
Advances in the Application of Acetonitrile in Organic Synthesis since 2018

{kind=link}
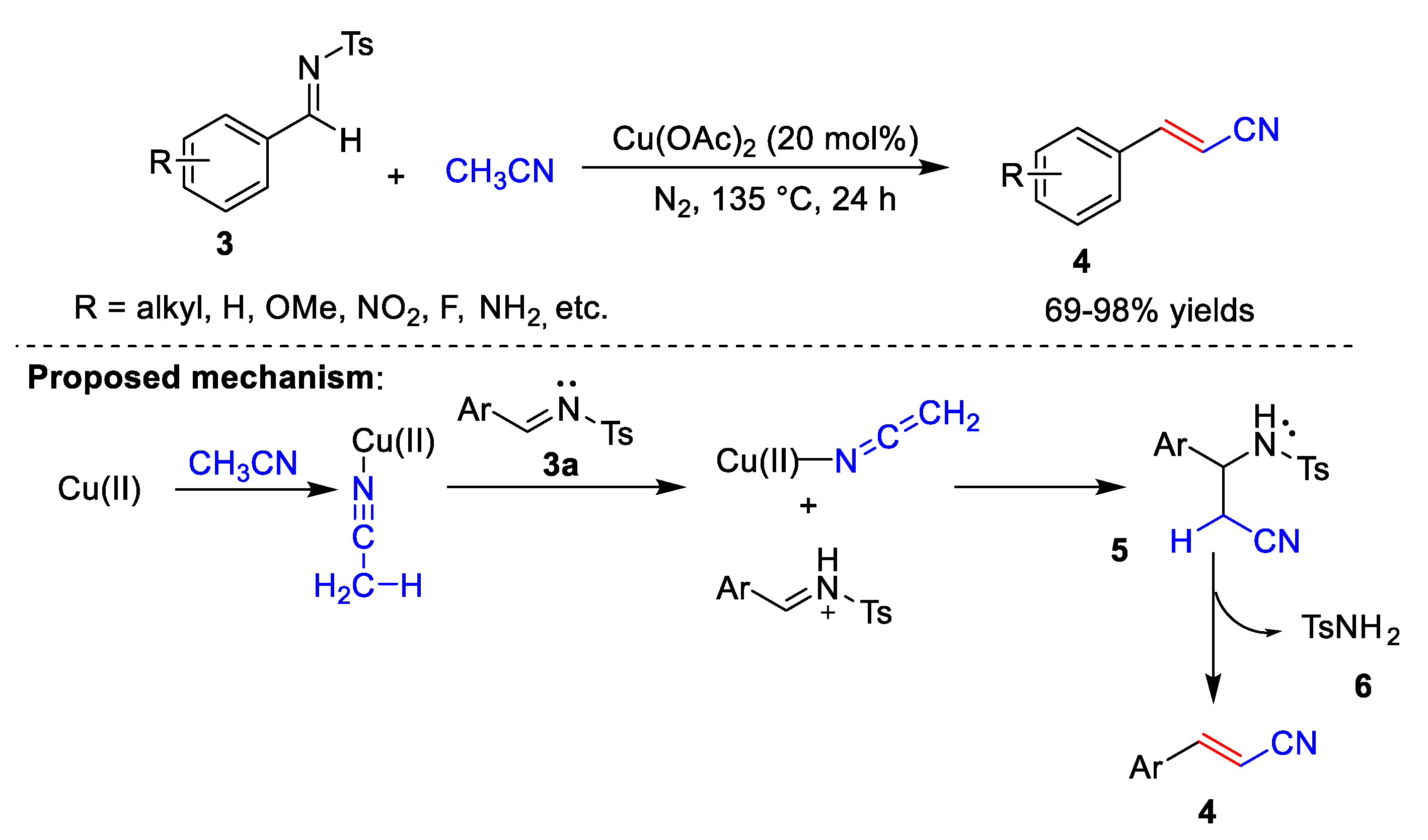{kind=link}
{kind=link}
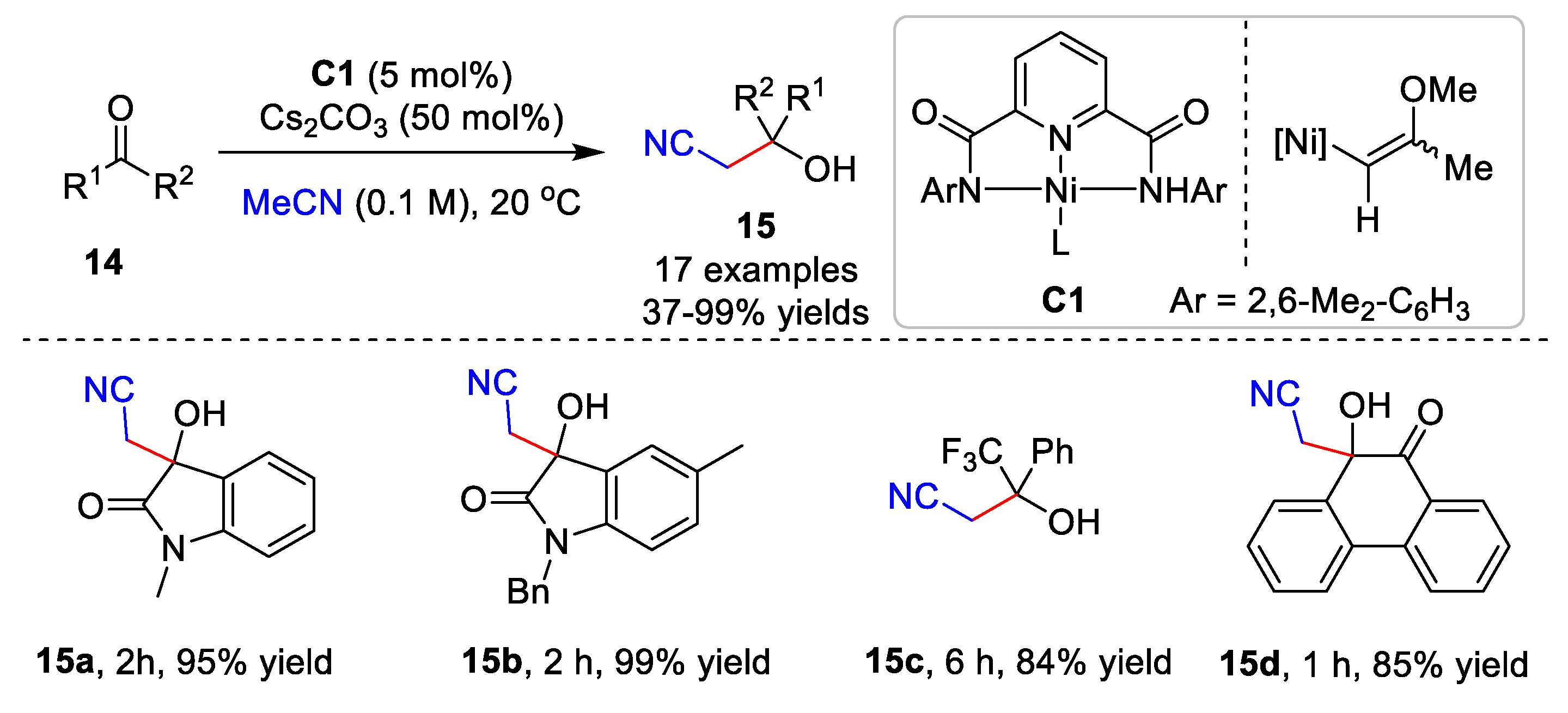{kind=link}
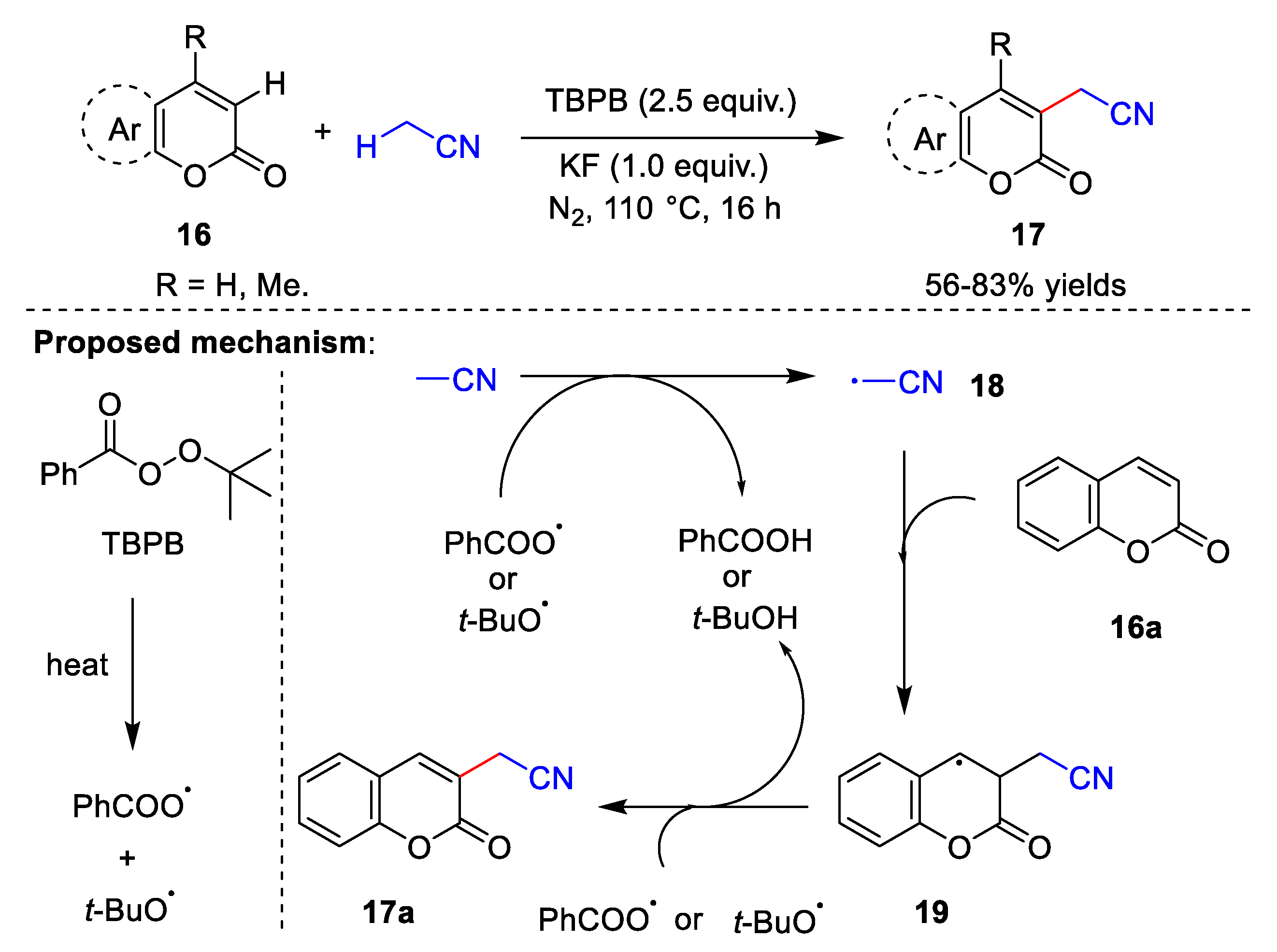{kind=link}
{kind=link}
{kind=link}
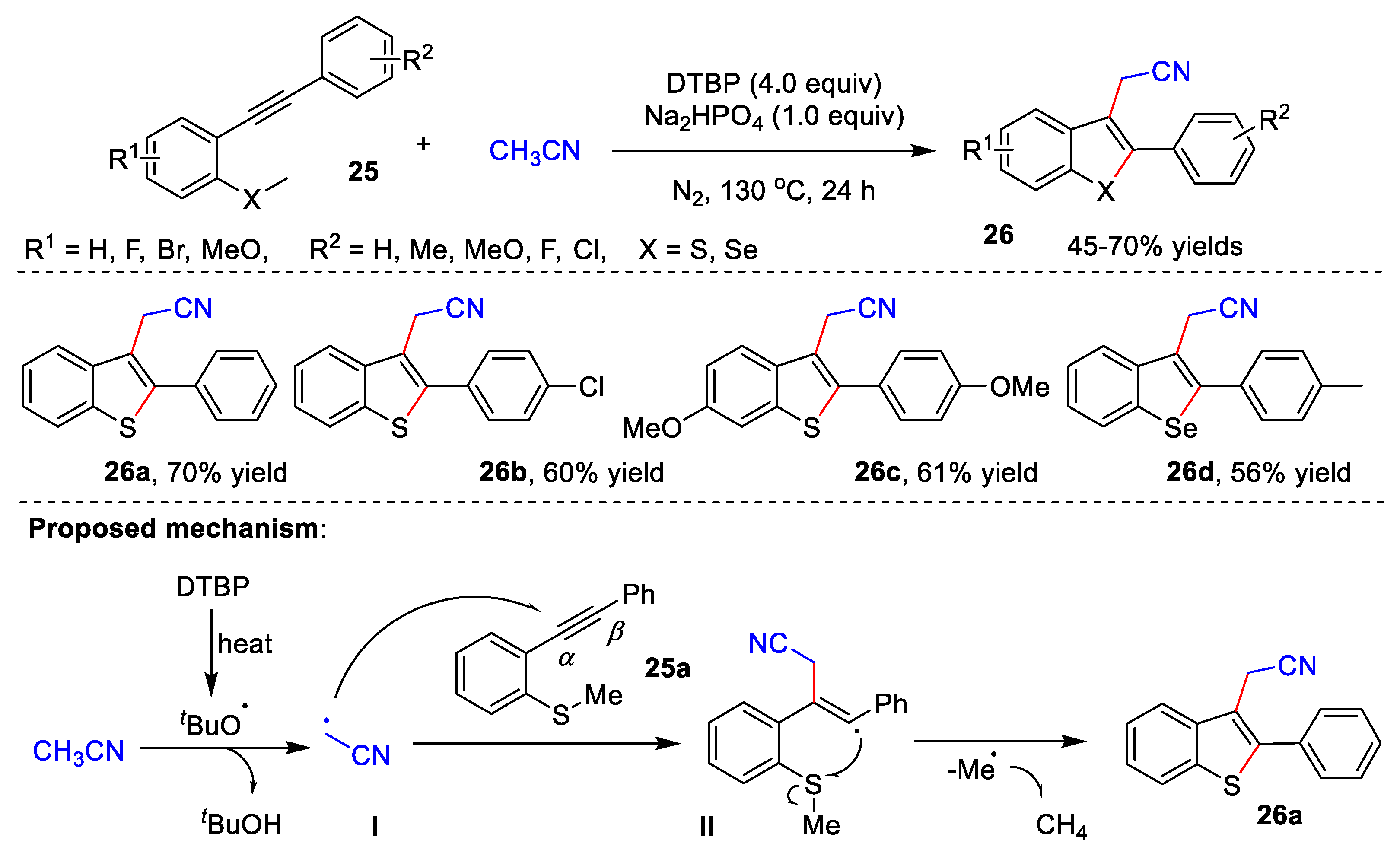{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cyanomethylation Reaction
2.1. Metal-Catalyzed Cyanomethylation
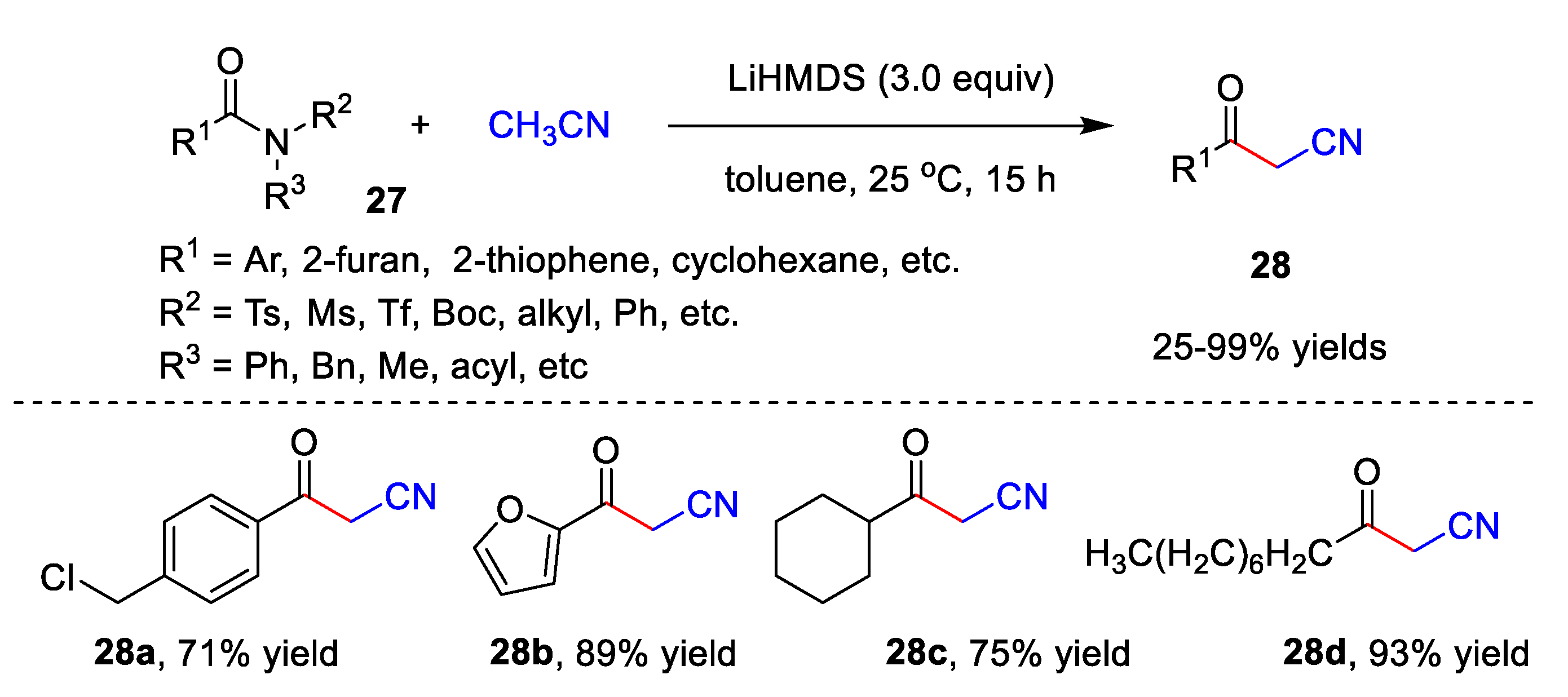
2.2. Transition Metal-Free Cyanomethylation
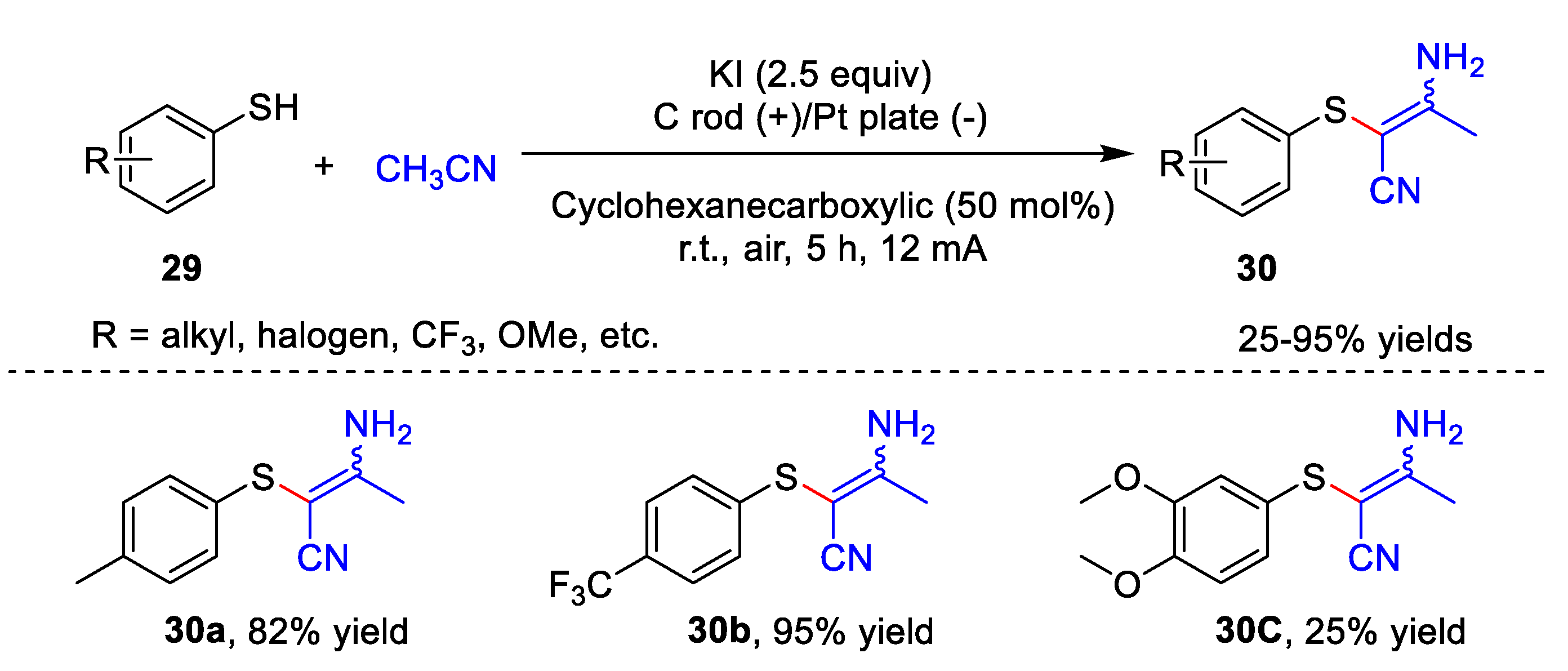
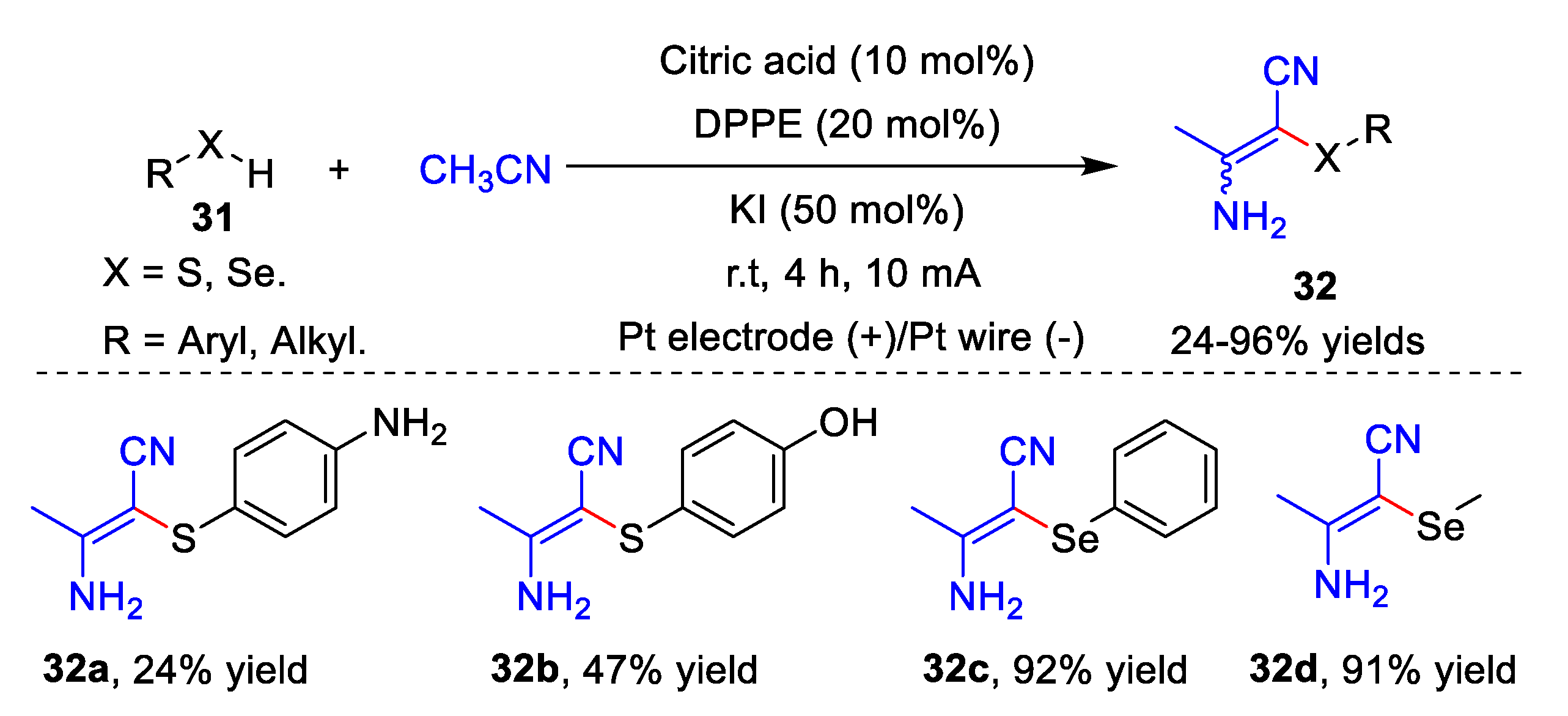
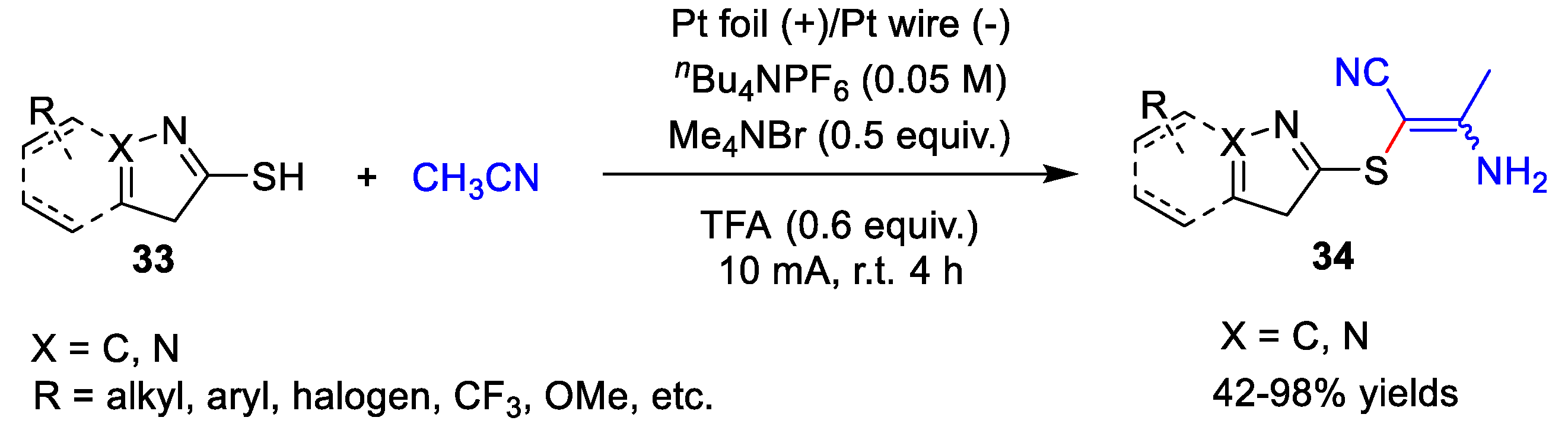
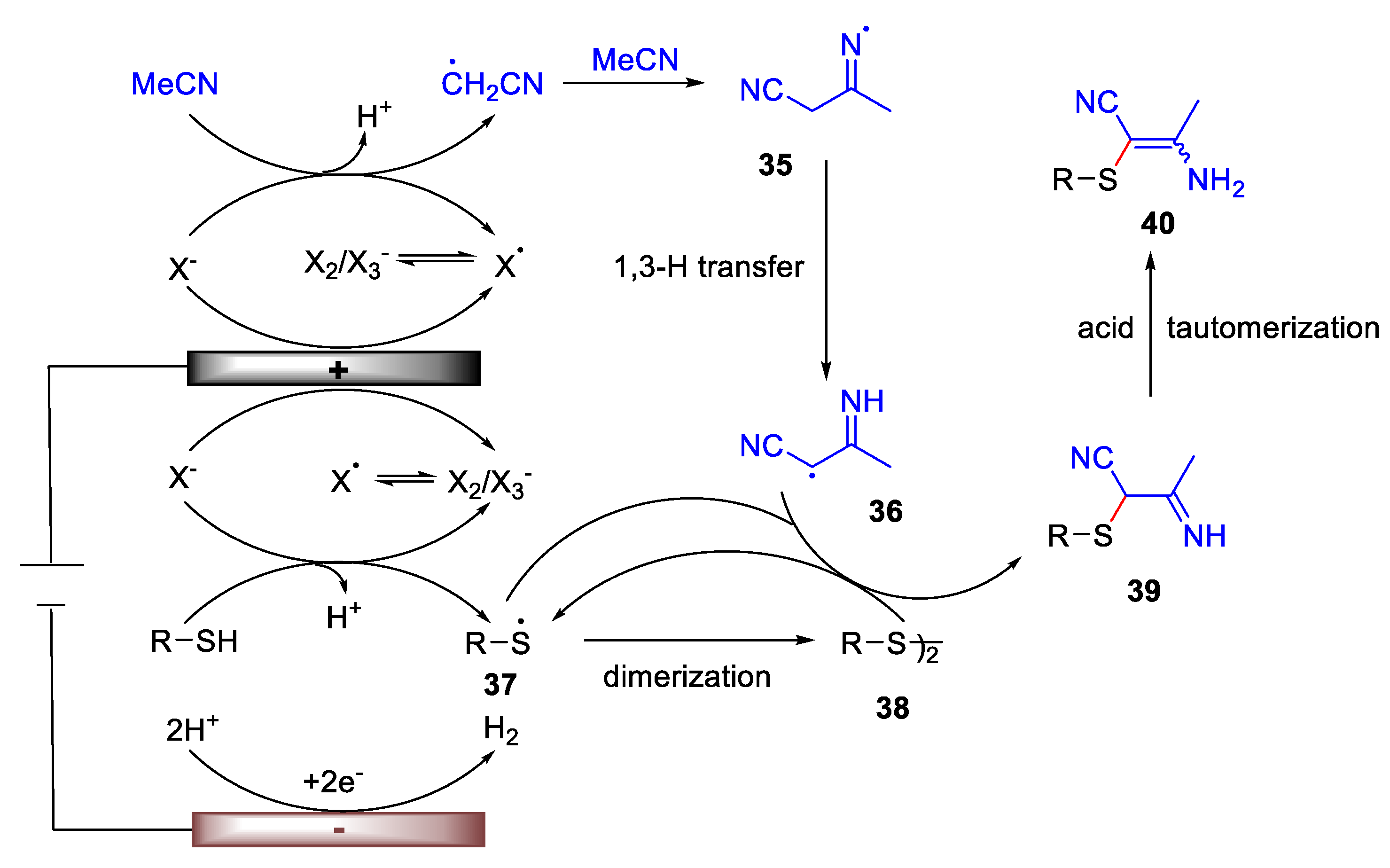
3. Electrochemical Oxidative Cross-Coupling Reaction
4. Cyclization Reaction
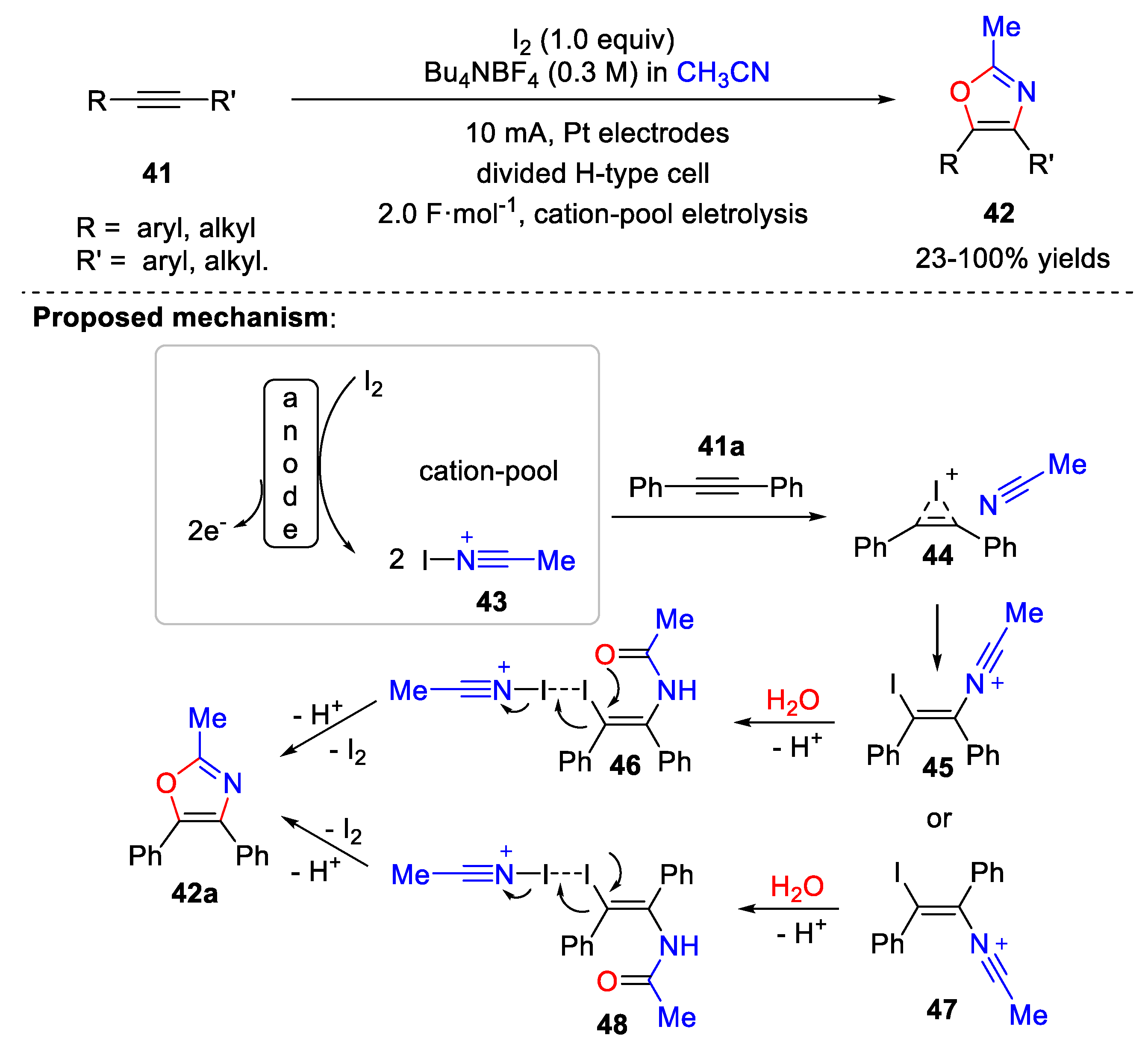
4.1. Synthesis of Oxazole
4.2. Synthesize Dihydroimidazole and 2-Oxazoline
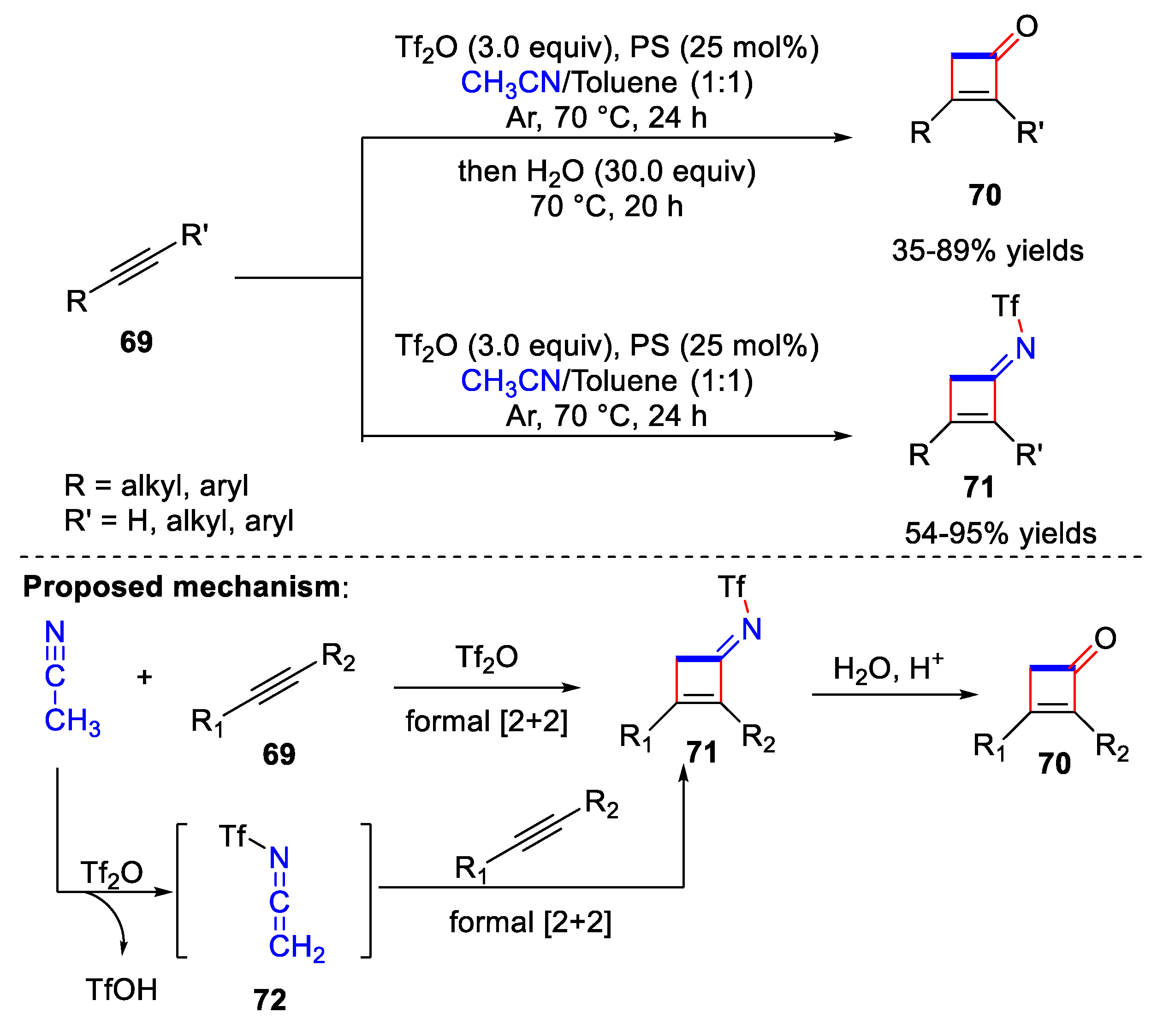
4.3. Synthesis of Cyclobutenone
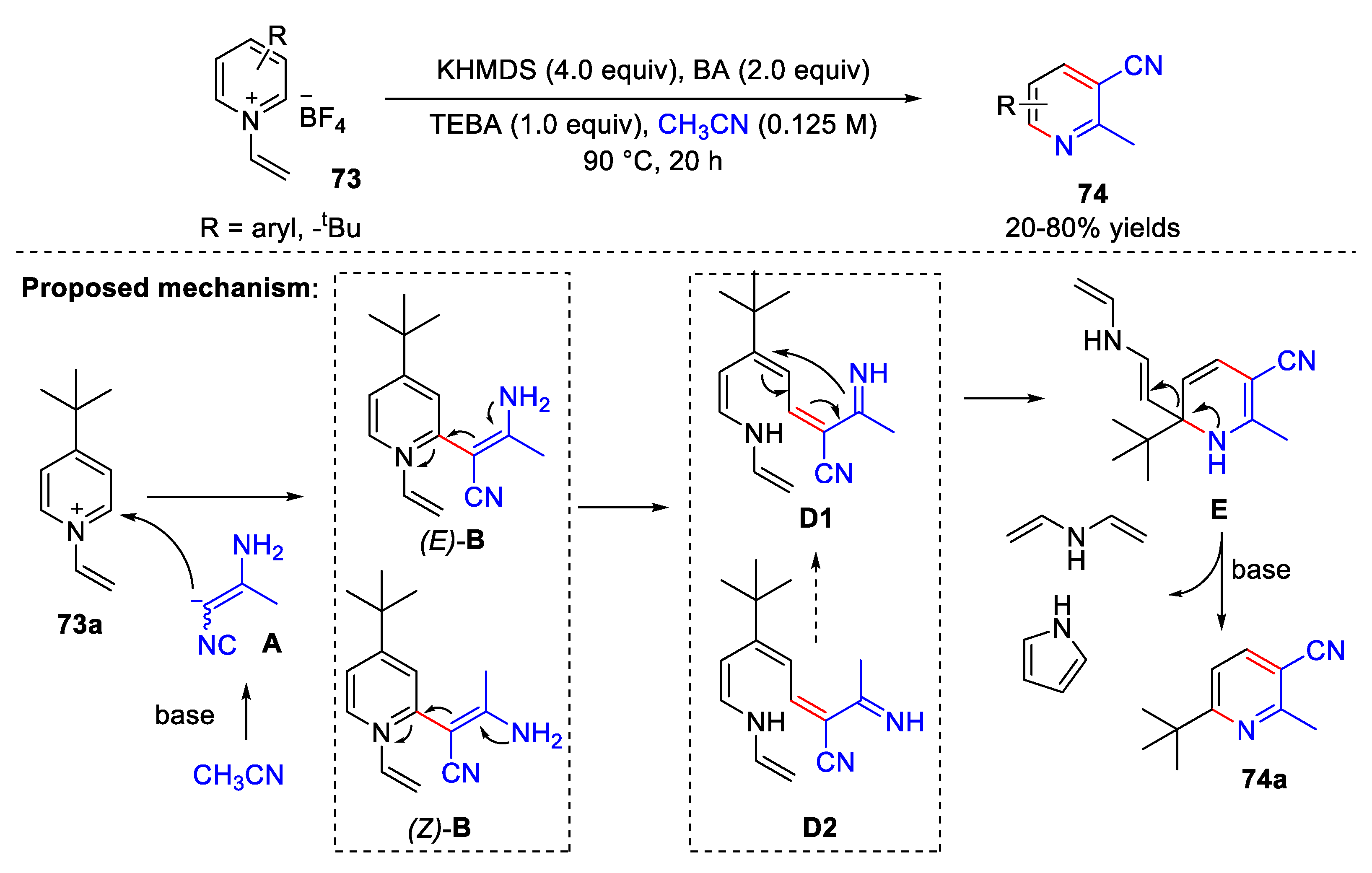
4.4. Synthesis of 3-Cyanopyridine
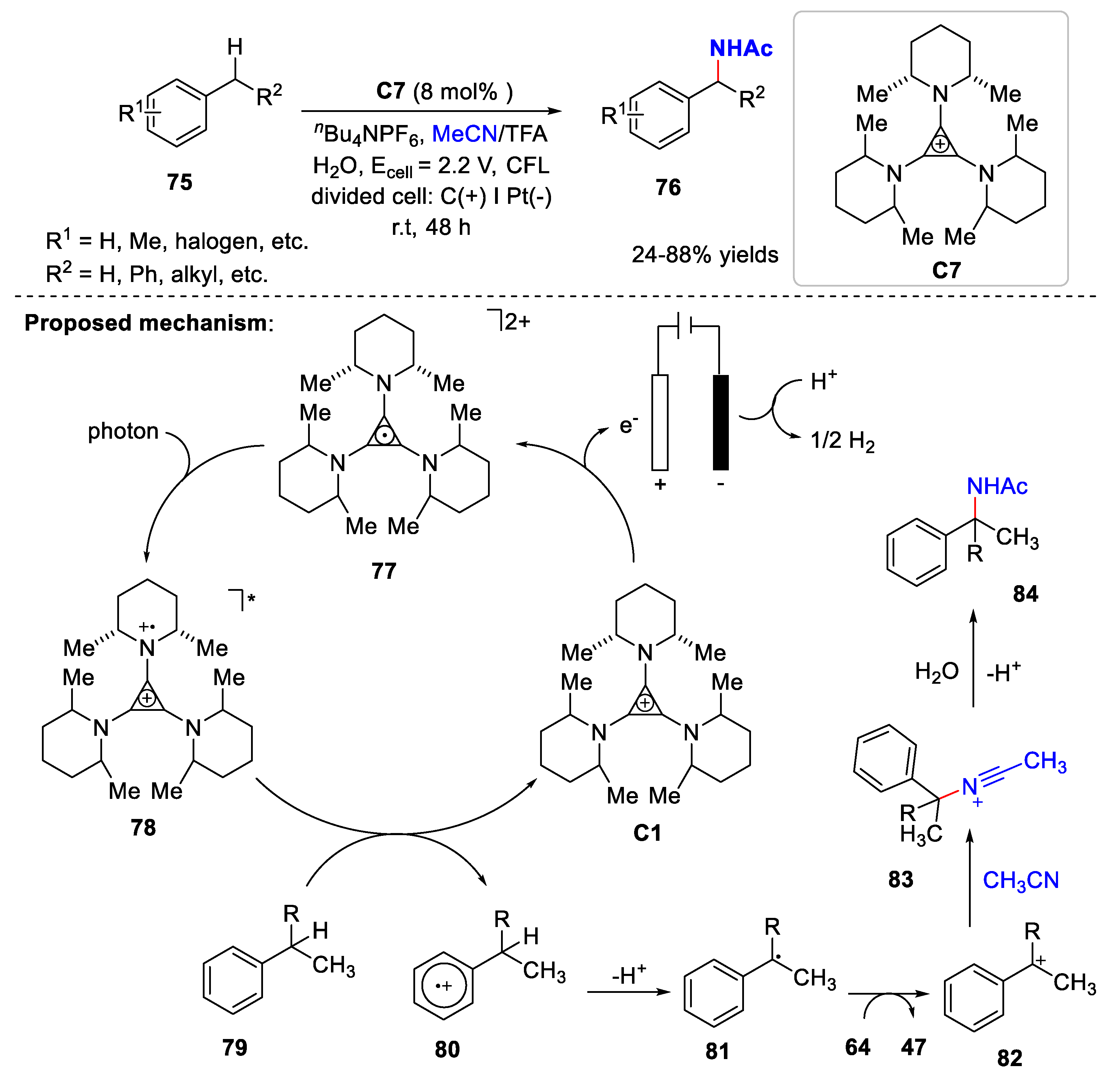
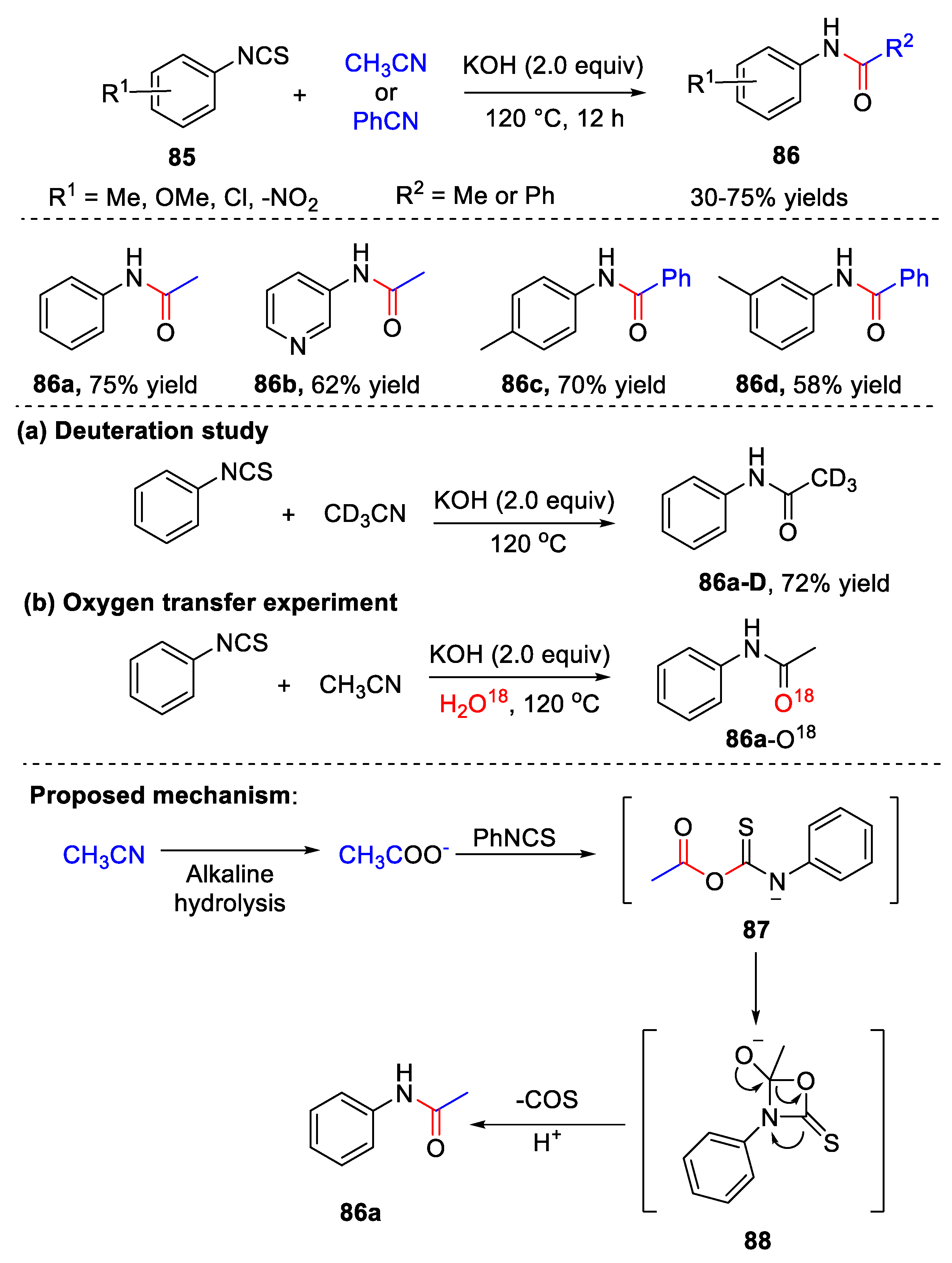
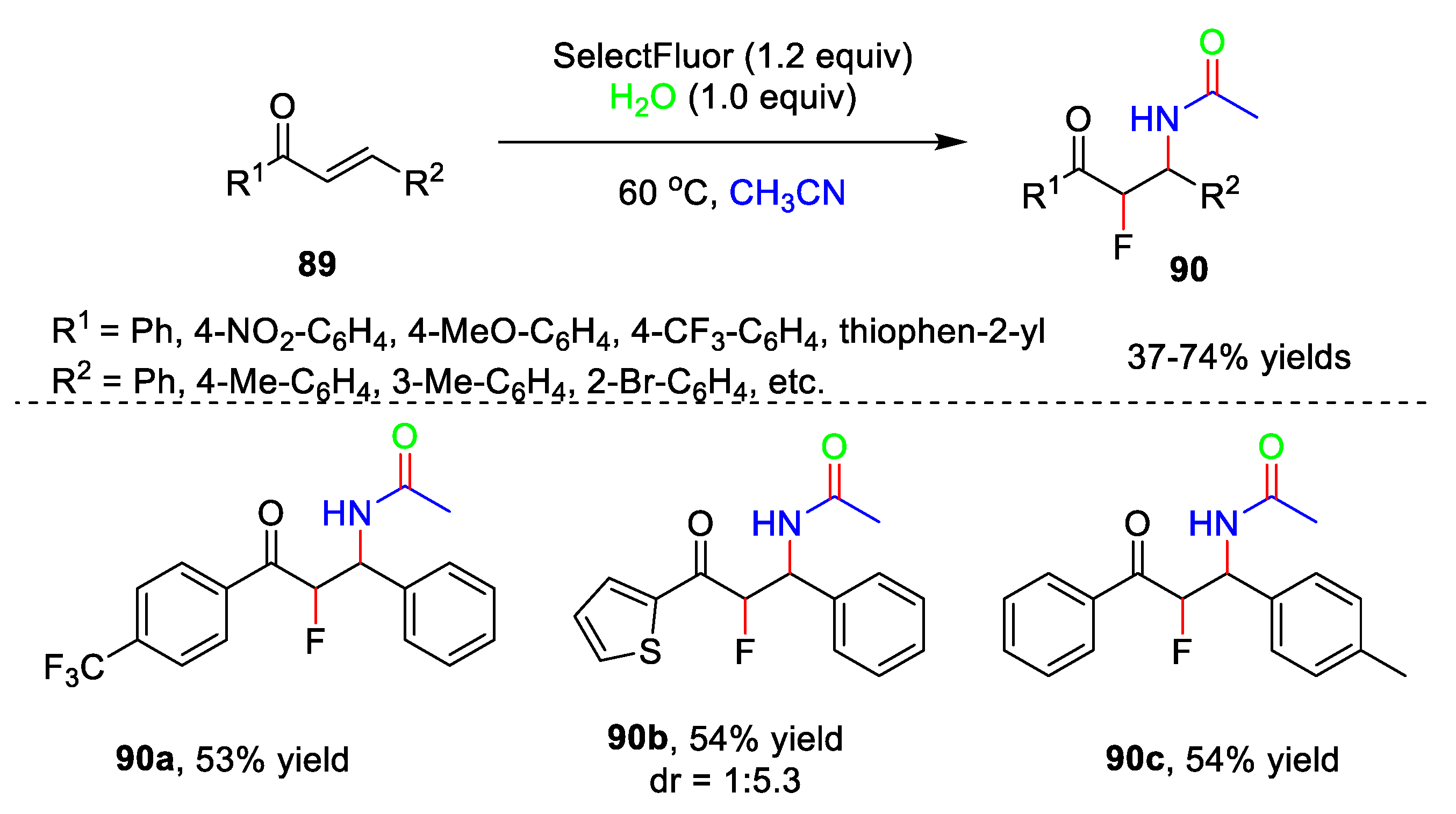
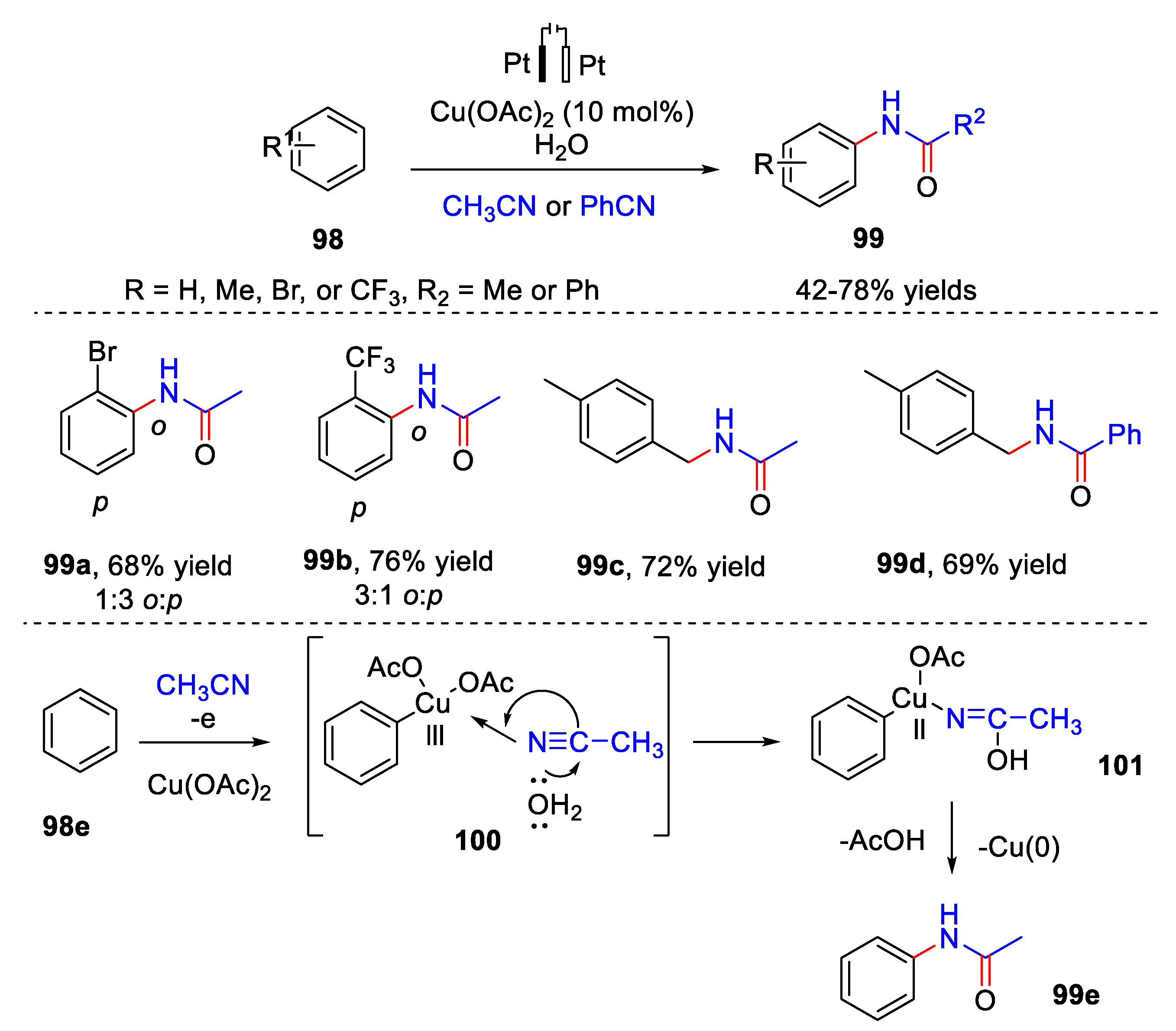
5. Amidation Reaction
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kütt, A.; Tshepelevitsh, S.; Saame, J.; Lõkov, M.; Kaljurand, I.; Selberg, S.; Leito, I. Strengths of acids in acetonitrile. Eur. J. Org. Chem. 2021, 2021, 1407. [Google Scholar] [CrossRef]
- Moran, S.; Ellis, H.B., Jr.; DeFrees, D.J.; McLean, A.D.; Ellison, G.B. Carbanion spectroscopy: Cyanomethide anion (CH2CN-). J. Am. Chem. Soc. 1987, 109, 5996. [Google Scholar] [CrossRef]
- Ashfold, M.N.R.; Simons, J.P. Vacuum ultraviolet photodissociation spectroscopy of CH3CN, CD3CN, CF3CN and CH3NC. J. Chem. Soc. Faraday Trans. 1978, 74, 1263. [Google Scholar] [CrossRef]
- Batanero, B.; Sánchez-Sánchez, C.M.; Montiel, V.; Aldaz, A.; Barba, F. Electrochemical synthesis of 3-phenylcinnamonitrile by reduction of benzophenone in acetonitrile. Electrochem. Commun. 2003, 5, 349. [Google Scholar] [CrossRef]
