
In Silico Structural and Functional Analysis of the Mitochondrial Malate Transporters in Oleaginous Fungus Mucor circinelloides WJ11


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
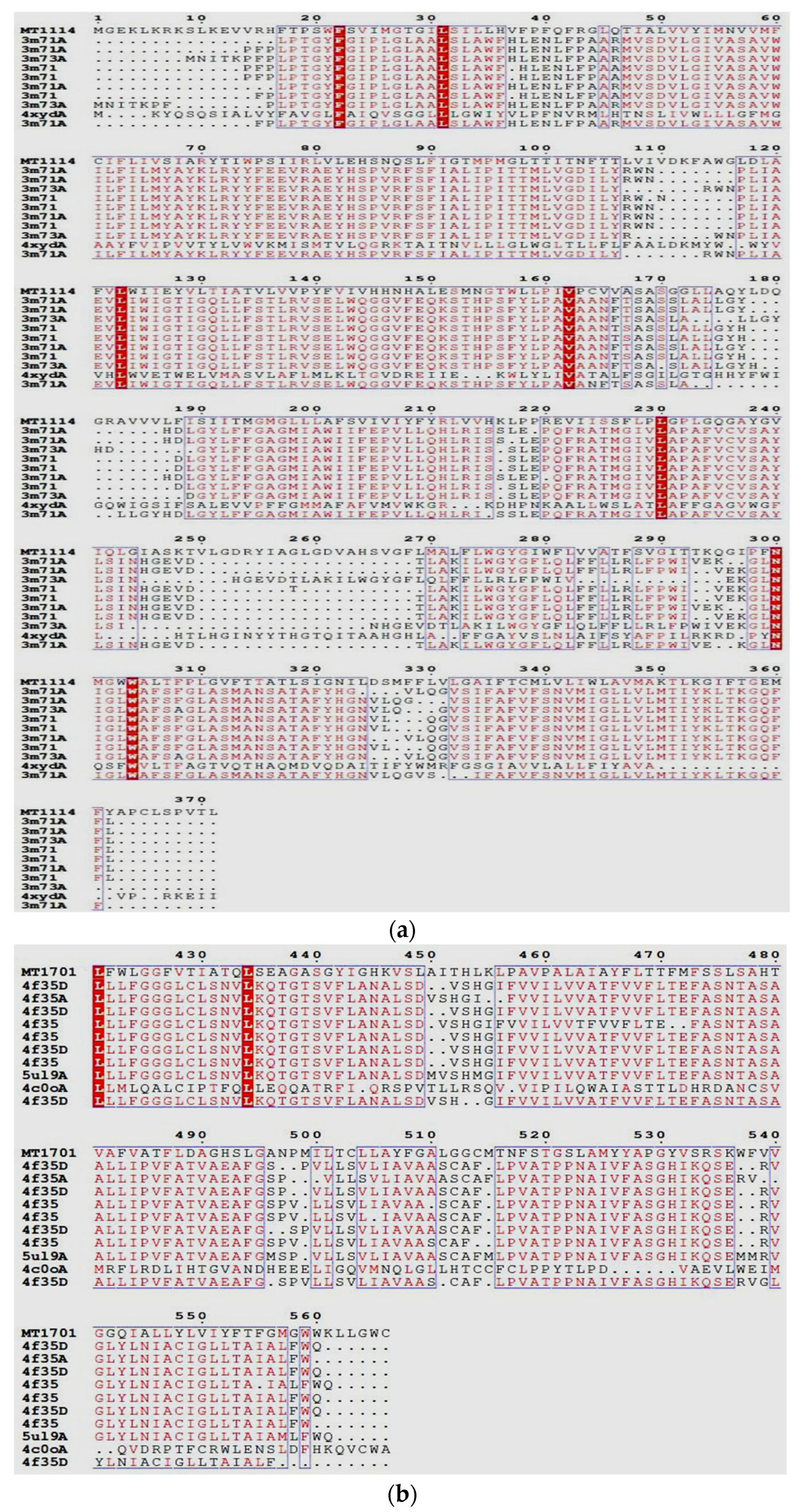
2.1. Nucleotide Sequence and Phylogenetic Analysis
2.2. Physico-Chemical Properties of Target Proteins
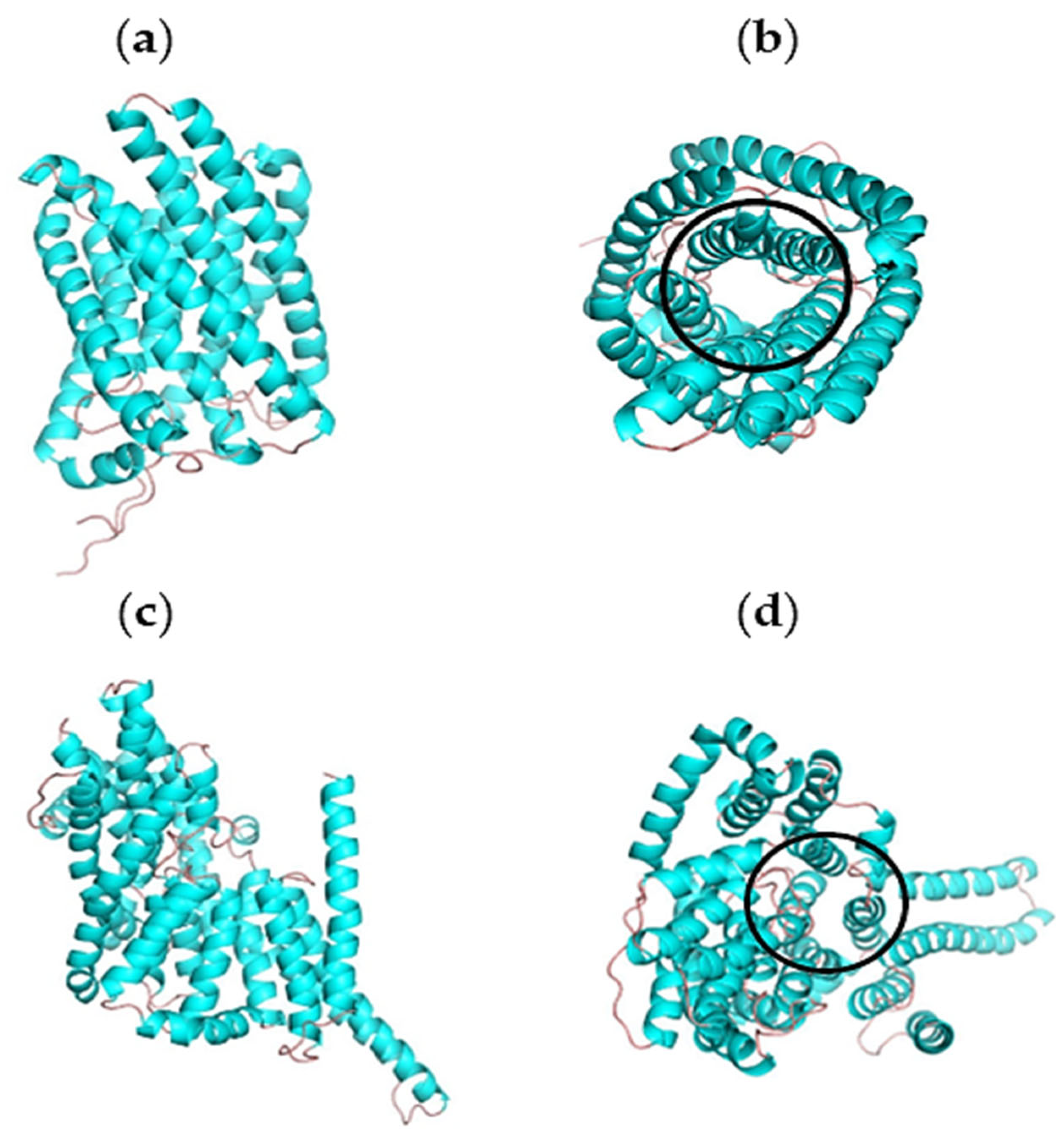
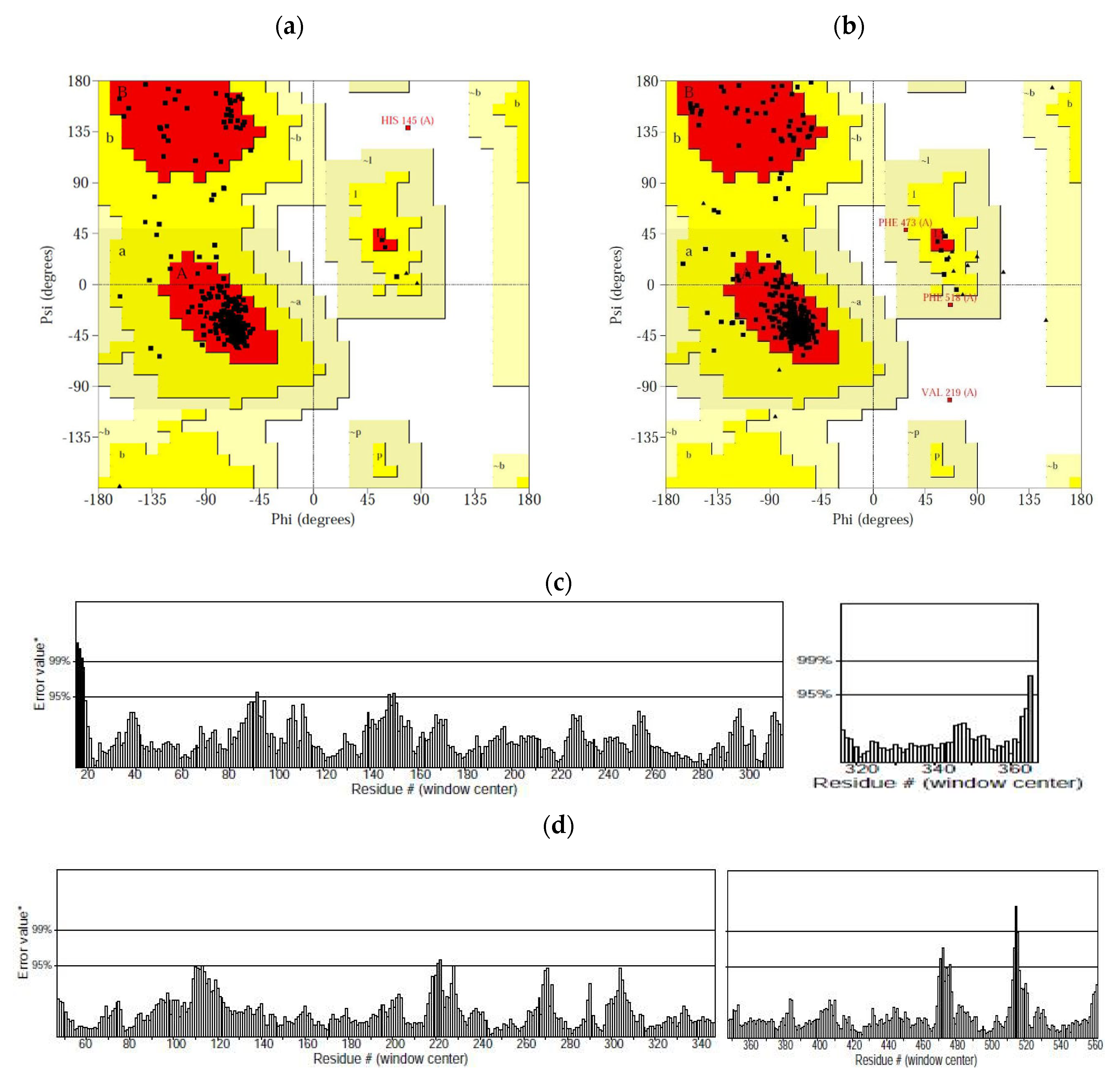
2.3. Homology Modelling and Validation
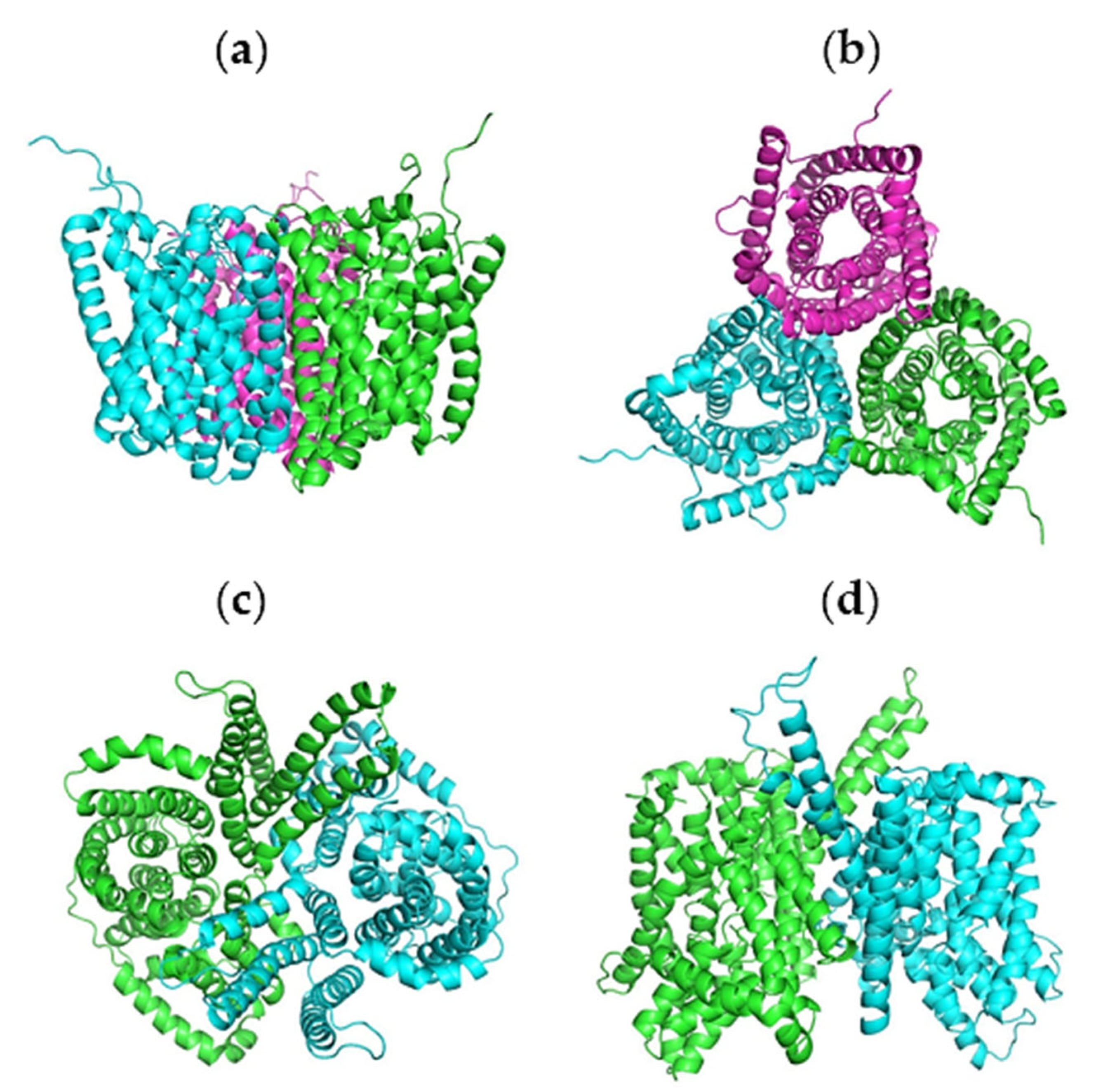
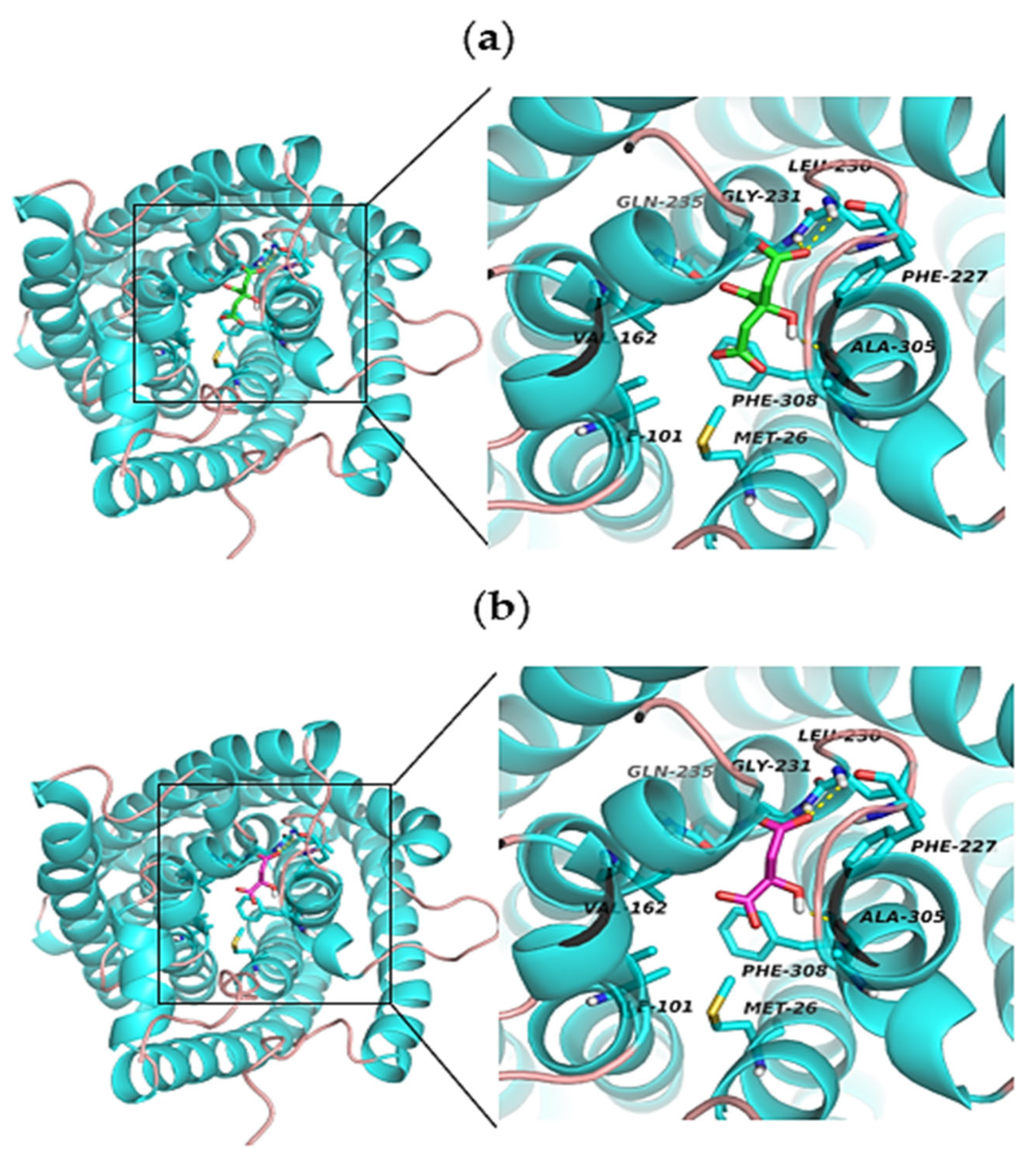
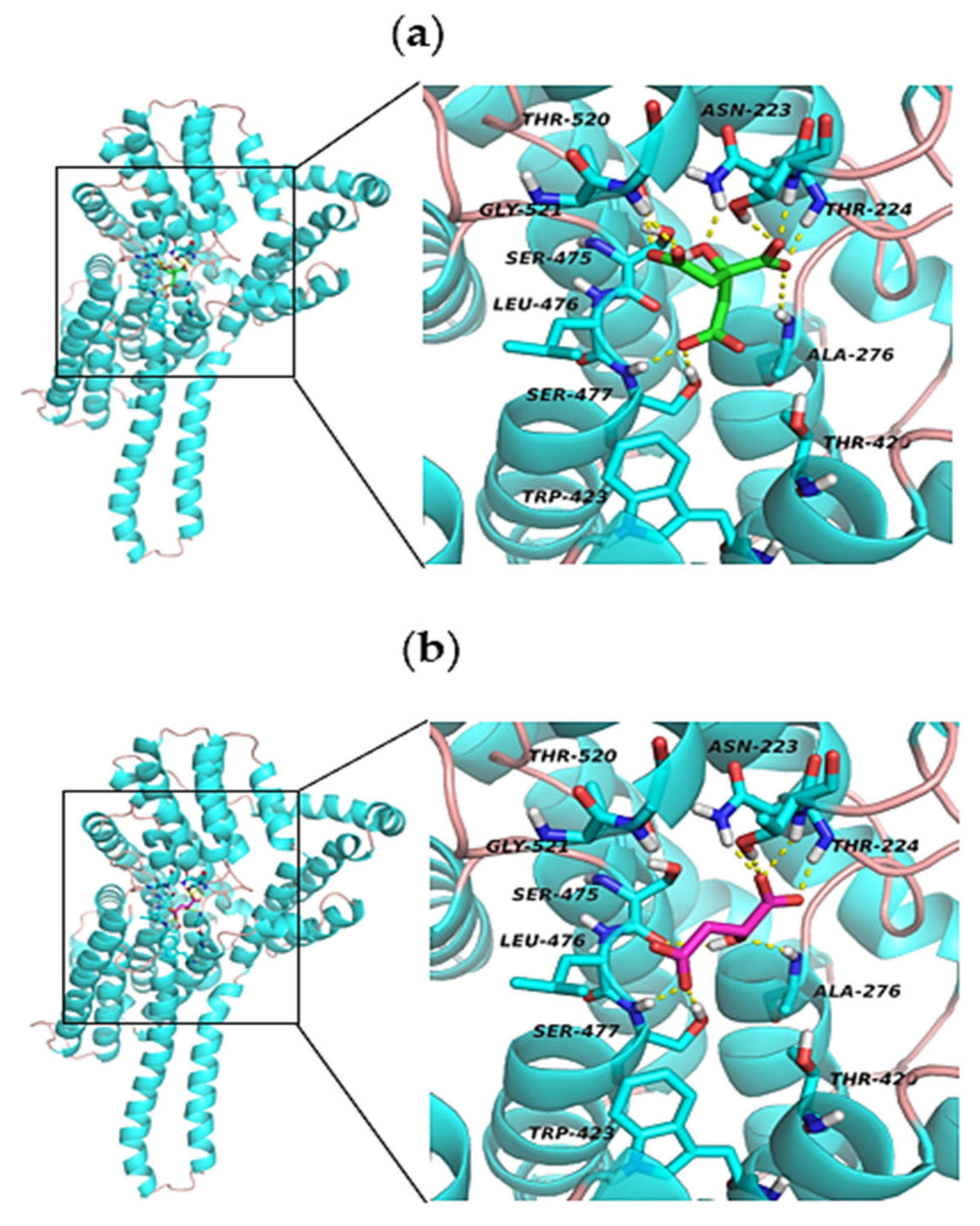
2.4. Molecular Docking Analysis
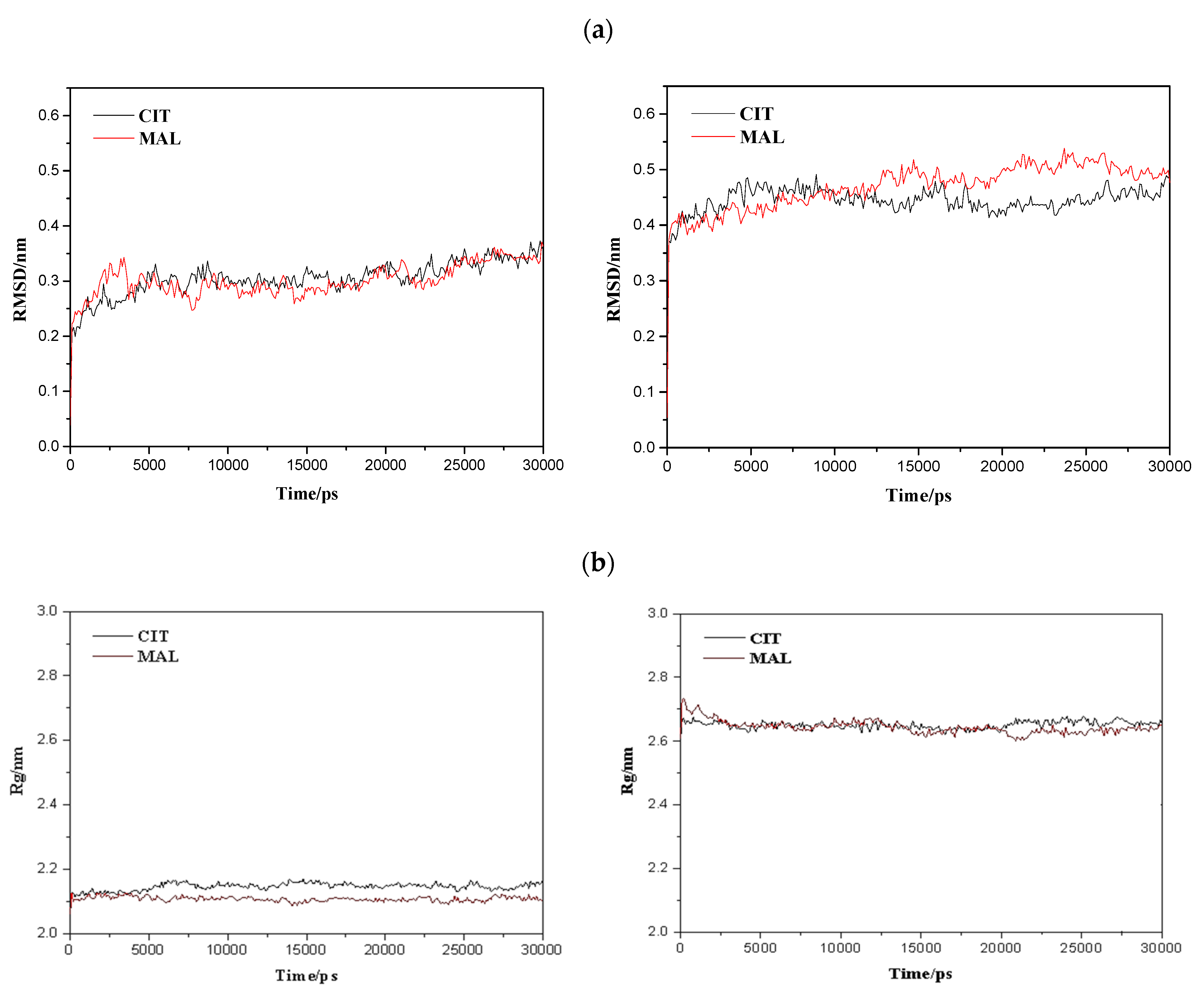
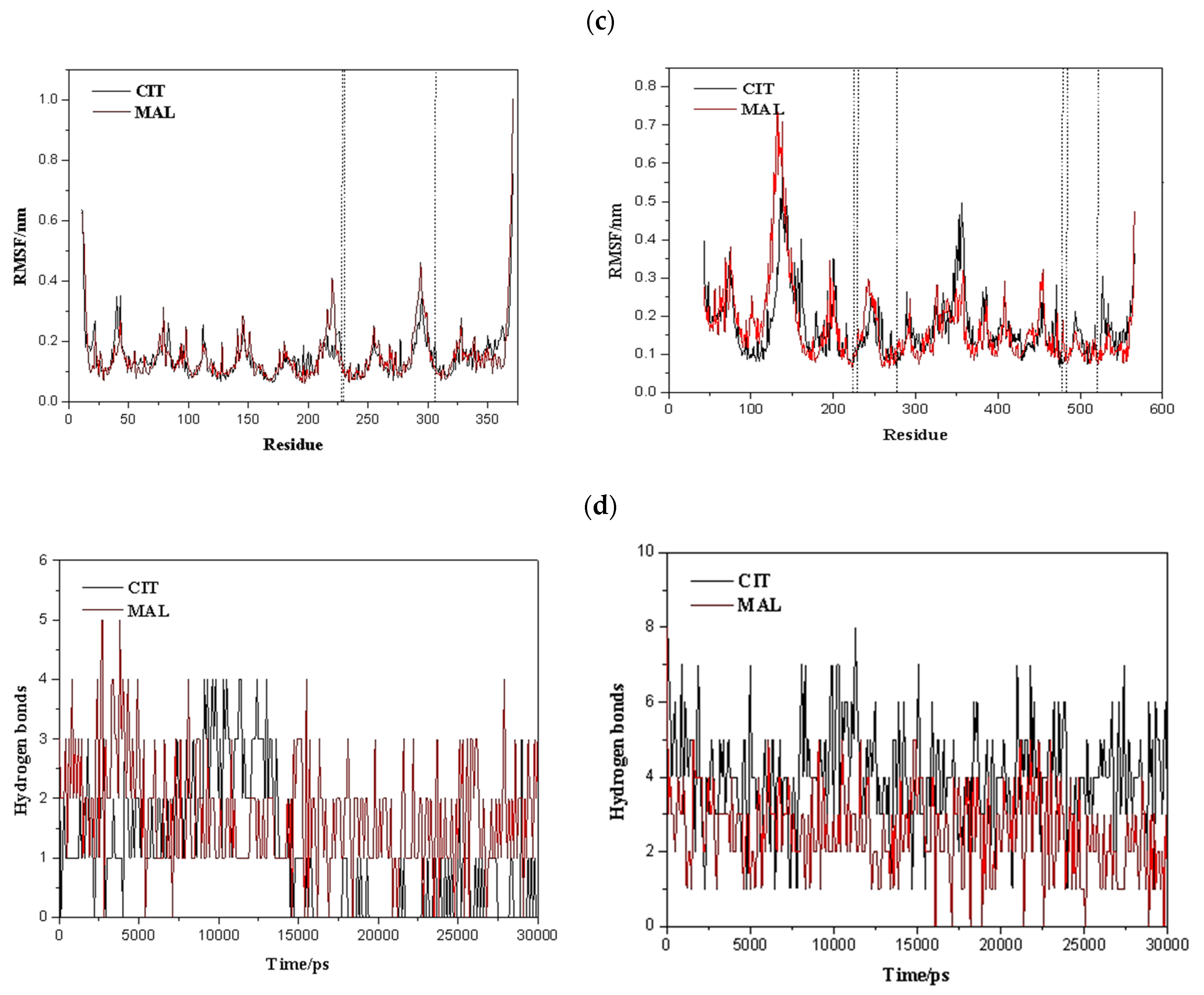
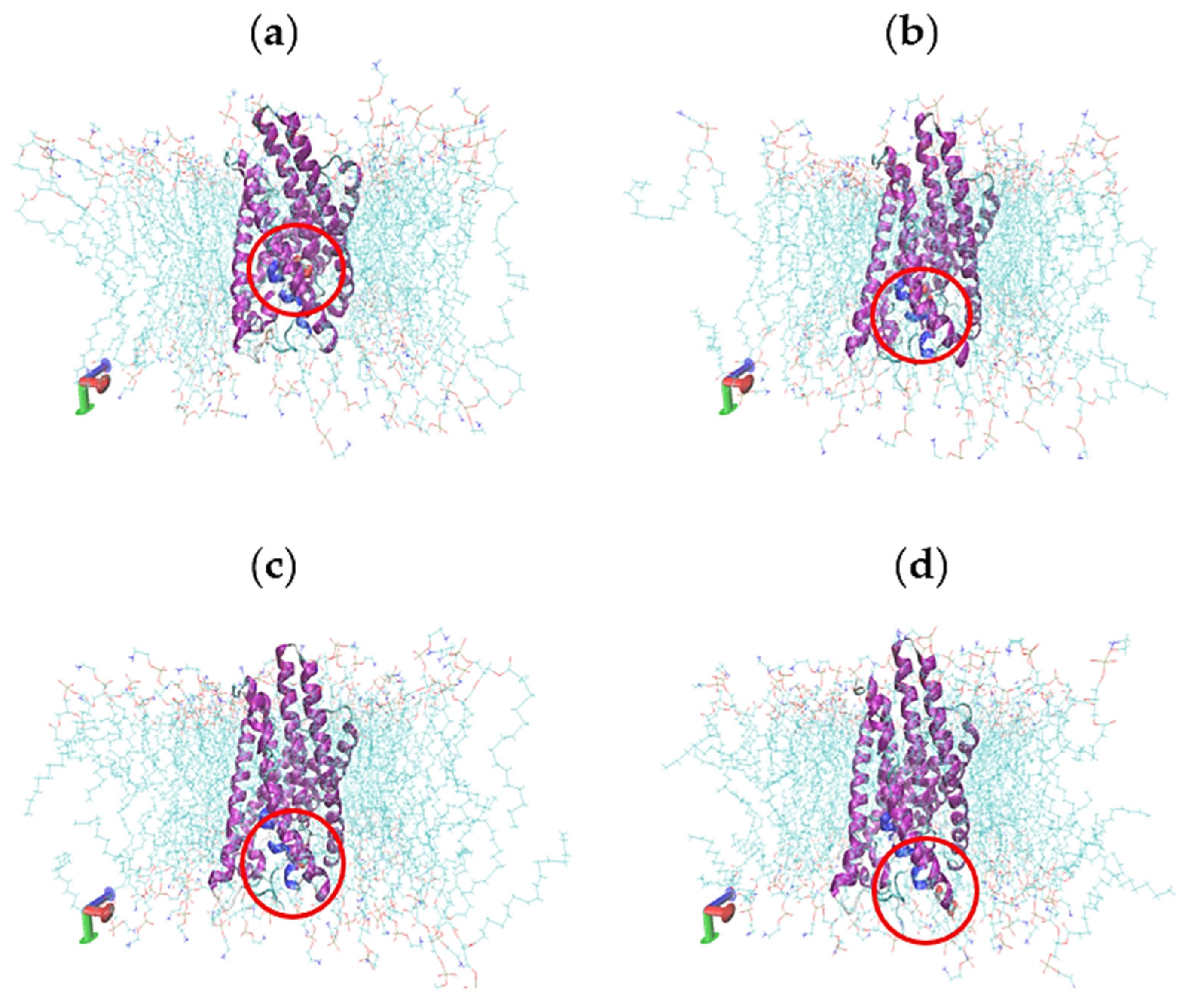
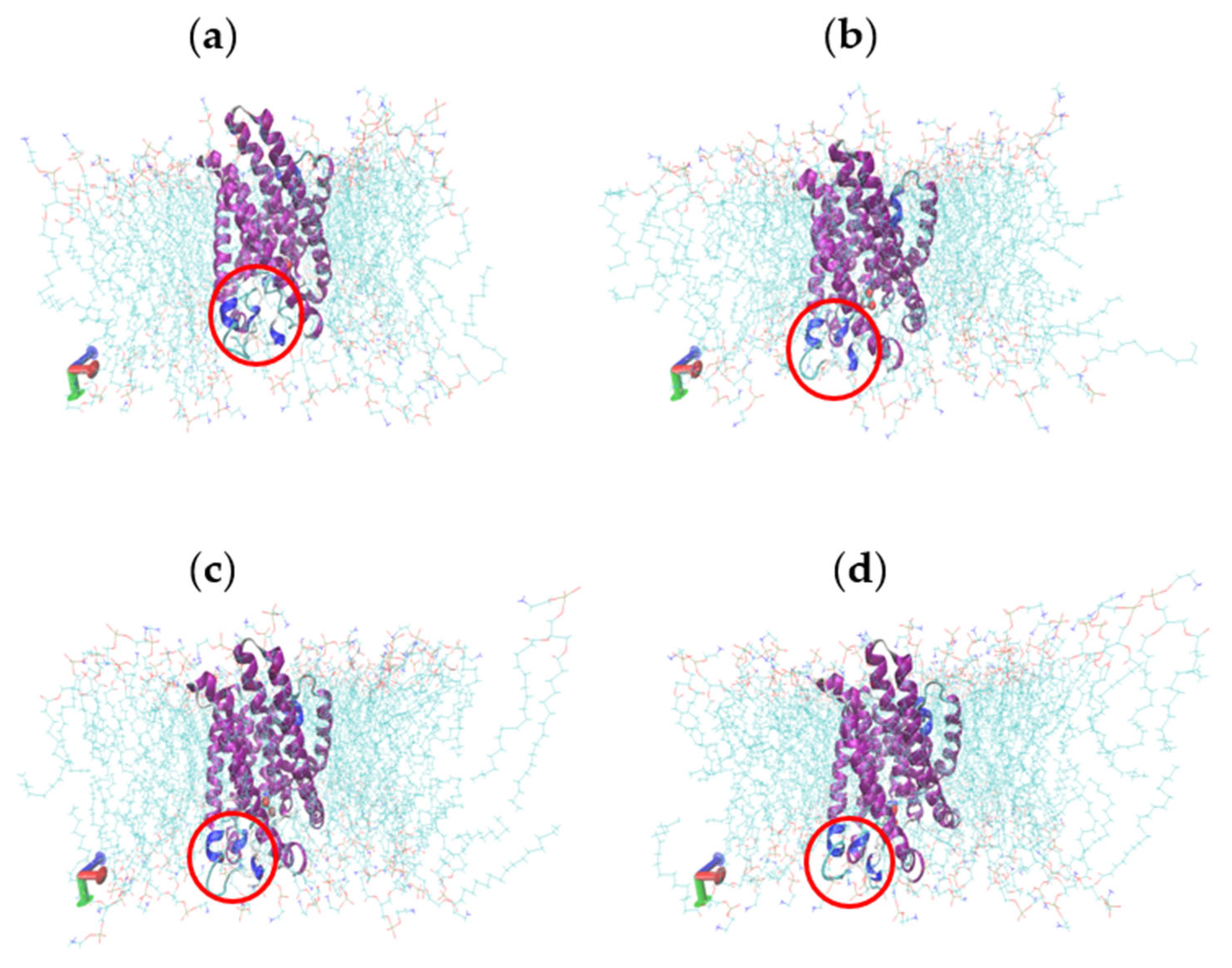
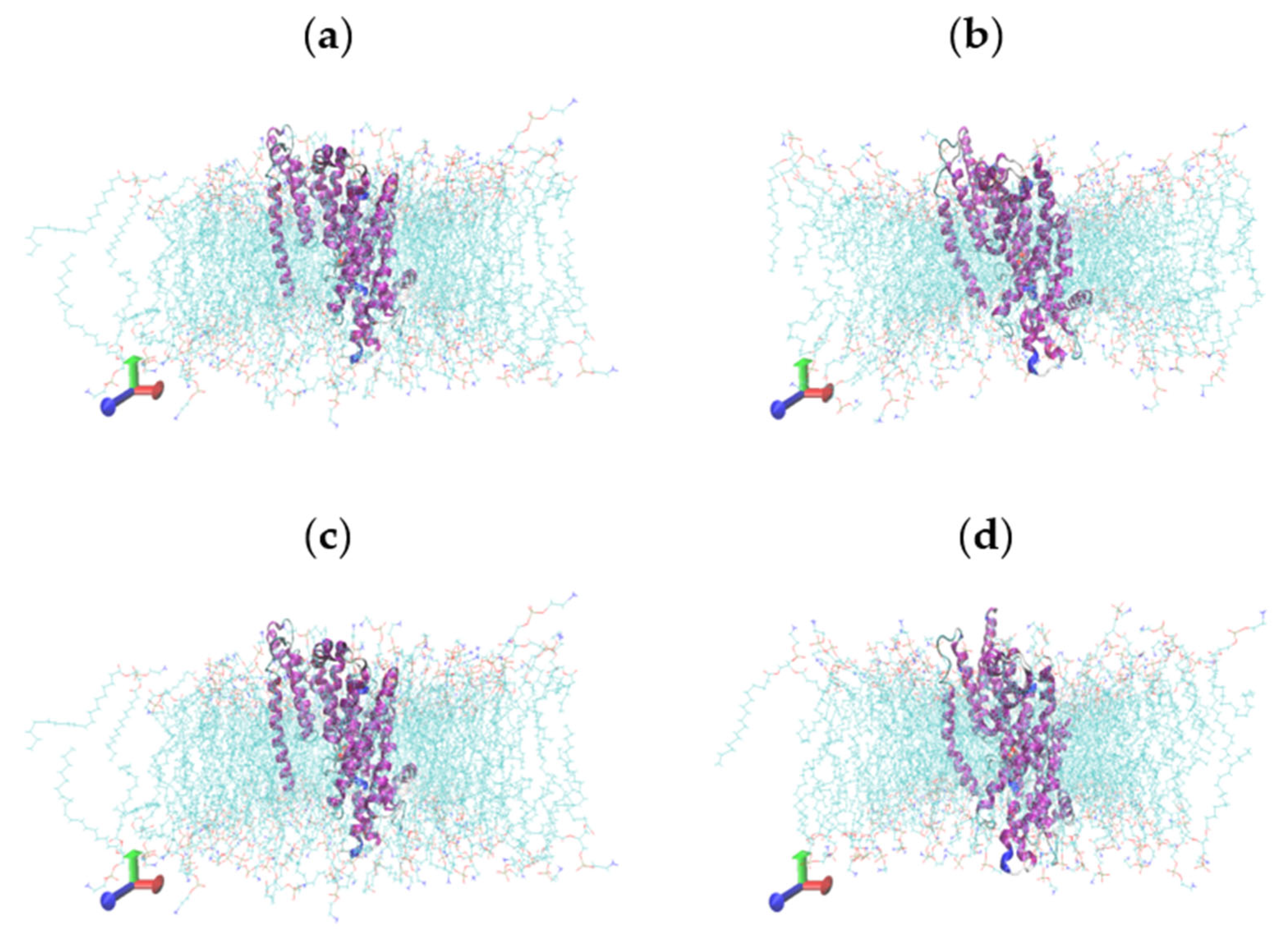
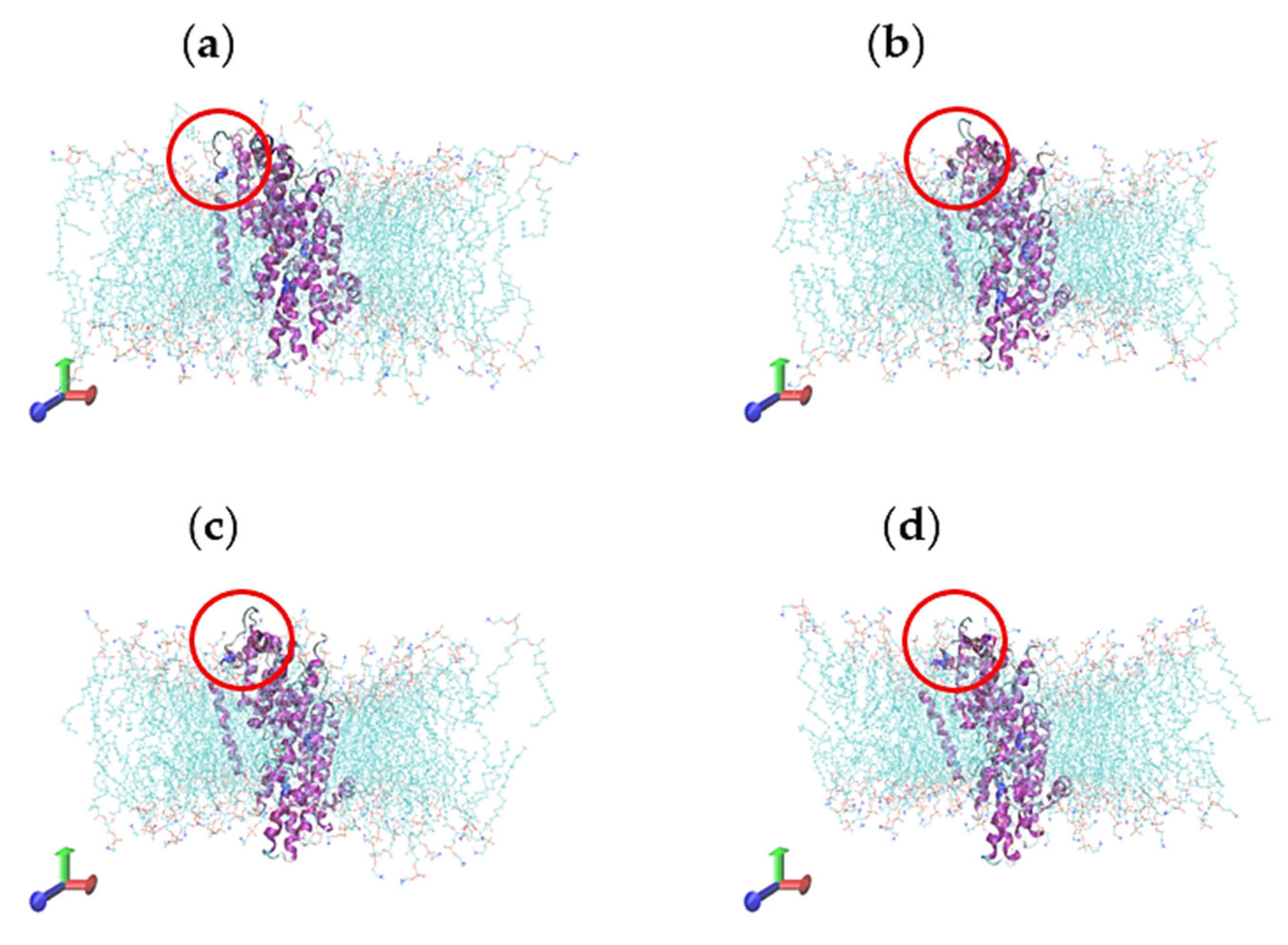
2.5. Molecular Dynamics Simulations
2.6. Characterisation of Conformational Study in Selected Proteins
2.7. Discussion
3. Materials and Methods
3.1. Nucleotide Sequences and Phylogenetic Analysis
3.2. Physico-Chemical Properties and Sequence Analysis
3.3. Homology Modelling and Validation
3.4. Molecular Docking
3.5. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ochsenreither, K.; Glück, C.; Stressler, T.; Fischer, L.; Syldatk, C. Production strategies and applications of microbial single cell oils. Front. Microbiol. 