76]. Based on the available data, ML algorithms allow finding patterns, which represent points with several characteristics or descriptors, e.g., enzyme sequences, their secondary and tertiary structures, substitutions, physicochemical properties of amino acids, etc. These properties usually range from tens to thousands in number, and are thus hard and extremely time-consuming to handle using conventional approaches.
ML can be implemented through unsupervised and supervised learning. Unsupervised learning reduces high-dimensional data to a smaller number of dimensions or identifies patterns from data. In turn, in supervised learning, algorithms use data labelled in advance (designated as a training set) to learn how to classify new, testing data. Labelled data thus consist of a set of training examples, where each example is composed of an input and a sought-after output value. Thus, major features or combinations of features are obtained, and can henceforth improve the label accuracy in the training set and further use the gathered information for future input labelling. To put it another way, one or several target characteristics, e.g., enzyme activity, specificity or stability, can be designated as labels. The goal is to design a predictor that will return labels for unseen data points based on their descriptors using a properly tagged training dataset. Supervised and unsupervised methods can be combined under specific conditions to yield semi-supervised learning [77,78].
Supervised learning is by far the preferred approach in enzyme engineering, as the focus is on improving one or more properties of the enzyme [78]. Overall, the process flow of machine learning can be divided in three stages (Figure 1). Stage 1, which involves data collection, recording and preparation of the input to be fed to the algorithm, is often considered the most laborious phase. The databases BRENDA, EnzymeML, IntEnzyDB, PDB Protein Data Bank and UniProtKB (these and further examples given in [78,79,80,81]) are by far the preferred sources for acquiring information. However, to extract useful information from the retrieved dataset, the data must be adequately pre-processed or cleaned, e.g., managing errors and missing data, detecting and removing duplicates, outliers and irrelevant information, as the quality of data heavily influences the precision of the final outcome [82]. Within this scope, and with the aim to facilitate the use of information throughout multiple scientific areas, steps towards the standardization of data and of semantics have been recently achieved [83]. In stage 2, algorithms process the data that is to be fed to the selected model. The final stage involves model validation using test data. Between stages 1 and 2, the available experimental data are split into two parts: part of the data are used for training subsets and adjusting the parameters of a predictor (stage 2); the remaining data are diverted to stage 3 for the final evaluation [78,84]. The algorithm also has to learn how to classify input data, e.g., assign a label such as spam/not spam. In terms of classification steps with binary labels or labels with a finite number of options, this evaluation is usually based on the number of true confusion matrices: positive and negative true/false. Here, a confusion matrix can be described as a summary of prediction results for a classification problem [78,85]. Classification is evaluated based on the sensitivity and specificity of the results [77]. For the regression steps, where the relationship between independent variables or features and a continuous dependent variable or outcome is addressed, the quality of the prediction is typically evaluated using root mean square deviation [77,78]. The final assessment (stage 3) is carried out on the test dataset. This is paramount as the goal is to ensure the robustness of a model through its successful application to datasets other than those used for training. In enzyme engineering, the occurrence of sequence similarities within training and testing datasets must be justified. Thus, an overrepresentation of a given enzyme family in the training set is likely to lead to a biased predictor that identifies patterns for that sole family. Additionally, similarity between sequences’ training and testing datasets is prone to produce overoptimistic results when the performance is evaluated in stage 3 [78].
Stage 2, which involves either adjusting a predictor or selecting a predictor among several possibilities, is often performed during the training step through K-fold validation. Thus, the training data are subdivided into equally sized K subsets, and then each of the subsets is used for testing while K-1 subsets are used for training. This process is repeated for K times, and finally an average of the performance scores from each subset is determined for each array of hyperparameters evaluated. K-fold validation is intended to mitigate both underfitting (usually high bias and low variance) and overfitting (usually high variance and low bias). Underfitting often takes place when a predictor fails to seize the underlying pattern of the training data and thus is unable to generalize; overfitting takes place when the predictor picks up the details and noise from the training data too well; hence, it is unable to generalize when exposed to unseen data. Underfitting may result from a lack of noise and variance or a too-short training duration. Overfitting is likely to occur due to excessive noise, irrelevant or missing data, data bias or poor quality [78,86,87].
2.2. Building a Machine Learning Sequence–Function Model
In the training stage of a machine learning model, the goal is to tune its parameters to optimize its predictive activity. Accordingly, training aims at the accurate prediction of labels for inputs unseen during training; thus, model performance evaluation must be carried out using data that are absent from the training set (e.g., 20% of the data should be saved for performance evaluation). Besides the parameters, values that are learned directly from the training data and are estimated by the algorithm with no manual contribution, building an ML model requires hyperparameters. These are values required to establish the complexity of the model. Oppositely to parameters, hyperparameters (e.g., the number of layers in a neural network or the scaling of input and/or output data) cannot be learned directly from the training data; thus, they have to be set by the practitioner either by hand or more typically using procedures such as Bayesian optimization, grid search and random search [49,87,88,89]. The selection of proper hyperparameters values is critical since even minor changes can significantly impact model accuracy [90]. Hence, the optimization of hyperparameter values is typically computationally intensive as for each hyperparameter, training a new model is required [49]. In practice, the selection of hyperparameters involves splitting the data remaining after selection of the test set into a training set and a validation set. The former is used to learn parameters, while the latter is used to select hyperparameters that are validated through a proper estimate of the test error. K-fold cross-validation, as described in Section 2.1, is often used, although it requires significant training time. Alternatively, a constant validation set may be used at the risk of a poorer estimate of test error [49,87]. The proper selection of hyperparameters is considered paramount to ensure the success of neural networks, since it determines the correct number of hidden layers, neurons per hidden layer and activation function. Different strategies have been proposed, from a basic random search of hyperparameters to more advanced techniques, such as Bayesian optimization. Irrespectively of the method, the successful implementation of a neural network model depends on the correct selection of hyperparameters, albeit this is often not given proper attention/importance [2].
2.3. A Brief Overview of ML Algorithms
ML relies on the use of algorithms, programs that receive and analyse data (input) and predict values (output) values within a suitable span. As new data are fed to the algorithm, it learns, enhances its operation and concomitantly performs better over time. Algorithms encompass accurate, probabilistic techniques that enable a computer to take a given reference point and identify patterns from vast and/or intricate datasets [91,92]. Different algorithms fostering different ways to achieve this goal have been developed. For instance, the simplest machine learning models apply linear transformation to the input features, such as using an amino acid at each position or the presence or absence of a mutation [93] or blocks of sequences in a library of chimeric proteins made through recombination [94]. Linear models were commonly used as baseline predictors prior to the development of more powerful models. On a different level of complexity and concept, neural networks stack multiple linear layers connected by nonlinear activation functions, which allows the extraction of high-level features from structured inputs. Neural networks are hence well suited for tasks with large, labelled datasets.
Irrespective of their intrinsic nature, MLMs display both merits and limitations. Among the former, MLMs can mine intricate functions and relationships and therefore efficiently model underlying processes; are able to take on extensive datasets, e.g., protein databanks, data from analytical methods such as LC-MS, GC-MS or MALDI-TOF MS, which have been paramount within the scope of multiomics research, offering insights into enzymes’ roles and structure; can find hidden structures and patterns from data and identify novel critical process parameters and control those. This is paramount to warrant validated ranges of critical quality attributes of bioproducts, which determine the value ranges that must be met for the bioproduct to be released. Among the demerits of MLMs, the requirement of large datasets for proper model training, the need for high computational power and the complexity of the set up and concomitant risk of faulty design may be suggested as the major shortcomings [2,16,95,96,97].
Multiple ML algorithms have already been applied to enzyme engineering. Random forests, for example, are used to predict protein solubility and thermostability, while support vector machines have been used to predict protein thermostability, enantioselectivity and membrane protein expression. K-nearest-neighbour classifiers have been applied to predict enzyme function and mechanisms, and various scoring and clustering algorithms have been employed for rapid functional sequence marks [78]. Like Gaussian processes, they have been used to predict thermostability, enzyme–substrate compatibility, fluorescence, membrane localization and channel rhodopsin photo-properties. Deep learning models, also known as neural networks, are well suited for tasks involving large, labelled datasets with examples from many protein families, such as protein–nucleic acid binding, protein–MHC binding, binding site prediction, protein–ligand binding, solubility, thermostability, subcellular localization, secondary structure, functional class and even 3D structure. Deep learning networks are also particularly useful in metabolic pathway optimization and genome annotation [49].
Further examples of ML algorithms, details and illustrative applications are given in Table 2.
3. Some Illustrative Applications of Machine Learning within Biocatalysis
ML enables the timely analysis and processing of big data, hence its application in the growing field of omics (e.g., genomics, proteomics, transcriptomics, metabolomics, lipidomics, single-cell omics, radiomics) [186,187,188,189]. As major outcomes of this intertwining within the scope of biocatalysis, tools have been implemented that expedite enzyme screening [53,55,157,190,191,192] and discovery of novel enzymes [193,194], enzyme engineering [26,49,190,192,195,196,197], design of artificial enzymes [198,199,200], assessment of enzyme function [201,202,203,204], biocatalyst formulation [58,205,206] and in the planning of biocatalysis by predicting suitable reaction pathways [207,208,209]. A schematic representation of the use of MLMs to improve biocatalysis is depicted in Figure 2.
In the following subsections, some representative examples of MLMs in the field of biocatalysis and related fields will be presented and discussed to complement the information given in Table 2.
3.1. Machine Learning Applications in Protein Engineering
Protein engineering focuses on the modification of existing proteins through either changes in amino acids in the protein sequence, typically through replacement, insertion or deletion of nucleotides in the encoding gene, or the design of new proteins. Machine learning has also been used to assist directed evolution for protein engineering, allowing protein functions to be optimized with no requirement of prior knowledge of the underlying physics or the biological pathways [49]. Briefly, MLMs speed up directed evolution by gaining information on the properties, namely the sequence–function relationship, of thoroughly characterized proteins that are available in databases. Once an adequately accurate algorithm is selected to model the structure–function relationship, it is trained, so that the parameters are tuned to maximize its predictive capacity, and evaluated to assess its performance. Afterwards, model-based optimization can proceed for the selection of the sequence or sequences that optimize the function. For instance, models of the mutational effects can be used that ultimately classify the mutations as positive, negative or neutral, or the model may identify a combination of mutations with a high likelihood of improving the function (Figure 3). Thus, ML enables a shift from tedious, repetitive and costly wet-lab cycles of mutation/screening where the best variant in each cycle is selected and used in a new cycle until the intended goal is achieved, to a dry-lab environment, which is less demanding in terms of costs and resources [49,67,210,211,212].
In a recent example, an unsupervised neural network model, MutCompute, was used to predict mutations in two previously engineered poly(ethyleneterephthalate) (PET) hydrolases, aiming to improve their overall catalytic activity at mild temperatures [213]. More specifically, the model can predict positions within a protein where the amino acids can be optimized considering the microenvironment surrounding them. The predictions ultimately suggested three mutations, which, in addition to two previous mutations in the protein scaffold, led to a final mutated enzyme with an increased hydrolytic activity as high as ~140-fold that of the wild type and the scaffold mutants. Moreover, the resulting mutated enzyme outperformed other PET hydrolases when used to process different commercial polyester products. Finally, the mutated enzyme was successfully used in a full, closed cycle of enzymatic degradation/chemical repolymerization, starting with tinted postconsumer plastic waste and ending in a clear, virgin PET film after a few days. Jia and co-workers relied on a PLS approach to predict the thermostability of 64 mutants of an ω-transaminase, using experimental data from six single-point mutated enzymes that were used as a learning dataset and using the half-life (t1/2) of the enzyme as a target result [214]. The authors were able to predict that an enzyme mutated at four specific positions with the suggested residues would result in an ~8-fold increase in t1/2 when compared with the wild type and an over 2-fold increase in t1/2 when compared with the most stable single-point mutated enzyme. The observed pattern was related to both the physical–chemical properties of the residues involved and their position in the protein. The work also highlighted the feasibility of using MLMs to achieve good prediction of enzyme behaviour with a reduced set of experimental data, cutting down on operational costs. The same broad conclusions were reported by Yoshida and co-workers [215] while screening ~8000 enzymes for a promising mutant Burkholderia cepacia lipase with improved thermostability. Using data from ~200 selected mutants, the authors relied on multivariate analysis to decrease the number of possible combinations to 20 candidates. This was based on the relationship between each residue and a set of physical chemical properties, which was used to establish explanatory valuables and train the model with the resulting thermostability activities as objective variables. The data were then split into two and designated as improved and non-improved. From the 20 candidates, which were experimentally prepared, a triple mutant emerged with significantly higher initial and residual activity upon incubation at 60 °C when compared with the wild type as well as the other mutant candidates. MLMs were also recently used to engineer halogenase WelO5* to alter the selectivity and activity of the enzyme to produce mutants able to functionalize sorophens A and C [216]. These macrolides, which have an anti-fungal role, are not substrates of the wild type. Using Gaussian processes, the authors picked up a double-mutated WelO5*active over sorophen A and narrowed down the number of variants to be screened for improved activity and selectivity to ultimately predict and experimentally obtain a mutant that displayed increases of two and three orders of magnitude in the apparent catalytic constant and total turnover number, respectively, when compared with the initial hit. Again, the authors highlighted the role of MLMs as a swift and cost-saving approach to produce effective biocatalysts. Further representative examples of the use of MLMs for enzyme engineering can be found in recently published reviews [195,217].
3.2. Process Optimization (Enzyme Synthesis)
The optimization of microbial enzyme production has traditionally been performed with the one-factor-at-a-time approach (OFAT), which is time-consuming and fails to identify interactions among variables. Hence, it has been gradually replaced with statistically designed experiments, e.g., response surface methodology (RSM), where several factors are varied simultaneously using empirical multivariate models [218,219]. More recently, MLMs have been gaining relevance, namely to address situations where detailed insight into the process is missing or the formulation of the reaction mechanism is not feasible [219,220]. Thus, MLMs have been used to determine the correlation between operational (input) conditions (e.g., pH, temperature, substrate concentration and nature, flow rates) and (output) microbial metabolism (e.g., enzyme synthesis) and then predict the output for given inputs. This is typically carried out through supervised learning algorithms, whereas the identification of hidden underlying patterns in data, outlier detection and dimensionality reduction are left to unsupervised learning algorithms, which have been used for various applications, e.g., process control. Overall, the algorithms used enable the prediction of relevant output variables, e.g., enzyme yield, or the most adequate parameter for scaling up or down [2,31,95,221,222] (Figure 4).
As recent example, an ANN was recently selected among other MLM methods to improve the fermentative production of a cellulolytic complex through optimization of nitrogen content (titre of yeast extract), inoculum size and duration of fermentation [223]. The ANN was singled out based on the improved accuracy of the predicted outcome, as illustrated by having the lowest mean square error, as compared to other MLM tested. Moreover, under ANN optimization, cellulase productivity improved ~2.8-fold, which bested the ~2.4-fold increase in cellulase productivity achieved when RSM was used in either case as compared to the cellulase productivity reported after initial screening studies. Additionally, the ANN model exhibited a coefficient of determination much closer to 1 than the RSM model, again highlighting the superior predictability of the former.
