

Air-Stable Efficient Nickel Catalyst for Hydrogenation of Organic Compounds
 , , , and
, , , and
Abstract
:1. Introduction
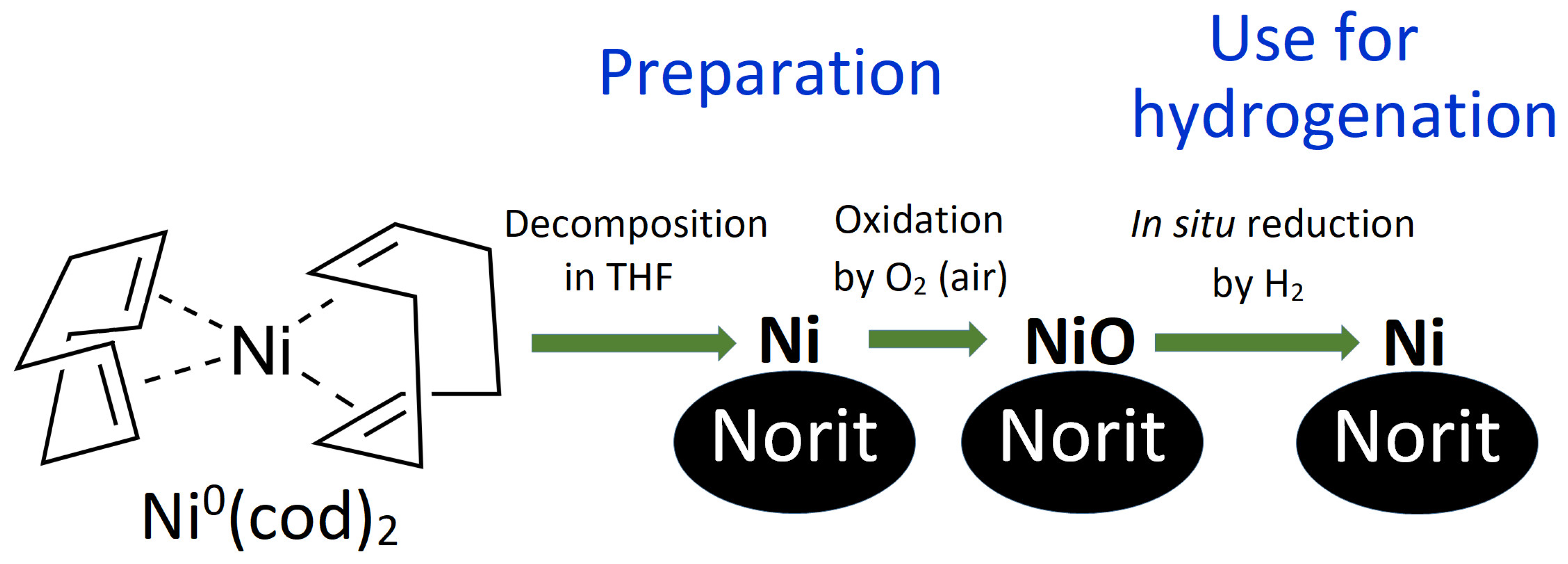
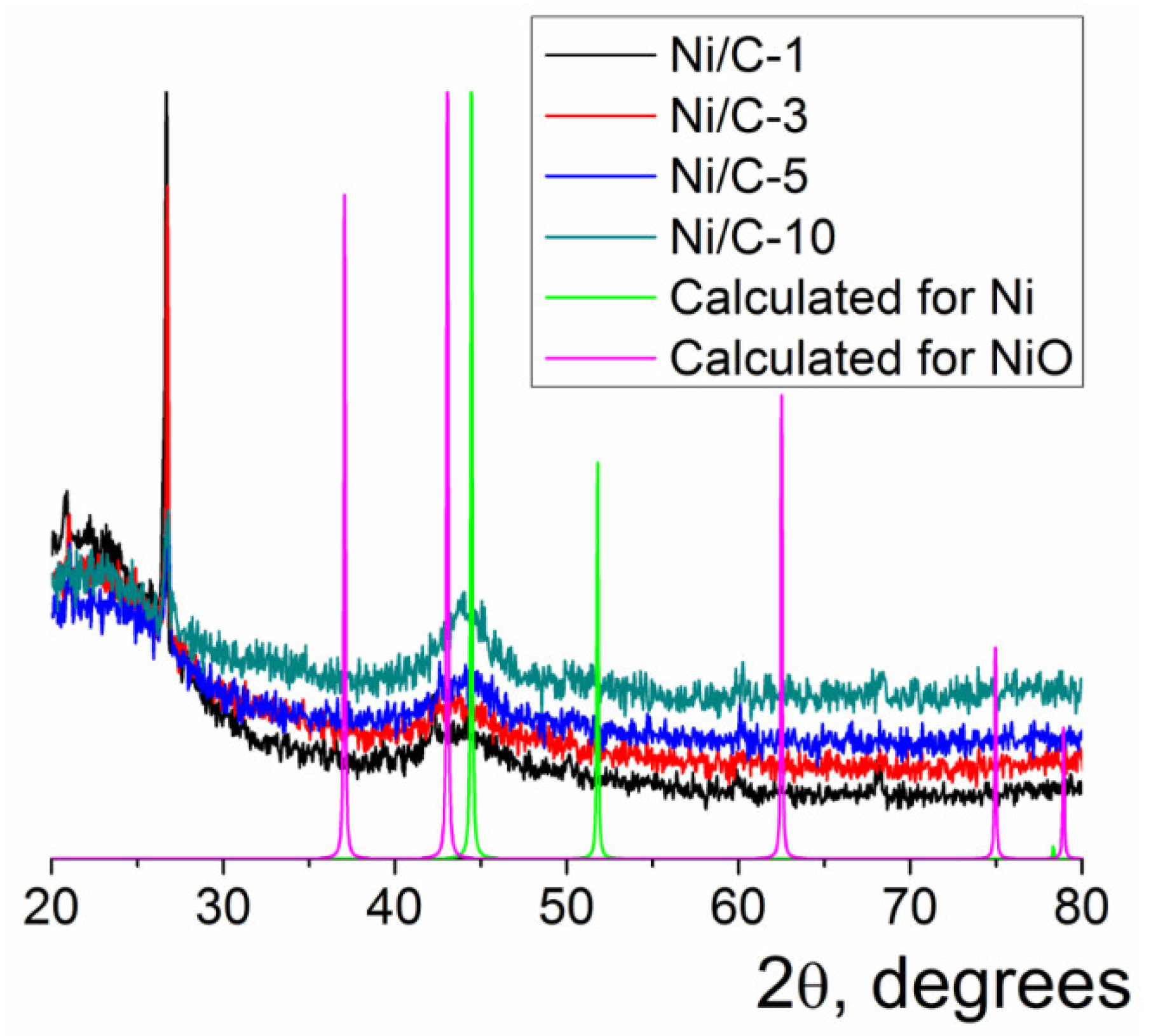
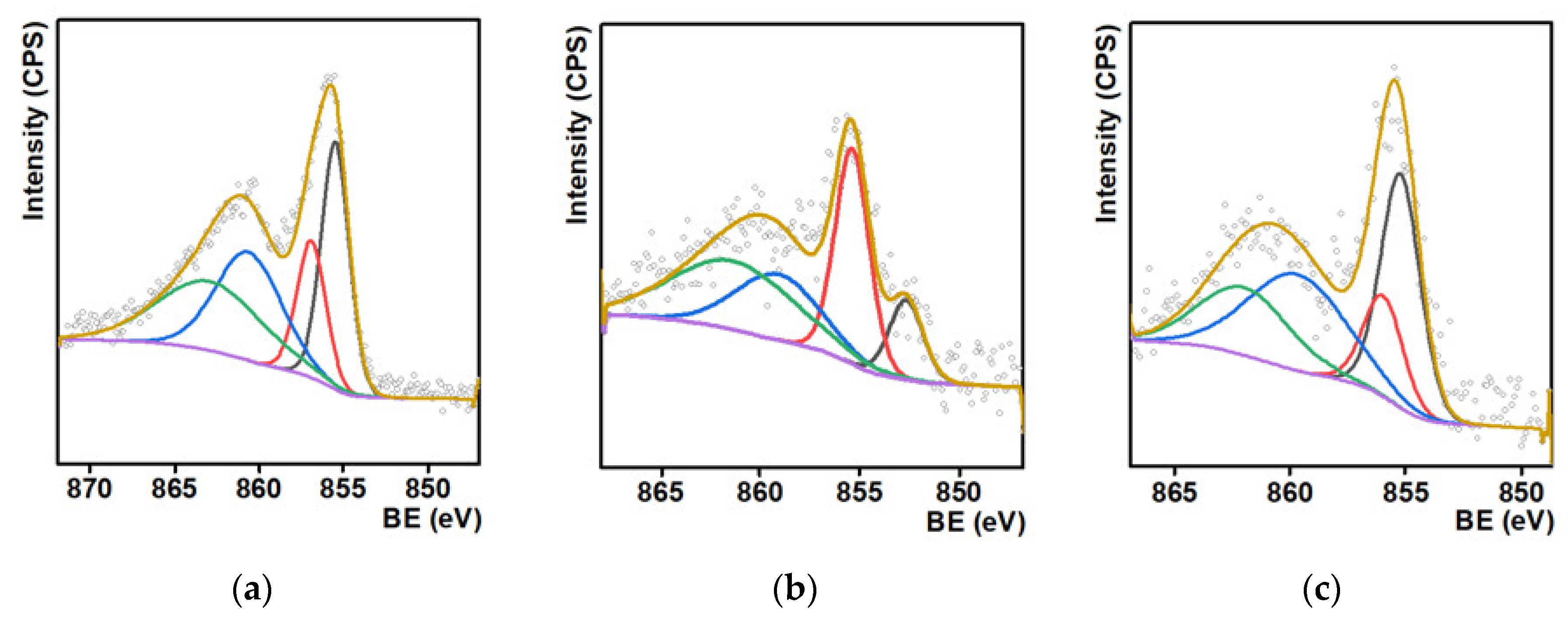
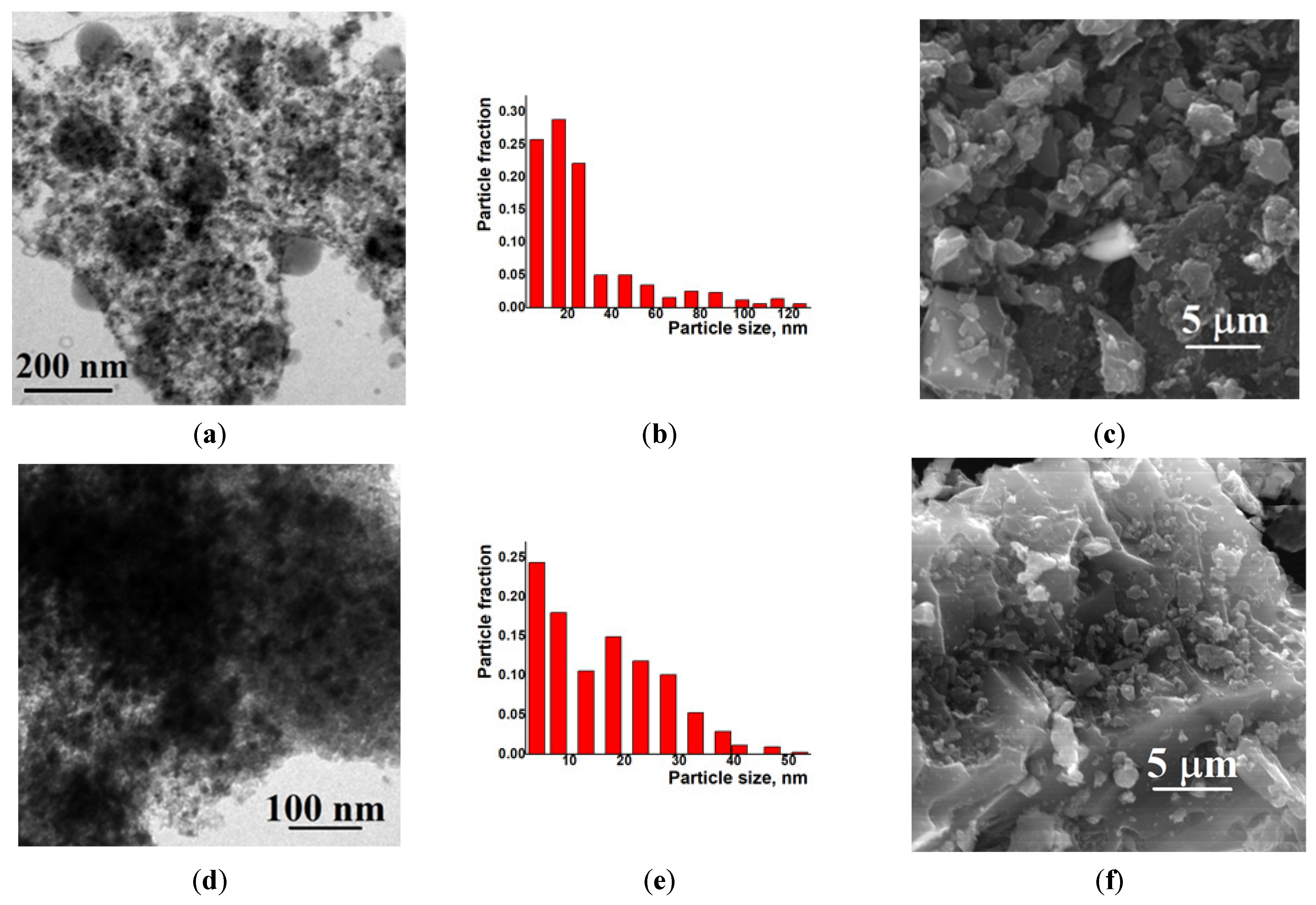
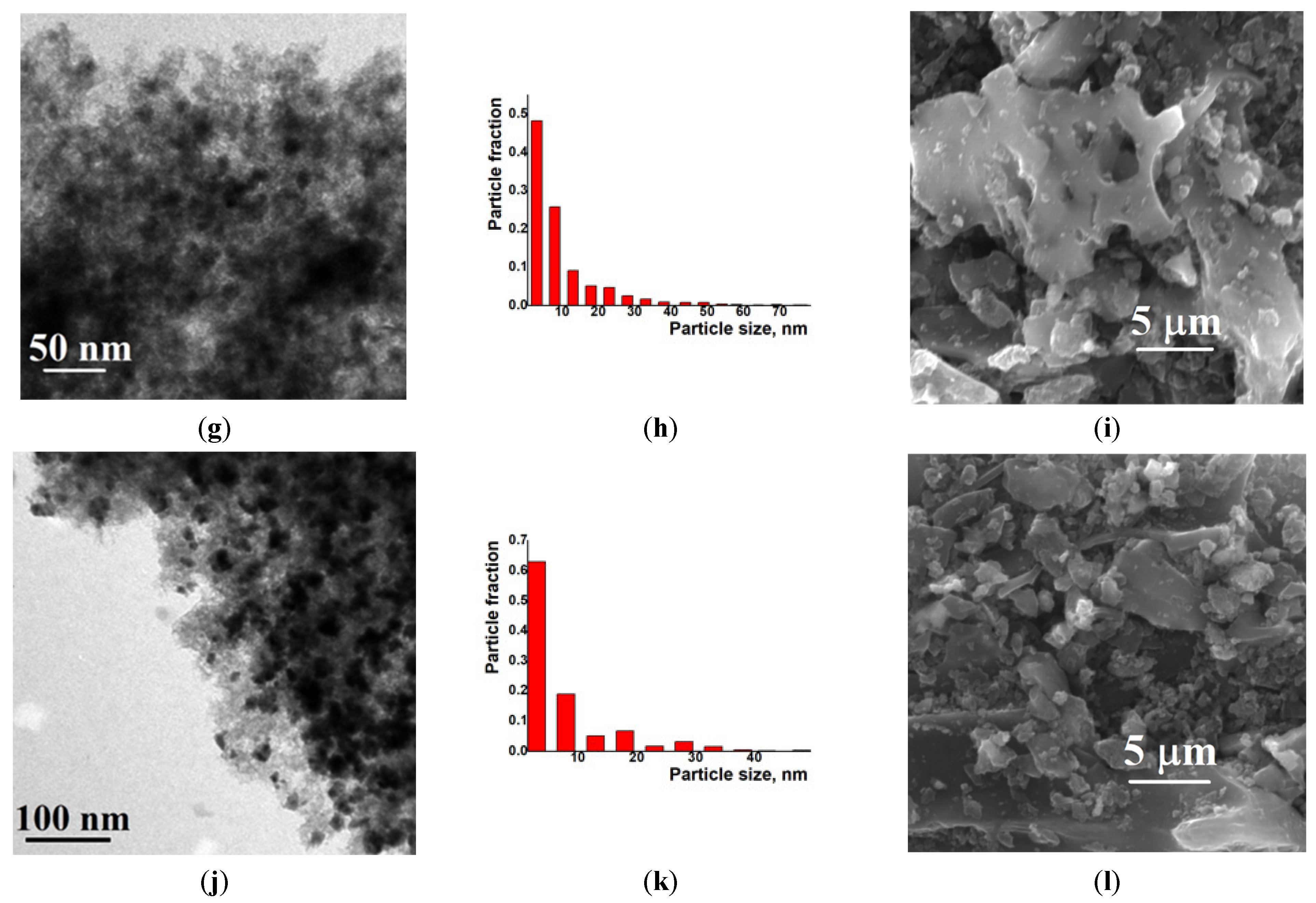
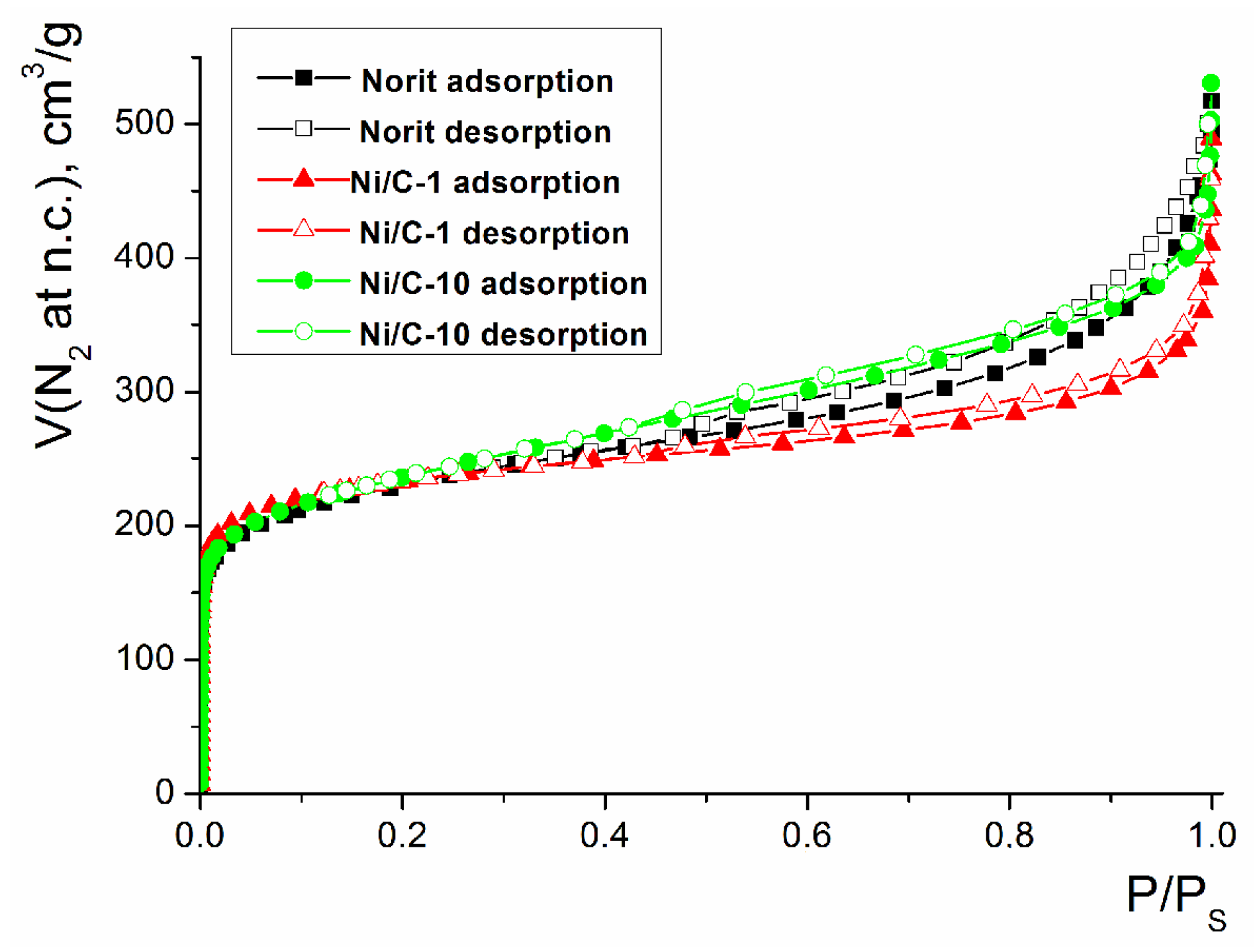
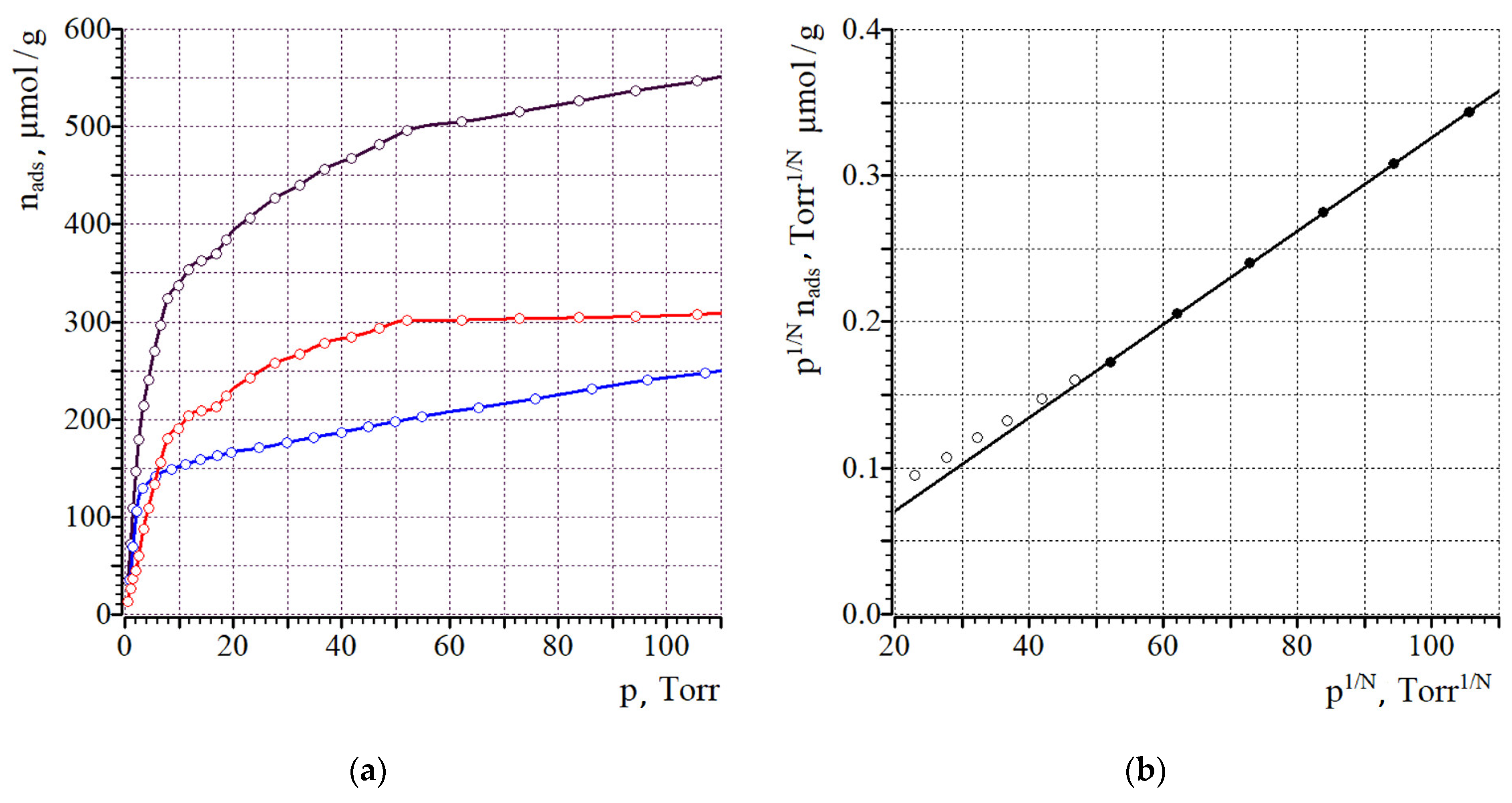
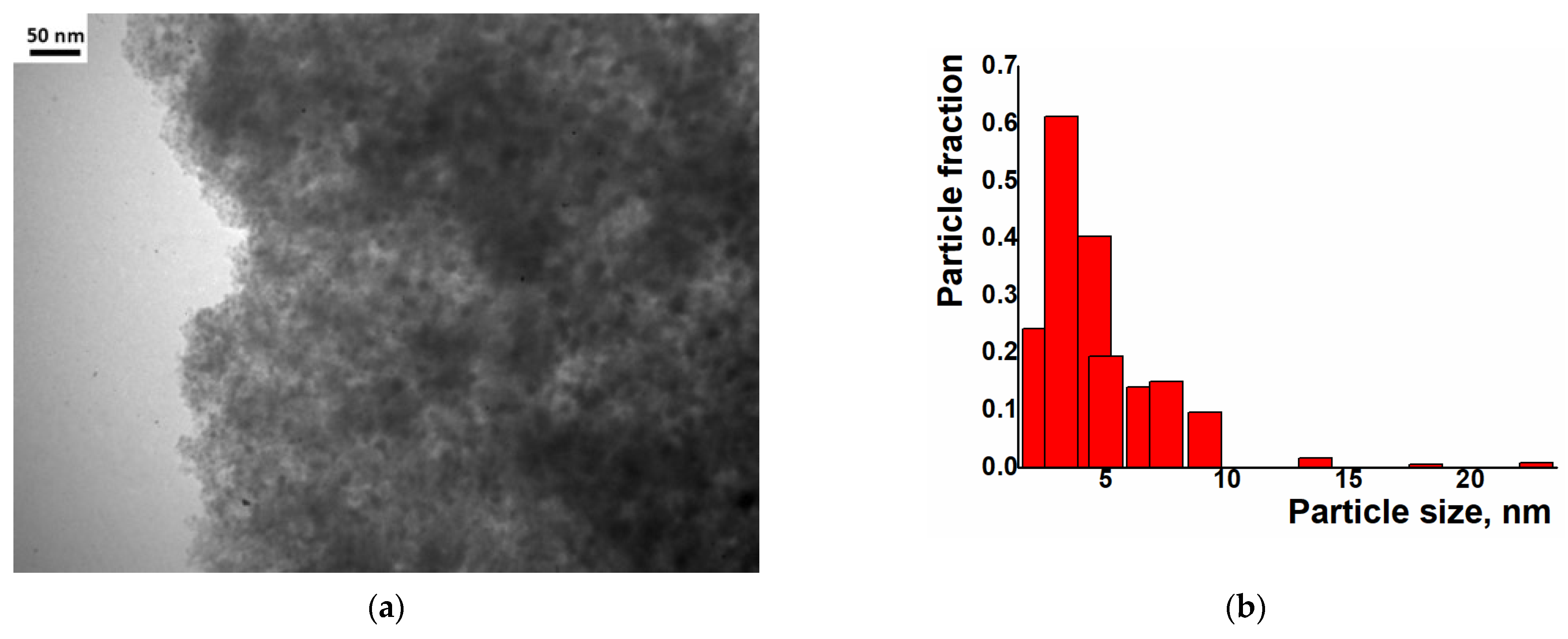
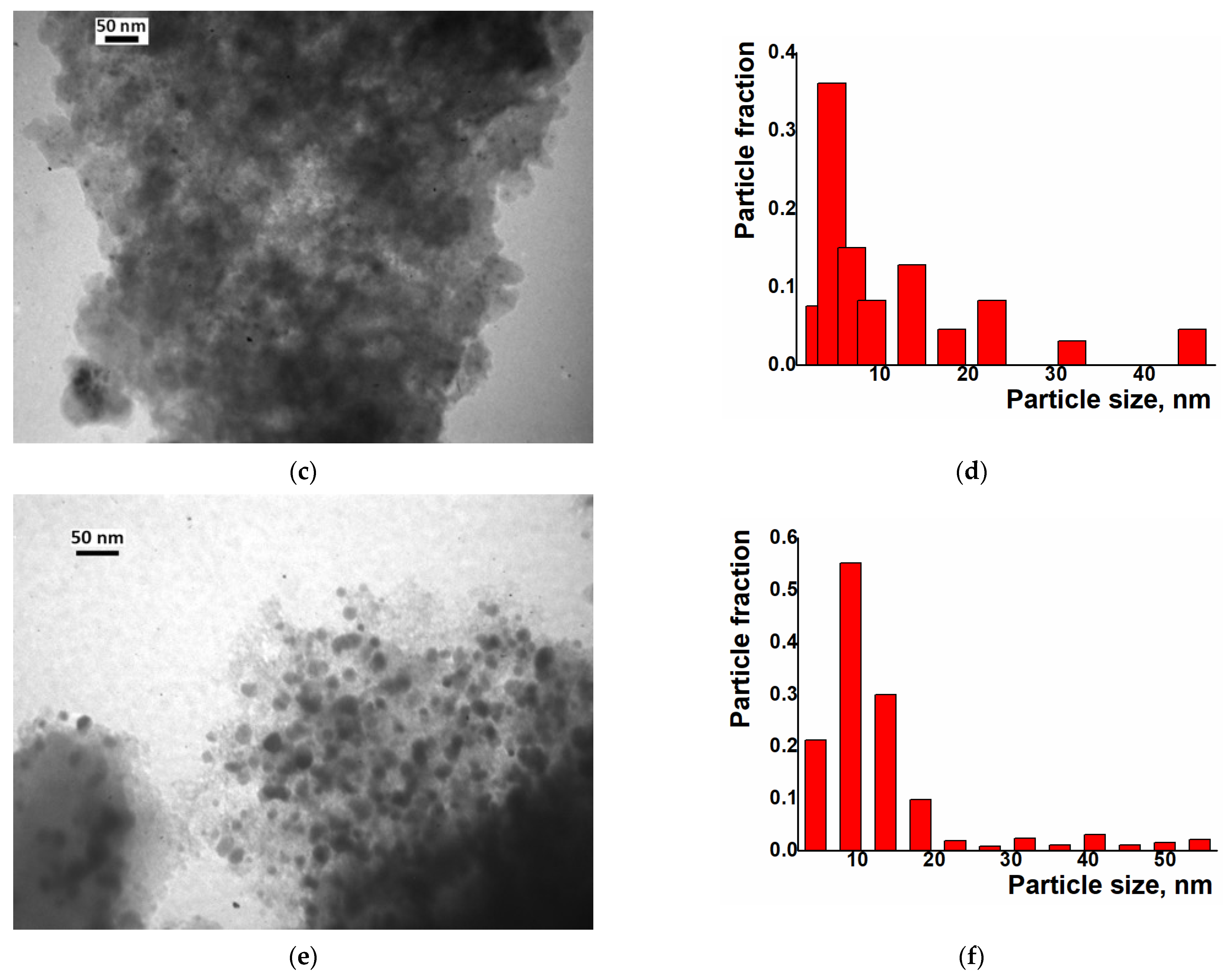
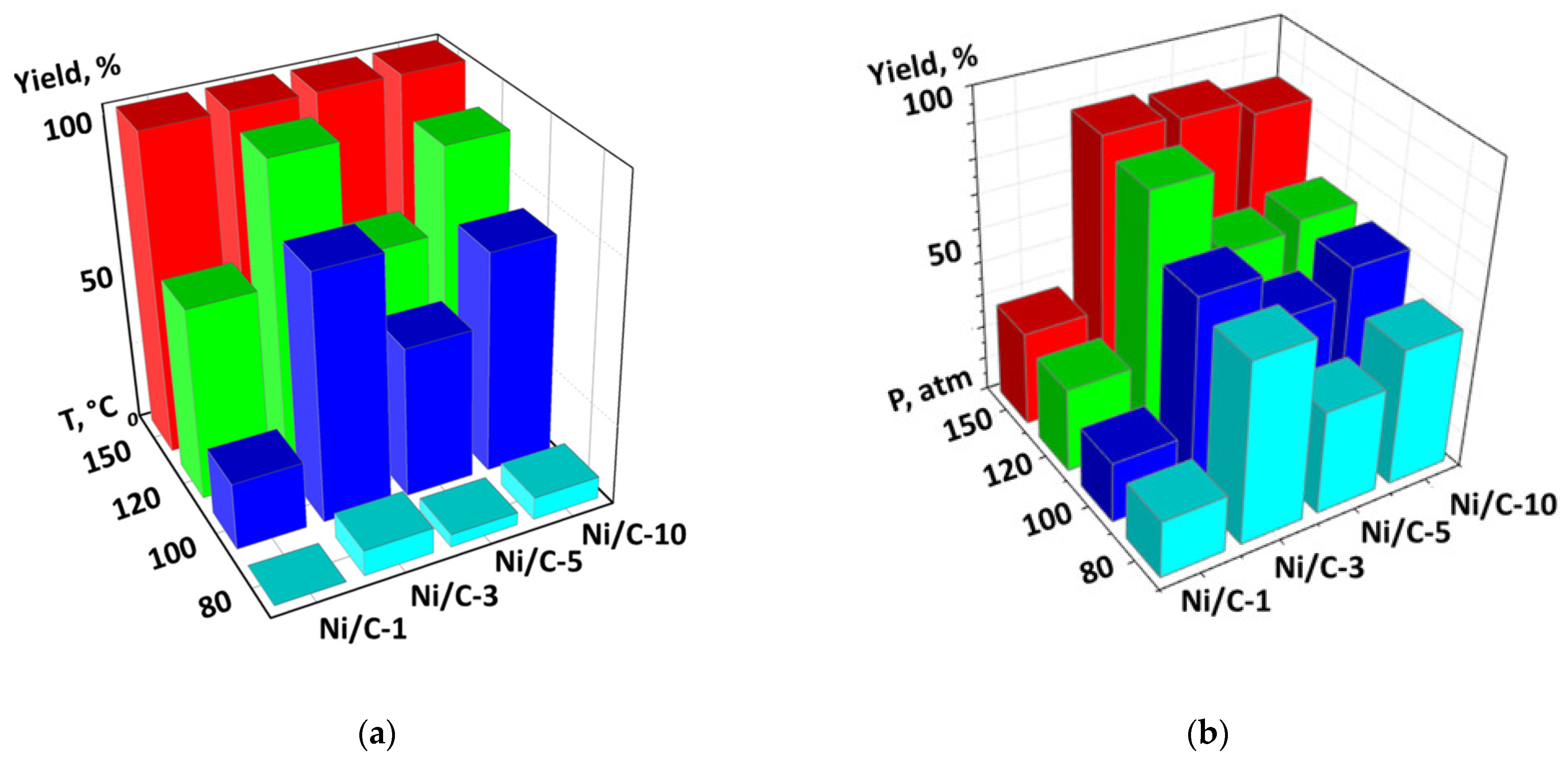
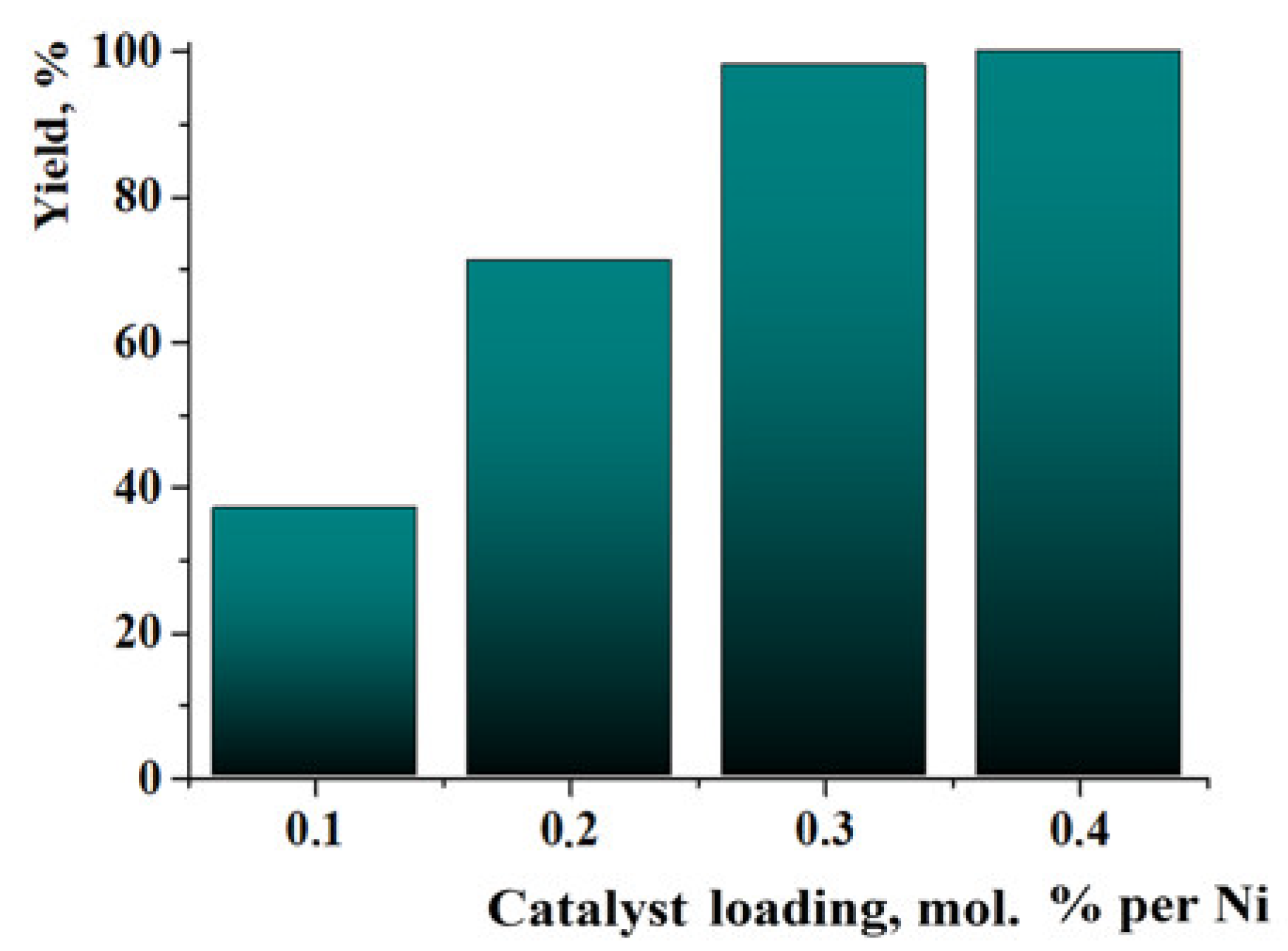
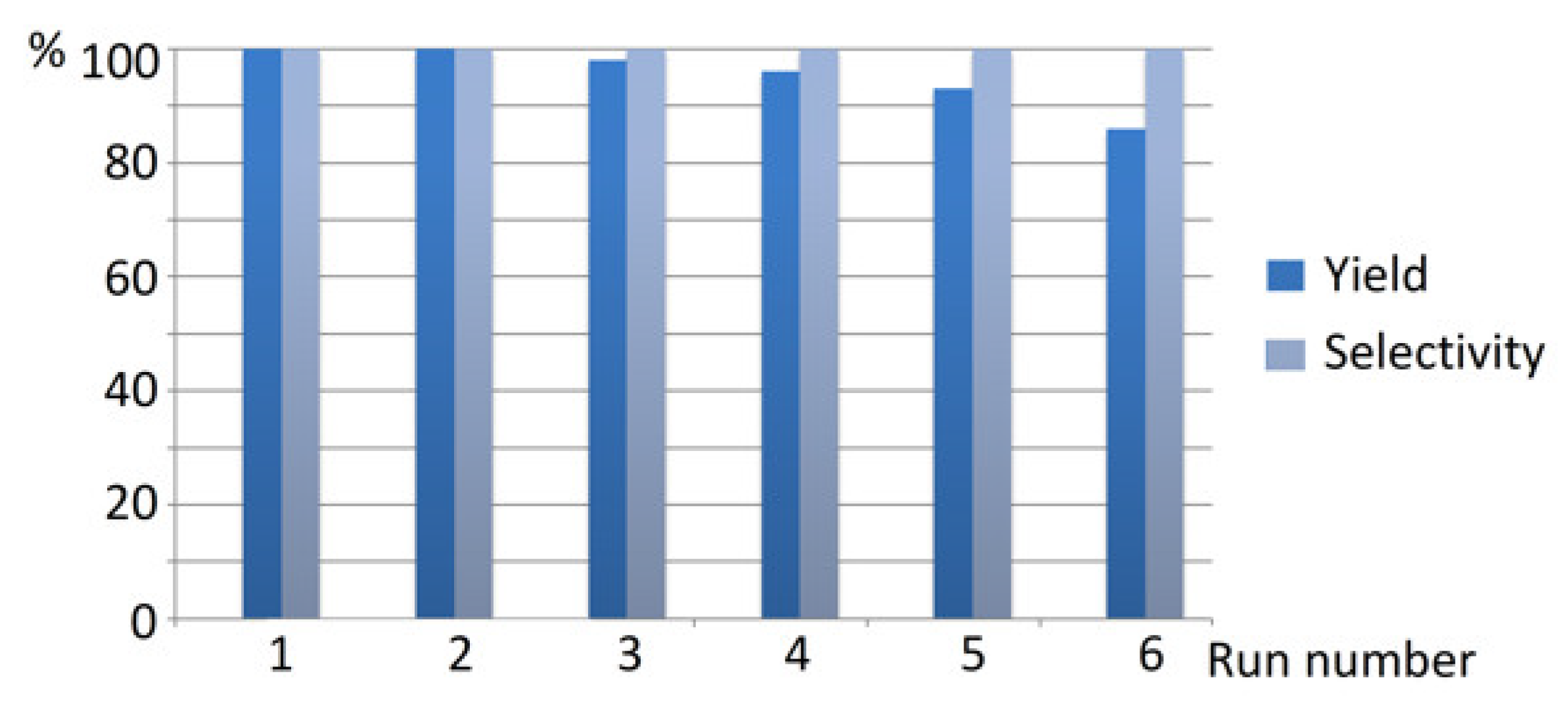
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nishimura, S. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, 1st ed.; Wiley-Interscience: New York, NY, USA, 2001; pp. 7–19. [Google Scholar]
- Rylander, P.N. Hydrogenation and Dehydrogenation. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Zhao, X.; Chang, Y.; Chen, W.-J.; Wu, Q.; Pan, X.; Chen, K.; Weng, B. Recent Progress in Pd-Based Nanocatalysts for Selective Hydrogenation. ACS Omega 2022, 7, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, D.M.; Hammond, G.B. The history and future challenges associated with the hydrogenation of vinyl fluorides Author links open overlay panel. J. Fluor. Chem. 2018, 207, 45–58. [Google Scholar] [CrossRef]
- Stoffels, M.A.; Klauck, F.J.R.; Hamadi, T.; Glorius, F.; Leker, J. Technology Trends of Catalysts in Hydrogenation Reactions: A Patent Landscape Analysis. Adv. Synth. Catal. 2020, 362, 1258–1274. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhou, M.; Wang, A.; Zhang, T. Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms. Chem. Rev. 2020, 120, 683–733. [Google Scholar] [CrossRef] [PubMed]