2016, 7, 1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, H.; El-Shanawany, A.R.; Shah, A.M.; Nazir, Y.; Naz, T.; Ullah, S.; Mustafa, K.; Song, Y. Comparative Analysis of Different Isolated Oleaginous Mucoromycota Fungi for Their γ-Linolenic Acid and Carotenoid Production. Biomed. Res. Int. 2020, 2020, 3621543. [Google Scholar] [CrossRef] [PubMed]
- Mhlongo, S.I.; Ezeokoli, O.T.; Roopnarain, A.; Ndaba, B.; Sekoai, P.T.; Habimana, O.; Pohl, C.H. The Potential of Single-Cell Oils Derived from Filamentous Fungi as Alternative Feedstock Sources for Biodiesel Production. Front. Microbiol. 2021, 12, 637381. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, S.; Kabir Khan, M.A.; Garre, V.; Vongsangnak, W.; Song, Y. Increased Lipid Accumulation in Mucor circinelloides by Overexpression of Mitochondrial Citrate Transporter Genes. Ind. Eng. Chem. Res. 2019, 58, 2125–2134. [Google Scholar] [CrossRef]
- Dong, X.; Chen, X.; Qian, Y.; Wang, Y.; Wang, L.; Qiao, W.; Liu, L. Metabolic engineering of Escherichia coli W3110 to produce L-malate. J. Biotechnol. Bioeng. 2017, 114, 656–664. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, H.; Zhang, Y.; Song, Y. 13C metabolic flux analysis on roles of malate transporter in lipid accumulation of Mucor circinelloides. Microb. Cell Fact. 2019, 18, 154. [Google Scholar] [CrossRef]
- Zhu, B.H.; Zhang, R.H.; Lv, N.N.; Yang, G.P.; Wang, Y.S.; Pan, K.H. The Role of Malic Enzyme on Promoting Total Lipid and Fatty Acid Production in Phaeodactylum tricornutum. Front. Plant Sci. 2018, 9, 826. [Google Scholar] [CrossRef] [Green Version]