Sarmah and co-workers singled out a GB method among other MLMs to model the growth kinetics of Candida antarctica (currently Moesziomyces antarcticus) for lipase production [224]. The selection was based on the high predictability of the said method as compared to the remaining ones, as illustrated by their low root mean square error, which was selected as an objective function. The selected GB method was shown to require less than half of the number of samples as compared to a fully conventional experimental approach to deliver a predictive Monod growth model that translates into noticeable savings. Additionally, the kinetic model parameters proved highly accurate, as the coefficient of determination was close to 1. The authors identified the peak of enzyme activity production, although no further comparison was performed.
Despite the acknowledged limitations of the OFAT, this approach can be advantageously used if combined with a suitable MLM, as exemplified by Das and Negi [225]. These authors generated data through the OFAT to identify the operational parameters—including carbon source (hexadecane) concentration, metal ion concentration, incubation time, inoculum size, pH and temperature of incubation—that improved alkane hydroxylase productivity in a submerged fermentation. The dataset was then divided and used to train, validate and test an ANN model to further optimize operational conditions, ultimately achieving a ~1.8-fold increase in enzyme productivity as compared with the best result obtained with the OFAT. A similar strategy that combined the OFAT and an ANN was employed by Kumar and co-workers to model and predict xylanase production by Penicillium citrinum xym2 [226]. Thus, a dataset was generated using the OFAT method that comprised the optimization of incubation pH, temperature and time, nitrogen source and titre and additional carbon source titre using xylanase activity as an output variable. The dataset was again used to train, validate and test two ANN models for different input conditions. Single hidden layers and double hidden layers were developed that enabled the accurate prediction of xylanase activity, as reflected by the low mean square errors of ~0.0046 and ~0.0022, respectively, and provided a good correlation between the observed and predicted data, as exemplified by the correlation coefficients of ~0.938 and ~0944, respectively. Not surprisingly, the double-layered ANN allowed for more accurate prediction of the actual values of enzyme activity.
Although submerged fermentation is the most widely used approach for microbial enzyme production, solid-state fermentation has been gaining relevance, particularly when filamentous fungi are used as the producing strain. Accordingly, Silva and co-workers evaluated ANN and SVM models to establish the effect of several operational parameters, e.g., incubation time, inducer concentration, inoculum concentration, moisture titre and pH of the nutrient solution, on alginate lyase, the output variable, with Cunninghamella echinulata as the enzyme producer [227]. Both models ruled out inoculum concentration as a relevant variable within the range of parameters tested. The ANN, as a predictive model, slightly outperformed the SVM, as the former led to a marginally higher coefficient of determination. This was somehow unexpected, since SVMs are considered more suitable when relatively small datasets are available, as was the case in the study considered.
A different approach to the enhancement of enzyme production using an MLM is illustrated in the work of Beier and co-workers, who used an MLM to associate a network of six selected genes with the induction of cellulase produced by Trichoderma reesei and thus establish the basis for overexpression of cellulase [228].
3.3. Biocatalyst Formulation
Enzyme immobilization, e.g., confinement of the enzyme in a restricted region of space, is known to potentiate the use of the enzyme, since it enhances stability and resistance to environmental factors, improves the control of the reaction and enables reusability [229]. However, despite being consistently used for several decades, enzyme immobilization is still vastly empiric, relying on tedious and time-consuming trial-and-error methodologies [230]. The use of rational design procedures is seen as a way to decrease the costs of immobilization protocols, but their implementation requires knowledge on several physical, chemical and biological variables related to the method of immobilization, the carrier and the enzyme itself and how to correlate these with output variables illustrative of immobilization efficacy, e.g., expressed activity, which is also known as activity recovery [230,231]. Thus, MLMs are an attractive way to deal with the vast combination of variables. MLMs can be used to retrieve information from databases that harbour information on enzyme structure and function and the physical–chemical features of carriers and infer key properties of the immobilized biocatalyst, e.g., activity and stability [232]. Additionally, MLMs can be used to process the huge number of data generated when a multidisciplinary approach combining physical–chemical and biological properties is used to determine the optimal immobilization procedure. Accordingly, Ralbovsky and Smith used multivariate analysis to process a complex dataset on pantothenate kinase immobilization into two commercial acrylamide- and methacrylate-based carriers that was generated through Raman hyperspectral imaging [205]. The latter can provide information on the chemicals involved in enzyme immobilization and their spatial distribution. Processing the image dataset with multivariate analysis and PCA allowed the researchers to identify and spatially resolve six diverse elements critical for the successful immobilization of the enzyme and to quantify the coverage of the surfaces by the enzyme. Ultimately, the authors proposed that the output variable quantitation of enzyme coverage can be used as a metric to evaluate enzyme immobilization. For the case study, the authors established that the acrylamide carrier allowed 1.3-fold higher enzyme coverage than the methacrylate carrier, hence rendering the former more effective for panthotenate kinase immobilization. The authors assumed that the often-observed correlation between enzyme loading and enzyme activity held, but no experimental validation was performed. Notwithstanding, the strategy presented provides a direct quantitative metric with which to evaluate enzyme immobilization efficacy. In a follow-up of this work, Ralbovsky and Smith established that the combined use of Raman hyperspectral imaging and PLS could be used to identify and classify samples of immobilized panthotenate kinase formulations. As an example, the set up developed allowed the identification of the carrier to which the enzyme was bound. The authors suggested that the work could be further expanded to identify and gain insight into how other carriers interact with the enzyme [233].
Chai and co-workers also opted for an MLM to predict the immobilization of several enzymes onto metal–organic frameworks (MOFs) [58]. The authors used as input variables properties of metals (e.g., metal ion concentration), ligands (e.g., ligand precursor functional group) and enzymes (e.g., dominant amino acid group), generating up to 12 input variables; as output variables, activity retention, enzyme loading, immobilization yield and reusability were used. However, the method of enzyme immobilization was not included in the model inputs. The dataset was obtained through a literature survey. RF and GP algorithms were used to handle the vast combination of available information, with the former emerging as the most adequate to predict enzyme loading, immobilization yield and reusability. However, neither model was able to accurately predict activity retention (coefficients of determination ~0.6). This is understandably the most difficult property to predict, and the poor fit was attributed by the authors to the lack of information regarding input parameters such as orientation and degree of exposure of the active site of the enzyme upon immobilization. Again, this outcome highlights the need for complex (and often costly and thus not widely available) techniques and equipment for the detailed characterization of the enzyme (and eventually remaining components of the immobilized biocatalyst) to implement reliable predictive models for rational enzyme immobilization.
3.4. Enzyme Screening
Screening for enzyme activity using databases and/or data from laboratory experiments involves properly handling a vast array of data, and accordingly ML techniques have been developed/adapted for such a role [190]. There are some limitations still, since not all databases are machine-learning-friendly databases, and some are misannotated or not frequently maintained, which makes choosing adequate databases for the use of MLMs critical. In addition, some of the databases are misannotated, populated with disproved results or no longer maintained. Hence, choosing the right datasets for machine learning is critical to avoid feeding inaccurate data to the model. The more a database complies with FAIR (findable, accessible, interoperable and reusable) guiding principles for scientific data management and stewardship, the better the data usage and lower the efforts required for data cleaning [2,78,212,234]. Some recent examples of the application of MLMs for enzyme screening are presented. Poly(ethylene terephthalate) (PET) is among the most frequently discarded plastics and thus presents a major environmental concern; hence, strategies for its biodegradation have been sought after. These include PET hydrolases that are able to depolymerize PET to its monomers close to its glass transition temperature, ~65–70 °C, in an aqueous environment [235]. PET hydrolase activity is considered scarce in nature; thus, Erickson and co-workers focused on expanding the range of putative thermotolerant PET hydrolases [236]. Upon examining several databases, e.g., the NCBI and BacDive databases, and selecting features, e.g., physical–chemical and residues, the authors tested several MLMs to identify thermophilicity. Ultimately, an SVM method [237] was chosen, which enabled the identification of 74 putative thermotolerant PET hydrolases, which were later evaluated for activity assessment. Roughly half of these proved active over amorphous PET, which highlights the success of the approach.
The concept of absolute enzyme specificity is a textbook example, yet the search for promiscuous enzymes is currently a hot research topic [237]. Enzyme promiscuity is related to the ability of an enzyme to catalyse reactions other than its native one [238]. Usually, an enzyme’s secondary activity is significantly lower than its primary activity under natural conditions [239], but this can be tuned using suitable engineering approaches [240,241]. Broad substrate acceptance by a given enzyme is most appealing from a biotechnological standpoint as this widens the range of applications without the added costs of producing multiple enzymes [237,242]. Esterases have wide applications in industry, e.g., as detergents, food and pharmaceuticals [243]. In a recent study, Xiang and co-workers presented a bioprospecting strategy for the identification of promiscuous esterases from sequences in databases through the combination of different MLMs [53]. The method ultimately led to the identification of ten sequences that were experimentally tested and validated as promiscuous esterases, thus establishing the validity of the proposed strategy.
Prediction of substrate scope for bacterial nitrilases using a structure-based approach was developed by Mou and co-workers [244]. The different MLMs evaluated by the authors to predict the substrate scope performed similarly, although the RF method provided marginally higher precision and sensitivity. The authors thus suggest that their RF approach could be used to predict substrate scope for other enzyme classes.
Many methods used for determining enzyme activity from databases rely on the use of amino acid sequences (either annotated or unannotated, the latter being favoured, as they foster the identification of novel enzyme functions) to retrieve Enzyme Commission (EC) numbers and enzyme family [79,245,246,247,248,249,250]. However, methods solely based on amino acid sequences have been deemed unable to predict more elaborate enzyme reactions and to establish the function of non-characterized enzymes with poor homology to annotated sequences [249]. An alternative method used to predict enzyme activity relies on knowledge of the chemical structure of substrates and products [251,252,253,254,255]. However, in some of these methods, information about the enzyme sequence is needed to elucidate the chemical structure of the compound involved. In a recent work, Watanabe and co-workers presented an approach that combines the two previously described strategies [249]. Briefly, upon the collection of information on the enzyme sequence and substrate/product chemical structure, the authors tested several MLMs to predict both EC numbers and full enzyme reactions. Ultimately, an RF-based model emerged as the most adequate as the area under the receiver (AUC), one of the metrics chosen to compare the models, presented a score of 0.94 (out of a possible maximum score of 1).
4. Conclusions
The impact of bioprocess engineering on our daily life has steadily increased, a trend that has been reinforced in recent years and is expected to proceed similarly due to public awareness of the need for sustainable and environmentally friendly production processes. Still, for bio-based production methods to thrive, they must be technically sound, economically competitive and sustainable, which requires the optimization of current processes or the introduction of novel processes and goods. This places a huge burden on the bio-based industry, as in established methods of bioprocess development, the translation of lab research to industrial reality is slow and computational power is only used to a limited degree. To effectively handle and process the huge number of data gathered by the bioprocessing community and identify and establish relationships, the use of MLMs has gained relevance. MLMs rely on algorithms that collect, pre-process and analyse data to predict a given output, identify hidden patterns and foster probabilistic techniques that allow patterns to be established from a reference point. Algorithms with differentiated structures and complexities that are succinctly described in this review have been developed and effectively implemented in bioprocess engineering fields, e.g., monitoring/control and analytical roles, downstream processing, prediction of metabolic pathways and of protein structure–function, among others. In the specific field of biocatalysis, notable developments have been recently reported in enzyme synthesis and engineering, screening for enzyme activity and biocatalyst formulation, fostering an increasingly rational approach towards the intended goals, namely improved activity and stability and substrate diversity. These have been achieved through the use of adequate algorithms and interaction with duly constructed databases. Still, for MLMs to be further disseminated, some challenges must be resolved, namely the need for further open-source methodology and databases as well as the proper storage and management of data. Additionally, MLMs require significant energy to generate a precise label with a set of particular data; hence, other learning methods are being looked into. The interpretability of MLMs is often questionable; hence, they should be made more explainable to support decision making during operations. This is typically exacerbated as the structure of the MLM method becomes more complex, hence the need for a user-friendly MLM that can identify the proper structure for a given bioprocess.