- Zhao, Q.; Ren, L.; Hou, J.; Yu, W.; Chang, J. Annulation Reactions of In-Situ-Generated N-(Het)aroyldiazenes with Isothiocyanates Leading to 2-Imino-1,3,4-oxadiazolines. Org. Lett. 2019, 21, 210. [Google Scholar] [CrossRef] [PubMed]
- Alanthadka, A.; Maheswari, C.U. N-Heterocyclic carbene-catalyzed oxidative amidation of aldehydes with amines. Adv. Synth. Catal. 2015, 357, 1199. [Google Scholar] [CrossRef]
- Deshidi, R.; Rizvi, M.A.; Shah, B.A. Highly efficient dehydrogenative cross-coupling of aldehydes with amines and alcohols. RSC Adv. 2015, 5, 90521. [Google Scholar] [CrossRef]
- Mohamadighader, N.; Saraei, M.; Nematollahi, D.; Goljani, H. Electrochemical study of 4-chloroaniline in a water/acetonitrile mixture. A new method for the synthesis of 4-chloro-2-(phenylsulfonyl)aniline and N-(4-chlorophenyl)benzenesulfonamide. RSC Adv. 2020, 10, 31563. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.M.F.; Farrag, A.M.; Harras, M.F.; Ibrahim, M.H.; Mehany, A.B.M. Apoptosis: A target for anticancer therapy with novel cyanopyridines. Bioorgan. Chem. 2020, 94, 103481. [Google Scholar] [CrossRef]
- Hu, P.; Chai, J.; Duan, Y.; Liu, Z.; Cui, G.; Chen, L. Progress in nitrile-based polymer electrolytes for high performance lithium batteries. J. Mater. Chem. A 2016, 4, 10070. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, W.; Shen, Z. Cu-catalyzed cyanation of indoles with acetonitrile as a cyano source. J. Org. Chem. 2015, 80, 8868. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Y.; Gong, M.; Wu, Q.; Zhang, J.; Kim, J.K.; Huang, M.; Wu, Y. Method for direct synthesis of α-cyanomethyl-β-dicarbonyl compounds with acetonitrile and 1,3-dicarbonyls. Org. Lett. 2016, 18, 4151. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Wang, L.; Rao, H.; Xu, H. Iron-catalyzed dehydrogenative sp3–sp2 Coupling via Direct Oxidative C–H Activation of Acetonitrile. Org. Lett. 2017, 19, 2226. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhuang, S.; Liu, P.; Sun, P. Cyanomethylation and cyclization of aryl alkynoates with acetonitrile under transition-metal-free conditions: Synthesis of 3-cyanomethylated coumarins. J. Org. Chem. 2016, 81, 11489. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, B.; Zhang, H.; Ju, C.-W.; Qin, Y.; Xue, X.-S.; Zhao, D. A ring expansion strategy towards diverse azaheterocycles. Nat. Chem. 2021, 13, 1006. [Google Scholar] [CrossRef]