- Hao, G.; Chen, H.; Du, K.; Huang, X.; Song, Y.; Gu, Z.; Wang, L.; Zhang, H.; Chen, W.; Chen, Y.Q. Increased fatty acid unsaturation and production of arachidonic acid by homologous over-expression of the mitochondrial malic enzyme in Mortierella alpina. Biotechnol. Lett. 2014, 36, 1827–1834. [Google Scholar] [CrossRef]
- Zhang, Y.; Adams, I.P.; Ratledge, C. Malic enzyme: The controlling activity for lipid production? Overexpression of malic enzyme in Mucor circinelloides leads to a 2.5-fold increase in lipid accumulation. Microbiology 2007, 153, 2013–2025. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Cánovas-Márquez, J.T.; Tang, X.; Chen, H.; Chen, Y.Q.; Chen, W.; Garre, V.; Song, Y.; Ratledge, C. Role of malate transporter in lipid accumulation of oleaginous fungus Mucor circinelloides. Appl. Microbiol. Biotechnol. 2016, 100, 1297–1305. [Google Scholar] [CrossRef]
- Huypens, P.; Pillai, R.; Sheinin, T.; Schaefer, S.; Huang, M.; Odegaard, M.L.; Ronnebaum, S.M.; Wettig, S.D.; Joseph, J.W. The dicarboxylate carrier plays a role in mitochondrial malate transport and in the regulation of glucose-stimulated insulin secretion from rat pancreatic beta cells. Diabetologia 2011, 54, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Khan MA, K.; Zhang, H.; Zhang, Y.; Certik, M.; Garre, V.; Song, Y. Mitochondrial Citrate Transport System in the Fungus Mucor circinelloides: Identification, Phylogenetic Analysis, and Expression Profiling During Growth and Lipid Accumulation. Curr. Microbiol. 2020, 77, 220–231. [Google Scholar] [CrossRef]