Author Contributions
Conceptualization, P.S.S. and P.F.; writing—original draft preparation, P.S.S.; writing—review and editing, P.S.S. and P.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Markov, S.A. Bioprocess Engineering. In Applied Science; Franceschetti, D.R., Ed.; Salem Press: New York, NY, USA, 2012; Volume 1, pp. 240–245. [Google Scholar]
- Mowbray, M.; Savage, T.; Wu, C.; Song, Z.; Cho, B.A.; Del Rio-Chanona, E.A.; Zhang, D. Machine Learning for Biochemical Engineering: A Review. Biochem. Eng. J. 2021, 172, 108054. [Google Scholar] [CrossRef]
- Singh, R.S. Industrial Biotechnology: An Overview. In Advances in Industrial Biotechnology; Singh, R.S., Pandey, A., Larroche, C., Eds.; International Publishing House Pvt. Ltd.: New Delhi, India, 2014; pp. 1–35. [Google Scholar]
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. MRNA Vaccines Manufacturing: Challenges and Bottlenecks. Vaccine 2021, 39, 2190–2200. [Google Scholar] [CrossRef] [PubMed]
- Danielson, N.; McKay, S.; Bloom, P.; Dunn, J.; Jakel, N.; Bauer, T.; Hannon, J.; Jewett, M.C.; Shanks, B. Industrial Biotechnology—An Industry at an Inflection Point. Ind. Biotechnol. 2020, 16, 321–332. [Google Scholar] [CrossRef]
- Schürrle, K. History, Current State, and Emerging Applications of Industrial Biotechnology. In Sustainability and Life Cycle Assessment in Industrial Biotechnology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 13–51. [Google Scholar]
- Harun, I.; del Rio-Chanona, E.A.; Wagner, J.L.; Lauersen, K.J.; Zhang, D.; Hellgardt, K. Photocatalytic Production of Bisabolene from Green Microalgae Mutant: Process Analysis and Kinetic Modeling. Ind. Eng. Chem. Res. 2018, 57, 10336–10344. [Google Scholar] [CrossRef]
- Mears, L.; Stocks, S.M.; Albaek, M.O.; Sin, G.; Gernaey, K.V. Mechanistic Fermentation Models for Process Design, Monitoring, and Control. Trends Biotechnol. 2017, 35, 914–924. [Google Scholar] [CrossRef]
- Almquist, J.; Cvijovic, M.; Hatzimanikatis, V.; Nielsen, J.; Jirstrand, M. Kinetic Models in Industrial Biotechnology—Improving Cell Factory Performance. Metab. Eng. 2014, 24, 38–60. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. Methods and Advances in Metabolic Flux Analysis: A Mini-Review. J. Ind. Microbiol. Biotechnol. 2015, 42, 317–325. [Google Scholar] [CrossRef]
- González-Figueredo, C.; Flores-Estrella, R.A.; Rojas-Rejón, O.A. Fermentation: Metabolism, Kinetic Models, and Bioprocessing. In Current Topics in Biochemical Engineering; Shiomi, N., Ed.; IntechOpen: Rijeka, Croatia, 2018; pp. 11–48. ISBN 978-1-83881-210-2. [Google Scholar]
- Mandenius, C.-F.; Brundin, A. Bioprocess Optimization Using Design-of-Experiments Methodology. Biotechnol. Prog. 2008, 24, 1191–1203. [Google Scholar] [CrossRef]
- Kumar, V.; Bhalla, A.; Rathore, A.S. Design of Experiments Applications in Bioprocessing: Concepts and Approach. Biotechnol. Prog. 2014, 30, 86–99. [Google Scholar] [CrossRef]
- Baumann, P.; Hubbuch, J. Downstream Process Development Strategies for Effective Bioprocesses: Trends, Progress, and Combinatorial Approaches. Eng. Life Sci. 2016, 17, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Lischeske, J.J.; Stickel, J.J. A Two-Phase Substrate Model for Enzymatic Hydrolysis of Lignocellulose: Application to Batch and Continuous Reactors. Biotechnol. Biofuels 2019, 12, 299. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Myint, M.; Nguyen-Khuong, T.; Ho, Y.S.; Ng, S.K.; Lakshmanan, M. Harnessing the Potential of Machine Learning for Advancing “Quality by Design” in Biomanufacturing. MAbs 2022, 14, 2013593. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hoshan, L.; Jiang, R.; Gupta, B.; Brodean, E.; O’Neill, K.; Seamans, T.C.; Bowers, J.; Chen, H. A Practical Approach in Bioreactor Scale-up and Process Transfer Using a Combination of Constant P/V and Vvm as the Criterion. Biotechnol. Prog. 2017, 33, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Marroquín-Fandiño, J.E.; Ramírez-Acosta, C.M.; Luna-Wandurraga, H.J.; Valderrama-Rincón, J.A.; Cruz, J.C.; Reyes, L.H.; Valderrama-Rincon, J.D. Novel External-Loop-Airlift Milliliter Scale Bioreactors for Cell Growth Studies: Low Cost Design, CFD Analysis and Experimental Characterization. J. Biotechnol. 2020, 324, 71–82. [Google Scholar] [CrossRef]
- Krychowska, A.; Kordas, M.; Konopacki, M.; Grygorcewicz, B.; Musik, D.; Wójcik, K.; Jędrzejczak-Silicka, M.; Rakoczy, R. Mathematical Modeling of Hydrodynamics in Bioreactor by Means of CFD-Based Compartment Model. Processes 2020, 8, 1301. [Google Scholar] [CrossRef]
- del Rio-Chanona, E.A.; Wagner, J.L.; Ali, H.; Fiorelli, F.; Zhang, D.; Hellgardt, K. Deep Learning-Based Surrogate Modeling and Optimization for Microalgal Biofuel Production and Photobioreactor Design. AIChE J. 2019, 65, 915–923. [Google Scholar] [CrossRef]
- Forte, M.B.S.; Taviot-Guého, C.; Leroux, F.; Rodrigues, M.I.; Maugeri Filho, F. Clavulanic Acid Separation on Fixed Bed Columns of Layered Double Hydroxides: Optimization of Operating Parameters Using Breakthrough Curves. Process Biochem. 2016, 51, 509–516. [Google Scholar] [CrossRef]
- Khanal, O.; Lenhoff, A.M. Developments and Opportunities in Continuous Biopharmaceutical Manufacturing. MAbs 2021, 13, 1903664. [Google Scholar] [CrossRef]
- Sequeiros-Borja, C.E.; Surpeta, B.; Brezovsky, J. Recent Advances in User-Friendly Computational Tools to Engineer Protein Function. Brief. Bioinform. 2021, 22, bbaa150. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Enzyme Models—From Catalysis to Prodrugs. Molecules 2021, 26, 3248. [Google Scholar] [CrossRef]
- Mignon, D.; Druart, K.; Michael, E.; Opuu, V.; Polydorides, S.; Villa, F.; Gaillard, T.; Panel, N.; Archontis, G.; Simonson, T. Physics-Based Computational Protein Design: An Update. J. Phys. Chem. A 2020, 124, 10637–10648. [Google Scholar] [CrossRef] [PubMed]
- Monza, E.; Gil, V.; Lucas, M.F. Computational Enzyme Design at Zymvol. In Enzyme Engineering: Methods and Protocols; Magnani, F., Marabelli, C., Paradisi, F., Eds.; Springer US: New York, NY, USA, 2022; pp. 249–259. ISBN 978-1-0716-1826-4. [Google Scholar]
- Sirin, S.; Pearlman, D.A.; Sherman, W. Physics-Based Enzyme Design: Predicting Binding Affinity and Catalytic Activity. Proteins Struct. Funct. Bioinform. 2014, 82, 3397–3409. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gilmour, S.G.; Mylona, K.; Goos, P. Optimal Design of Experiments for Hybrid Nonlinear Models, with Applications to Extended Michaelis–Menten Kinetics. J. Agric. Biol. Environ. Stat. 2020, 25, 601–616. [Google Scholar] [CrossRef]
- Vasić-Rački, D.; Findrik, Z.; Presečki, A.V. Modelling as a Tool of Enzyme Reaction Engineering for Enzyme Reactor Development. Appl. Microbiol. Biotechnol. 2011, 91, 845–856. [Google Scholar] [CrossRef]
- Jiménez, A.; Castillo, A.; Mahn, A. Kinetic Study and Modeling of Wild-Type and Recombinant Broccoli Myrosinase Produced in E. coli and S. cerevisiae as a Function of Substrate Concentration, Temperature, and PH. Catalysts 2022, 12, 683. [Google Scholar] [CrossRef]
- Du, Y.-H.; Wang, M.-Y.; Yang, L.-H.; Tong, L.-L.; Guo, D.-S.; Ji, X.-J. Optimization and Scale-Up of Fermentation Processes Driven by Models. Bioengineering 2022, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Passi, A.; Tibocha-Bonilla, J.D.; Kumar, M.; Tec-Campos, D.; Zengler, K.; Zuniga, C. Genome-Scale Metabolic Modeling Enables In-Depth Understanding of Big Data. Metabolites 2022, 12, 14. [Google Scholar] [CrossRef]
- Flevaris, K.; Chatzidoukas, C. Facilitating the Industrial Transition to Microbial and Microalgal Factories through Mechanistic Modelling within the Industry 4.0 Paradigm. Curr. Opin. Chem. Eng. 2021, 33, 100713. [Google Scholar] [CrossRef]
- Shi, Z.; Liu, P.; Liao, X.; Mao, Z.; Zhang, J.; Wang, Q.; Sun, J.; Ma, H.; Ma, Y. Data-Driven Synthetic Cell Factories Development for Industrial Biomanufacturing. BioDesign Res. 2022, 2022, 9898461. [Google Scholar] [CrossRef]
- Wu, Y.; Jameel, A.; Xing, X.-H.; Zhang, C. Advanced Strategies and Tools to Facilitate and Streamline Microbial Adaptive Laboratory Evolution. Trends Biotechnol. 2022, 40, 38–59. [Google Scholar] [CrossRef]
- Mey, F.; Clauwaert, J.; van Huffel, K.; Waegeman, W.; de Mey, M. Improving the Performance of Machine Learning Models for Biotechnology: The Quest for Deus Ex Machina. Biotechnol. Adv. 2021, 53, 107858. [Google Scholar] [CrossRef] [PubMed]
- Khaleghi, M.K.; Savizi, I.S.P.; Lewis, N.E.; Shojaosadati, S.A. Synergisms of Machine Learning and Constraint-Based Modeling of Metabolism for Analysis and Optimization of Fermentation Parameters. Biotechnol. J. 2021, 16, 2100212. [Google Scholar] [CrossRef] [PubMed]
- Bruce, S.O.; Onyegbule, F.A. Biosynthesis of Natural Products. In Bioactive Compounds; Zepka, L.Q., Nascimento, T.C., do Jacob-Lopes, E., Eds.; IntechOpen: Rijeka, Croatia, 2021; pp. 51–68. ISBN 978-1-83969-270-3. [Google Scholar]
- Tibrewal, N.; Tang, Y. Biocatalysts for Natural Product Biosynthesis. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 347–366. [Google Scholar] [CrossRef]
- Sturm, N.; Quinn, R.J.; Kellenberger, E. Structural Searching of Biosynthetic Enzymes to Predict Protein Targets of Natural Products. Planta Med. 2018, 84, 304–310. [Google Scholar] [CrossRef]
- Bar-Even, A.; Noor, E.; Savir, Y.; Liebermeister, W.; Davidi, D.; Tawfik, D.S.; Milo, R. The Moderately Efficient Enzyme: Evolutionary and Physicochemical Trends Shaping Enzyme Parameters. Biochemistry 2011, 50, 4402–4410. [Google Scholar] [CrossRef]
- Goldsmith, M.; Tawfik, D.S. Enzyme Engineering: Reaching the Maximal Catalytic Efficiency Peak. Curr. Opin. Struct. Biol. 2017, 47, 140–150. [Google Scholar] [CrossRef]
- Wackett, L.P.; Robinson, S.L. The Ever-Expanding Limits of Enzyme Catalysis and Biodegradation: Polyaromatic, Polychlorinated, Polyfluorinated, and Polymeric Compounds. Biochem. J. 2020, 477, 2875–2891. [Google Scholar] [CrossRef]
- Yang, H.; Li, J.; Shin, H.; Du, G.; Liu, L.; Chen, J. Molecular Engineering of Industrial Enzymes: Recent Advances and Future Prospects. Appl. Microbiol. Biotechnol. 2014, 98, 23–29. [Google Scholar] [CrossRef]
- Dev, A.; Srivastava, A.K.; Karmakar, S. New Generation Hybrid Nanobiocatalysts. In Handbook of Nanomaterials for Industrial Applications; Elsevier: Amsterdam, The Netherlands, 2018; pp. 217–231. [Google Scholar]
- Rocha, R.A.; Speight, R.E.; Scott, C. Engineering Enzyme Properties for Improved Biocatalytic Processes in Batch and Continuous Flow. Org. Process. Res. Dev. 2022, 26, 1914–1924. [Google Scholar] [CrossRef]
- Galanie, S.; Entwistle, D.; Lalonde, J. Engineering Biosynthetic Enzymes for Industrial Natural Product Synthesis. Nat. Prod. Rep. 2020, 37, 1122–1143. [Google Scholar] [CrossRef]
- Gado, J.E.; Harrison, B.E.; Sandgren, M.; Ståhlberg, J.; Beckham, G.T.; Payne, C.M. Machine Learning Reveals Sequence-Function Relationships in Family 7 Glycoside Hydrolases. J. Biol. Chem. 2021, 297, 100931. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-Learning-Guided Directed Evolution for Protein Engineering. Nat. Methods 2019, 16, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Dong, X.; Li, X.; Liu, Z.; Liu, H. Prediction of Disulfide Bond Engineering Sites Using a Machine Learning Method. Sci. Rep. 2020, 10, 10330. [Google Scholar] [CrossRef]
- Xie, W.J.; Asadi, M.; Warshel, A. Enhancing Computational Enzyme Design by a Maximum Entropy Strategy. Proc. Natl. Acad. Sci. USA 2022, 119, e2122355119. [Google Scholar] [CrossRef] [PubMed]