- Zhang, H.-Z.; Zhao, Z.-L.; Zhou, C.-H. Recent advance in oxazole-based medicinal chemistry. Eur. J. Med. Chem. 2018, 144, 444. [Google Scholar] [CrossRef]
- Facchinetti, V.; Gomes, C.R.B.; de Souza, M.V.N. Application of nitriles on the synthesis of 1,3-oxazoles, 2-oxazolines, and oxadiazoles: An update from 2014 to 2021. Tetrahedron 2021, 102, 132544. [Google Scholar] [CrossRef]
- McIver, A.; Young, D.D.; Deiters, A. A general approach to triphenylenes and azatriphenylenes: Total synthesis of dehydrotylophorine and tylophorine. Chem. Commun. 2008, 2008, 4750. [Google Scholar] [CrossRef]
- Yagyu, T.; Takemoto, Y.; Yoshimura, A.; Zhdankin, V.V.; Saito, A. Iodine(III)-catalyzed formal [2+2+1] cycloaddition reaction for metal-free construction of oxazoles. Org. Lett. 2017, 19, 2506. [Google Scholar] [CrossRef]
- Hajra, S.; Sinha, D.; Bhowmick, M. Metal triflate catalyzed reactions of alkenes, NBS, nitriles, and TMSN3: synthesis of 1,5-disubstituted tetrazoles. J. Org. Chem. 2007, 72, 1852. [Google Scholar] [CrossRef]
- Hoff, B. Acetonitrile as a building block and reactant. Synthesis 2018, 50, 2824. [Google Scholar] [CrossRef]
- Kumar Verma, P.; Vishwakarma, R.A.; Sawant, S.D. Reaction medium as the installing reservoir for key functionalities in the molecules. Asian J. Org. Chem. 2019, 8, 777. [Google Scholar] [CrossRef]
- Chen, H.; Dai, W.; Chen, Y.; Xu, Q.; Chen, J.; Yu, L.; Zhao, Y.; Ye, M.; Pan, Y. Efficient and selective nitrile hydration reactions in water catalyzed by an unexpected dimethylsulfinyl anion generated in situ from CsOH and DMSO. Green Chem. 2014, 16, 2136. [Google Scholar] [CrossRef]
- Ibrahim, A.D.; Entsminger, S.W.; Fout, A.R. Insights into a chemoselective cobalt catalyst for the hydroboration of alkenes and nitriles. ACS Catal. 2017, 7, 3730. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Liu, J.; Wan, X.; Xu, Q. Clean synthesis of primary to tertiary carboxamides by CsOH-catalyzed aminolysis of nitriles in water. Green Chem. 2016, 18, 4865. [Google Scholar] [CrossRef]