- Xie, Z.R.; Hwang, M.J. Methods for Predicting Protein–Ligand Binding Sites. In Molecular Modeling of Proteins; Kukol, A., Ed.; Methods in Molecular Biology, (Methods and Protocols); Humana Press: New York, NY, USA, 2015; Volume 1215. [Google Scholar] [CrossRef]
- Ykema, A.; Bakels, R.H.A.; Verwoert, I.I.G.S.; Smit, H.; van Verseveld, H.W. Growth yield, maintenance requirements and lipid formation in the oleaginous yeast Apiotrichum curvatum. Biotechnol. Bioeng. 1989, 34, 1268–1276. [Google Scholar] [CrossRef]
- Shah, A.M.; Hassan, M.; Zichen, Z.; Yuanda, S. Isolation, characterization and fatty acid analysis of Gilbertella persicaria DSR1: A potential new source of high value single-cell oil. Biomass Bioenergy 2021, 151, 106156. [Google Scholar] [CrossRef]
- Chutrakul, C.; Jeennor, S.; Panchanawaporn, S.; Cheawchanlertfa, P.; Suttiwattanakul, S.; Veerana, M.; Laoteng, K. Metabolic engineering of long chain-polyunsaturated fatty acid biosynthetic pathway in oleaginous fungus for dihomo-gamma linolenic acid production. J. Biotechnol. 2016, 218, 85–93. [Google Scholar] [CrossRef]
- Anantram, A.; Janve, M.; Degani, M.; Singhal, R.; Kundaikar, H. Homology modelling of human divalent metal transporter (DMT): Molecular docking and dynamic simulations for duodenal iron transport. J. Mol. Graph. Model. 2018, 85, 145–152. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef]
- Ikai, A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar] [CrossRef]