- Pertusi, D.A.; Moura, M.E.; Jeffryes, J.G.; Prabhu, S.; Walters Biggs, B.; Tyo, K.E.J. Predicting Novel Substrates for Enzymes with Minimal Experimental Effort with Active Learning. Metab. Eng. 2017, 44, 171–181. [Google Scholar] [CrossRef]
- Xiang, R.; Fernandez-Lopez, L.; Robles-Martín, A.; Ferrer, M.; Guallar, V. EP-Pred: A Machine Learning Tool for Bioprospecting Promiscuous Ester Hydrolases. Biomolecules 2022, 12, 1529. [Google Scholar] [CrossRef]
- Voutilainen, S.; Heinonen, M.; Andberg, M.; Jokinen, E.; Maaheimo, H.; Pääkkönen, J.; Hakulinen, N.; Rouvinen, J.; Lähdesmäki, H.; Kaski, S.; et al. Substrate Specificity of 2-Deoxy-D-Ribose 5-Phosphate Aldolase (DERA) Assessed by Different Protein Engineering and Machine Learning Methods. Appl. Microbiol. Biotechnol. 2020, 104, 10515–10529. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.; Das, R.; Yang, K.K.; Coley, C.W. Machine Learning Modeling of Family Wide Enzyme-Substrate Specificity Screens. PLoS Comput. Biol. 2022, 18, e1009853. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Perez-Ortiz, G.; Peate, J.; Barry, S.M. Redesigning Enzymes for Biocatalysis: Exploiting Structural Understanding for Improved Selectivity. Front. Mol. Biosci. 2022, 9, 908285. [Google Scholar] [CrossRef]
- Velez Rueda, A.J.; Palopoli, N.; Zacarías, M.; Sommese, L.M.; Parisi, G. ProtMiscuity: A Database of Promiscuous Proteins. Database 2019, 2019, baz103. [Google Scholar] [CrossRef]
- Chai, M.; Moradi, S.; Erfani, E.; Asadnia, M.; Chen, V.; Razmjou, A. Application of Machine Learning Algorithms to Estimate Enzyme Loading, Immobilization Yield, Activity Retention, and Reusability of Enzyme–Metal–Organic Framework Biocatalysts. Chem. Mater. 2021, 33, 8666–8676. [Google Scholar] [CrossRef]
- Roura Padrosa, D.; Marchini, V.; Paradisi, F. CapiPy: Python-Based GUI-Application to Assist in Protein Immobilization. Bioinformatics 2021, 37, 2761–2762. [Google Scholar] [CrossRef]
- Meng, C.; Hu, Y.; Zhang, Y.; Guo, F. PSBP-SVM: A Machine Learning-Based Computational Identifier for Predicting Polystyrene Binding Peptides. Front. Bioeng. Biotechnol. 2020, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.D.; Kim, G.B.; Kim, Y.; Lee, S.Y. Applications of Artificial Intelligence to Enzyme and Pathway Design for Metabolic Engineering. Curr. Opin. Biotechnol. 2022, 73, 101–107. [Google Scholar] [CrossRef]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 Is a Deep Unsupervised Language Model for Protein Design. Nat. Commun. 2022, 13, 4348. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Alva, J.; Ochoa-Toledo, L.; Villalobos-Alva, M.J.; Aliseda, A.; Pérez-Escamirosa, F.; Altamirano-Bustamante, N.F.; Ochoa-Fernández, F.; Zamora-Solís, R.; Villalobos-Alva, S.; Revilla-Monsalve, C.; et al. Protein Science Meets Artificial Intelligence: A Systematic Review and a Biochemical Meta-Analysis of an Inter-Field. Front. Bioeng. Biotechnol. 2022, 10, 788300. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Kortemme, T. Recent Advances in de Novo Protein Design: Principles, Methods, and Applications. J. Biol. Chem. 2021, 296, 100558. [Google Scholar] [CrossRef]
- Singh, N.; Malik, S.; Gupta, A.; Srivastava, K.R. Revolutionizing Enzyme Engineering through Artificial Intelligence and Machine Learning. Emerg Top Life Sci. 2021, 5, 113–125. [Google Scholar] [CrossRef]
- Cadet, X.F.; Gelly, J.C.; van Noord, A.; Cadet, F.; Acevedo-Rocha, C.G. Learning Strategies in Protein Directed Evolution. In Directed Evolution: Methods and Protocols; Currin, A., Swainston, N., Eds.; Springer US: New York, NY, USA, 2022; pp. 225–275. ISBN 978-1-0716-2152-3. [Google Scholar]
- Saito, Y.; Oikawa, M.; Sato, T.; Nakazawa, H.; Ito, T.; Kameda, T.; Tsuda, K.; Umetsu, M. Machine-Learning-Guided Library Design Cycle for Directed Evolution of Enzymes: The Effects of Training Data Composition on Sequence Space Exploration. ACS Catal. 2021, 11, 14615–14624. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the Sequence Specificities of DNA- and RNA-Binding Proteins by Deep Learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef]
- Hui, S.; Xing, X.; Bader, G.D. Predicting PDZ Domain Mediated Protein Interactions from Structure. BMC Bioinform. 2013, 14, 27. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A Universal SNP and Small-Indel Variant Caller Using Deep Neural Networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Dias-Audibert, F.L.; Navarro, L.C.; de Oliveira, D.N.; Delafiori, J.; Melo, C.F.O.R.; Guerreiro, T.M.; Rosa, F.T.; Petenuci, D.L.; Watanabe, M.A.E.; Velloso, L.A.; et al. Combining Machine Learning and Metabolomics to Identify Weight Gain Biomarkers. Front. Bioeng. Biotechnol. 2020, 8, 6. [Google Scholar] [CrossRef]
- Erban, A.; Fehrle, I.; Martinez-Seidel, F.; Brigante, F.; Más, A.L.; Baroni, V.; Wunderlin, D.; Kopka, J. Discovery of Food Identity Markers by Metabolomics and Machine Learning Technology. Sci. Rep. 2019, 9, 9697. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, M.H.; Jahanbekam, A.; Sadri, H.; Schuh, K.; Dusel, G.; Prehn, C.; Adamski, J.; Koch, C.; Sauerwein, H. Metabolomics Meets Machine Learning: Longitudinal Metabolite Profiling in Serum of Normal versus Overconditioned Cows and Pathway Analysis. J. Dairy Sci. 2019, 102, 11561–11585. [Google Scholar] [CrossRef]
- Liebal, U.W.; Phan, A.N.T.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites 2020, 10, 243. [Google Scholar] [CrossRef]
- Heinemann, D. (Ed.) Praxiskommentar Transparenzgesetz (LTranspG RLP), 1st ed.; Springer Fachmedien Wiesbaden: Wiesbaden, Germany, 2019; ISBN 978-3-658-18436-0. [Google Scholar]
- Helmy, M.; Smith, D.; Selvarajoo, K. Systems Biology Approaches Integrated with Artificial Intelligence for Optimized Metabolic Engineering. Metab. Eng. Commun. 2020, 11, e00149. [Google Scholar] [CrossRef] [PubMed]
- Cuperlovic-Culf, M. Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling. Metabolites 2018, 8, 4. [Google Scholar] [CrossRef]
- Mazurenko, S.; Prokop, Z.; Damborsky, J. Machine Learning in Enzyme Engineering. ACS Catal. 2020, 10, 1210–1223. [Google Scholar] [CrossRef]
- Yan, B.; Ran, X.; Gollu, A.; Cheng, Z.; Zhou, X.; Chen, Y.; Yang, Z.J. IntEnzyDB: An Integrated Structure–Kinetics Enzymology Database. J. Chem. Inf. Model. 2022, 62, 5841–5848. [Google Scholar] [CrossRef]
- Pleiss, J. Standardized Data, Scalable Documentation, Sustainable Storage—EnzymeML as A Basis for FAIR Data Management In Biocatalysis. ChemCatChem 2021, 13, 3909–3913. [Google Scholar] [CrossRef]
- Minkiewicz, P.; Darewicz, M.; Iwaniak, A.; Bucholska, J.; Starowicz, P.; Czyrko, E. Internet Databases of the Properties, Enzymatic Reactions, and Metabolism of Small Molecules—Search Options and Applications in Food Science. Int. J. Mol. Sci. 2016, 17, 2039. [Google Scholar] [CrossRef]
- Chicco, D.; Oneto, L.; Tavazzi, E. Eleven Quick Tips for Data Cleaning and Feature Engineering. PLoS Comput. Biol. 2022, 18, e1010718. [Google Scholar] [CrossRef] [PubMed]
- Menke, M.J.; Behr, A.S.; Rosenthal, K.; Linke, D.; Kockmann, N.; Bornscheuer, U.T.; Dörr, M. Development of an Ontology for Biocatalysis. Chem. Ing. Tech. 2022, 94, 1827–1835. [Google Scholar] [CrossRef]
- Bur, A.M.; Shew, M.; New, J. Artificial Intelligence for the Otolaryngologist: A State of the Art Review. Otolaryngol. Head Neck Surg. 2019, 160, 603–611. [Google Scholar] [CrossRef]
- Niroula, A.; Vihinen, M. Variation Interpretation Predictors: Principles, Types, Performance, and Choice. Hum. Mutat. 2016, 37, 579–597. [Google Scholar] [CrossRef]
- Sharma, A.; Mishra, P.K. State-of-the-Art in Performance Metrics and Future Directions for Data Science Algorithms. J. Sci. Res. 2020, 64, 221–238. [Google Scholar] [CrossRef]
- Badillo, S.; Banfai, B.; Birzele, F.; Davydov, I.I.; Hutchinson, L.; Kam-Thong, T.; Siebourg-Polster, J.; Steiert, B.; Zhang, J.D. An Introduction to Machine Learning. Clin. Pharmacol. Ther. 2020, 107, 871–885. [Google Scholar] [CrossRef]
- Cai, Z.; Long, Y.; Shao, L. Classification Complexity Assessment for Hyper-Parameter Optimization. Pattern Recognit. Lett. 2019, 125, 396–403. [Google Scholar] [CrossRef]
- Abbott, A.S.; Turney, J.M.; Zhang, B.; Smith, D.G.A.; Altarawy, D.; Schaefer, H.F. PES-Learn: An Open-Source Software Package for the Automated Generation of Machine Learning Models of Molecular Potential Energy Surfaces. J. Chem. Theory Comput. 2019, 15, 4386–4398. [Google Scholar] [CrossRef]
- Hoopes, A.; Hoffmann, M.; Fischl, B.; Guttag, J.; Dalca, A.V. HyperMorph: Amortized Hyperparameter Learning for Image Registration. In International Conference on Information Processing in Medical Imaging; Springer: Berlin/Heidelberg, Germany, 2021; pp. 3–17. [Google Scholar]
- Basha, S.M.; Rajput, D.S. Survey on Evaluating the Performance of Machine Learning Algorithms: Past Contributions and Future Roadmap. In Deep Learning and Parallel Computing Environment for Bioengineering Systems; Elsevier: Amsterdam, The Netherlands, 2019; pp. 153–164. [Google Scholar]
- Abraham, G.K.; Jayanthi, V.S.; Bhaskaran, P. Convolutional Neural Network for Biomedical Applications. In Computational Intelligence and Its Applications in Healthcare; Elsevier: Amsterdam, The Netherlands, 2020; pp. 145–156. [Google Scholar]
- Fox, R.J.; Davis, S.C.; Mundorff, E.C.; Newman, L.M.; Gavrilovic, V.; Ma, S.K.; Chung, L.M.; Ching, C.; Tam, S.; Muley, S.; et al. Improving Catalytic Function by ProSAR-Driven Enzyme Evolution. Nat. Biotechnol. 2007, 25, 338–344. [Google Scholar] [CrossRef]
- Li, Y.; Drummond, D.A.; Sawayama, A.M.; Snow, C.D.; Bloom, J.D.; Arnold, F.H. A Diverse Family of Thermostable Cytochrome P450s Created by Recombination of Stabilizing Fragments. Nat. Biotechnol. 2007, 25, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Helleckes, L.M.; Hemmerich, J.; Wiechert, W.; von Lieres, E.; Grünberger, A. Machine Learning in Bioprocess Development: From Promise to Practice. Trends Biotechnol. 2022, 41, 817–835. [Google Scholar] [CrossRef]
- Mowbray, M.; Vallerio, M.; Perez-Galvan, C.; Zhang, D.; Del Rio Chanona, A.; Navarro-Brull, F.J. Industrial Data Science—A Review of Machine Learning Applications for Chemical and Process Industries. React. Chem. Eng. 2022, 7, 1471–1509. [Google Scholar] [CrossRef]
- Lim, S.J.; Son, M.; Ki, S.J.; Suh, S.-I.; Chung, J. Opportunities and Challenges of Machine Learning in Bioprocesses: Categorization from Different Perspectives and Future Direction. Bioresour. Technol. 2023, 370, 128518. [Google Scholar] [CrossRef] [PubMed]
- Presnell, K.V.; Alper, H.S. Systems Metabolic Engineering Meets Machine Learning: A New Era for Data-Driven Metabolic Engineering. Biotechnol. J. 2019, 14, 1800416. [Google Scholar] [CrossRef]
- Mondal, P.P.; Galodha, A.; Verma, V.K.; Singh, V.; Show, P.L.; Awasthi, M.K.; Lall, B.; Anees, S.; Pollmann, K.; Jain, R. Review on machine learning-based bioprocess optimization, monitoring, and control systems. Bioresour. Technol. 2023, 370, 128523. [Google Scholar] [CrossRef] [PubMed]
- Duong-Trung, N.; Born, S.; Woo Kim, J.; Schermeyer, M.-T.; Paulick, K.; Borisyak, M.; Cruz-Bournazou, M.N.; Werner, T.; Scholz, R.; Schmidt-Thieme, L.; et al. When bioprocess engineering meets machine learning: A survey from the perspective of automated bioprocess development. Biochem. Eng. J. 2023, 190, 108764. [Google Scholar] [CrossRef]
- Applied Multivariate Statistical Analysis; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2007; ISBN 978-3-540-72243-4.