- Veisi, H.; Maleki, B.; Hamelian, M.; Ashrafi, S.S. Chemoselective hydration of nitriles to amides using hydrated ionic liquid (IL) tetrabutylammonium hydroxide (TBAH) as a green catalyst. RSC Adv. 2015, 5, 6365. [Google Scholar] [CrossRef]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010, 53, 7902. [Google Scholar] [CrossRef]
- Ha, T.M.; Guo, W.; Wang, Q.; Zhu, J. Copper-catalyzed cyanoalkylative aziridination of N-sulfonyl allylamines. Adv. Synth. Catal. 2020, 362, 2205. [Google Scholar] [CrossRef]
- Ahmad, M.S.; Ahmad, A. Cu-catalyzed cyanomethylation of imines and alpha, beta-alkenes with acetonitrile and its derivatives. RSC Adv. 2021, 11, 5427. [Google Scholar] [CrossRef]
- Yao, H.; Zhong, X.; Wang, B.; Lin, S.; Yan, Z. Cyanomethylation of the benzene rings and pyridine rings via direct oxidative cross-dehydrogenative coupling with acetonitrile. Org. Lett. 2022, 24, 2030. [Google Scholar] [CrossRef]
- Saito, A.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric addition of acetonitrile to aldimines. Org. Lett. 2019, 21, 8187. [Google Scholar] [CrossRef]
- Saito, A.; Adachi, S.; Kumagai, N.; Shibasaki, M. Direct catalytic asymmetric addition of alkylnitriles to aldehydes with designed nickel-carbene complexes. Angew. Chem. Int. Ed. 2021, 60, 8739. [Google Scholar] [CrossRef] [PubMed]
- Coffinet, A.; Levacher, V.; Gillaizeau, I.; Brière, J.-F.; Oudeyer, S. Nickel-catalyzed cyanoalkylation of ketone derivatives. Adv. Synth. Catal. 2023, 365, 156. [Google Scholar] [CrossRef]
- Csákÿ, A. Metal-free organocatalysis. Catalysts 2018, 8, 195. [Google Scholar] [CrossRef]
- Zhang, R.; Jin, S.; Liu, Q.; Lin, S.; Yan, Z. Transition metal-free cross-dehydrogenative coupling reaction of coumarins with acetonitrile or acetone. J. Org. Chem. 2018, 83, 13030. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xia, Z.; Xiao, Y.; Yu, Y.; Yu, L.; Song, Z.; Wu, Q.; Zhang, J.; Tan, Z. Transition-metal-free cross-dehydrogenative couplings of 8-aminoquinoline amides at C5 position with acetonitrile, ethers or acetone. Eur. J. Org. Chem. 2021, 2021, 5012. [Google Scholar] [CrossRef]