- Westerhaus, F.A.; Jagadeesh, R.V.; Wienhofer, G.; Pohl, M.-M.; Radnik, J.; Surkus, A.-E.; Rabeah, J.; Junge, K.; Junge, H.; Nielsen, M.; et al. Heterogenized cobalt oxide catalysts for nitroarene reduction by pyrolysis of molecularly defined complexes. Nat. Chem. 2013, 5, 537–543. [Google Scholar] [CrossRef]
- Chen, F.; Surkus, A.-E.; He, L.; Pohl, M.-M.; Radnik, J.; Topf, C.; Junge, K.; Beller, M. Selective Catalytic Hydrogenation of Heteroarenes with N-Graphene-Modified Cobalt Nanoparticles (Co3O4–Co/NGr@α-Al2O3). J. Am. Chem. Soc. 2015, 137, 11718–11724. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, R.V.; Surkus, A.-E.; Junge, H.; Pohl, M.-M.; Radnik, J.; Rabeah, J.; Huan, H.; Schünemann, V.; Brückner, A.; Beller, M. Nanoscale Fe2O3-Based Catalysts for Selective Hydrogenation of Nitroarenes to Anilines. Science 2013, 342, 1073–1076. [Google Scholar] [CrossRef]
- Morales, M.V.; Conesa, J.M.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. Tunable selectivity of Ni catalysts in the hydrogenation reaction of 5-hydroxymethylfurfural in aqueous media: Role of the carbon supports. Carbon 2021, 182, 265–275. [Google Scholar] [CrossRef]
- Palladium Prices—Interactive Historical Chart. Available online: https://www.macrotrends.net/2542/palladium-prices-historical-chart-data (accessed on 27 February 2023).
- Lipshutz, B.H. Development of Nickel-on-Charcoal as a “Dirt-Cheap’’ Heterogeneous Catalyst: A Personal Account. Adv. Synth. Catal. 2001, 343, 515–526. [Google Scholar] [CrossRef]
