
Advances in Catalysts for Water–Gas Shift Reaction Using Waste-Derived Synthesis Gas


Abstract
:
1. Introduction
2. Overview
2.1. Brief History of Water–Gas Shift Reaction and Catalysts
2.2. The Composition of Waste-Derived Synthesis Gas
3. Overview of Catalyst Results
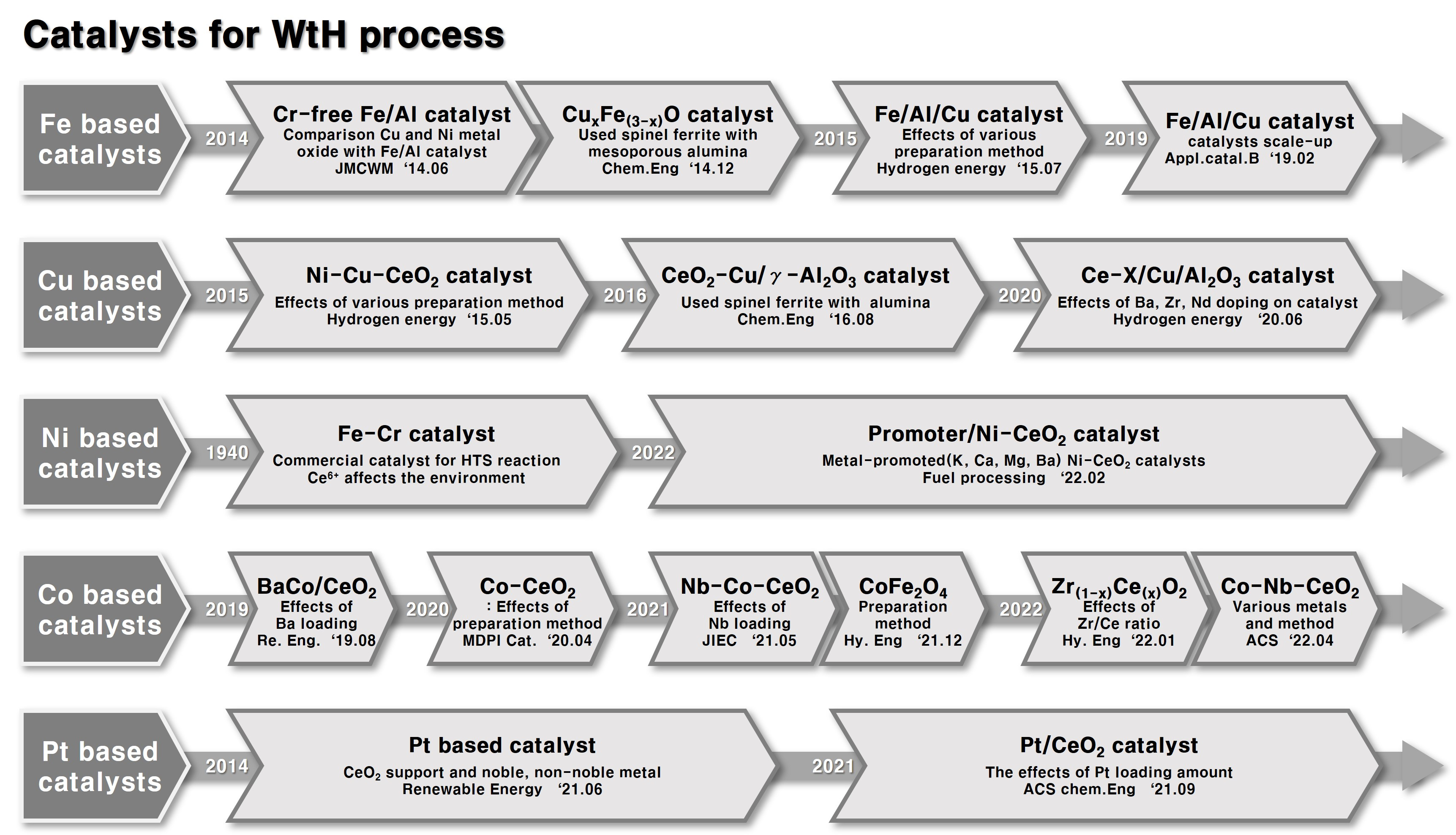
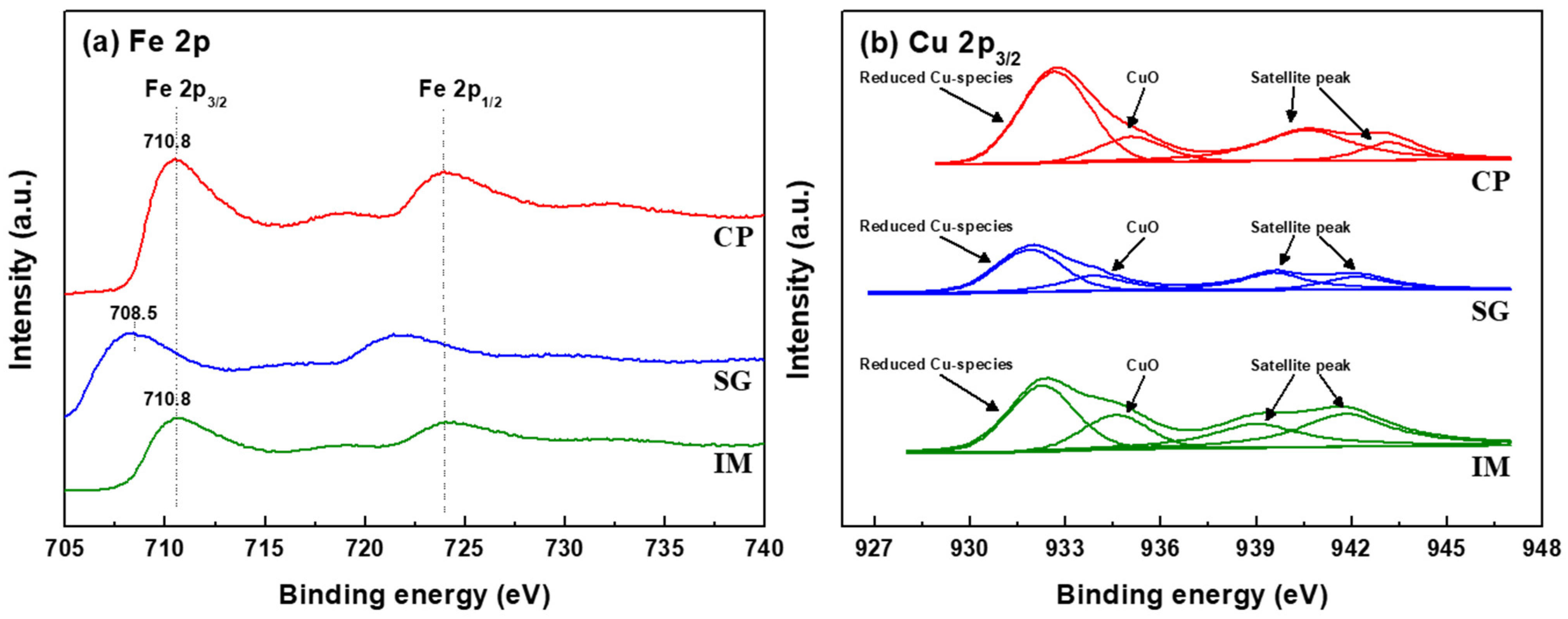
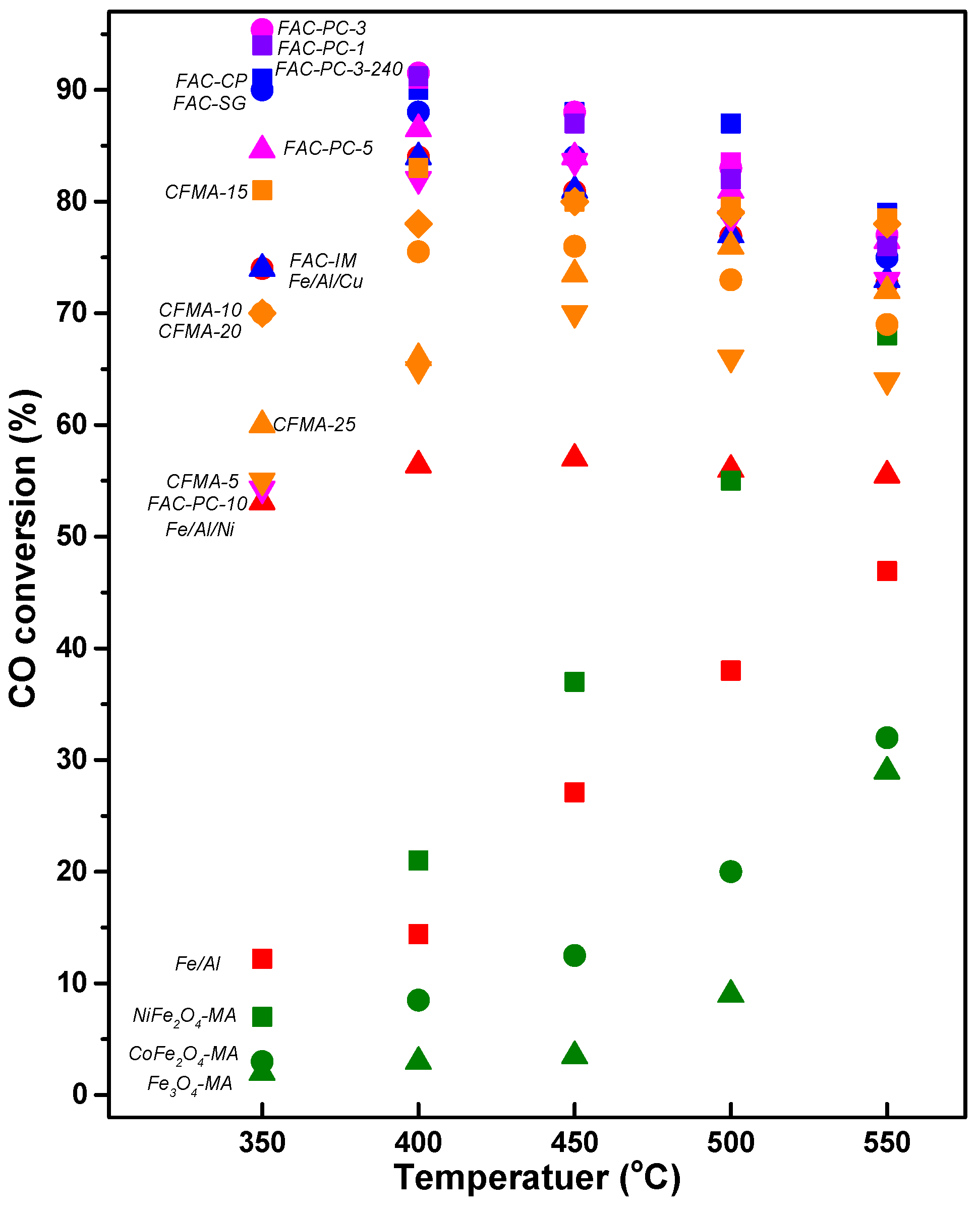
3.1. Fe-Based Catalyst (Easy Reducibility of Fe2O3)
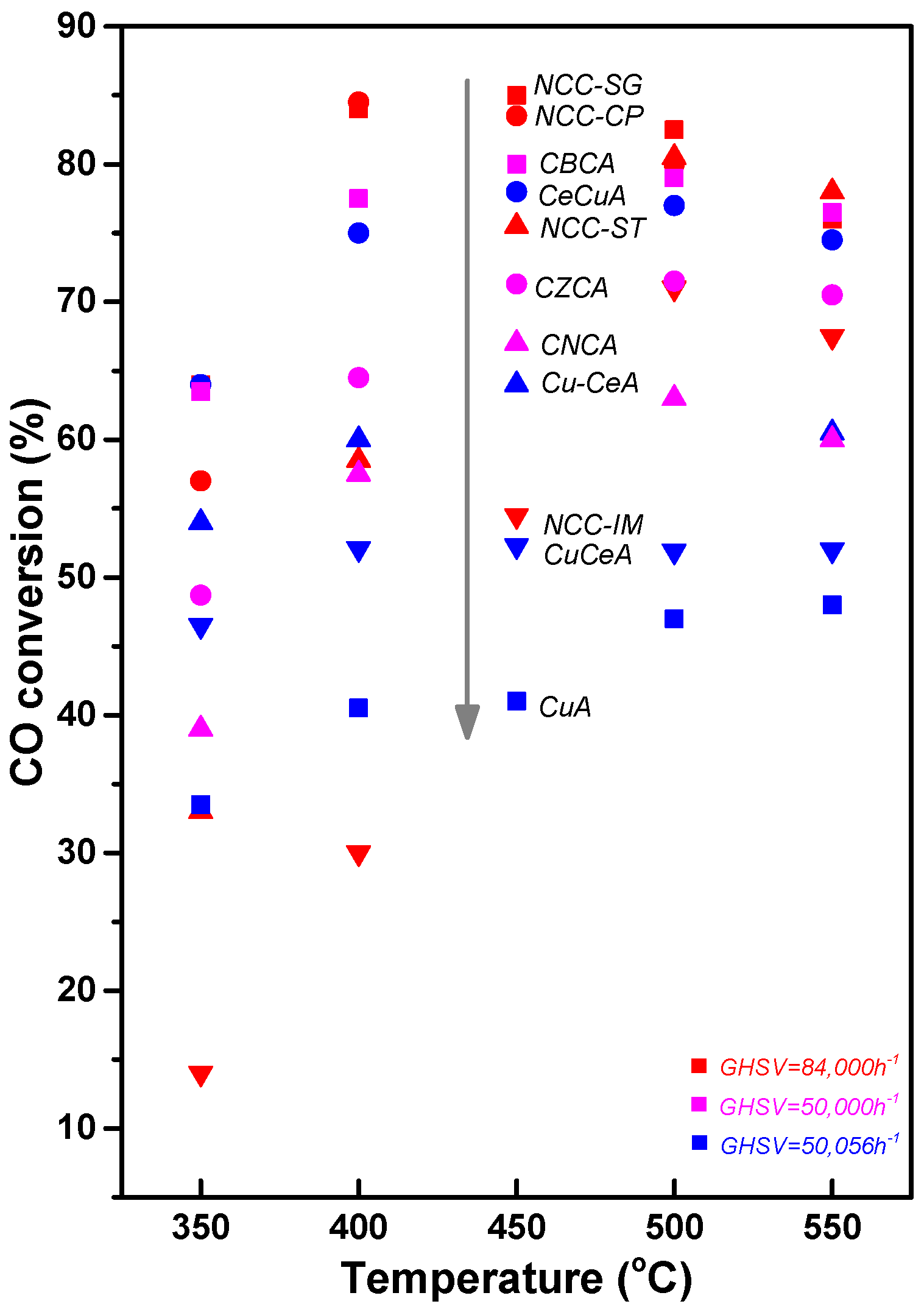
3.2. Cu-Based Catalysts (Excellent Redox Ability of Cu)
3.3. Ni-Based Catalyst (High OSC)
3.4. Co-Based Catalyst
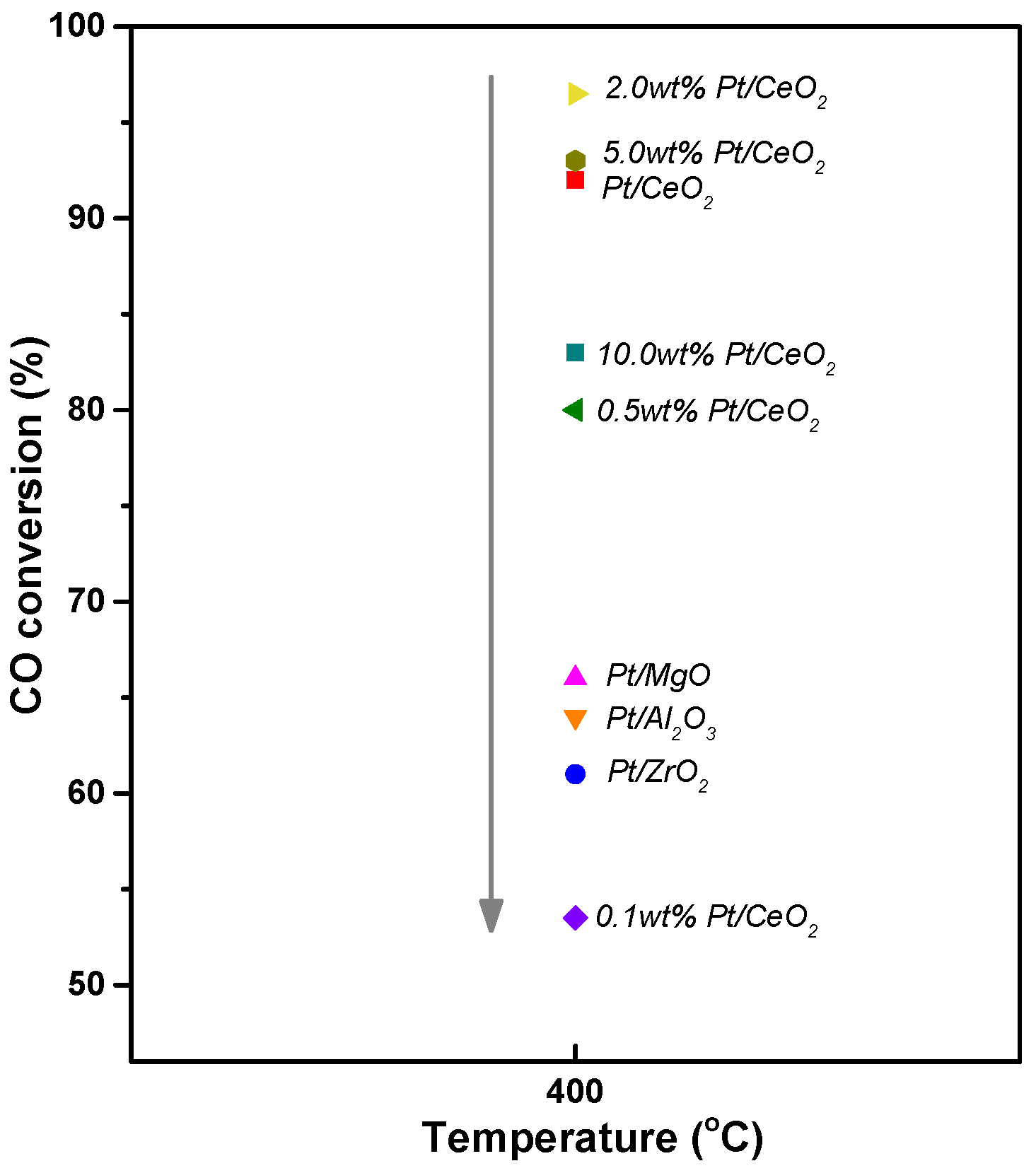
3.5. Pt-Based Catalyst
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lee, Y.L.; Kim, K.J.; Jang, W.J.; Shim, J.O.; Jeon, K.W.; Na, H.S.; Kim, H.M.; Bae, J.W.; Nam, S.C.; Jeon, B.H.; et al. Increase in stability of BaCo/CeO2 catalyst by optimizing the loading amount of Ba promoter for high-temperature water-gas shift reaction using waste-derived synthesis gas. Renew. Energy 2020, 145, 2715–2722. [Google Scholar] [CrossRef]
- Ellabban, O.; Abu-Rub, H.; Blaabjerg, F. Renewable energy resources: Current status, future prospects and their enabling technology. Renew. Sustain. Energy Rev. 2014, 39, 748–764. [Google Scholar] [CrossRef]
- Gallastegi-Villa, M.; Aranzabal, A.; González-Marcos, J.A.; González-Velasco, J.R. Tailoring dual redox-acid functionalities in VOx/TiO2/ZSM5 catalyst for simultaneous abatement of PCDD/Fs and NOx from municipal solid waste incineration. Appl. Catal. B 2017, 205, 310–318. [Google Scholar] [CrossRef]
- Jeong, D.W.; Subramanian, V.; Shim, J.O.; Jang, W.J.; Seo, Y.C.; Roh, H.S.; Gu, J.H.; Lim, Y.T. High-Temperature Water Gas Shift Reaction Over Fe/Al/Cu Oxide Based Catalysts Using Simulated Waste-Derived Synthesis Gas. Catal. Lett. 2013, 143, 438–444. [Google Scholar] [CrossRef]
- Arena, U. Process and technological aspects of municipal solid waste gasification. A review. Waste Manag. 2012, 32, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, Y.L.; Na, H.S.; Ahn, S.Y.; Shim, J.O.; Jeon, B.H.; Roh, H.S. Efficient Waste to Energy Conversion Based on Co-CeO2 Catalyzed Water-Gas Shift Reaction. Catalysts 2020, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.S.; Kim, K.J.; Cho, E.H.; Park, H.R.; Roh, H.S.; Ko, C.H. Enhanced Fe–Cr dispersion on mesoporous silica support using surfactant-assisted melt-infiltration for the water-gas shift reaction in waste-to-hydrogen processes. Int. J. Hydrogen Energy 2022, Article in press. [Google Scholar] [CrossRef]
- Choi, T.Y.; Kim, H.M.; Park, M.J.; Jeong, C.H.; Jeong, D.W. Effect of ZrO2 on the performance of Co–CeO2 catalysts for hydrogen production from waste-derived synthesis gas using high-temperature water gas shift reaction. Int. J. Hydrogen Energy 2022, 47, 14294–14303. [Google Scholar] [CrossRef]
- Abdel-Shafy, H.I.; Mansour, M.S.M. Solid waste issue: Sources, composition, disposal, recycling, and valorization. Egypt. J. Pet. 2018, 27, 1275–1290. [Google Scholar] [CrossRef]
- Vijayan, D.S.; Parthiban, D. Effect of Solid waste based stabilizing material for strengthening of Expansive soil—A review. Environ. Technol. Innov. 2020, 20, 101108. [Google Scholar] [CrossRef]
- Na, H.S.; Jeong, D.W.; Jang, W.J.; Shim, J.O.; Roh, H.S. The effect of preparation method on Fe/Al/Cu oxide-based catalyst performance for high temperature water gas shift reaction using simulated waste-derived synthesis gas. Int. J. Hydrogen Energy 2015, 40, 12268–12274. [Google Scholar] [CrossRef]
- Jeong, D.W.; Jang, W.J.; Shim, J.O.; Han, W.B.; Jeon, K.W.; Seo, Y.C.; Roh, H.S.; Gu, J.H.; Lim, Y.T. A comparison study on high-temperature water–gas shift reaction over Fe/Al/Cu and Fe/Al/Ni catalysts using simulated waste-derived synthesis gas. J. Mater. Cycles Waste Manag 2014, 16, 650–656. [Google Scholar] [CrossRef]