- Hong, G.; Nahide, P.D.; Kozlowski, M.C. Cyanomethylation of substituted fluorenes and oxindoles with alkyl nitriles. Org. Lett. 2020, 22, 1563. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, Z.-Q. Free radical addition of nitrile, ketone, and ester to alkyne and the selectivity discussion. Org. Lett. 2019, 21, 8810. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhong, T.; Zhang, L.; Chen, J.; Chen, Z.; Jiang, X.; Yu, C. Radical-triggered cyclization of methylthio-substituted alkynones: Synthesis of diverse 3-alkylthiochromones. Eur. J. Org. Chem. 2020, 2020, 4534. [Google Scholar] [CrossRef]
- Cai, T.; Shen, F.; Ni, Y.; Xu, H.; Shen, R.; Gao, Y. Cascade radical annulation of 2-alkynylthio(seleno)anisoles with acetone or acetonitrile: Synthesis of 3-acetomethyl- or cyanomethyl-substituted benzothio(seleno)phenes. J. Org. Chem. 2021, 86, 1002. [Google Scholar] [CrossRef]
- Liu, H.; Yang, Z.; Huang, G.; Yu, J.-T.; Pan, C. Cyanomethylative cyclization of unactivated alkenes with nitriles for the synthesis of cyano-containing ring-fused quinazolin-4(3H)-ones. New J. Chem. 2022, 46, 1347. [Google Scholar] [CrossRef]
- Park, M.S.; Lee, S. Transition-metal-catalyst-free reaction of amides and acetonitriles: Synthesis of β-ketonitriles. Org. Chem. Front. 2022, 9, 5178. [Google Scholar] [CrossRef]
- Qian, P.; Zha, Z.; Wang, Z. Recent advances in C−H functionalization with electrochemistry and various iodine-containing reagents. ChemElectroChem 2020, 7, 2527. [Google Scholar] [CrossRef]
- Horn, E.J.; Rosen, B.R.; Baran, P.S. Synthetic organic electrochemistry: An enabling and innately sustainable method. ACS Cent. Sci. 2016, 2, 302. [Google Scholar] [CrossRef]
- Rockl, J.L.; Pollok, D.; Franke, R.; Waldvogel, S.R. A decade of electrochemical dehydrogenative C-C coupling of aryls. Acc. Chem. Res. 2020, 53, 45. [Google Scholar] [CrossRef]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 2017, 117, 13230. [Google Scholar] [CrossRef] [PubMed]
- He, W.-B.; Zhao, S.-J.; Chen, J.-Y.; Jiang, J.; Chen, X.; Xu, X.; He, W.-M. External electrolyte-free electrochemical one-pot cascade synthesis of 4-thiocyanato-1H-pyrazoles. Chin. Chem. Lett. 2023, 34, 107640. [Google Scholar] [CrossRef]