- Asaula, V.M.; Lytvynenko, A.S.; Mishura, A.M.; Gavrilenko, K.S.; Ryabukhin, S.V.; Volochnyuk, D.M.; Kolotilov, S.V. In-situ formation of Ni nanoparticles supported by MIL-101 porous coordination polymer for catalytic hydrogenation of quinoline. Inorg. Chem. Comm. 2020, 121, 108203. [Google Scholar] [CrossRef]
- Asaula, V.M.; Shvets, O.V.; Pariiska, O.O.; Bur’yanov, V.V.; Ryabukhin, S.V.; Volochnyuk, D.M.; Kolotilov, S.V. Composites Based on Nanodispersed Nickel, Graphene-Like Carbon, and Aerosil for Catalytic Hydrogenation of Furfural and Quinoline. Theor. Experim. Chem. 2020, 56, 261–267. [Google Scholar] [CrossRef]
- Asaula, V.M.; Buryanov, V.V.; Solod, B.Y.; Tryus, D.M.; Pariiska, O.O.; Kotenko, I.E.; Volovenko, Y.M.; Volochnyuk, D.M.; Ryabukhin, S.V.; Kolotilov, S.V. Catalytic Hydrogenation of Substituted Quinolines on Co–Graphene Composites. Eur. J. Org. Chem. 2021, 2021, 6616–6625. [Google Scholar] [CrossRef]
- Hahn, G.; Kunnas, P.; de Jonge, N.; Kempe, R. General synthesis of primary amines via reductive amination employing a reusable nickel catalyst. Nat. Catal. 2019, 2, 71–77. [Google Scholar] [CrossRef]
- Miroshnikova, A.V.; Kazachenko, A.S.; Tarabanko, V.E.; Sychev, V.V.; Skripnikov, A.M.; Mikhlin, Y.L.; Kosivtsov, Y.; Chudina, A.I.; Taran, O.P. Hydrogenation of Flax Shives in Ethanol over a Ni/C Catalyst. Catalysts 2022, 12, 1177. [Google Scholar] [CrossRef]
- Murugesan, K.; Beller, M.; Jagadeesh, R.V. Reusable nickel nanoparticles-catalyzed reductive amination for selective syn-thesis of primary amines. Angew. Chem. Int. Ed. 2019, 131, 5118–5122. [Google Scholar] [CrossRef]