- Guruprasad, K.; Reddy, B.V.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef]
- Gill, S.C.; von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Bachmair, A.; Finley, D.; Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986, 234, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Gholampour-Faroji, N.; Farazmand, R.; Hemmat, J.; Haddad-Mashadrizeh, A. Modeling, stability and the activity assessment of glutathione reductase from Streptococcus Thermophilus; Insights from the in-silico simulation study. Comput. Biol. Chem. 2019, 83, 107121. [Google Scholar] [CrossRef]
- Kumar, P.N.; Swapna, T.H.; Khan, M.Y.; Daddam, J.R.; Hameeda, B. Molecular dynamics and protein interaction studies of lipopeptide (Iturin A) on α- amylase of Spodoptera litura. J. Theor. Biol. 2017, 415, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Jayasimha, R.; Basha, S.; Kotha, P.; Katike, U. Designing, docking and molecular dynamics simulation studies of novel cloperastine analogues as anti-allergic agents: Homology modeling and active site prediction for the human histamine H1 receptor. RSC Adv. 2020, 10, 4745–4754. [Google Scholar] [CrossRef] [Green Version]
- Xun, S.; Jiang, F.; Wu, Y.-D. Significant refinement of protein structure models using a residue-specific force field. J. Chem. Theory Comput. 2015, 11, 1949–1956. [Google Scholar] [CrossRef]