- Johnson, R.A.; Wichern, D.W. Applied Multivariate Statistical Analysis, 6th ed.; Pearson Education, Inc.: Hoboken, NJ, USA, 2007. [Google Scholar]
- do Carmo Nicoletti, M.; Jain, L.C. (Eds.) Computational Intelligence Techniques for Bioprocess Modelling, Supervision and Control; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2009; Volume 218, ISBN 978-3-642-01887-9. [Google Scholar]
- De Carvalho, C.C.; Da Fonseca, M.M.R. Principal Component Analysis Applied to Bacterial Cell Behaviour in the Presence of Organic Solvents. Biocatal. Biotransformat. 2004, 22, 203–214. [Google Scholar] [CrossRef]
- Nucci, E.R.; Cruz, A.J.G.; Giordano, R.C. Monitoring Bioreactors Using Principal Component Analysis: Production of Penicillin G Acylase as a Case Study. Bioprocess Biosyst. Eng. 2010, 33, 557–564. [Google Scholar] [CrossRef]
- Hans, S.; Ulmer, C.; Narayanan, H.; Brautaset, T.; Krausch, N.; Neubauer, P.; Schäffl, I.; Sokolov, M.; Cruz Bournazou, M.N. Monitoring Parallel Robotic Cultivations with Online Multivariate Analysis. Processes 2020, 8, 582. [Google Scholar] [CrossRef]
- Wang, B.; Kennedy, M.A. Principal Components Analysis of Protein Sequence Clusters. J. Struct. Funct. Genom. 2014, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Palla, M.; Punthambaker, S.; Stranges, B.; Vigneault, F.; Nivala, J.; Wiegand, D.; Ayer, A.; Craig, T.; Gremyachinskiy, D.; Franklin, H.; et al. Multiplex Single-Molecule Kinetics of Nanopore-Coupled Polymerases. ACS Nano 2021, 15, 489–502. [Google Scholar] [CrossRef]
- Ribeiro da Cunha, B.; Fonseca, L.P.; Calado, C.R.C. A Phenotypic Screening Bioassay for Escherichia Coli Stress and Antibiotic Responses Based on Fourier-Transform Infrared (FTIR) Spectroscopy and Multivariate Analysis. J. Appl. Microbiol. 2019, 127, 1776–1789. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, P.S.; Soares, A.; Castanho, A.; Almeida, A.S.; Oliveira, J.; Brites, C. Optimization of Rice Amylose Determination by NIR-Spectroscopy Using PLS Chemometrics Algorithms. Food Chem. 2018, 242, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.-M. Multiple Linear Regression for Protein Secondary Structure Prediction. Proteins Struct. Funct. Genet. 2001, 43, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Janairo, G.I.B.; Yu, D.E.C.; Janairo, J.I.B. A Machine Learning Regression Model for the Screening and Design of Potential SARS-CoV-2 Protease Inhibitors. Netw. Model. Anal. Health Inform. Bioinform. 2021, 10, 51. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, X.; He, B.; Guo, J.; Zhao, B.; Zhang, Y.; Zhou, Z.; Zhou, X.; Zhang, R.; Abliz, Z. The Impact of Chronic Environmental Metal and Benzene Exposure on Human Urinary Metabolome among Chinese Children and the Elderly Population. Ecotoxicol. Environ. Saf. 2019, 169, 232–239. [Google Scholar] [CrossRef]
- Stubbs, S.; Zhang, J.; Morris, J. Chapter 10—BioProcess Performance Monitoring Using Multiway Interval Partial Least Squares. In Computer Aided Chemical Engineering; Singh, R., Yuan, Z., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 41, pp. 243–259. ISBN 1570-7946. [Google Scholar]
- Duran-Villalobos, C.A.; Goldrick, S.; Lennox, B. Multivariate Statistical Process Control of an Industrial-Scale Fed-Batch Simulator. Comput. Chem. Eng. 2020, 132, 106620. [Google Scholar] [CrossRef]
- Freire, R.S.; Ferreira, M.M.C.; Durán, N.; Kubota, L.T. Dual Amperometric Biosensor Device for Analysis of Binary Mixtures of Phenols by Multivariate Calibration Using Partial Least Squares. Anal. Chim. Acta 2003, 485, 263–269. [Google Scholar] [CrossRef]
- Tsanaktsidou, E.; Karavasili, C.; Zacharis, C.K.; Fatouros, D.G.; Markopoulou, C.K. Partial Least Square Model (PLS) as a Tool to Predict the Diffusion of Steroids Across Artificial Membranes. Molecules 2020, 25, 1387. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.I.; Rhee, C.; Cho, K.H.; Shin, S.G. Comparison of Different Machine Learning Algorithms to Estimate Liquid Level for Bioreactor Management. Environ. Eng. Res. 2022, 28, 220037. [Google Scholar] [CrossRef]
- Xu, Y.; Verma, D.; Sheridan, R.P.; Liaw, A.; Ma, J.; Marshall, N.M.; McIntosh, J.; Sherer, E.C.; Svetnik, V.; Johnston, J.M. Deep Dive into Machine Learning Models for Protein Engineering. J. Chem. Inf. Model. 2020, 60, 2773–2790. [Google Scholar] [CrossRef]
- Li, W.; Li, C.; Wang, T. Application of Machine Learning Algorithms in MBR Simulation under Big Data Platform. Water Pract. Technol. 2020, 15, 1238–1247. [Google Scholar] [CrossRef]
- Afify, H.M.; Abdelhalim, M.B.; Mabrouk, M.S.; Sayed, A.Y. Protein Secondary Structure Prediction (PSSP) Using Different Machine Algorithms. Egypt. J. Med. Hum. Genet. 2021, 22, 54. [Google Scholar] [CrossRef]
- Liu, B.; Wang, X.; Lin, L.; Tang, B.; Dong, Q.; Wang, X. Prediction of Protein Binding Sites in Protein Structures Using Hidden Markov Support Vector Machine. BMC Bioinform. 2009, 10, 381. [Google Scholar] [CrossRef]
- Meng, C.; Jin, S.; Wang, L.; Guo, F.; Zou, Q. AOPs-SVM: A Sequence-Based Classifier of Antioxidant Proteins Using a Support Vector Machine. Front. Bioeng. Biotechnol. 2019, 7, 224. [Google Scholar] [CrossRef]
- Cavalcanti, A.B.S.; Barros, R.P.C.; Costa, V.C.d.O.; da Silva, M.S.; Tavares, J.F.; Scotti, L.; Scotti, M.T. Computer-Aided Chemotaxonomy and Bioprospecting Study of Diterpenes of the Lamiaceae Family. Molecules 2019, 24, 3908. [Google Scholar] [CrossRef]
- Landon, S.; Chalkley, O.; Breese, G.; Grierson, C.; Marucci, L. Understanding Metabolic Flux Behaviour in Whole-Cell Model Output. Front. Mol. Biosci. 2021, 8, 732079. [Google Scholar] [CrossRef]
- Wu, S.G.; Wang, Y.; Jiang, W.; Oyetunde, T.; Yao, R.; Zhang, X.; Shimizu, K.; Tang, Y.J.; Bao, F.S. Rapid Prediction of Bacterial Heterotrophic Fluxomics Using Machine Learning and Constraint Programming. PLoS Comput. Biol. 2016, 12, e1004838. [Google Scholar] [CrossRef]
- Waqas, S.; Harun, N.Y.; Sambudi, N.S.; Arshad, U.; Nordin, N.A.H.M.; Bilad, M.R.; Saeed, A.A.H.; Malik, A.A. SVM and ANN Modelling Approach for the Optimization of Membrane Permeability of a Membrane Rotating Biological Contactor for Wastewater Treatment. Membranes 2022, 12, 821. [Google Scholar] [CrossRef] [PubMed]
- Agatonovic-Kustrin, S.; Beresford, R. Basic Concepts of Artificial Neural Network (ANN) Modeling and Its Application in Pharmaceutical Research. J. Pharm. Biomed. Anal. 2000, 22, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Rowland, Z.; Lazaroiu, G.; Podhorská, I. Use of Neural Networks to Accommodate Seasonal Fluctuations When Equalizing Time Series for the CZK/RMB Exchange Rate. Risks 2020, 9, 1. [Google Scholar] [CrossRef]
- Cybenko, G. Approximation by Superpositions of a Sigmoidal Function. Math. Control. Signals Syst. 1989, 2, 303–314. [Google Scholar] [CrossRef]
- Heidari, A.A.; Faris, H.; Mirjalili, S.; Aljarah, I.; Mafarja, M. Ant Lion Optimizer: Theory, Literature Review, and Application in Multi-Layer Perceptron Neural Networks. In Nature-Inspired Optimizers; Springer: Berlin/Heidelberg, Germany, 2020; pp. 23–46. [Google Scholar]
- Amidi, A.; Amidi, S.; Vlachakis, D.; Megalooikonomou, V.; Paragios, N.; Zacharaki, E.I. EnzyNet: Enzyme Classification Using 3D Convolutional Neural Networks on Spatial Representation. PeerJ 2018, 6, e4750. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Oubounyt, M.; Louadi, Z.; Tayara, H.; Chong, K.T. DeePromoter: Robust Promoter Predictor Using Deep Learning. Front. Genet. 2019, 10, 286. [Google Scholar] [CrossRef]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified Rational Protein Engineering with Sequence-Based Deep Representation Learning. Nat. Methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef]
- Han, S.; Kim, T.; Kim, D.; Park, Y.-L.; Jo, S. Use of Deep Learning for Characterization of Microfluidic Soft Sensors. IEEE Robot. Autom. Lett. 2018, 3, 873–880. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep Learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Ding, W.; Nakai, K.; Gong, H. Protein Design via Deep Learning. Brief. Bioinform. 2022, 23, bbac102. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Protein Structure Prediction Using Multiple Deep Neural Networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins Struct. Funct. Bioinform. 2019, 87, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Kothakota, A.; Pandiselvam, R.; Siliveru, K.; Pandey, J.P.; Sagarika, N.; Srinivas, C.H.S.; Kumar, A.; Singh, A.; Prakash, S.D. Modeling and Optimization of Process Parameters for Nutritional Enhancement in Enzymatic Milled Rice by Multiple Linear Regression (MLR) and Artificial Neural Network (ANN). Foods 2021, 10, 2975. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Li, H.; Xu, Z.; Hou, S.; Yang, D. User-Friendly Optimization Approach of Fed-Batch Fermentation Conditions for the Production of Iturin a Using Artificial Neural Networks and Support Vector Machine. Electron. J. Biotechnol. 2015, 18, 273–280. [Google Scholar] [CrossRef]
- Zhu, P.; Kang, X.; Zhao, Y.; Latif, U.; Zhang, H. Predicting the Toxicity of Ionic Liquids toward Acetylcholinesterase Enzymes Using Novel QSAR Models. Int. J. Mol. Sci. 2019, 20, 2186. [Google Scholar] [CrossRef]
- Hopf, T.A.; Colwell, L.J.; Sheridan, R.; Rost, B.; Sander, C.; Marks, D.S. Three-Dimensional Structures of Membrane Proteins from Genomic Sequencing. Cell 2012, 149, 1607–1621. [Google Scholar] [CrossRef]
- Gelman, S.; Fahlberg, S.A.; Heinzelman, P.; Romero, P.A.; Gitter, A. Neural Networks to Learn Protein Sequence–Function Relationships from Deep Mutational Scanning Data. Proc. Natl. Acad. Sci. USA 2021, 118, e2104878118. [Google Scholar] [CrossRef]
- Rezaeenour, J.; Yari Eili, M.; Roozbahani, Z.; Ebrahimi, M. Prediction of Protein Thermostability by an Efficient Neural Network Approach. Health Manag. Inf. Sci. 2016, 3, 102–110. [Google Scholar]
- Fang, X.; Huang, J.; Zhang, R.; Wang, F.; Zhang, Q.; Li, G.; Yan, J.; Zhang, H.; Yan, Y.; Xu, L. Convolution Neural Network-Based Prediction of Protein Thermostability. J. Chem. Inf. Model. 2019, 59, 4833–4843. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of Protein Subcellular Localization Using Deep Learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Szalkai, B.; Grolmusz, V. Near Perfect Protein Multi-Label Classification with Deep Neural Networks. Methods 2018, 132, 50–56. [Google Scholar] [CrossRef]
- Khurana, S.; Rawi, R.; Kunji, K.; Chuang, G.-Y.; Bensmail, H.; Mall, R. DeepSol: A Deep Learning Framework for Sequence-Based Protein Solubility Prediction. Bioinformatics 2018, 34, 2605–2613. [Google Scholar] [CrossRef]
- Ajjolli Nagaraja, A.; Charton, P.; Cadet, X.F.; Fontaine, N.; Delsaut, M.; Wiltschi, B.; Voit, A.; Offmann, B.; Damour, C.; Grondin-Perez, B.; et al. A Machine Learning Approach for Efficient Selection of Enzyme Concentrations and Its Application for Flux Optimization. Catalysts 2020, 10, 291. [Google Scholar] [CrossRef]
- Staszak, M.; Staszak, K.; Wieszczycka, K.; Bajek, A.; Roszkowski, K.; Tylkowski, B. Machine Learning in Drug Design: Use of Artificial Intelligence to Explore the Chemical Structure–Biological Activity Relationship. WIREs Comput. Mol. Sci. 2021, 12, e1568. [Google Scholar] [CrossRef]
- Seeger, M. Gaussian Processes for Machine Learning. Int. J. Neural Syst. 2004, 14, 69–106. [Google Scholar] [CrossRef]
- Rasmussen, C.E.; Williams, C.K.I. Gaussian Processes for Machine Learning; MIT Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Wang, K.A.; Pleiss, G.; Gardner, J.R.; Tyree, S.; Weinberger, K.Q.; Wilson, A.G. Exact Gaussian Processes on a Million Data Points. In Proceedings of the 33rd International Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 8–14 December 2019. [Google Scholar]
- Liu, H.; Ong, Y.-S.; Shen, X.; Cai, J. When Gaussian Process Meets Big Data: A Review of Scalable GPs. IEEE Trans. Neural Netw. Learn. Syst. 2020, 31, 4405–4423. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. MCSM: Predicting the Effects of Mutations in Proteins Using Graph-Based Signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef]
- Mellor, J.; Grigoras, I.; Carbonell, P.; Faulon, J.-L. Semisupervised Gaussian Process for Automated Enzyme Search. ACS Synth. Biol. 2016, 5, 518–528. [Google Scholar] [CrossRef]
- Saito, Y.; Oikawa, M.; Nakazawa, H.; Niide, T.; Kameda, T.; Tsuda, K.; Umetsu, M. Machine-Learning-Guided Mutagenesis for Directed Evolution of Fluorescent Proteins. ACS Synth. Biol. 2018, 7, 2014–2022. [Google Scholar] [CrossRef] [PubMed]