- Subotin, V.V.; Asaula, V.M.; Lishchenko, Y.L.; Ivanytsya, M.O.; Pariiska, O.O.; Ryabukhin, S.V.; Volochnyuk, D.M.; Kolotilov, S.V. Catalytic Reductive Amination of Aromatic Aldehydes on Co-Containing Composites. Chemistry 2023, 5, 281–293. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, Y.; Mei, D.; Bullock, R.M.; Gutiérrez, O.Y.; Camaioni, D.M.; Lercher, J.A. The Critical Role of Reductive Steps in the Nickel-Catalyzed Hydrogenolysis and Hydrolysis of Aryl Ether C−O Bonds. Angew. Chem. Int. Ed. 2020, 59, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, A.G.; Hartwig, J.F. Selective, Nickel-Catalyzed Hydrogenolysis of Aryl Ethers. Science 2011, 332, 439–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friend, C.M.; Xu, B. Heterogeneous Catalysis: A Central Science for a Sustainable Future. Acc. Chem. Res. 2017, 50, 517–521. [Google Scholar] [CrossRef]
- Ivanytsya, M.O.; Ryabukhin, S.V.; Volochnyuk, D.M.; Kolotilov, S.V. Modern Approaches to the Creation of Immobilized Metal-Complex Catalysts for Hydrogenation, Alkene Metathesis, and Cross-Coupling Processes: A Review. Theor. Exp. Chem. 2020, 56, 283–308. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Tasler, S.; Chrisman, W.; Spliethoff, B.; Tesche, B. On the Nature of the ‘Heterogeneous’ Catalyst: Nickel-on-Charcoal. J. Org. Chem. 2003, 68, 1177–1189. [Google Scholar] [CrossRef]
- Urushibara, Y.; Nishimura, S. A Method for the Preparation of the Raney Nickel Catalyst with a Greater Activity. Bull. Chem. Soc. Jpn. 1957, 30, 199. [Google Scholar]
- Subotin, V.V.; Vashchenko, B.V.; Asaula, V.M.; Verner, E.V.; Ivanytsya, M.O.; Shvets, O.; Ostapchuk, E.N.; Grygorenko, O.O.; Ryabukhin, S.V.; Volochnyuk, D.M.; et al. Screening of Palladium/Charcoal Catalysts for Hydrogenation of Diene Carboxylates with Isolated-Rings (Hetero)aliphatic Scaffold. Molecules 2023, 28, 1201. [Google Scholar] [CrossRef]