- Pasandideh, R.; Dadmanesh, M.; Khalili, S. In silico molecular modeling and docking studies on the Leishmania mitochondrial iron transporter-1 (LMIT1). Comp. Clin. Pathol. 2020, 29, 115–125. [Google Scholar] [CrossRef]
- Boyd, A.; Ciufo, L.F.; Barclay, J.W.; Graham, M.E.; Haynes, L.P.; Doherty, M.K.; Riesen, M.; Burgoyne, R.D.; Morgan, A. A random mutagenesis approach to isolate dominant-negative yeast sec1 mutants reveals a functional role for domain 3a in yeast and mammalian Sec1/Munc18 proteins. Genetics 2008, 180, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Brender, J.R.; Zhang, J.; Zhang, Y. Protein structure prediction provides comparable performance to crystallographic structures in docking-based virtual screening. Methods 2015, 71, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Jianyi, Y.; Yang, Z. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Skolnick, J.; Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, R.; Jain, S.K.; Shukla, P.K. In-silico characterization of β-(1,3)-endoglucanase (ENGL1) from Aspergillus fumigatus by homology modeling and docking studies. Bioinformation 2013, 9, 802–807. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.L.; MacArthur, M.W.; Hutchinson, E.G.; Thornton, J.M. Stereochemical quality of protein structure coordinates. Proteins Struct. Funct. Bioinf. 1992, 12, 345–364. [Google Scholar] [CrossRef]
- Sariyer, E.; Yakarsonmez, S.; Danis, O.; Turgut-Balik, D. A study of Bos taurus muscle specific enolase; biochemical characterization, homology modelling and investigation of molecular interaction using molecular docking and dynamics simulations. Int. J. Biol. Macromol. 2018, 120, 2346–2353. [Google Scholar] [CrossRef]