- Ding, L.; Niu, K.; Liu, Y.; Wang, Q. Electro-reductive C-H cyanoalkylation of quinoxalin-2(1H)-ones. Chin. Chem. Lett. 2022, 33, 4057. [Google Scholar] [CrossRef]
- Pappo, D.; Kashman, Y. β-Turn mimetic: synthesis of cyclic thioenamino peptides. Org. Lett. 2006, 8, 1177. [Google Scholar] [CrossRef]
- Kraft, S.; Ryan, K.; Kargbo, R.B. Recent advances in asymmetric hydrogenation of tetrasubstituted olefins. J. Am. Chem. Soc. 2017, 139, 11630. [Google Scholar] [CrossRef]
- Lu, F.; Yang, Z.; Wang, T.; Wang, T.; Zhang, Y.; Yuan, Y.; Lei, A. Electrochemical oxidative Csp3-H/S-H cross-coupling with hydrogen evolution for synthesis of tetrasubstituted olefins. Chin. J. Chem. 2019, 37, 547. [Google Scholar] [CrossRef]
- He, T.-J.; Ye, Z.; Ke, Z.; Huang, J.-M. Stereoselective synthesis of sulfur-containing β-enaminonitrile derivatives through electrochemical Csp3–H bond oxidative functionalization of acetonitrile. Nat. Commum. 2019, 10, 833. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.L.; Huang, J.M. Bromide-catalyzed electrochemical Csp3−H oxidation of acetonitrile: Stereoselective synthesis of heteroaryl vinyl sulfides. Adv. Synth. Catal. 2022, 364, 2648. [Google Scholar] [CrossRef]
- Igarashi, E.; Sakamoto, K.; Yoshimura, T.; Matsuo, J. Formal [4+2] cycloaddition of 3-phenylcyclobutanones with nitriles. Tetrahedron Lett. 2019, 60, 13. [Google Scholar] [CrossRef]
- Sattler, L.E.; Hilt, G. Iodonium cation-pool electrolysis for the three-component synthesis of 1,3-oxazoles. Chem. Eur. J. 2021, 27, 605. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Liu, C.; Li, W.; Yu, J.; Wang, M.; Zhang, Y. Electrochemical synthesis of polysubstituted oxazoles from ketones and acetonitrile. Org. Lett. 2022, 24, 5762. [Google Scholar] [CrossRef]
- Shen, T.; Lambert, T.H. Electrophotocatalytic diamination of vicinal C-H bonds. Science 2021, 371, 620. [Google Scholar] [CrossRef]
- Qin, Q.; Luo, X.; Wei, J.; Zhu, Y.; Wen, X.; Song, S.; Jiao, N. Acetonitrile activation: An effective two-carbon unit for cyclization. Angew. Chem. Int. Ed. 2019, 58, 4376. [Google Scholar] [CrossRef]
- Neff, R.K.; Su, Y.-L.; Liu, S.; Rosado, M.; Zhang, X.; Doyle, M.P. Generation of Halomethyl Radicals by Halogen Atom Abstraction and Their Addition Reactions with Alkenes. J. Am. Chem. Soc. 2019, 141, 16643. [Google Scholar] [CrossRef]