- Jeong, D.W.; Jha, A.; Jang, W.J.; Han, W.B.; Roh, H.S. Performance of spinel ferrite catalysts integrated with mesoporous Al2O3 in the high temperature water–gas shift reaction. Chem. Eng. J. 2015, 265, 100–109. [Google Scholar] [CrossRef]
- Jeong, D.W.; Potdar, H.S.; Roh, H.S. Comparative Study on Nano-Sized 1 wt% Pt/Ce0.8Zr0.2O2 and 1 wt% Pt/Ce0.2Zr0.8O2 Catalysts for a Single Stage Water Gas Shift Reaction. Catal. Lett. 2012, 142, 439–444. [Google Scholar] [CrossRef]
- Roh, H.S.; Jeong, D.W.; Kim, K.S.; Eum, I.H.; Koo, K.Y.; Yoon, W.L. Single Stage Water–Gas Shift Reaction Over Supported Pt Catalysts. Catal. Lett. 2011, 141, 95–99. [Google Scholar] [CrossRef]
- Newsome, D.S. The Water–Gas Shift Reaction. Catal. Rev. 1980, 21, 275–318. [Google Scholar] [CrossRef]
- Hutchings, G.J.; Copperthwaitet, R.G.; Gottschalk, F.M.; Hunter, R.; Mellor, J.; Orchard, S.W.; Sangiorgio, T. A comparative evaluation of cobalt chromium oxide, cobalt manganese oxide, and copper manganese oxide as catalysts for the water–gas shift reaction. J. Catal. 1992, 137, 408–422. [Google Scholar] [CrossRef]
- Jeong, D.W.; Potdar, H.S.; Shim, J.O.; Jang, W.J.; Roh, H.S. H2 production from a single stage water–gas shift reaction over Pt/CeO2, Pt/ZrO2, and Pt/Ce(1−x)Zr(x)O2 catalysts. Int. J. Hydrogen Energy 2013, 38, 4502–4507. [Google Scholar] [CrossRef]
- Subramanian, V.; Potdar, H.S.; Jeong, D.W.; Shim, J.O.; Jang, W.J.; Roh, H.S.; Jung, U.H.; Yoon, W.L. Synthesis of a Novel Nano-Sized Pt/ZnO Catalyst for Water Gas Shift Reaction in Medium Temperature Application. Catal. Lett. 2012, 142, 1075–1081. [Google Scholar] [CrossRef]
- Thomas, J.M.; Thomas, W.J. Principles and Practice of Heterogeneous Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 1997. [Google Scholar]
- Jeong, D.W.; Potdar, H.S.; Kim, K.S.; Roh, H.S. The effect of sodium in activity enhancement of nano-sized Pt/CeO2 catalyst for water gas shift reaction at low temperature. Bull. Korean Chem. Soc. 2011, 32, 3557–3558. [Google Scholar] [CrossRef] [Green Version]
- Jeong, D.W.; Jang, W.J.; Shim, J.O.; Han, W.B.; Roh, H.S.; Jung, U.H.; Yoon, W.L. Low-temperature water–gas shift reaction over supported Cu catalysts. Renew. Energy 2014, 65, 102–107. [Google Scholar] [CrossRef]
- Lee, D.W.; Lee, M.S.; Lee, J.Y.; Kim, S.M.; Eom, H.J.; Moon, D.J.; Lee, K.Y. Hydrogen production by the water-gas shift reaction using CuNi/Fe2O3 catalyst. Catal. Sci. Technol. 2015, 5, 2752. [Google Scholar] [CrossRef]
- Jeong, D.W.; Na, H.S.; Shim, J.O.; Jang, W.J.; Roh, H.S.; Jung, U.H.; Yoon, W.L. Hydrogen production from low temperature WGS reaction on co-precipitated Cu–CeO2 catalysts: An optimization of Cu loading. Int. J. Hydrogen Energy 2014, 39, 9135–9142. [Google Scholar] [CrossRef]
- Li, L.; Song, L.; Chen, C.; Zhang, Y.; Zhan, Y.; Lin, X.; Zheng, Q.; Wang, H.; Ma, H.; Ding, L.; et al. Modified precipitation processes and optimized copper content of CuO–CeO2 catalysts for water–gas shift reaction. Int. J. Hydrogen Energy 2014, 39, 19570–19582. [Google Scholar] [CrossRef]
- Jha, A.; Jeong, D.W.; Jang, W.J.; Lee, Y.L.; Roh, H.S. Hydrogen production from water–gas shift reaction over Ni–Cu–CeO2 oxide catalyst: The effect of preparation methods. Int. J. Hydrogen Energy 2015, 40, 9209–9216. [Google Scholar] [CrossRef]
- Shim, J.O.; Na, H.S.; Jha, A.; Jang, W.J.; Jeong, D.W.; Nah, I.W.; Jeon, B.H.; Roh, H.S. Effect of preparation method on the oxygen vacancy concentration of CeO2-promoted Cu/γ-Al2O3 catalysts for HTS reactions. Chem. Eng. J. 2016, 306, 908–915. [Google Scholar] [CrossRef]
- Jang, W.J.; Shim, J.O.; Jeon, K.W.; Na, H.S.; Kim, H.M.; Lee, Y.L.; Roh, H.S.; Jeong, D.W. Design and scale-up of a Cr-free Fe-Al-Cu catalyst for hydrogen production from waste-derived synthesis gas. Appl. Catal. B 2019, 249, 72–81. [Google Scholar] [CrossRef]