- Skariyachan, S.; Ravishankar, R.; Gopal, D.; Muddebihalkar, A.G.; Uttarkar, A.; Praveen PK, U.; Niranjan, V. Response regulator GacA and transcriptional activator RhlR proteins involved in biofilm formation of Pseudomonas aeruginosa are prospective targets for natural lead molecules: Computational modelling, molecular docking and dynamic simulation studies. Infect. Genet. Evol. 2020, 85, 104448. [Google Scholar] [CrossRef]
- Kopp, J.; Schwede, T. Automated protein structure homology modeling: A progress report. Pharmacogenomics 2004, 5, 405–416. [Google Scholar] [CrossRef]
- Floudas, C.; Fung, H.; McAllister, S.; Mönnigmann, M.; Rajgaria, R. Advances in protein structure prediction and de novo protein design: A review. Chem. Eng. Sci. 2006, 61, 966–988. [Google Scholar] [CrossRef]
- Ankita, S.; Ravi, K.G.; Chikati, R.; Medicherla, V.J. Homology modeling and molecular docking of heme peroxidase from Euphorbia tirucalli: Substrate specificity and thiol inhibitor interactions. J. Mole. Liqu. 2016, 220, 383–394. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Haspel, N.; Moll, M.; Baker, M.L.; Chiu, W.; Kavraki, L.E. Tracing conformational changes in proteins. BMC Str. Biol. 2010, 10, S1. [Google Scholar] [CrossRef] [Green Version]
- Kolb, P.; Irwin, J.J. Docking screens: Right for the right reasons? Curr. Top. Med. Chem. 2009, 9, 755–770. [Google Scholar] [CrossRef] [Green Version]