- Alsalme, A.; Toraba, M.A.; Khan, M.; Alzaqri, N.A.; Alshammari, S.G.; Alotaibic, M.A.; Siddiqui, M.R.H. Facile synthesis of nickel based nanostructures from Ni[EMIM]Cl2 ionic liquid precursor: Effects of thermal and chemical methods on the properties of nanoparticles. RSC Adv. 2016, 6, 86340–86345. [Google Scholar] [CrossRef]
- Adil, S.F.; Ashraf, M.; Khan, M.; Assal, M.E.; Shaik, M.R.; Kuniyil, M.; Al-Warthan, A.; Siddiqui, M.R.H.; Tremel, W.; Tahir, M.N. Advances in graphene/inorganic nanoparticle composites for catalytic applications. Chem. Rec. 2022, 22, e202100274. [Google Scholar] [CrossRef]
- Kumar, M.; Xiong, X.; Sun, Y.; Yu, I.K.M.; Tsang, D.C.W.; Hou, D.; Gupta, J.; Bhaskar, T.; Pandey, A. Critical review on biochar-supported catalysts for pollutant degradation and sustainable biorefinery. Adv. Sustain. Syst. 2020, 4, 1900149. [Google Scholar] [CrossRef]
- Raney, M. Method of Producing Finely-Divided Nickel. U.S. Patent 1,628,190, 14 May 1926. [Google Scholar]
- Harry, B.; Homer, A. Catalyst, Raney Nickel, W6 (with high contents of aluminum and adsorbed hydrogen). Org. Synth. 1949, 29, 24. [Google Scholar]
- Augustine, R.L. Heterogeneous Catalysis for the Synthetic Chemist; CRC Press: Boca Raton, FL, USA, 1996; pp. 248–249. [Google Scholar]
- Yakukhnov, S.A.; Pentsak, E.O.; Galkin, K.I.; Mironenko, R.M.; Drozdov, V.A.; Likholobov, V.A.; Ananikov, V.P. Rapid “Mix-and-Stir” Preparation of Well-Defined Palladium on Carbon Catalysts for Efficient Practical Use. ChemCatChem 2018, 10, 1869–1873. [Google Scholar] [CrossRef]