- Grevatt, P.C. Toxicological Review of Hexavalent Chromium. In Summary Information on the Integrated Risk Information System; Environmental Protection Agency: Washington, DC, USA, 1998. [Google Scholar]
- Del Rey, I.; Ayuso, J.; Galvín, A.P.; Jiménez, J.R.; López, M.; García-Garrido, M.L. Analysis of chromium and sulphate origins in construction recycled materials based on leaching test results. Waste Manag. 2015, 46, 278–286. [Google Scholar] [CrossRef]
- Twigg, M.V. Catalyst Handbook; Wolfe Scientific Books; Routledge: New York, NY, USA, 1989. [Google Scholar] [CrossRef]
- Reddy, G.K.; Boolchand, P.; Smirniotis, P.G. Unexpected Behavior of Copper in Modified Ferrites during High Temperature WGS Reaction—Aspects of Fe3+ ↔ Fe2+ Redox Chemistry from Mössbauer and XPS Studies. J. Phys. Chem. C. 2012, 116, 11019–11031. [Google Scholar] [CrossRef]
- Martos, C.; Dufour, J.; Ruiz, A. Synthesis of Fe3O4-based catalysts for the high-temperature water gas shift reaction. Int. J. Hydrogen Energy 2009, 34, 4475–4481. [Google Scholar] [CrossRef]
- Natesakhawat, S.; Wang, X.; Zhang, L.; Ozkan, U.S. Development of chromium-free iron-based catalysts for high-temperature water-gas shift reaction. J. Mol. Catal. A Chem. 2006, 260, 82–94. [Google Scholar] [CrossRef]
- Meshkani, F.; Rezaei, M. Preparation of mesoporous nanocrystalline iron-based catalysts for high temperature water gas shift reaction: Effect of preparation factors. Chem. Eng. J. 2015, 260, 107–116. [Google Scholar] [CrossRef]
- Zhu, M.; Rocha, T.C.R.; Lunkenbein, T.; Knop-Gericke, A.; Schlögl, R.; Wachs, I.E. Promotion Mechanisms of Iron Oxide-Based High Temperature Water–Gas Shift Catalysts by Chromium and Copper. ACS Catal. 2016, 6, 4455–4464. [Google Scholar] [CrossRef]
- Zhu, M.; Wachs, I.E. Iron-Based Catalysts for the High-Temperature Water–Gas Shift (HT-WGS) Reaction: A Review. ACS Catal. 2016, 6, 722–732. [Google Scholar] [CrossRef]
- Keturakis, C.J.; Zhu, M.; Gibson, E.K.; Daturi, M.; Tao, F.; Frenkel, A.I.; Wachs, I.E. Dynamics of CrO3–Fe2O3 Catalysts during the High-Temperature Water-Gas Shift Reaction: Molecular Structures and Reactivity. ACS Catal. 2016, 6, 4786–4798. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Yalçın, Ö.; Wachs, I.E. Revealing structure-activity relationships in chromium free high temperature shift catalysts promoted by earth abundant elements. Appl. Catal. B 2018, 232, 205–212. [Google Scholar] [CrossRef]
- Sun, Y.; Hla, S.S.; Duffy, G.J.; Cousins, A.J.; French, D.; Morpeth, L.D.; Edwards, J.H.; Roberts, D.G. Effect of Ce on the structural features and catalytic properties of La(0.9−x)CexFeO3 perovskite-like catalysts for the high temperature water–gas shift reaction. Int. J. Hydrogen Energy 2011, 36, 79–86. [Google Scholar] [CrossRef]
- Jha, A.; Jeong, D.W.; Lee, Y.L.; Jang, W.J.; Shim, J.O.; Jeon, K.W.; Rode, C.V.; Roh, H.S. Chromium free high temperature water–gas shift catalyst for the production of hydrogen from waste derived synthesis gas. Appl. Catal. A Gen. 2016, 522, 21–31. [Google Scholar] [CrossRef]
- Jeong, D.W.; Jang, W.J.; Jha, A.; Han, W.B.; Jeon, K.W.; Kim, S.H.; Roh, H.S. The Effect of Metal on Catalytic Performance over MFe2O4 Catalysts for High Temperature Water-Gas Shift Reaction. J. Nanoelectron. Optoe. 2015, 10, 530–534. [Google Scholar] [CrossRef]
- Lang, C.; Sécordel, X.; Kiennemann, A.; Courson, C. Water gas shift catalysts for hydrogen production from biomass steam gasification. Fuel Process. Technol. 2017, 156, 246–252. [Google Scholar] [CrossRef]
- Lee, D.W.; Lee, M.S.; Lee, J.Y.; Kim, S.M.; Eom, H.J.; Moon, D.J.; Lee, K.Y. The review of Cr-free Fe-based catalysts for high-temperature water-gas shift reactions. Catal. Today 2013, 210, 2–9. [Google Scholar] [CrossRef]