- Bedbrook, C.N.; Yang, K.K.; Rice, A.J.; Gradinaru, V.; Arnold, F.H. Machine Learning to Design Integral Membrane Channelrhodopsins for Efficient Eukaryotic Expression and Plasma Membrane Localization. PLoS Comput. Biol. 2017, 13, e1005786. [Google Scholar] [CrossRef]
- Bedbrook, C.N.; Yang, K.K.; Robinson, J.E.; Mackey, E.D.; Gradinaru, V.; Arnold, F.H. Machine Learning-Guided Channelrhodopsin Engineering Enables Minimally Invasive Optogenetics. Nat. Methods 2019, 16, 1176–1184. [Google Scholar] [CrossRef]
- Tulsyan, A.; Khodabandehlou, H.; Wang, T.; Schorner, G.; Coufal, M.; Undey, C. Spectroscopic Models for Real-time Monitoring of Cell Culture Processes Using Spatiotemporal Just-in-time Gaussian Processes. AIChE J. 2021, 67, e17210. [Google Scholar] [CrossRef]
- He, F.; Stumpf, M.P.H. Quantifying Dynamic Regulation in Metabolic Pathways with Nonparametric Flux Inference. Biophys. J. 2019, 116, 2035–2046. [Google Scholar] [CrossRef]
- Polikar, R. Ensemble Based Systems in Decision Making. IEEE Circuits Syst. Mag. 2006, 6, 21–45. [Google Scholar] [CrossRef]
- Sagi, O.; Rokach, L. Ensemble Learning: A Survey. WIREs Data Min. Knowl. Discov. 2018, 8, e1249. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, Y. (Eds.) Ensemble Machine Learning; Springer US: Boston, MA, USA, 2012; ISBN 978-1-4419-9325-0. [Google Scholar]
- Muller, A.C.; Guido, S. Introduction to Machine Learning with Python: A Guide for Data Scientists; O’Reilly Media, Incorporated: Sebastopol, CA, USA, 2018; ISBN 9789352134571. [Google Scholar]
- Li, H.; Leung, K.-S.; Wong, M.-H.; Ballester, P.J. Substituting Random Forest for Multiple Linear Regression Improves Binding Affinity Prediction of Scoring Functions: Cyscore as a Case Study. BMC Bioinform. 2014, 15, 291. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, C.; Mehrotra, D.; Misra, N.K. Predicting the Protein Structure Using Random Forest Approach. Procedia Comput. Sci. 2018, 132, 1654–1662. [Google Scholar] [CrossRef]
- Hakala, K.; Kaewphan, S.; Bjorne, J.; Mehryary, F.; Moen, H.; Tolvanen, M.; Salakoski, T.; Ginter, F. Neural Network and Random Forest Models in Protein Function Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 1772–1781. [Google Scholar] [CrossRef]
- Reimann, R.; Zeng, B.; Jakopec, M.; Burdukiewicz, M.; Petrick, I.; Schierack, P.; Rödiger, S. Classification of Dead and Living Microalgae Chlorella vulgaris by Bioimage Informatics and Machine Learning. Algal. Res. 2020, 48, 101908. [Google Scholar] [CrossRef]
- Whitmore, L.S.; Davis, R.W.; McCormick, R.L.; Gladden, J.M.; Simmons, B.A.; George, A.; Hudson, C.M. BioCompoundML: A General Biofuel Property Screening Tool for Biological Molecules Using Random Forest Classifiers. Energy Fuels 2016, 30, 8410–8418. [Google Scholar] [CrossRef]
- Yadav, S.K.; Tiwari, A.K. Classification of Enzymes Using Machine Learning Based Approaches: A Review. Mach. Learn. Appl. Int. J. 2015, 2, 30–49. [Google Scholar] [CrossRef]
- Barati Farimani, A.; Heiranian, M.; Aluru, N.R. Identification of Amino Acids with Sensitive Nanoporous MoS2: Towards Machine Learning-Based Prediction. NPJ 2D Mater. Appl. 2018, 2, 14. [Google Scholar] [CrossRef]
- Long, F.; Fan, J.; Xu, W.; Liu, H. Predicting the Performance of Medium-Chain Carboxylic Acid (MCCA) Production Using Machine Learning Algorithms and Microbial Community Data. J. Clean. Prod. 2022, 377, 134223. [Google Scholar] [CrossRef]
- Toprak-Cavdur, T.; Anis, P.; Bakir, M.; Sebatli-Saglam, A.; Cavdur, F. Dyeing Behavior of Enzyme and Chitosan-Modified Polyester and Estimation of Colorimetry Parameters Using Random Forests. Fibers Polym. 2023, 24, 221–241. [Google Scholar] [CrossRef]
- Kroll, A.; Engqvist, M.K.M.; Heckmann, D.; Lercher, M.J. Deep Learning Allows Genome-Scale Prediction of Michaelis Constants from Structural Features. PLoS Biol. 2021, 19, e3001402. [Google Scholar] [CrossRef]
- Asgharzadeh, P.; Birkhold, A.I.; Trivedi, Z.; Özdemir, B.; Reski, R.; Röhrle, O. A NanoFE Simulation-Based Surrogate Machine Learning Model to Predict Mechanical Functionality of Protein Networks from Live Confocal Imaging. Comput. Struct. Biotechnol. J. 2020, 18, 2774–2788. [Google Scholar] [CrossRef] [PubMed]
- Sutton, R.S.; Barto, A.G. Reinforcement Learning: An Introduction; A Bradford Book: Cambridge, MA, USA, 2018; ISBN 0262039249. [Google Scholar]
- Li, D.; Qian, L.; Jin, Q.; Tan, T. Reinforcement Learning Control with Adaptive Gain for a Saccharomyces Cerevisiae Fermentation Process. Appl. Soft Comput. 2011, 11, 4488–4495. [Google Scholar] [CrossRef]
- Chai, W.Y.; Teo, K.T.K.; Tan, M.K.; Tham, H.J. Fermentation Process Control and Optimization. Chem. Eng. Technol. 2022, 45, 1731–1747. [Google Scholar] [CrossRef]
- Treloar, N.J.; Fedorec, A.J.H.; Ingalls, B.; Barnes, C.P. Deep Reinforcement Learning for the Control of Microbial Co-Cultures in Bioreactors. PLoS Comput. Biol. 2020, 16, e1007783. [Google Scholar] [CrossRef]
- Mowbray, M.R.; Wu, C.; Rogers, A.W.; Del Rio-Chanona, E.A.; Zhang, D. A Reinforcement Learning-based Hybrid Modeling Framework for Bioprocess Kinetics Identification. Biotechnol. Bioeng. 2023, 120, 154–168. [Google Scholar] [CrossRef]
- Sabzevari, M.; Szedmak, S.; Penttilä, M.; Jouhten, P.; Rousu, J. Strain Design Optimization Using Reinforcement Learning. PLoS Comput. Biol. 2022, 18, e1010177. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Duigou, T.; Faulon, J.-L. Reinforcement Learning for Bioretrosynthesis. ACS Synth. Biol. 2020, 9, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, Y.; Zhang, Y.; Li, K.; Lin, M.; Pan, F.; Wu, W.; Zhang, J. A Reinforcement Learning Approach for Protein–Ligand Binding Pose Prediction. BMC Bioinform. 2022, 23, 368. [Google Scholar] [CrossRef] [PubMed]
- de Jongh, R.P.H.; van Dijk, A.D.J.; Julsing, M.K.; Schaap, P.J.; de Ridder, D. Designing Eukaryotic Gene Expression Regulation Using Machine Learning. Trends Biotechnol. 2020, 38, 191–201. [Google Scholar] [CrossRef]
- Erfanian, N.; Heydari, A.A.; Iañez, P.; Derakhshani, A.; Ghasemigol, M.; Farahpour, M.; Nasseri, S.; Safarpour, H.; Sahebkar, A. Deep Learning Applications in Single-Cell Omics Data Analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Amer, B.; Baidoo, E.E.K. Omics-Driven Biotechnology for Industrial Applications. Front. Bioeng. Biotechnol. 2021, 9, 613307. [Google Scholar] [CrossRef]
- Li, R.; Li, L.; Xu, Y.; Yang, J. Machine Learning Meets Omics: Applications and Perspectives. Brief. Bioinform. 2021, 23, bbab460. [Google Scholar] [CrossRef]
- Vasina, M.; Velecký, J.; Planas-Iglesias, J.; Marques, S.M.; Skarupova, J.; Damborsky, J.; Bednar, D.; Mazurenko, S.; Prokop, Z. Tools for Computational Design and High-Throughput Screening of Therapeutic Enzymes. Adv. Drug Deliv. Rev. 2022, 183, 114143. [Google Scholar] [CrossRef]
- Hon, J.; Borko, S.; Stourac, J.; Prokop, Z.; Zendulka, J.; Bednar, D.; Martinek, T.; Damborsky, J. EnzymeMiner: Automated Mining of Soluble Enzymes with Diverse Structures, Catalytic Properties and Stabilities. Nucleic Acids Res. 2020, 48, W104–W109. [Google Scholar] [CrossRef]
- Vanella, R.; Kovacevic, G.; Doffini, V.; Fernández de Santaella, J.; Nash, M.A. High-Throughput Screening, next Generation Sequencing and Machine Learning: Advanced Methods in Enzyme Engineering. Chem. Commun. 2022, 58, 2455–2467. [Google Scholar] [CrossRef]
- Robinson, S.L.; Piel, J.; Sunagawa, S. A Roadmap for Metagenomic Enzyme Discovery. Nat. Prod. Rep. 2021, 38, 1994–2023. [Google Scholar] [CrossRef]
- Foroozandeh Shahraki, M.; Ariaeenejad, S.; Fallah Atanaki, F.; Zolfaghari, B.; Koshiba, T.; Kavousi, K.; Salekdeh, G.H. MCIC: Automated Identification of Cellulases from Metagenomic Data and Characterization Based on Temperature and PH Dependence. Front. Microbiol. 2020, 11, 567863. [Google Scholar] [CrossRef] [PubMed]
- Siedhoff, N.E.; Schwaneberg, U.; Davari, M.D. Machine Learning-Assisted Enzyme Engineering. Methods Enzymol. 2020, 643, 281–315. [Google Scholar]
- Giessel, A.; Dousis, A.; Ravichandran, K.; Smith, K.; Sur, S.; McFadyen, I.; Zheng, W.; Licht, S. Therapeutic Enzyme Engineering Using a Generative Neural Network. Sci. Rep. 2022, 12, 1536. [Google Scholar] [CrossRef] [PubMed]
- Alonso, S.; Santiago, G.; Cea-Rama, I.; Fernandez-Lopez, L.; Coscolín, C.; Modregger, J.; Ressmann, A.K.; Martínez-Martínez, M.; Marrero, H.; Bargiela, R.; et al. Genetically Engineered Proteins with Two Active Sites for Enhanced Biocatalysis and Synergistic Chemo- and Biocatalysis. Nat. Catal. 2020, 3, 319–328. [Google Scholar] [CrossRef]
- Roda, S.; Fernandez-Lopez, L.; Benedens, M.; Bollinger, A.; Thies, S.; Schumacher, J.; Coscolín, C.; Kazemi, M.; Santiago, G.; Gertzen, C.G.W.; et al. A Plurizyme with Transaminase and Hydrolase Activity Catalyzes Cascade Reactions. Angew. Chem. Int. Ed. 2022, 61, e202207344. [Google Scholar] [CrossRef]
- Hu, Q.; Jayasinghe-Arachchige, V.M.; Sharma, G.; Serafim, L.F.; Paul, T.J.; Prabhakar, R. Mechanisms of Peptide and Phosphoester Hydrolysis Catalyzed by Two Promiscuous Metalloenzymes (Insulin Degrading Enzyme and Glycerophosphodiesterase) and Their Synthetic Analogues. WIREs Comput. Mol. Sci. 2020, 10, e1466. [Google Scholar] [CrossRef]
- Vornholt, T.; Christoffel, F.; Pellizzoni, M.M.; Panke, S.; Ward, T.R.; Jeschek, M. Systematic Engineering of Artificial Metalloenzymes for New-to-Nature Reactions. Sci. Adv. 2021, 7, eabe4208. [Google Scholar] [CrossRef]
- Feehan, R.; Franklin, M.W.; Slusky, J.S.G. Machine Learning Differentiates Enzymatic and Non-Enzymatic Metals in Proteins. Nat. Commun. 2021, 12, 3712. [Google Scholar] [CrossRef]
- Amidi, A.; Amidi, S.; Vlachakis, D.; Paragios, N.; Zacharaki, E.I. A Machine Learning Methodology for Enzyme Functional Classification Combining Structural and Protein Sequence Descriptors. In Bioinformatics and Biomedical Engineering; Springer: Berlin/Heidelberg, Germany, 2016; pp. 728–738. [Google Scholar]
- Zou, Z.; Tian, S.; Gao, X.; Li, Y. MlDEEPre: Multi-Functional Enzyme Function Prediction with Hierarchical Multi-Label Deep Learning. Front. Genet. 2019, 9, 714. [Google Scholar] [CrossRef]
- Romero, P.A.; Tran, T.M.; Abate, A.R. Dissecting Enzyme Function with Microfluidic-Based Deep Mutational Scanning. Proc. Natl. Acad. Sci. USA 2015, 112, 7159–7164. [Google Scholar] [CrossRef]
- Ralbovsky, N.M.; Smith, J.P. Machine Learning and Chemical Imaging to Elucidate Enzyme Immobilization for Biocatalysis. Anal. Chem. 2021, 93, 11973–11981. [Google Scholar] [CrossRef]
- Han, X.; Ning, W.; Ma, X.; Wang, X.; Zhou, K. Improving Protein Solubility and Activity by Introducing Small Peptide Tags Designed with Machine Learning Models. Metab. Eng. Commun. 2020, 11, e00138. [Google Scholar] [CrossRef]
- Probst, D.; Manica, M.; Nana Teukam, Y.G.; Castrogiovanni, A.; Paratore, F.; Laino, T. Biocatalysed Synthesis Planning Using Data-Driven Learning. Nat. Commun. 2022, 13, 964. [Google Scholar] [CrossRef]
- Finnigan, W.; Hepworth, L.J.; Flitsch, S.L.; Turner, N.J. RetroBioCat as a Computer-Aided Synthesis Planning Tool for Biocatalytic Reactions and Cascades. Nat. Catal. 2021, 4, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Kreutter, D.; Schwaller, P.; Reymond, J.-L. Predicting Enzymatic Reactions with a Molecular Transformer. Chem. Sci. 2021, 12, 8648–8659. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, B.J.; Johnston, K.E.; Wu, Z.; Arnold, F.H. Advances in Machine Learning for Directed Evolution. Curr. Opin. Struct. Biol. 2021, 69, 11–18. [Google Scholar] [CrossRef]
- Li, G.; Dong, Y.; Reetz, M.T. Can Machine Learning Revolutionize Directed Evolution of Selective Enzymes? Adv. Synth. Catal. 2019, 361, 2377–2386. [Google Scholar] [CrossRef]
- Tatta, E.R.; Imchen, M.; Moopantakath, J.; Kumavath, R. Bioprospecting of Microbial Enzymes: Current Trends in Industry and Healthcare. Appl. Microbiol. Biotechnol. 2022, 106, 1813–1835. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Diaz, D.J.; Czarnecki, N.J.; Zhu, C.; Kim, W.; Shroff, R.; Acosta, D.J.; Alexander, B.R.; Cole, H.O.; Zhang, Y.; et al. Machine Learning-Aided Engineering of Hydrolases for PET Depolymerization. Nature 2022, 604, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Sun, T.; Wang, Y.; Shen, Y. A Machine Learning Study on the Thermostability Prediction of (R)-ω-Selective Amine Transaminase from Aspergillus Terreus. Biomed Res. Int. 2021, 2021, 2593748. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Kawai, S.; Fujitani, M.; Koikeda, S.; Kato, R.; Ema, T. Enhancement of Protein Thermostability by Three Consecutive Mutations Using Loop-Walking Method and Machine Learning. Sci. Rep. 2021, 11, 11883. [Google Scholar] [CrossRef] [PubMed]