- Ul-Haq, Z.; Saeed, M.; Halim, S.A.; Khan, W. 3D Structure Prediction of Human β1-Adrenergic Receptor via Threading-Based Homology Modeling for Implications in Structure-Based Drug Designing. PLoS ONE 2015, 10, e0122223. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, 320–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptors | Binding Energy with Citrate (Binding Energy, kcal/mol) | Binding Energy with Malate (Binding Energy, kcal/mol) |
|---|---|---|
| MT | −3.44 | −2.85 |
| Sodit-a | −7.27 | −6.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, W.; Mohamed, H.; Shah, A.M.; Zhang, H.; Pang, S.; Shi, W.; Xue, F.; Song, Y. In Silico Structural and Functional Analysis of the Mitochondrial Malate Transporters in Oleaginous Fungus Mucor circinelloides WJ11. Catalysts 2023, 13, 705. https://doi.org/10.3390/catal13040705
Yang W, Mohamed H, Shah AM, Zhang H, Pang S, Shi W, Xue F, Song Y. In Silico Structural and Functional Analysis of the Mitochondrial Malate Transporters in Oleaginous Fungus Mucor circinelloides WJ11. Catalysts. 2023; 13(4):705. https://doi.org/10.3390/catal13040705
Chicago/Turabian StyleYang, Wu, Hassan Mohamed, Aabid Manzoor Shah, Huaiyuan Zhang, Shuxian Pang, Wenyue Shi, Futing Xue, and Yuanda Song. 2023. "In Silico Structural and Functional Analysis of the Mitochondrial Malate Transporters in Oleaginous Fungus Mucor circinelloides WJ11" Catalysts 13, no. 4: 705. https://doi.org/10.3390/catal13040705