- Park, P.W.; Ledford, J.S. The influence of surface structure on the catalytic activity of cerium promoted copper oxide catalysts on alumina: Oxidation of carbon monoxide and methane. Catal. Lett. 1998, 50, 41–48. [Google Scholar] [CrossRef]
- Reina, T.R.; Ivanova, S.; Delgado, J.J.; Ivanov, I.; Idakiev, V.; Tabakova, T.; Centeno, M.A.; Odriozola, J.A. Viability of Au/CeO2–ZnO/Al2O3 Catalysts for Pure Hydrogen Production by the Water–Gas Shift Reaction. ChemCatChem 2014, 6, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Shiau, C.Y.; Ma, M.W.; Chuang, C.S. CO oxidation over CeO2-promoted Cu/γ-Al2O3 catalyst: Effect of preparation method. Appl. Catal. A Gen 2006, 302, 89–95. [Google Scholar] [CrossRef]
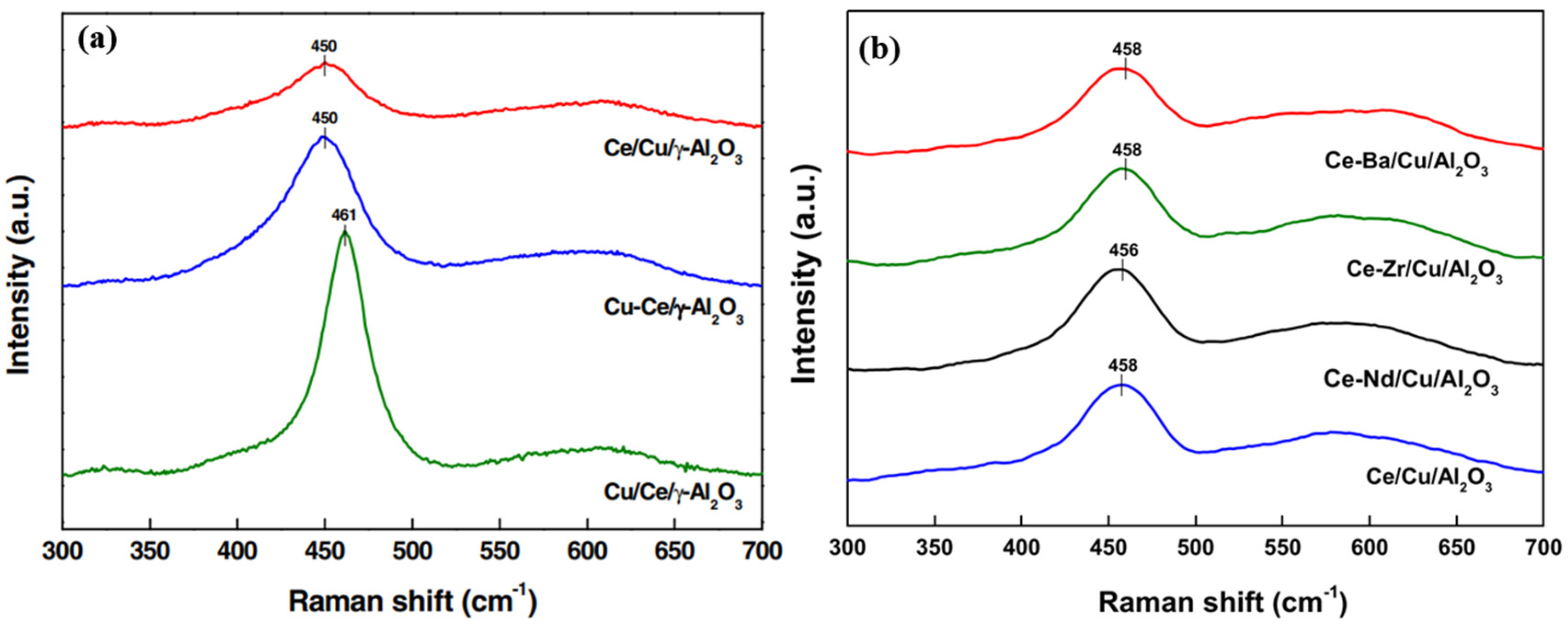
- Na, H.S.; Ahn, S.Y.; Lee, Y.L.; Kim, K.J.; Jang, W.J.; Shim, J.O.; Roh, H.S. The role of additives (Ba, Zr, and Nd) on Ce/Cu/Al2O3 catalyst for water-gas shift reaction. Int. J. Hydrogen Energy 2020, 45, 24726–24737. [Google Scholar] [CrossRef]
- Liotta, L.F.; Ousmane, M.; Carlo, G.D.; Pantaleo, G.; Deganello, G.; Marcì, G.; Retailleau, L.; Giroir-Fendler, A. Total oxidation of propene at low temperature over Co3O4–CeO2 mixed oxides: Role of surface oxygen vacancies and bulk oxygen mobility in the catalytic activity. Appl. Catal. A Gen. 2008, 347, 81–88. [Google Scholar] [CrossRef] [Green Version]
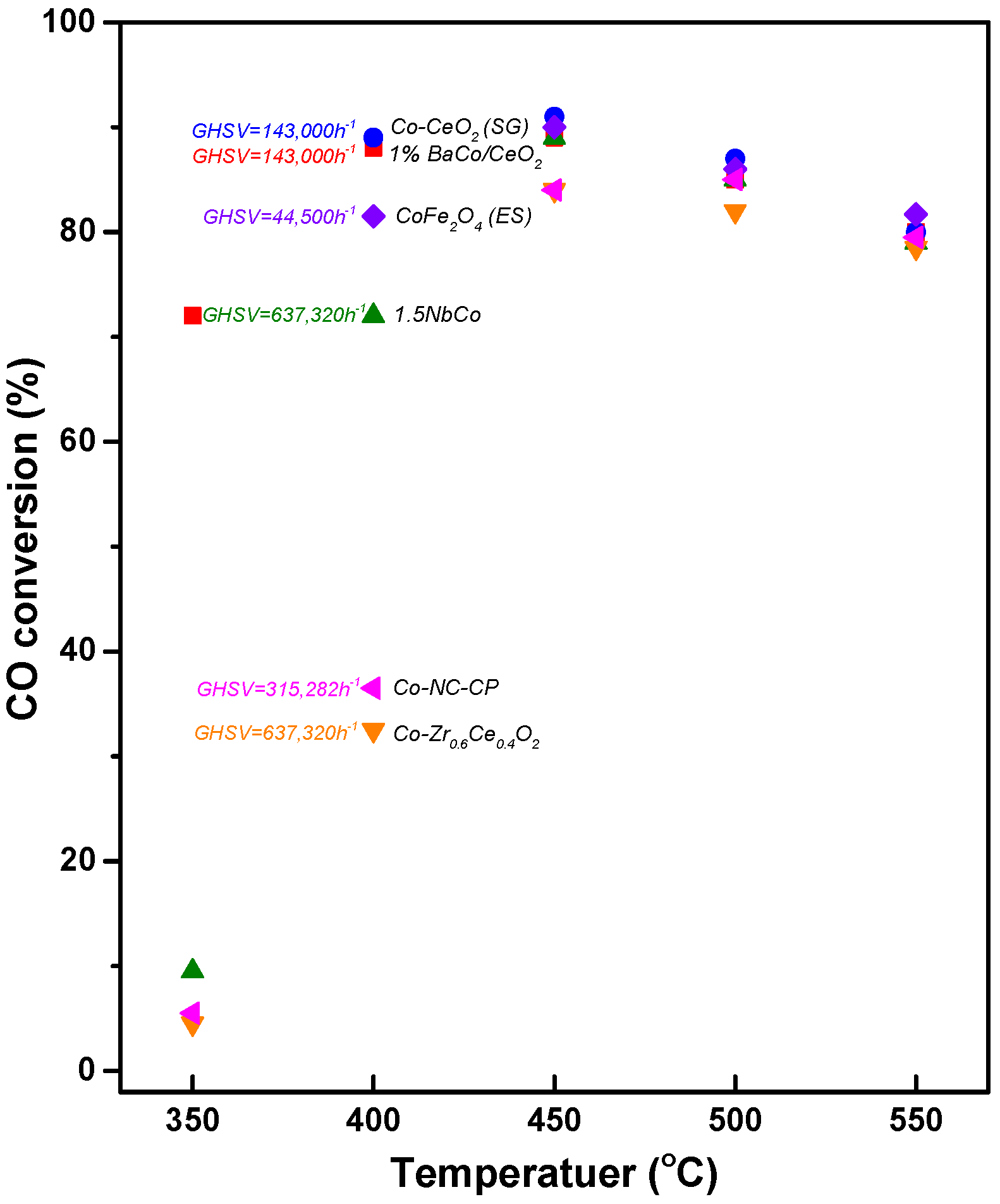
- Lee, Y.L.; Jha, A.; Jang, W.J.; Shim, J.O.; Rode, C.V.; Jeon, B.H.; Bae, J.W.; Roh, H.S. Effect of alkali and alkaline earth metal on Co/CeO2 catalyst for the water-gas shift reaction of waste derived synthesis gas. Appl. Catal. A Gen. 2018, 551, 63–70. [Google Scholar] [CrossRef]
- Jha, A.; Jeong, D.W.; Lee, Y.L.; Nah, I.W.; Roh, H.S. Enhancing the catalytic performance of cobalt oxide by doping on ceria in the high temperature water–gas shift reaction. RSC Adv. 2015, 5, 103023–103029. [Google Scholar] [CrossRef]
- Jeong, C.H.; Jeon, K.W.; Byeon, H.J.; Choi, T.Y.; Kim, H.M.; Jeong, D.W. Effects of niobium addition on active metal and support in Co–CeO2 catalyst for the high temperature water gas shift reaction. J. Ind. Eng. Chem 2021, 100, 149–158. [Google Scholar] [CrossRef]
- Silva, L.P.C.; Terra, L.E.; Coutinho, A.C.S.L.S.; Passos, F.B. Sour water–gas shift reaction over Pt/CeZrO2 catalysts. J. Catal. 2016, 341, 1–12. [Google Scholar] [CrossRef]
- Vaezi, M.; Passandideh-Fard, M.; Moghiman, M.; Charmchi, M. Gasification of heavy fuel oils: A thermochemical equilibrium approach. Fuel 2011, 90, 878–885. [Google Scholar] [CrossRef]
- Breault, R.W. Gasification Processes Old and New: A Basic Review of the Major Technologies. Energies 2010, 3, 216–240. [Google Scholar] [CrossRef] [Green Version]
- Bhadra, B.N.; Jhung, S.H. Oxidative desulfurization and denitrogenation of fuels using metal-organic framework-based/-derived catalysts. Appl. Catal. B 2019, 259, 118021. [Google Scholar] [CrossRef]
- Lee, Y.L.; Kim, K.J.; Hong, G.R.; Ahn, S.Y.; Kim, B.J.; Park, H.R.; Yun, S.J.; Bae, J.W.; Jeon, B.H.; Roh, H.S. Sulfur-Tolerant Pt/CeO2 Catalyst with Enhanced Oxygen Storage Capacity by Controlling the Pt Content for the Waste-to-Hydrogen Processes. ACS Sustain. Chem. Eng. 2021, 9, 15287–15293. [Google Scholar] [CrossRef]
- Hulteberg, C. Sulphur-tolerant catalysts in small-scale hydrogen production, a review. Int. J. Hydrogen Energy 2012, 37, 3978–3992. [Google Scholar] [CrossRef]
- Lee, Y.L.; Kim, K.J.; Hong, G.R.; Ahn, S.Y.; Kim, B.J.; Shim, J.O.; Roh, H.S. Highly sulfur tolerant and regenerable Pt/CeO2 catalyst for waste to energy. Renew. Energy 2021, 178, 334–343. [Google Scholar] [CrossRef]
- Hiller, H.; Reimert, R.; Stönner, H.M. Gas Production, 1. Introduction. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar] [CrossRef]
- Tamaru, K. The History of the Development of Ammonia Synthesis. In Catalytic Ammonia Synthesis; Springer: New York, NY, USA, 1991; pp. 1–18. [Google Scholar] [CrossRef]