- Büchler, J.; Malca, S.H.; Patsch, D.; Voss, M.; Turner, N.J.; Bornscheuer, U.T.; Allemann, O.; le Chapelain, C.; Lumbroso, A.; Loiseleur, O.; et al. Algorithm-Aided Engineering of Aliphatic Halogenase WelO5* for the Asymmetric Late-Stage Functionalization of Soraphens. Nat. Commun. 2022, 13, 371. [Google Scholar] [CrossRef]
- Feehan, R.; Montezano, D.; Slusky, J.S.G. Machine Learning for Enzyme Engineering, Selection and Design. Protein Eng. Des. Sel. 2021, 34, gzab019. [Google Scholar] [CrossRef] [PubMed]
- Czitrom, V. One-Factor-at-a-Time versus Designed Experiments. Am. Stat. 1999, 53, 126. [Google Scholar] [CrossRef]
- Kumar, R.; Nair, A.; Rao, A.S.; Veena, S.M.; Muddapur, U.; Anantharaju, K.S.; More, S.S. Reforming Process Optimization of Enzyme Production Using Artificial Intelligence and Machine Learning. In Optimization of Sustainable Enzymes Production; Chapman and Hall/CRC: Boca Raton, FL, USA, 2022; pp. 75–97. [Google Scholar]
- Lahiri, D.; Nag, M.; Mukherjee, D.; Garai, S.; Banerjee, R.; Ray, R.R. Recent Trends in Approaches for Optimization of Process Parameters for the Production of Microbial Cellulase from Wastes. Environ. Sustain. 2021, 4, 273–284. [Google Scholar] [CrossRef]
- Schweidtmann, A.M.; Esche, E.; Fischer, A.; Kloft, M.; Repke, J.; Sager, S.; Mitsos, A. Machine Learning in Chemical Engineering: A Perspective. Chem. Ing. Tech. 2021, 93, 2029–2039. [Google Scholar] [CrossRef]
- Solle, D.; Hitzmann, B.; Herwig, C.; Pereira Remelhe, M.; Ulonska, S.; Wuerth, L.; Prata, A.; Steckenreiter, T. Between the Poles of Data-Driven and Mechanistic Modeling for Process Operation. Chem. Ing. Tech. 2017, 89, 542–561. [Google Scholar] [CrossRef]
- Singhal, A.; Kumari, N.; Ghosh, P.; Singh, Y.; Garg, S.; Shah, M.P.; Jha, P.K.; Chauhan, D.K. Optimizing Cellulase Production from Aspergillus Flavus Using Response Surface Methodology and Machine Learning Models. Environ. Technol. Innov. 2022, 27, 102805. [Google Scholar] [CrossRef]
- Sarmah, N.; Mehtab, V.; Bugata, L.S.P.; Tardio, J.; Bhargava, S.; Parthasarathy, R.; Chenna, S. Machine Learning Aided Experimental Approach for Evaluating the Growth Kinetics of Candida Antarctica for Lipase Production. Bioresour. Technol. 2022, 352, 127087. [Google Scholar] [CrossRef]
- Das, S.; Negi, S. Enhanced Production of Alkane Hydroxylase from Penicillium Chrysogenum SNP5 (MTCC13144) through Feed-Forward Neural Network and Genetic Algorithm. AMB Express 2022, 12, 28. [Google Scholar] [CrossRef]
- Kumar, G.; Saha, S.P.; Ghosh, S.; Mondal, P.K. Artificial Neural Network-Based Modelling of Optimized Experimental Study of Xylanase Production by Penicillium Citrinum Xym2. Proc. Inst. Mech. Eng. Part E J. Process. Mech. Eng. 2022, 236, 1340–1348. [Google Scholar] [CrossRef]
- De Farias Silva, C.E.; Costa, G.Y.S.C.M.; Ferro, J.V.; de Oliveira Carvalho, F.; da Gama, B.M.V.; Meili, L.; dos Santos Silva, M.C.; Almeida, R.M.R.G.; Tonholo, J. Application of Machine Learning to Predict the Yield of Alginate Lyase Solid-State Fermentation by Cunninghamella Echinulata: Artificial Neural Networks and Support Vector Machine. React. Kinet. Mech. Catal. 2022, 135, 3155–3171. [Google Scholar] [CrossRef]
- Beier, S.; Stiegler, M.; Hitzenhammer, E.; Schmoll, M. Screening for Genes Involved in Cellulase Regulation by Expression under the Control of a Novel Constitutive Promoter in Trichoderma Reesei. Curr. Res. Biotechnol. 2022, 4, 238–246. [Google Scholar] [CrossRef]
- Almeida, F.L.C.; Prata, A.S.; Forte, M.B.S. Enzyme Immobilization: What Have We Learned in the Past Five Years? Biofuels Bioprod. Biorefining 2022, 16, 587–608. [Google Scholar] [CrossRef]
- Sastre, D.E.; Reis, E.A.; Marques Netto, C.G.C. Strategies to Rationalize Enzyme Immobilization Procedures. Methods Enzymol. 2020, 630, 81–110. [Google Scholar] [PubMed]
- Boudrant, J.; Woodley, J.M.; Fernandez-Lafuente, R. Parameters Necessary to Define an Immobilized Enzyme Preparation. Process Biochem. 2020, 90, 66–80. [Google Scholar] [CrossRef]
- Pei, X.; Luo, Z.; Qiao, L.; Xiao, Q.; Zhang, P.; Wang, A.; Sheldon, R.A. Putting Precision and Elegance in Enzyme Immobilisation with Bio-Orthogonal Chemistry. Chem. Soc. Rev. 2022, 51, 7281–7304. [Google Scholar] [CrossRef] [PubMed]
- Ralbovsky, N.M.; Smith, J.P. Machine Learning for Prediction, Classification, and Identification of Immobilized Enzymes for Biocatalysis. Pharm. Res. 2023; Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Kawai, F.; Furushima, Y.; Mochizuki, N.; Muraki, N.; Yamashita, M.; Iida, A.; Mamoto, R.; Tosha, T.; Iizuka, R.; Kitajima, S. Efficient Depolymerization of Polyethylene Terephthalate (PET) and Polyethylene Furanoate by Engineered PET Hydrolase Cut190. AMB Express 2022, 12, 134. [Google Scholar] [CrossRef]
- Erickson, E.; Gado, J.E.; Avilán, L.; Bratti, F.; Brizendine, R.K.; Cox, P.A.; Gill, R.; Graham, R.; Kim, D.-J.; König, G.; et al. Sourcing Thermotolerant Poly(Ethylene Terephthalate) Hydrolase Scaffolds from Natural Diversity. Nat. Commun. 2022, 13, 7850. [Google Scholar] [CrossRef]
- Martínez-Martínez, M.; Coscolín, C.; Santiago, G.; Chow, J.; Stogios, P.J.; Bargiela, R.; Gertler, C.; Navarro-Fernández, J.; Bollinger, A.; Thies, S.; et al. Determinants and Prediction of Esterase Substrate Promiscuity Patterns. ACS Chem. Biol. 2018, 13, 225–234. [Google Scholar] [CrossRef]
- Singla, P.; Bhardwaj, R.D. Enzyme Promiscuity—A Light on the “Darker” Side of Enzyme Specificity. Biocatal. Biotransformation 2020, 38, 81–92. [Google Scholar] [CrossRef]
- Gupta, R.D. Recent Advances in Enzyme Promiscuity. Sustain. Chem. Process. 2016, 4, 2. [Google Scholar] [CrossRef]
- McDonald, A.D.; Bruffy, S.K.; Kasat, A.T.; Buller, A.R. Engineering Enzyme Substrate Scope Complementarity for Promiscuous Cascade Synthesis of 1,2-Amino Alcohols. Angew. Chem. Int. Ed. 2022, 61, e202212637. [Google Scholar] [CrossRef]
- Giunta, C.I.; Cea-Rama, I.; Alonso, S.; Briand, M.L.; Bargiela, R.; Coscolín, C.; Corvini, P.F.-X.; Ferrer, M.; Sanz-Aparicio, J.; Shahgaldian, P. Tuning the Properties of Natural Promiscuous Enzymes by Engineering Their Nano-Environment. ACS Nano 2020, 14, 17652–17664. [Google Scholar] [CrossRef] [PubMed]
- Arora, B.; Mukherjee, J.; Gupta, M.N. Enzyme Promiscuity: Using the Dark Side of Enzyme Specificity in White Biotechnology. Sustain. Chem. Process. 2014, 2, 25. [Google Scholar] [CrossRef]
- Rafeeq, H.; Hussain, A.; Safdar, A.; Shabbir, S.; Bilal, M.; Sher, F.; Franco, M.; Iqbal, H.M.N. Esterases and Their Industrial Applications. In Industrial Applications of Microbial Enzymes; CRC Press: Boca Raton, FL, USA, 2022; pp. 169–190. [Google Scholar]
- Mou, Z.; Eakes, J.; Cooper, C.J.; Foster, C.M.; Standaert, R.F.; Podar, M.; Doktycz, M.J.; Parks, J.M. Machine Learning-based Prediction of Enzyme Substrate Scope: Application to Bacterial Nitrilases. Proteins Struct. Funct. Bioinform. 2021, 89, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, M.; Stam, M.; Médigue, C.; Lespinet, O.; Vallenet, D. Profiling the Orphan Enzymes. Biol. Direct 2014, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Sarker, B.; Ritchie, D.W.; Aridhi, S. GrAPFI: Predicting Enzymatic Function of Proteins from Domain Similarity Graphs. BMC Bioinform. 2020, 21, 168. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Umarov, R.; Xie, B.; Fan, M.; Li, L.; Gao, X. DEEPre: Sequence-Based Enzyme EC Number Prediction by Deep Learning. Bioinformatics 2018, 34, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.; Bileschi, M.L.; Belanger, D.; Colwell, L.J. ProteInfer, Deep Neural Networks for Protein Functional Inference. Elife 2023, 12, e80942. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Murata, M.; Ogawa, T.; Vavricka, C.J.; Kondo, A.; Ogino, C.; Araki, M. Exploration and Evaluation of Machine Learning-Based Models for Predicting Enzymatic Reactions. J. Chem. Inf. Model. 2020, 60, 1833–1843. [Google Scholar] [CrossRef]
- Schaller, K.S.; Molina, G.A.; Kari, J.; Schiano-di-Cola, C.; Sørensen, T.H.; Borch, K.; Peters, G.H.J.; Westh, P. Virtual Bioprospecting of Interfacial Enzymes: Relating Sequence and Kinetics. ACS Catal. 2022, 12, 7427–7435. [Google Scholar] [CrossRef]
- Yu, M.-S.; Lee, H.-M.; Park, A.; Park, C.; Ceong, H.; Rhee, K.-H.; Na, D. In Silico Prediction of Potential Chemical Reactions Mediated by Human Enzymes. BMC Bioinform. 2018, 19, 207. [Google Scholar] [CrossRef]
- Matsuta, Y.; Ito, M.; Tohsato, Y. ECOH: An Enzyme Commission Number Predictor Using Mutual Information and a Support Vector Machine. Bioinformatics 2013, 29, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Mu, F.; Unkefer, C.J.; Unkefer, P.J.; Hlavacek, W.S. Prediction of Metabolic Reactions Based on Atomic and Molecular Properties of Small-Molecule Compounds. Bioinformatics 2011, 27, 1537–1545. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tian, S.; Allen, D.; Oler, E.; Peters, H.; Lui, V.W.; Gautam, V.; Djoumbou-Feunang, Y.; Greiner, R.; Metz, T.O. BioTransformer 3.0—A Web Server for Accurately Predicting Metabolic Transformation Products. Nucleic Acids Res. 2022, 50, W115–W123. [Google Scholar] [CrossRef]
- Tian, S.; Djoumbou-Feunang, Y.; Greiner, R.; Wishart, D.S. CypReact: A Software Tool for in Silico Reactant Prediction for Human Cytochrome P450 Enzymes. J. Chem. Inf. Model. 2018, 58, 1282–1291. [Google Scholar] [CrossRef]
Figure 1.
Schematics of the overall machine learning methodology considering the 3 stages involved. The first stage involves raw data collection and their cleaning. The cleaned data are split into two sets, one providing training data (Stage 2) and the other testing data (Stage 3). In the second stage, several algorithms are evaluated to find the one that best fits the matter at hand. In stage 3, the model selected in stage 2 is tested again with a new set of data to establish how it performs. Depending on the outcome, the model may require adjustments, so going back to previous stages may be required.
Figure 1.
Schematics of the overall machine learning methodology considering the 3 stages involved. The first stage involves raw data collection and their cleaning. The cleaned data are split into two sets, one providing training data (Stage 2) and the other testing data (Stage 3). In the second stage, several algorithms are evaluated to find the one that best fits the matter at hand. In stage 3, the model selected in stage 2 is tested again with a new set of data to establish how it performs. Depending on the outcome, the model may require adjustments, so going back to previous stages may be required.

Figure 2.
MLMs in biocatalysis: selection of the most adequate enzyme from information retrieved from databases and processed by an adequate algorithm to improve enzyme efficiency.
Figure 2.
MLMs in biocatalysis: selection of the most adequate enzyme from information retrieved from databases and processed by an adequate algorithm to improve enzyme efficiency.

Figure 3.
Protein engineering assisted by machine learning. Information on sequence–function relationship of proteins is retrieved from databases and curated, and an algorithm is used to identify the sequences most likely to display the desired property, which is then used to synthesize the optimized recombinant protein.
Figure 3.
Protein engineering assisted by machine learning. Information on sequence–function relationship of proteins is retrieved from databases and curated, and an algorithm is used to identify the sequences most likely to display the desired property, which is then used to synthesize the optimized recombinant protein.

Figure 4.
A schematic overview of the use of MLMs in fermentation optimization and fermentation scale-up. Operational conditions (e.g., temperature, pH, medium composition) are provided, or scale-up criteria are selected and a target value (e.g., productivity) is defined. The selected algorithm processes the information to identify the conditions that optimize the target value or enable its reproduction throughout different process scales.
Figure 4.
A schematic overview of the use of MLMs in fermentation optimization and fermentation scale-up. Operational conditions (e.g., temperature, pH, medium composition) are provided, or scale-up criteria are selected and a target value (e.g., productivity) is defined. The selected algorithm processes the information to identify the conditions that optimize the target value or enable its reproduction throughout different process scales.