- Gao, F.; Webb, J.D.; Hartwig, J.F. Chemo- and Regioselective Hydrogenolysis of Diaryl Ether C-O Bonds by a Robust Heterogeneous Ni/C Catalyst: Applications to the Cleavage of Complex Lignin-Related Fragments. Angew. Chem. Int. Ed. 2016, 55, 1474–1478. [Google Scholar] [CrossRef]
- Afanasenko, A.; Elangovan, S.; Stuart, M.C.A.; Bonura, G.; Frusteri, F.; Barta, K. Efficient nickel-catalysed N-alkylation of amines with alcohols. Catal. Sci. Technol. 2018, 8, 5498–5505. [Google Scholar] [CrossRef] [Green Version]
- Sergeev, A.G.; Webb, J.D.; Hartwig, J.F. A Heterogeneous Nickel Catalyst for the Hydrogenolysis of Aryl Ethers without Arene Hydrogenation. J. Am. Chem. Soc. 2012, 134, 20226–20229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noack, K.; Zbinden, H.; Schlögl, R. Identification of the state of palladium in various hydrogenation catalysts by XPS. Catal. Lett. 1990, 4, 145–156. [Google Scholar] [CrossRef]
- Teschner, D.; Révay, Z.; Borsodi, J.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R.; Milroy, D.; Jackson, S.D.; Torres, D.; Sautet, P. Understanding Palladium Hydrogenation Catalysts: When the Nature of the Reactive Molecule Controls the Nature of the Catalyst Active Phase. Angew. Chem. Int. Ed. 2008, 47, 9274–9278. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y.; Nitta, Y.; Imanaka, T.; Teranishi, S. Surface state, catalytic activity and selectivity of nickel catalysts in hydrogenation reactions. Part 2; Surface characterization of Raney nickel and Urushibara nickel catalysts by X-ray photoelectron spectroscopy. J. Chem. Soc. Faraday Trans. 1 1980, 76, 998–1007. [Google Scholar] [CrossRef]