- Ambrosi, A.; Denmark, S.E. Harnessing the Power of the Water-Gas Shift Reaction for Organic Synthesis. Angew. Chem. Int. Ed. 2016, 55, 12164–12189. [Google Scholar] [CrossRef]
- Smith, C.; Hill, A.K.; Torrente-Murciano, L. Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13, 331–344. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. A review of recent advances in water-gas shift catalysis for hydrogen production. Emergent. Mater. 2020, 3, 881–917. [Google Scholar] [CrossRef]
- Gradisher, L.; Dutcher, B.; Fan, M. Catalytic hydrogen production from fossil fuels via the water gas shift reaction. Appl. Energy 2015, 139, 335–349. [Google Scholar] [CrossRef]
- Boretti, A.; Banik, B.K. Advances in Hydrogen Production from Natural Gas Reforming. Adv. Energy. Sustain. Res. 2021, 2, 2100097. [Google Scholar] [CrossRef]
- Turner, J.A. Sustainable Hydrogen Production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Franchi, G.; Capocelli, M.; De Falco, M.; Piemonte, V.; Barba, D. Hydrogen Production via Steam Reforming: A Critical Analysis of MR and RMM Technologies. Membranes 2020, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Ertl, G.; Knözinger, H.; Weitkamp, J. Handbook of Heterogeneous Catalysis; Wiley VCH: Weinheim, Germany, 1997. [Google Scholar]
- Subramanian, V.; Jeong, D.W.; Han, W.B.; Jang, W.J.; Shim, J.O.; Roh, H.S. H2 production from high temperature shift of the simulated waste derived synthesis gas over magnetite catalysts prepared by citric acid assisted direct synthesis method. Int. J. Hydrogen Energy 2013, 38, 8699–8703. [Google Scholar] [CrossRef]
- Ariëns, M.I.; Chlan, V.; Novák, P.; van de Water, L.G.A.; Dugulan, A.I.; Brück, E.; Hensen, E.J.M. The role of chromium in iron-based high-temperature water-gas shift catalysts under industrial conditions. Appl. Catal. B 2021, 297, 120465. [Google Scholar] [CrossRef]
- Mendes, D.; Mendes, A.; Madeira, L.M.; Iulianelli, A.; Sousa, J.M.; Basile, A. The water-gas shift reaction: From conventional catalytic systems to Pd-based membrane reactors—A review. Asia-Pac. J. Chem. Eng. 2009, 5, 111–137. [Google Scholar] [CrossRef]
- Li, J.L.; Inui, T. Characterization of precursors of methanol synthesis catalysts, copper/zinc/aluminum oxides, precipitated at different pHs and temperatures. Appl. Catal. A Gen. 1996, 137, 105–117. [Google Scholar] [CrossRef]
- Shim, J.O.; Na, H.S.; Ahn, S.Y.; Jeon, K.W.; Jang, W.J.; Jeon, B.H.; Roh, H.S. An important parameter for synthesis of Al2O3 supported Cu-Zn catalysts in low-temperature water-gas shift reaction under practical reaction condition. Int. J. Hydrogen Energy 2019, 44, 14853–14860. [Google Scholar] [CrossRef]
- Kondrat, S.A.; Smith, P.J.; Lu, L.; Bartley, J.K.; Taylor, S.H.; Spencer, M.S.; Kelly, G.J.; Park, C.W.; Kiely, C.J.; Hutchings, G.J. Preparation of a highly active ternary Cu-Zn-Al oxide methanol synthesis catalyst by supercritical CO2 anti-solvent precipitation. Catal. Today 2013, 317, 12–20. [Google Scholar] [CrossRef]
- Smith RJ, B.; Loganathan, M.; Shantha, M.S. A Review of the Water Gas Shift Reaction Kinetics. Int. J. Chem. React. Eng. 2010, 8. [Google Scholar] [CrossRef]
- Etim, U.J.; Song, Y.; Zhong, Z. Improving the Cu/ZnO-Based Catalysts for Carbon Dioxide Hydrogenation to Methanol, and the Use of Methanol as a Renewable Energy Storage Media. Front. Energy Res. 2020, 8, 545431. [Google Scholar] [CrossRef]
- Lucarelli, C.; Molinari, C.; Faure, R.; Fornasari, G.; Gary, D.; Schiaroli, N.; Vaccari, A. Novel Cu-Zn-Al catalysts obtained from hydrotalcite-type precursors for middle-temperature water-gas shift applications. Appl. Clay Sci. 2018, 155, 103–110. [Google Scholar] [CrossRef]