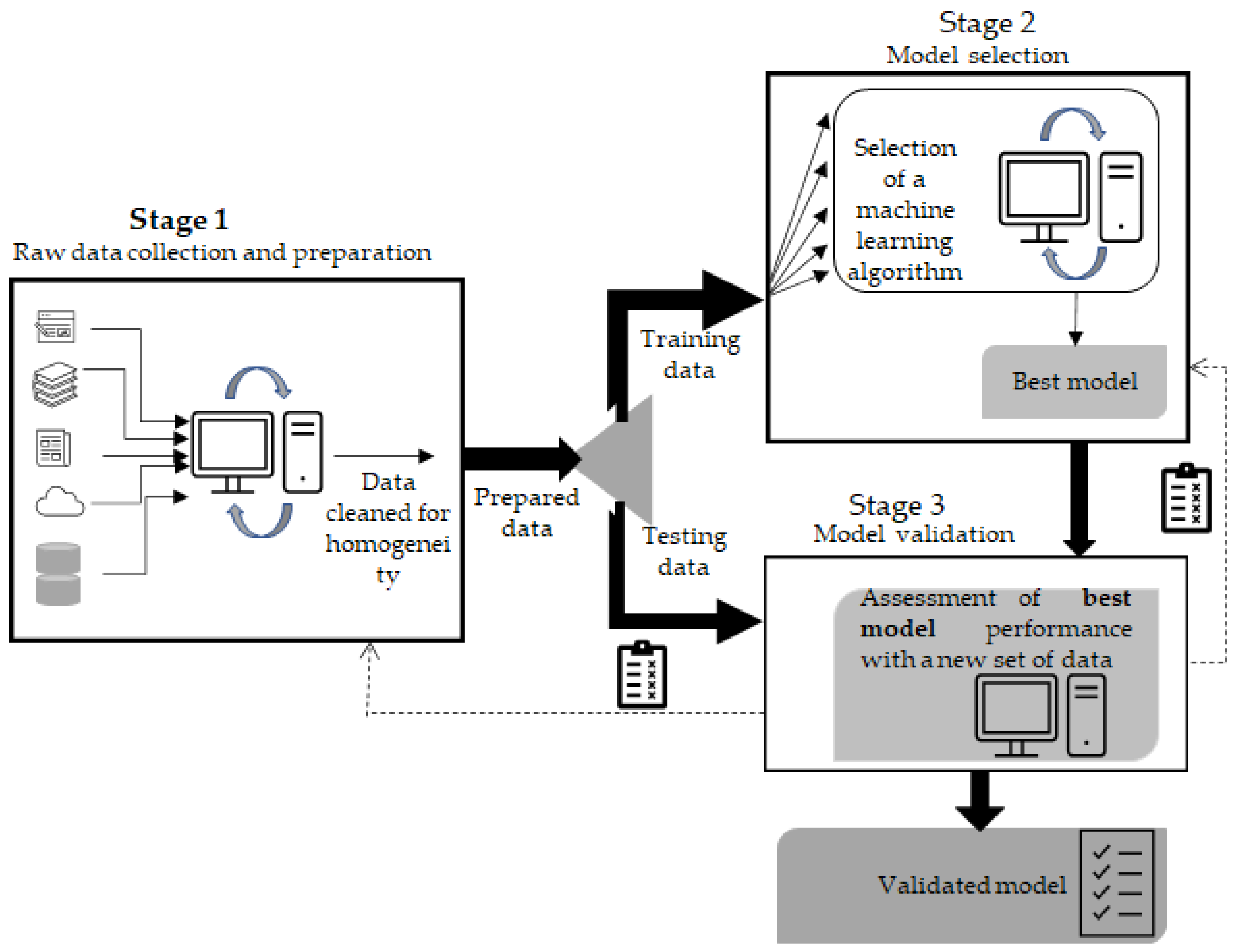{kind=link}
{kind=link}
{kind=link}
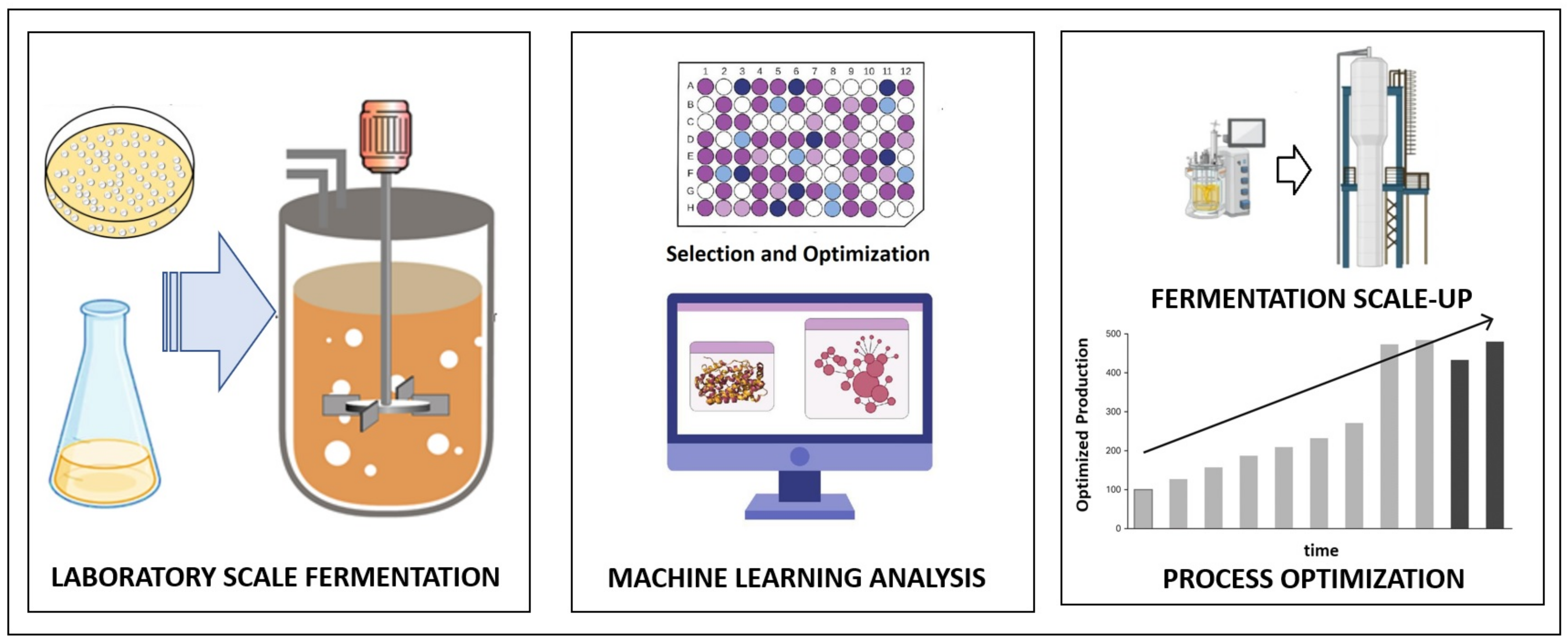{kind=link}
| Chronology | Milestones | Comments |
|---|---|---|
| Pre-20th century | Brewing, baking, dairy, wine alcohol fermentation | Production methods based on empiricism, mostly related to the food sector |
| Pre-WWII | Acetone–butanol–ethanol (ABE) fermentation, production of amino acids and citric acid | Early steps in: (a) the production of biofuels (ABE) 1,2; (b) fermentative production of small molecules 1 |
| 1940s to 1980s | Large scale production of: (a) antibiotics, steroids and other small molecules, (b) enzymes, single-cell protein and other macromolecules | Introduction of large-scale aerated bioreactors operating under aseptic conditions Insight into the molecular machinery of bacteria and cells, e.g., DNA structure, mechanisms and control of protein synthesis |
| 1980s henceforth | Production of recombinant biopharmaceuticals, e.g., monoclonal antibodies, interferons, vaccines Production of biopolymers (e.g., bioplastics), biofuels (biohydrogen) and messenger RNA vaccines Process design in a circular economy perspective | Introduction of recombinant DNA technology; rational design and directed evolution of proteins. Overall creation of biomolecular diversity. Enhanced therapeutic proteins and enzymes Trends in automation, process integration and data-driven process optimization |
1 Significant improvements in hardware, microbial sources, standardization and modelling throughout the XX and XXI centuries; 2 trend revived from 1970s henceforth (fuel crisis and environmental concerns).
Table 2.
An overview of ML algorithms: basic aspects and illustrative applications. Specific issues and comprehensive details are out of the scope of this review and can be found in recent publications [2,91,95,98,99,100].
| Algorithm and Key Issues | Examples of Applications |
|---|---|
| Multivariate analysis Abridges a set of machine learning algorithms, such as principal component analysis (PCA), linear regression (LR) and multiple linear regression (MLR) and partial least-square regression (PLS) [2,101,102]. Largely used and still dominant as ML tools in the bioprocessing industry since their inception in the late XX century [2]. PCA is an unsupervised method that reduces the size of a dataset, allowing new uncorrelated variables (i.e., latent variables) and a respective maximization of their variation. It can be used to discriminate components, find hidden patterns and identify abnormalities, etc. MLR is a supervised method that uses several independent variables to predict the outcome of a single dependent variable when a single independent variable is used to predict the outcome of the dependent variable MLR reduces to LR. PLS is a supervised algorithm related to dimensionality reduction that can directly relate an input dataset and a corresponding output dataset, establishing a linear correlation between the input and output variables within their latent space [103]. | PCA: bacterial cell behaviour in the presence of organic solvents [104], bioreactor monitoring [105,106], protein sequence clusters [107], enzyme screening [108], mode of action of antibiotics and discovery of new bioactive compounds [109] and analysis of cereals [110] MLR: prediction of secondary protein structure [111], screening of protease inhibitors [112]; LR: effect of active metabolites in a population [113], effect of linear transformation on the input features, as achieved via placing an amino acid at each position or the presence or absence of a mutation [93]; effects of blocks of sequence in a library of chimeric proteins made through recombination [94]. PLS: monitoring [114] and control of bioreactors [115]; development of a biosensor device for analysis of binary mixtures of phenols [116]; and prediction of steroid diffusion across artificial membranes [117]. |
| Support vector machines (SVMs) A supervised algorithm that can be used for both classification and regression purposes (more commonly the former). Targets the finding of a hyperplane that optimally divides a dataset into two classes. Able to extract complex nonlinear relationships, as typically observed within bio-applications. Has been used in bioprocessing since the late XX century. Limited use in the presence of large datasets, questionable model interpretability and lack of uncertainty disclosure associated with prediction hamper further dissemination. Has been gradually replaced in several settings by other methods, e.g., artificial neural networks and random forests, as these also provide more accurate predictions [2,118,119,120]. | SVMs: prediction of the secondary structure of a protein [121], prediction of protein binding sites [122], identification of antioxidant proteins [123], chemotaxonomy studies based on secondary metabolites (diterpenes) [124], analysis of metabolic fluxes in microbial cells [125,126] and optimization of the permeability of a membrane used in a bioreactor for wastewater treatment [127]. |
| Artificial neural networks (ANNs) Mimic the way brain cells process information. Used in either supervised or unsupervised learning. An ANN is a topological structure formed by processing elements (artificial neurons) connected with coefficients (weights) and organized in layers [128,129]. ANNs provide a flexible regression structure to predict the relationship between inputs and outputs and can estimate any function [130]. By providing this specific flexible model structure and a set of input and output data, the parameters of the neural network can be changed iteratively so that the inputs match their correct output and estimates become closer and closer to the training data [2]. Roughly, ANNs can be presented as single-layer perceptron (SL) and multi-layer (ML) networks. The SL contains only two layers (input and output) yet fails to handle complex patterns; hence, more layers (ML), termed hidden layers, can be introduced [131]. To vary the weights to approximate an underlying function, the derivative of the error between the training output and the predicted response with respect to the weights of the network is determined, allowing gradient-dependent optimization solvers to minimize the error [2]. Several network structures have been proposed, e.g., convolutional neural networks, which enable a matrix or tensor of inputs such as an image [20,132,133]; recurrent neural networks, which use so-called internal memory [134,135,136]; deep neural networks, where many hidden layers facilitate the modelling of intricate underlying functions due to the large number of parameters [137,138,139] and clearly embrace the deep learning concept since more than three layers are involved. ANNs are gradually replacing PCA and PLS methods due to their relatively poor accuracy when simulating nonlinear biochemical reaction systems [2]. | ANNs: modelling and optimization of enzymatic treatment for nutritional enhancement of rice [140], optimization of fermentation conditions for production of lipopeptide antibiotic [141], optimization of algal biofuel production [20], prediction of the toxicity of ionic liquids towards enzyme activity [142], liquid level control for bioreactor management [118], classification of 3D enzyme structure [132], prediction of protein structure [49,133,139,143], recognition of amino acids in protein engineering [135], de novo protein design [138], learning of protein function–structure relationship [144], protein thermostability [145,146], protein subcellular localization [147], protein functional class [148], protein solubility [149], recognition of promoter sequences [134], calibration of biosensors [136], prediction of flux in metabolic pathways given enzyme concentrations [150], tapping into the relationship between the chemical structure of given molecules and their biological activity for drug design [151] |
| Gaussian processes (GPs) A probabilistic machine learning algorithm in which the estimates obtained are probability distributions as opposed to scalar values. Can be used in both supervised and unsupervised learning. Usually defined as a class of machine learning interpolation techniques with no assumed measurement noise, a GP will provide an exact fit to the dataset. Estimates are typically made based on the weighted sum of the output data, weighted by the distance of the predictions from the existing data in the input space. The resulting probability distributions provide insight into the uncertainty of a forecast. GP models are attractive given their flexible non-parametric nature and computational simplicity [2,152]. GP is a distribution that, instead of returning unique values, returns functions. The referred distribution is thus conditioned on the training data using Bayesian reasoning, ultimately leading to a predictive distribution [153]. The run time for exact GP regression scales with the cube of the number of training examples, which makes it unsuitable for large (>103) datasets, but fast and accurate approximations are currently available [154]. Gaussian process prediction is hampered by the inversion of a covariance matrix, which computationally scales with the number of data points. Alternative processes have thus been developed, namely sparse Gaussian processes that approximate the posterior predictive distribution or the precision matrix, which scales with exponentially larger datasets [155] | GPs: prediction of protein stability upon mutation [156], screening of Michaelis constant (KM), and hence substrate affinity, for a given enzyme–substrate pair [157], assistance in directed evolution in a model system where protein function is altered and green fluorescence is transformed into yellow fluorescence [158], identification of channelrhodopsins that express and localize to the plasma membrane and conversion of a channelrhodopsins unable to localize into one that localizes well to the plasma membrane [159], engineering channelrhodopsins to obtain a mutant with high light sensitivity and potential application in optogenetics [160], real-time monitoring of cell culture processes through prediction of glucose and lactate concentrations [161], determination of the dynamics of a metabolic pathway with no need for time-dependent flux measurements [162]. |
| Ensemble learning (EL) Abridges supervised learning methods by merging predictions from several inducers for a decision. Thus, errors of an individual inducer will be counterweighed by others. An inducer, also called a base learner, is an algorithm that relates input and output data. The often-improved predictive performance of ensemble learning methods prevents overfitting. This minimizes the risk of obtaining local optimal models and widens the search location to obtain an optimal fit [2,163]. EL methods are divided into dependent and independent frameworks, depending on the relationship between each inducer [164]. Random forests (RFs) are among the latter and rank as the most common EL method in biochemical engineering [2]. RFs encompass decision trees, a flowchart-like parallel structure where if–else statements on inputs estimate output predictions as inducers [2,164]. Gradient boosting (GB) encompasses a dependent framework, where the construction of each inducer depends on the previously trained predecessor. Typically requires over 103 trees, is memory-demanding and has a high computational cost [164]. Given their different structures, RFs and GB should be used primarily for classification and regression studies, respectively [165,166]. | RFs: prediction of protein–ligand docking affinity [167], prediction of flux in a membrane bioreactor [120], prediction of protein structure [168], protein function prediction [169], model for automatic classification of live and dead cells in Chlorella vulgaris [170], classification of compounds with key fuel properties [171], classification of enzymes [172], predictive models for drug combination therapy for tackling microbial infections, amino acid identification for health diagnostics [173], prediction of medium-chain carboxylic acid production from waste biomass [174], development of an environmentally friendly polyester dyeing process upon enzyme- and chitosan-driven surface modifications of the polyester [175]. GB: development of a broad KM predictive model from structural features [176], prediction of the mechanical functionality of protein networks [177]. |
| Reinforcement learning (RL) RL differs from supervised and unsupervised learning. RL fosters a trial-and-error approach where the algorithm learns continuously through iteration and feedback based on a reward and penalty strategy for each tested sequence. The obvious goal is for the algorithm to maximize the cumulative reward through a series of adequate decisions [178]. RL is a relative newcomer to biochemical engineering but has been present in the chemical process industry since at least the early 2000s. Its adaptability without the need for large-labelled datasets suggests it may be easily disseminated in the near future [2,96], particularly (but not exclusively) for fermentation process control and optimization [96,179,180,181] | RL: Identification of the structure of a kinetic model and prediction of the kinetic parameters of a microbial fermentation [182], tuning the metabolic enzyme levels to improve production in microbial fermentation (e.g., synthesis of l-tryptophan) through a model-free approach and with no knowledge of the microbial metabolic network or its regulation [183], search for pathways for the production of valuable compounds by using the bioretrosynthesis space [184], addressing protein–ligand docking [185]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sampaio, P.S.; Fernandes, P. Machine Learning: A Suitable Method for Biocatalysis. Catalysts 2023, 13, 961. https://doi.org/10.3390/catal13060961
AMA Style
Sampaio PS, Fernandes P. Machine Learning: A Suitable Method for Biocatalysis. Catalysts. 2023; 13(6):961. https://doi.org/10.3390/catal13060961
Chicago/Turabian StyleSampaio, Pedro Sousa, and Pedro Fernandes. 2023. "Machine Learning: A Suitable Method for Biocatalysis" Catalysts 13, no. 6: 961. https://doi.org/10.3390/catal13060961
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.