- Tomilin, D.N.; Sobenina, L.N.; Saliy, I.V.; Ushakov, I.A.; Belogolova, A.M.; Trofimov, B.A. Substituted pyrrolyl-cyanopyridines on the platform of acylethynylpyrroles via their 1 : 2 annulation with acetonitrile under the action of lithium metal. New J. Chem. 2022, 46, 13149. [Google Scholar] [CrossRef]
- Duan, X.; Sun, R.; Tang, J.; Li, S.; Yang, X.; Zheng, X.; Li, R.; Chen, H.; Fu, H.; Yuan, M. Facile synthesis of 2-methylnicotinonitrile through degenerate ring transformation of pyridinium salts. J. Org. Chem. 2022, 87, 7975. [Google Scholar] [CrossRef]
- Valeur, E.; Bradley, M. Amide bond formation: Beyond the myth of coupling reagents. Chem. Soc. Rev. 2009, 38, 606. [Google Scholar] [CrossRef] [PubMed]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827. [Google Scholar] [CrossRef]
- Nasser, J.; Zhang, L.; Lin, J.; Sodano, H. Aramid nanofiber reinforced polymer nanocomposites via amide–amide hydrogen bonding. ACS Appl. Polym. Mater. 2020, 2, 2934. [Google Scholar] [CrossRef]
- Yan, Z.; Sun, B.; Huang, P.; Zhao, H.; Ding, H.; Su, W.; Jin, C. Visible-light-promoted radical alkylation/cyclization of allylic amide with N-hydroxyphthalimide ester: Synthesis of oxazolines. Chin. Chem. Lett. 2022, 33, 1997. [Google Scholar] [CrossRef]
- Huang, Y.; Pi, C.; Tang, Z.; Wu, Y.; Cui, X. Cp*Co(III)-catalyzed C-H amidation of azines with dioxazolones. Chin. Chem. Lett. 2020, 31, 3237. [Google Scholar] [CrossRef]
- Shen, T.; Lambert, T.H. C-H Amination via electrophotocatalytic Ritter-type reaction. J. Am. Chem. Soc. 2021, 143, 8597. [Google Scholar] [CrossRef]
- Zhong, P.; Wu, J.; Wu, J.; Liu, K.; Wan, C.; Liu, J.-B. Solvent-controlled selective synthesis of amides and thioureas from isothiocyanates. Tetrahedron Lett. 2022, 107, 154099. [Google Scholar] [CrossRef]
- Zhou, J.; Fang, Y.; Wang, F.; Li, J. Catalyst-free regioselective hydroxyfluorination and aminofluorination of α,β-unsaturated ketones. Org. Biomol. Chem. 2019, 17, 4470. [Google Scholar] [CrossRef]
- Liu, S.; Klussmann, M. Acid promoted radical-chain difunctionalization of styrenes with stabilized radicals and (N,O)-nucleophiles. Chem. Commun. 2020, 56, 1557. [Google Scholar] [CrossRef]