- Houston, R.; Labbé, N.; Hayes, D.; Daw, C.S.; Abdoulmoumine, N. Intermediate Temperature Water-Gas Shift Kinetics for Hydrogen Production. React. Chem. Eng. 2019, 4, 1814–1822. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Gonzalez, I.D.; Navarro, R.M.; Fierro, J.L.G.; Efstathiou, A.M. Effects of Reaction Temperature and Support Composition on the Mechanism of Water–Gas Shift Reaction over Supported-Pt Catalysts. J. Phys. Chem. C 2011, 115, 11595–11610. [Google Scholar] [CrossRef]
- Mierczynski, P.; Maniukiewicz, W.; Maniecki, T.P. Comparative studies of Pd, Ru, Ni, Cu/ZnAl2O4 catalysts for the water gas shift reaction. Cent. Eur. J. Chem. 2013, 11, 912–919. [Google Scholar] [CrossRef]
- Alptekin, F.M.; Celiktas, M.S. Review on Catalytic Biomass Gasification for Hydrogen Production as a Sustainable Energy Form and Social, Technological, Economic, Environmental, and Political Analysis of Catalysts. ACS Omega 2022, 7, 24918–24941. [Google Scholar] [CrossRef] [PubMed]
- Bukur, D.B.; Todic, B.; Elbashir, N. Role of water-gas-shift reaction in Fischer–Tropsch synthesis on iron catalysts: A review. Catal. Today 2016, 275, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Eloffy, M.G.; Elgarahy, A.M.; Saber, A.N.; Hammad, A.; El-Sherif, D.M.; Shehata, M.; Mohsen, A.; Elwakeel, K.Z. Biomass-to-sustainable biohydrogen: Insights into the production routes, and technical challenges. Chem. Eng. J. Adv. 2022, 12, 100410. [Google Scholar] [CrossRef]
- Shin, J.E. Hydrogen Technology Development and Policy Status by Value Chain in South Korea. Energies 2022, 15, 8983. [Google Scholar] [CrossRef]
- Lee, D.; Kim, K. Research and Development Investment and Collaboration Framework for the Hydrogen Economy in South Korea. Sustainability 2021, 13, 10686. [Google Scholar] [CrossRef]
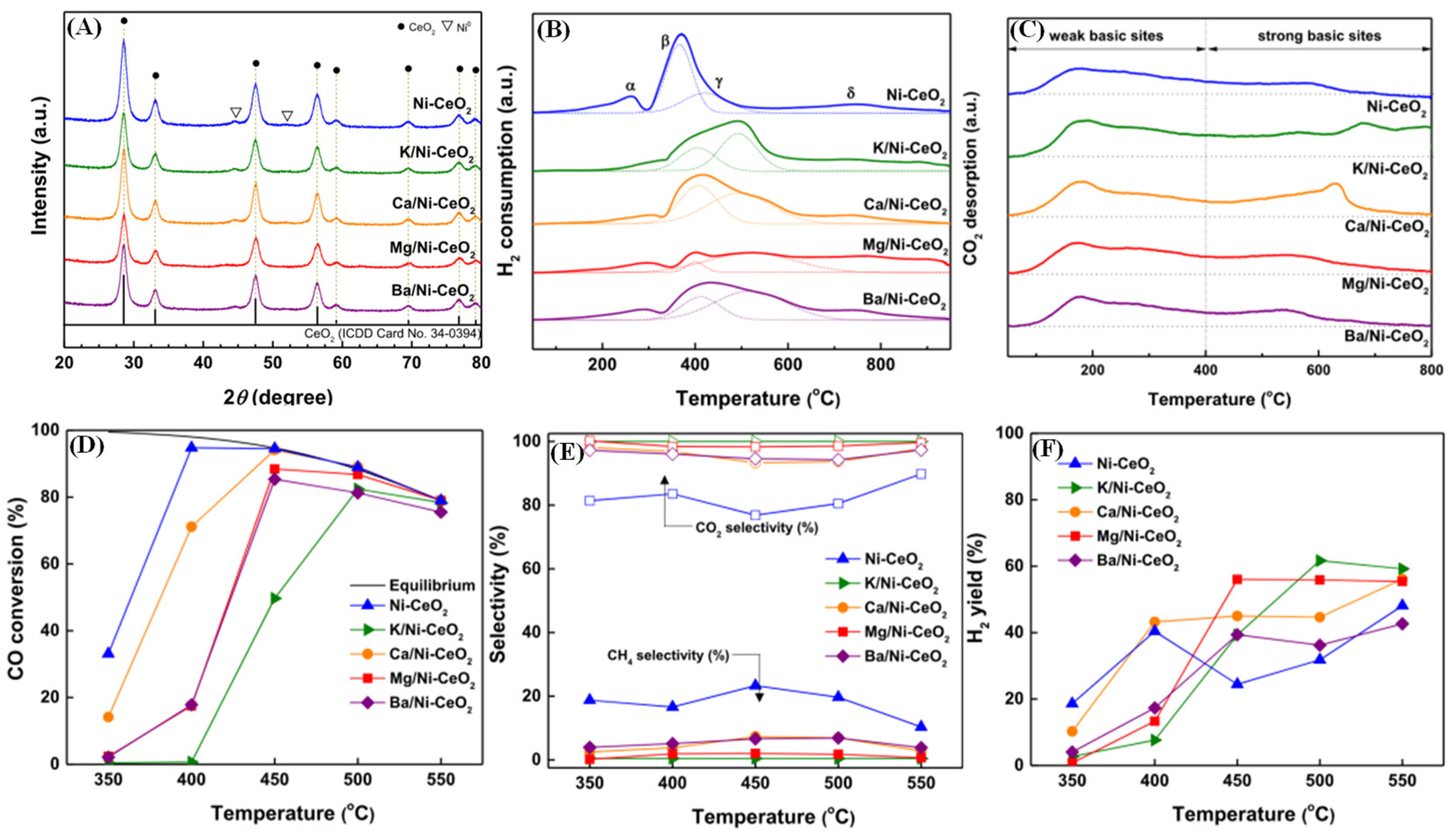
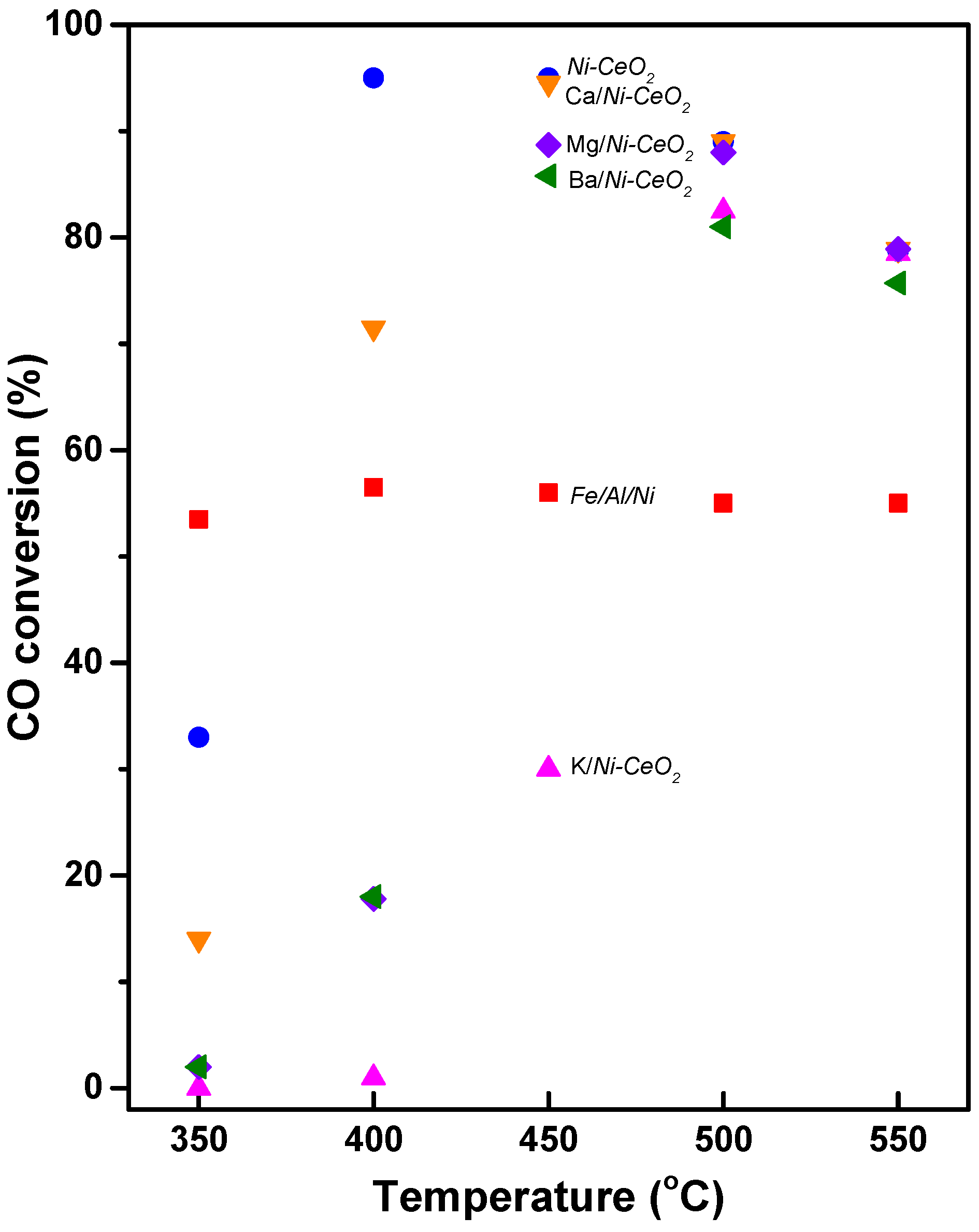
- Byeon, H.J.; Jeon, K.W.; Kim, H.M.; Lee, Y.H.; Heo, Y.S.; Park, M.J.; Jeong, D.W. Promotion of methanation suppression by alkali and alkaline earth metals in Ni-CeO2 catalysts for water–gas shift reaction using waste-derived synthesis gas. Fuel Process. Technol. 2022, 231, 107229. [Google Scholar] [CrossRef]
- Lee, Y.L.; Kim, H.Y.; Kim, K.J.; Hong, G.R.; Shim, J.O.; Ju, Y.W.; Roh, H.S. Nanofiber structured oxygen defective CoFe2O4-x catalyst for the water-gas shift reaction in waste-to-hydrogen processes. Int. J. Hydrogen Energy 2022, 47, 30950–30958. [Google Scholar] [CrossRef]
- Jeon, K.W.; Byeon, H.J.; Kim, H.M.; Choe, B.; Jeong, C.H.; Choi, T.Y.; Won, W.; Jeong, D.W. Development of Co–Nb–CeO2 Catalyst for Hydrogen Production from Waste-Derived Synthesis Gas Using Techno-Economic and Environmental Assessment. ACS Sustain. Chem. Eng. 2022, 10, 6289–6303. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Kim, K.J.; Kim, B.J.; Shim, J.O.; Jang, W.J.; Roh, H.S. Unravelling the active sites and structure-activity relationship on Cu–ZnO–Al2O3 based catalysts for water-gas shift reaction. Appl. Catal. B Environ. 2023, 325, 122320. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Jang, W.J.; Shim, J.O.; Jeon, B.H.; Roh, H.S. CeO2-based oxygen storage capacity materials in environmental and energy catalysis for carbon neutrality: Extended application and key catalytic properties. Catal. Rev. 2023, 1–84. [Google Scholar] [CrossRef]
- Lee, Y.L.; Kim, K.J.; Hong, G.R.; Roh, H.S. Target-oriented water–gas shift reactions with customized reaction conditions and catalysts. Chem. Eng. J. 2023, 458, 141422. [Google Scholar] [CrossRef]
- Valizadeh, S.; Hakimian, H.; Farooq, A.; Jeon, B.H.; Chen, W.H.; Lee, S.H.; Jung, S.C.; Seo, M.W.; Park, Y.K. Valorization of biomass through gasification for green hydrogen generation: A comprehensive review. Bioresour. Technol. 2022, 365, 128143. [Google Scholar] [CrossRef] [PubMed]