- Leineweber, A.; Jacobs, H.; Hull, S. Ordering of Nitrogen in Nickel Nitride Ni3N Determined by Neutron Diffraction. Inorg. Chem. 2001, 40, 5818–5822. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.L. The Scherrer formula for X-ray particle size determination. Phys. Rev. 1939, 56, 978–982. [Google Scholar] [CrossRef]
- D’Addato, S.; Grillo, V.; Altieri, S.; Tondi, R.; Valeri, S.; Frabboni, S. Structure and stability of nickel/nickel oxide core–shell nanoparticles. J. Condens. Matter Phys. 2011, 23, 175003. [Google Scholar] [CrossRef]
- Duan, Y.; Li, J. Structure study of nickel nanoparticles. Mater. Chem. Phys. 2004, 87, 452–454. [Google Scholar] [CrossRef]
- Railsback, J.G.; Johnston-Peck, A.C.; Wang, J.; Tracy, J.B. Size-Dependent Nanoscale Kirkendall Effect During the Oxidation of Nickel Nanoparticles. ACS Nano 2010, 4, 1913–1920. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Biesinger, M.C.; Smart, R.S.C.; McIntrye, N.S. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 2006, 600, 1771–1779. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. X-ray photoelectron spectroscopic chemical state quantification of mixed nickel metal, oxide and hydroxide systems. Surf. Interface Anal. 2009, 41, 324–332. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. The role of the Auger parameter in XPS studies of nickel metal, halides and oxides. Phys. Chem. Chem. Phys. 2012, 14, 2434–2442. [Google Scholar] [CrossRef]
- Nickel X-ray Photoelectron Spectra, Nickel Electron Configuration, and Other Elemental Information. Available online: https://www.thermofisher.com/ua/en/home/materials-science/learning-center/periodic-table/transition-metal/nickel.html (accessed on 30 March 2023).