- Liu, S.; Klussmann, M. Organo-redox-catalysis for the difunctionalization of alkenes and oxidative Ritter reactions by C–H functionalization. Org. Chem. Front. 2021, 8, 2932. [Google Scholar] [CrossRef]
- Guan, Y.-Q.; Min, X.-T.; He, G.-C.; Ji, D.-W.; Guo, S.-Y.; Hu, Y.-C.; Chen, Q.-A. The serendipitous effect of KF in Ritter reaction: Photo-induced amino-alkylation of alkenes. iScience 2021, 24, 102969. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yuan, Y.; Ye, K.-Y. Electrochemical synthesis of vicinal azidoacetamides. Chem. Commun. 2023, 59, 422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, M.; You, K.; Pang, Y.; Ma, B. Metal-free aminofluorination of α-diazo 2H-benzopyran-4-one: Convenient access to β-fluoramides. Org. Biomol. Chem. 2022, 20, 7027. [Google Scholar] [CrossRef]
- Strekalova, S.; Kononov, A.; Rizvanov, I.; Budnikova, Y. Acetonitrile and benzonitrile as versatile amino sources in copper-catalyzed mild electrochemical C-H amidation reactions. RSC Adv. 2021, 11, 37540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, W.; Wang, B.; Tan, H.; Jiao, N.; Song, S. Electrophilic amidomethylation of arenes with DMSO/MeCN reagents. Org. Chem. Front. 2022, 9, 2430. [Google Scholar] [CrossRef]
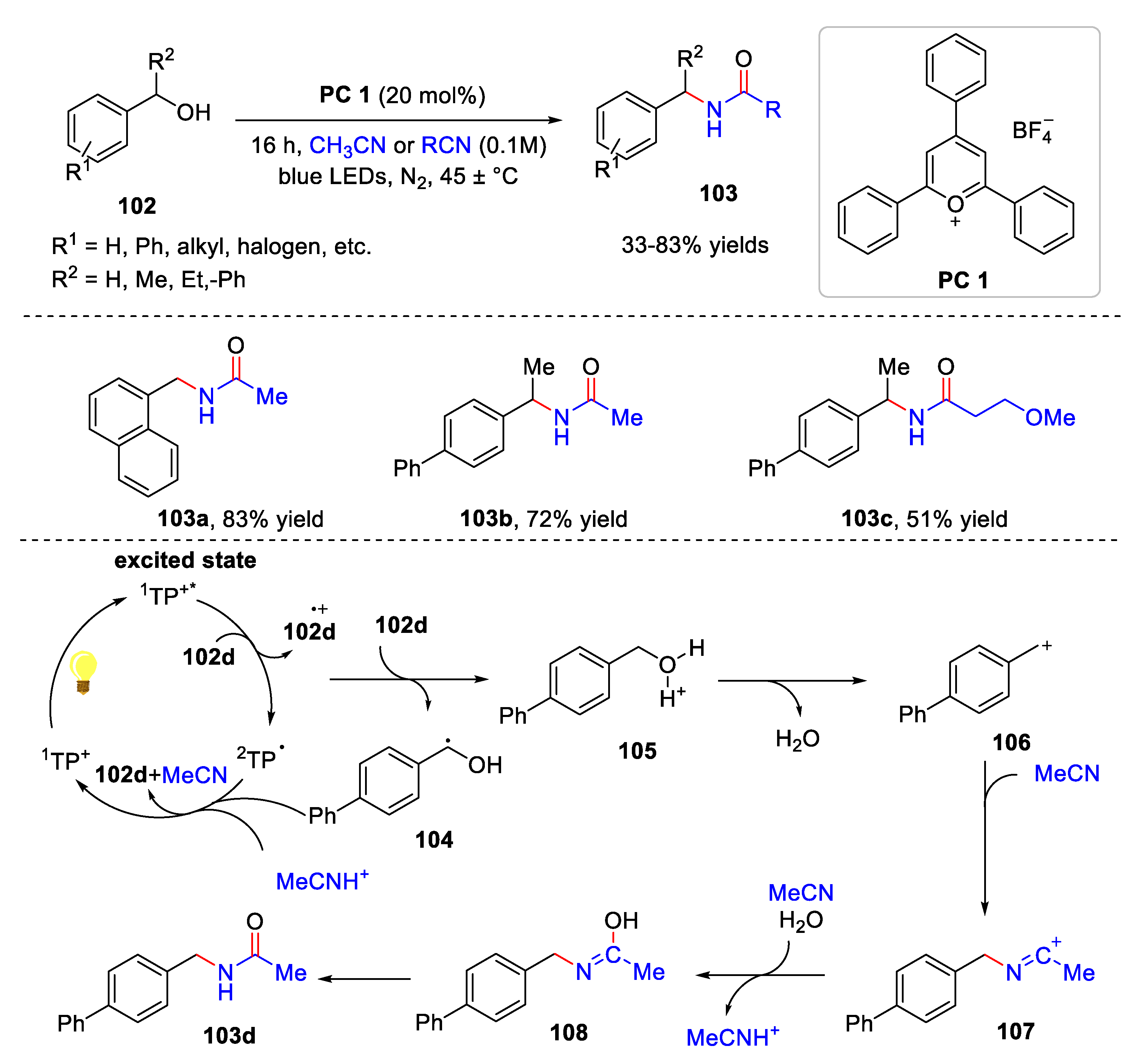
- Bao, L.; Zhang, B.-B.; Wang, Z.-X.; Chen, X.-Y. Photocatalytic dehydrations for the Ritter reaction. Org. Chem. Front. 2023, 10, 1375. [Google Scholar] [CrossRef]
























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, P.; Zhang, L.; Luo, N.; Liu, J. Advances in the Application of Acetonitrile in Organic Synthesis since 2018. Catalysts 2023, 13, 761. https://doi.org/10.3390/catal13040761
Zhong P, Zhang L, Luo N, Liu J. Advances in the Application of Acetonitrile in Organic Synthesis since 2018. Catalysts. 2023; 13(4):761. https://doi.org/10.3390/catal13040761
Chicago/Turabian StyleZhong, Pinyong, Linjun Zhang, Nianhua Luo, and Jinbiao Liu. 2023. "Advances in the Application of Acetonitrile in Organic Synthesis since 2018" Catalysts 13, no. 4: 761. https://doi.org/10.3390/catal13040761