- Valizadeh, S.; Jang, S.H.; Rhee, G.H.; Lee, J.; Show, P.L.; Khan, M.A.; Jeon, B.H.; Lin, K.Y.A.; Ko, C.H.; Chen, W.H.; et al. Biohydrogen production from furniture waste via catalytic gasification in air over Ni-loaded Ultra-stable Y-type zeolite. Chem. Eng. J. 2022, 433, 133793. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | CO (vol%) | CO2 (vol%) | CH4 (vol%) | H2 (vol%) | N2 (vol%) | H2O (vol%) | Ref. |
|---|---|---|---|---|---|---|---|
| Fe-based | 38.2 | 21.5 | 2.3 | 29.2 | 8.8 | - | [11,12,28] |
| 17.02 | 9.55 | 1.03 | 13.14 | 4.06 | 55.20 | [13] | |
| 37.99 | 21.32 | 2.30 | 29.33 | 9.06 | - | [13] | |
| Cu-based | 38.0 | 21.3 | 2.3 | 29.3 | 9.1 | - | [26,27] |
| 37.99 | 21.28 | 2.31 | 29.34 | 9.08 | - | [48] | |
| Co-based | 38.2 | 21.5 | 2.3 | 29.2 | 8.8 | - | [1] |
| 17.02 | 9.55 | 1.03 | 13.14 | 4.06 | 55.20 | [6,8,53] | |
| 37.99 | 21.32 | 2.30 | 29.33 | 9.06 | - | [6,8,53] | |
| 39.70 | 21.50 | 2.35 | 27.05 | 9.40 | - | [88] | |
| 37.87 | 21.47 | 2.30 | 29.31 | 9.05 | - | [89] | |
| Pt-based | 37.99 | 21.28 | 2.31 | 29.34 | 9.08 | - | [59] |
| 39.70 | 21.50 | 2.35 | 27.05 | 9.40 | - | [57] | |
| Ni-based | 37.87 | 21.47 | 2.30 | 29.31 | 9.05 | - | [87] |
| Catalyst | BET Surface Area (m2/g) a | Crystallite Size (nm) b | Ref. | ||
|---|---|---|---|---|---|
| Fresh | Used | Fresh (Fe2O3) | Used (Fe3O4) | ||
| Fe/Al | 56.6 | 12.5 | 18.4 | 24.4 | [12] |
| Fe/Al/Cu | 73.0 | 20.3 | 17.3 | 17.9 | [12] |
| Fe/Al/Ni | 81.4 | 11.2 | 14.9 | 20.6 | [12] |
| FAC-CP | 165.1 | 32.6 | N.A. c | 18.1 | [11] |
| FAC-SG | 104.9 | 20.1 | - | 23.0 | [11] |
| FAC-IM | 73.0 | 15.5 | 17.3 | 27.4 | [11] |
| FAC-PC-1 (Cu dispersion: 5.9%) | 168 | - | - | 13.4 | [28] |
| FAC-PC-3 (Cu dispersion: 5.7%) | 165 | - | - | 13.7 | [28] |
| FAC-PC-5 (Cu dispersion: 4.7%) | 132 | - | - | 16.6 | [28] |
| FAC-PC-10 (Cu dispersion: 2.8%) | 60 | - | - | 22.3 | [28] |
| FAC-PC-3-240 (Cu dispersion: 5.6%) | 166 | - | - | 15.9 | [28] |
| Catalyst | CuO to Cu0 (°C) | Fe2O3 to Fe3O4 (°C) | Fe3O4 to FeO (°C) | Ref. |
|---|---|---|---|---|
| Fe/Al | - | 381 | 580 | [12] |
| Fe/Al/Cu | 178 | 325 | 660 | |
| Fe/Al/Ni | - | 380 | 580 (560 Ni) | |
| FAC-CP | 154 | 177 | 600~700 | [11] |
| FAC-SG | 166 | 203 | 600~700 | |
| FAC-IM | 178 | 325 | 660 | |
| Bare Fe | - | 405 | 700 | [28] |
| FAC-PC-1 | 126 | 155 | - | [28] |
| FAC-PC-3 | 127 | 159 | - | |
| FAC-PC-5 | 153 | 185 | - | |
| FAC-PC-10 | 205 | 253 | - | |
| FAC-PC-3-240 | 128 | 158 | - |
| Catalyst | BET Surface Area (m2/g) | Crystallite Size (nm) | Crystallite Size of Metallic Cu (nm) b | ||
|---|---|---|---|---|---|
| Fresh | Used | Fresh a | Used a | ||
| CuFe2O4-MA | 163 | 110 | 7.8 | - | - |
| NiFe2O4-MA | 212 | 107 | 7.5 | - | - |
| CoFe2O4-MA | 188 | 59 | 3.7 | - | - |
| Fe3O4-MA | 176 | 91 | 10.6 | - | - |
| CFMA-5 | 176 | 82 | 8.0 | 12.6 | N.A. c |
| CFMA-10 | 163 | 110 | 7.8 | 12.3 | 18.2 |
| CFMA-15 | 97 | 72 | 9.4 | 12.2 | 19.1 |
| CFMA-20 | 42 | 41 | 11.3 | 13.8 | 23.7 |
| CFMA-25 | 36 | 23 | 17.3 | 19.6 | 26.1 |
| Catalyst | BET Surface Area (m2/g) a | Cu Surface Area (m2/g) b | Crystallite Size (nm) | Lattice Parameter | Cu Dispersion (%) b | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fresh a | Used a | Cu b | CuO c | CeO2 c | ||||||
| CeO2 | 81 | - | 9 | - | - | - | - | 5.45 | - | [26] |
| NCC-SG | 102 | - | 5 | 6 | - | - | - | 5.43 | - | [26] |
| NCC-CP | 96 | - | 8 | 8 | - | - | - | 5.39 | - | [26] |
| NCC-ST | 79 | - | 10 | 11 | - | - | - | 5.41 | - | [26] |
| NCC-IM | 14 | - | 13 | 13 | - | - | - | 5.34 | - | [26] |
| CuA | 163.4 | 2.5 | - | - | 34.4 | - | - | - | 2.9 | [27] |
| CeCuA | 124.5 | 2.2 | - | - | 50.7 | 27.4 | 9.6 | - | 1.7 | [27,48] |
| Cu-CeA | 114.0 | 2.0 | - | - | 56.3 | - | - | - | 1.5 | [27] |
| CuCeA | 116.1 | 1.5 | - | - | 75.4 | - | - | - | 1.2 | [27] |
| CBCA | 114.0 | - | - | - | - | 28.2 | 9.8 | - | 1.7 | [48] |
| CZCA | 111.9 | - | - | - | - | 29.4 | 10.2 | - | 1.5 | [48] |