- Kurmach, M.M.; Konysheva, K.M.; Pertko, O.P.; Yaremov, P.S.; Voloshyna, Y.G.; Shvets, O.V. Catalytic Properties of Nickel-Containing Hierarchical Zeolites in the Reaction of n-Hexane Hydroizomerization. Theor. Exp. Chem. 2023. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef] [Green Version]
- Hyward, D.O.; Trapnell, B.M.W. Chemisorption, 2nd ed.; Butterworths: London, UK, 1964; p. 159. [Google Scholar]
- Thomas, J.M.; Lambert, R.M. (Eds.) Characterisation of Catalysts; J. Wiley & Sons: New York, NY, USA, 1980. [Google Scholar]
- Anderson, J.R. Structure of Metallic Catalysts; Academic Press: London, UK, 1975; p. 360. [Google Scholar]
- Lim, B.; Jiang, M.; Yu, T.; Camargo, P.H.C.; Xia, Y. Nucleation and growth mechanisms for Pd-Pt bimetallic nanodendrites and their electrocatalytic properties. Nano Res. 2010, 3, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Simonov, P.A.; Troitskii, S.Y.; Likholobov, V.A. Preparation of the Pd/C catalysts: A molecular-level study of active site formation. Kinet. Catal. 2000, 41, 255–269. [Google Scholar] [CrossRef]
- Yurchenko, D.V.; Lytvynenko, A.S.; Abdullayev, E.N.; Peregon, N.V.; Gavrilenko, K.S.; Gorlova, A.; Ryabukhin, S.V.; Volochnyuk, D.M.; Kolotilov, S.V. Catalytic oxidation of benzoins by hydrogen peroxide on nanosized HKUST-1: Influence of substituents on the reaction rates and DFT modeling of the reaction path. Molecules 2023, 28, 747. [Google Scholar] [CrossRef]
- Reed-Berendt, B.G.; Polidano, K.; Morrill, L.C. Recent advances in homogeneous borrowing hydrogen catalysis using earth-abundant first row transition metals. Org. Biomol. Chem. 2019, 17, 1595–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pariiska, O.O.; Mazur, D.O.; Asaula, V.M.; Buryanov, V.V.; Socha, R.; Kurys, Y.I.; Kolotilov, S.V.; Koshechko, V.G.; Pokhodenko, V.D. Influence of the Structure of Nanocomposites Based on Co,N,S-Doped Carbon and Co9S8 on the Catalytic Properties in the Processes of Quinoline and Its Methyl Derivatives Hydrogenation. Theor. Exp. Chem. 2023. [Google Scholar] [CrossRef]
- Hyejin, C.; Fanni, T.; Béla, T. Selective Reduction of Condensed N-Heterocycles using Water as a Solvent and a Hydrogen Source. Org. Biomol. Chem. 2013, 11, 1209–1215. [Google Scholar]
- Du, H. A Kind of Preparation Method of Hydrogenated Quinoline. Patent CN103613539B, 18 November 2015. [Google Scholar]
- Ryabchuk, P.; Agostini, G.; Pohl, M.-M.; Lund, H.; Agapova, A.; Junge, H.; Junge, K.; Beller, M. Intermetallic Nickel Silicide Nanocatalyst—A Non-noble Metal–Based General Hydrogenation Catalyst. Sci. Adv. 2018, 4, eaat0761. [Google Scholar] [CrossRef] [Green Version]
- Mao, S.; Ryabchuk, P.; Dastgir, S.; Anwar, M.; Junge, K.; Beller, M. Silicon-Enriched Nickel Nanoparticles for Hydrogenation of N-Heterocycles in Aqueous Media. ACS Appl. Nano Mater. 2022, 5, 5625–5630. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, X.; Denga, Y.; Shi, F. Catalytic Hydrogenation of Aromatic Rings Catalyzed by Pd/NiO. RSC Adv. 2014, 4, 2729–2732. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, H.; Liu, K.; Chen, K.-J. Water-assisted One-pot Synthesis of N-Doped Carbon Supported Ru Catalysts for Heterogeneous Catalysis. Chem. Commun. 2020, 56, 11311–11314. [Google Scholar] [CrossRef]
- Fang, M.; Sánchez-Delgado, R.A. Ruthenium Nanoparticles Supported on Magnesium Oxide: A Versatile and Recyclable Dual-site Catalyst for Hydrogenation of Mono- and Poly-cyclic Arenes, N-heteroaromatics, and S-heteroaromatics. J. Catal. 2014, 311, 357–368. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 5th ed.; Elsevier: Oxford, UK, 2003. [Google Scholar]
- Krysan, D.J.; Mackenzie, P.B. A new, convenient preparation of bis (1,5-cyclooctadiene) nickel (0). J. Org. Chem. 1990, 55, 4229–4230. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET a, m2/g | VT, cm3/g | Vmicro, cm3/g |
|---|---|---|---|
| NORIT | 849 | 0.604 | 0.31 |
| Ni/C-1 | 885 | 0.498 | 0.35 |
| Ni/C-3 | 895 | 0.487 | 0.36 |
| Ni/C-5 | 866 | 0.475 | 0.32 |
| Ni/C-10 | 861 | 0.591 | 0.31 |
| Composite | Ni Content, % | SNi, m2/g (Ni) | SNi, m2/g (Composite) | Dispersion (a), % | dNi, nm |
|---|---|---|---|---|---|
| Ni/C-1 | 0.9 | 935 | 8.4 | 100 | 0.7 |
| Ni/C-3 | 2.8 | 397 | 11.1 | 60 | 1.7 |
| Ni/C-5 | 4.5 | 341 | 15.3 | 51 | 2.0 |
| Ni/C-10 | 9.1 | 91 | 8.3 | 14 | 7.4 |
| Composite | mol. % (Calc. Per Ni) | Yield, % | mol. % (Calc. Per Ni) | Yield, % |
|---|---|---|---|---|
| Ni/C-1 | 2.5 | 100 | 1 | 44 |
| Ni/C-3 | 2.5 | 100 | 1 | 96 |
| Ni/C-5 | 2.5 | 100 | 1 | 89 |
| Ni/C-10 | 2.5 | 100 | 1 | 88 |
| Entry | Substrate | Products, Yield (%) |
|---|---|---|
| Alkenes | ||
| 1 |  |  |
| 2 |  |  |
| 3 |  |  |
| 4 |  |  |
| Alkynes | ||
| 5 |  |  |
| 6 |  |  |
| 7 |  |  |
| 8 |  |  |
| Carbonyl compounds | ||
| 9 |  |  |
| 10 |  |  |
| 11 |  |  |
| Nitro-compounds | ||
| 12 |  |  |
| 13 |  |  |
| Heterocycles | ||
| 14 |  |  |
| 15 |  |  |
| 16 |  |  |
| 17 |  |  |
| 18 |  |  |
| 19 |  |  |
| 20 |  |  |
| 21 |  |  |
| 22 |  |  |
| 23 |  |  |
| 24 |  |  |
| 25 |  |  |
| 26 |  |  |
| 27 |  |  |
| 28 |  |  |
| 29 |  |  |
| 30 |  |  |
| No. | Catalyst | Solvent | Metal, mol. % | T, °C | P, atm. | t, h. | Yield, % | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Ni/C-3 | MeOH | 1 | 100 | 100 | 24 | 97 | This work |
| 2 | Ni/C-3 | MeOH | 0.3 | 100 | 100 | 72 | 95 | This work |
| 3 | Raney nickel (a) | MeOH | 40 | 100 | 100 | 4 | 100 | This work |
| 4 | Raney nickel | MeOH | 10 | 100 | 100 | 4 | 25 | This work |
| 5 | Raney nickel | MeOH | 1 | 100 | 100 | 4 | 0 | This work |
| 6 | Raney nickel | H2O | 500 | 110 | - (b) | 1 | 100 | [60] |
| 7 | Raney nickel | MeOH | 10 | 180 | 90 | 4 (c) | 75 (d) | [61] |
| 8 | Ni-Mel@SiO2 (e) | MeOH | 3 | 100 | 100 | 24 | 25 | [14] |
| 9 | Ni-phen@SiO2 (f) | MeOH/ H2O 1:1 | 4.5 | 120 | 50 | 20 | 87 | [62] |
| 10 | Ni-phen@SiO2 (f) | H2O | 4 | 120 | 30 | 16 | 90 | [63] |
| 11 | NiBx@MIL-101(Cr) (g) | MeOH | 8 | 150 | 50 | 24 | 65 | [13] |
| 12 | Co-Phen@SiO2 (f) | MeOH | 3 | 100 | 100 | 24 | 100 | [15] |
| 13 | Pd/NiO | n-hexane | 1.7 | 120 | 50 | 24 | 93 | [64] |
| 14 | Ru/N-doped carbon | EtOH | 0.3 | 80 | 20 | 2.5 | 95 | [65] |
| 15 | Ru/MgO | THF | 0.025 | 150 | 50 | 1.4 | 92 (h) | [66] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subotin, V.V.; Ivanytsya, M.O.; Terebilenko, A.V.; Yaremov, P.S.; Pariiska, O.O.; Akimov, Y.M.; Kotenko, I.E.; Sabov, T.M.; Kurmach, M.M.; Ryabukhin, S.V.; et al. Air-Stable Efficient Nickel Catalyst for Hydrogenation of Organic Compounds. Catalysts 2023, 13, 706. https://doi.org/10.3390/catal13040706
Subotin VV, Ivanytsya MO, Terebilenko AV, Yaremov PS, Pariiska OO, Akimov YM, Kotenko IE, Sabov TM, Kurmach MM, Ryabukhin SV, et al. Air-Stable Efficient Nickel Catalyst for Hydrogenation of Organic Compounds. Catalysts. 2023; 13(4):706. https://doi.org/10.3390/catal13040706
Chicago/Turabian StyleSubotin, Vladyslav V., Mykyta O. Ivanytsya, Anastasiya V. Terebilenko, Pavel S. Yaremov, Olena O. Pariiska, Yuri M. Akimov, Igor E. Kotenko, Tomash M. Sabov, Mykhailo M. Kurmach, Sergey V. Ryabukhin, and et al. 2023. "Air-Stable Efficient Nickel Catalyst for Hydrogenation of Organic Compounds" Catalysts 13, no. 4: 706. https://doi.org/10.3390/catal13040706