| CNCA | 122.0 | - | - | - | - | 30.2 | 9.9 | - | 1.4 | [48] |
| Catalyst | CuO Species Interacted with CeO2 (°C) | CuO Species Not Interacted with CeO2 (°C) | Bulk CuO (°C) | Ref. |
|---|---|---|---|---|
| NCC-SG | 169 | 257 | - | [26] |
| NCC-CP | 187 | - | - | |
| NCC-ST | 165 | 200 | 265 | |
| NCC-IM | 182 | 261 | 302 | |
| CuA | - | - | 197 | [27] |
| CeCuA | 136 | 158 | 183 | |
| Cu-CeA | 141 | - | 177 | |
| CuCeA | - | - | 188 | |
| CBCA | 170 | 186 | 215 | [48] |
| CZCA | 134 | 163 | 203 | |
| CNCA | 114 | 143 | 180 |
| Catalyst | BET Surface Area (m2/g) a | Crystallite Size (nm) | Ni0 Dispersion c (%) | Active Ni Site c (mol/gcat) | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Fresh | Used | Fresh b (Fe2O3) | Used b (Fe3O4) | Ni0 c | ||||
| Fe/Al/Ni | 81.4 | 11.2 | 14.9 | 20.6 | - | - | - | [12] |
| Ni-CeO2 | 131.29 | - | - | - | 21.03 | 4.00 | 1.37 × 10−6 | [87] |
| K/Ni-CeO2 | 110.34 | - | - | - | 26.71 | 3.16 | 1.08 × 10−6 | [87] |
| Ca/Ni-CeO2 | 112.45 | - | - | - | 25.61 | 3.28 | 1.13 × 10−6 | [87] |
| Mg/Ni-CeO2 | 109.85 | - | - | - | 61.13 | 1.39 | 3.16 × 10−6 | [87] |
| Ba/Ni-CeO2 | 118.83 | - | - | - | 26.33 | 3.20 | 1.14 × 10−6 | [87] |
| Catalysts | BET Surface Area (m2/g) a | Co0 Crystallite Size (nm) b | Lattice Parameter (A) b | Co Dispersion (%) c | Co0/ (Co0 + Co2+ + Co3+) (%) d | Ref. |
|---|---|---|---|---|---|---|
| BCC-1 | 60 | N.A. a | - | 0.63 | 49.8 | [1] |
| CC-SG | 30 | - | - | 1.61 | - | [6] |
| 1.5NbCo | 114 | 4.7 | 5.430 | 3.47 | 26 | [53] |
| CZ6C4 | 186.8 | 8.7 | 5.380 | 1.96 | 42.3 | [8] |
| CoFe2O4-ES | 5.8 | 35.3 | - | - | 34.4 | [88] |
| CNC-CP | 115.39 | - | 5.428 | 3.41 | 50.80 | [89] |
| Catalyst | Co3O4 to CoO (°C) | CoO to Co0 (°C) | Surface Oxygen Species of CeO2 (°C) | Ref. |
|---|---|---|---|---|
| BCC-0 | 327 | 376 | - | [1] |
| BCC-1 | 264 | 333 | - | |
| BCC-2 | 285 | 354 | - | |
| BCC-3 | 299 | 390 | - | |
| CC-SG | 224 | 414 | - | [6] |
| CC-IWI | 327 | 377 | - | |
| CC-CP | 295 | 367 | - | |
| CC-HT | 211 | 262 | - | |
| CZ2C8 | 335 | 430 | - | [8] |
| CZ4C6 | 326 | 411 | - | |
| CZ6C4 | 335 | 420 | - | |
| CZ8C2 | 326 | 411 | - | |
| C-Z | 308 | 353 | - | |
| C-C | 320 | 414 | 573 | |
| 0.5NbCo | 304 | 414 | 532 | [53] |
| 1.5NbCo | 304 | 414 | 532 | |
| 2.5NbCo | 304 | 414 | 532 | |
| CNC-CP | 304 | 414 | 532 | [89] |
| CNC-IWI | 333 | 385 | 424 | |
| CNC-SG | 302 | 354 | 491 | |
| CNC-HT | 322 | 436 | 768 |
| Catalyst | Fe2O3 to Fe3O4 (°C) | Ref. |
|---|---|---|
| CoFe2O4-ES | 351 | [88] |
| CoFe2O4-SG | 558 | |
| CoFe2O4-HT | 598 | |
| CoFe2O4-CP | 544 |
| Catalysts | BET Surface Area (m2/g) | Pt0 Dispersion (%) | OSC (10−4 gmol/gcat) | Ref. |
|---|---|---|---|---|
| Pt/CeO2 | 77 | 76.29 | 6.66 | [59] |
| Pt/ZrO2 | 284 | 59.14 | 2.04 | [59] |
| Pt/MgO | 167 | 76.18 | 0.86 | [59] |
| Pt/Al2O3 | 202 | 61.10 | 1.87 | [59] |
| CeO2 | 105 | - | 3.93 | [57] |
| 0.1 wt% Pt/CeO2 | 62 | 94.1 | 6.30 | [57] |
| 0.5 wt% Pt/CeO2 | 66 | 81.3 | 6.46 | [57] |
| 2.0 wt% Pt/CeO2 | 77 | 76.3 | 6.66 | [57] |
| 5.0 wt% Pt/CeO2 | 73 | 38.1 | 6.34 | [57] |
| 10.0 wt% Pt/CeO2 | 56 | 10.5 | 5.27 | [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, R.-R.; Jeon, I.-J.; Jang, W.-J.; Roh, H.-S.; Shim, J.-O. Advances in Catalysts for Water–Gas Shift Reaction Using Waste-Derived Synthesis Gas. Catalysts 2023, 13, 710. https://doi.org/10.3390/catal13040710
Lee R-R, Jeon I-J, Jang W-J, Roh H-S, Shim J-O. Advances in Catalysts for Water–Gas Shift Reaction Using Waste-Derived Synthesis Gas. Catalysts. 2023; 13(4):710. https://doi.org/10.3390/catal13040710
Chicago/Turabian StyleLee, Ru-Ri, I-Jeong Jeon, Won-Jun Jang, Hyun-Seog Roh, and Jae-Oh Shim. 2023. "Advances in Catalysts for Water–Gas Shift Reaction Using Waste-Derived Synthesis Gas" Catalysts 13, no. 4: 710. https://doi.org/10.3390/catal13040710