
Multifunctional Enzymes in Microbial Secondary Metabolic Processes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Multifunctional Synthases in the Synthesis of Secondary Metabolic Processes
2.1. Multifunctional Polyketide Synthases
2.2. Multifunctional Non-Ribosomal Peptide Synthase
2.3. Multifunctional Terpene Synthases
3. Multifunctional Post-Modifying Enzymes in the Synthesis of Secondary Metabolic Processes
3.1. Multifunctional Cytochrome P450
3.2. Other Oxygenases
3.3. Post-Modifying Enzymes in the Synthesis of Lanthipeptides
3.4. Other Multifunctional Modifying Enzymes
4. Future Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Stark, G.R. Multifunctional proteins: One gene—More than one enzyme. Trends Biochem. Sci. 1977, 2, 64–66. [Google Scholar] [CrossRef]
- Hopwood, D.A. Genetic contributions to understanding polyketide synthases. Chem. Rev. 1997, 97, 2465–2498. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Macdonald, T.L. Chemical mechanisms of catalysis by cytochromes-P-450-a unified view. Acc. Chem. Res. 1984, 17, 9–16. [Google Scholar] [CrossRef]
- Grunewald, J.; Marahiel, M.A. Chemoenzymatic and template-directed synthesis of bioactive macrocyclic peptides. Microbiol. Mol. Biol. Rev. 2006, 70, 121–146. [Google Scholar] [CrossRef] [Green Version]
- Velkov, T.; Lawen, A. Non-ribosomal peptide synthetases as technological platforms for the synthesis of highly modified peptide bioeffectors--Cyclosporin synthetase as a complex example. Biotechnol. Annu Rev. 2003, 9, 151–197. [Google Scholar] [CrossRef]
- Koglin, A.; Walsh, C.T. Structural insights into nonribosomal peptide enzymatic assembly lines. Nat. Prod. Rep. 2009, 26, 987–1000. [Google Scholar] [CrossRef] [Green Version]
- Christianson, D.W. Structural and chemical biology of terpenoid cyclases. Chem. Rev. 2017, 117, 11570–11648. [Google Scholar] [CrossRef] [Green Version]
- Shen, B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 2003, 7, 285–295. [Google Scholar] [CrossRef]
- Sultana, A.; Kallio, P.; Jansson, A.; Wang, J.S.; Niemi, J.; Mantsala, P.; Schneider, G. Structure of the polyketide cyclase SnoaL reveals a novel mechanism for enzymatic aldol condensation. EMBO J. 2004, 23, 1911–1921. [Google Scholar] [CrossRef] [Green Version]
- Chooi, Y.H.; Tang, Y. Navigating the fungal polyketide chemical space: From genes to molecules. J. Org. Chem. 2012, 77, 9933–9953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piel, J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat. Prod. Rep. 2010, 27, 996–1047. [Google Scholar] [CrossRef] [PubMed]
- Conti, E.; Stachelhaus, T.; Marahiel, M.A.; Brick, P. Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 1997, 16, 4174–4183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, J.J.; Kessler, N.; Marahiel, M.A.; Stubbs, M.T. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. USA 2002, 99, 12120–12125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachelhaus, T.; Mootz, H.D.; Marahiel, M.A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. CHEM Biol. 1999, 6, 493–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachelhaus, T.; Marahiel, M.A. Modular structure of peptide synthetases revealed by dissection of the multifunctional enzyme GrsA. J. Biol. Chem. 1995, 270, 6163–6169. [Google Scholar] [CrossRef] [Green Version]
- Weissman, K.J. The structural biology of biosynthetic megaenzymes. Nat. Chem. Biol. 2015, 11, 660–670. [Google Scholar] [CrossRef]
- Bevitt, D.J.; Cortes, J.; Haydock, S.F.; Leadlay, P.F. 6-deoxyerythronolide-b synthase-2 from Saccharopolyspora-erythraea-cloning of the structural gene, sequence-analysis and inferred domain-structure of the multifunctional enzyme. Eur. J. Biochem. 1992, 204, 39–49. [Google Scholar] [CrossRef]
- Cortes, J.; Haydock, S.F.; Roberts, G.A.; Bevitt, D.J.; Leadlay, P.F. An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora-erythraea. Nature 1990, 348, 176–178. [Google Scholar] [CrossRef]
- Donadio, S.; Katz, L. Organization of the enzymatic domains in the multifunctional polyketide synthase involved in erythromycin formation in Saccharopolyspora-erythraea. Gene 1992, 111, 51–60. [Google Scholar] [CrossRef]
- Donadio, S.; Staver, M.J.; McAlpine, J.B.; Swanson, S.J.; Katz, L. Modular organization of genes required for complex polyketide biosynthesis. Science 1991, 252, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Cane, D.E.; Khosla, C. Structure-based dissociation of a type I polyketide synthase module. CHEM Biol. 2007, 14, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nivina, A.; Yuet, K.P.; Hsu, J.; Khosla, C. Evolution and diversity of assembly-line polyketide synthases. Chem. Rev. 2019, 119, 12524–12547. [Google Scholar] [CrossRef] [PubMed]
- Stachelhaus, T.; Huser, A.; Marahiel, M.A. Biochemical characterization of peptides carrier protein (PCP), the thiolation domain of multifunctional peptide synthetases. CHEM Biol. 1996, 3, 913–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- vonDohren, H.; Keller, U.; Vater, J.; Zocher, R. Multifunctional peptide synthetases. Chem. Rev. 1997, 97, 2675–2705. [Google Scholar] [CrossRef]
- Stachelhaus, T.; Mootz, H.D.; Bergendahl, V.; Marahiel, M.A. Peptide bond formation in nonribosomal peptide biosynthesis-Catalytic role of the condensation domain. J. Biol. Chem. 1998, 273, 22773–22781. [Google Scholar] [CrossRef] [Green Version]
- Finking, R.; Marahiel, M.A. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 2004, 58, 453–488. [Google Scholar] [CrossRef]
- Takeda, K.; Kemmoku, K.; Satoh, Y.; Ogasawara, Y.; Shin-ya, K.; Dairi, T. N-phenylacetylation and nonribosomal peptide synthetases with substrate promiscuity for biosynthesis of heptapeptide variants, JBIR-78 and JBIR-95. ACS Chem. Biol. 2017, 12, 1813–1819. [Google Scholar] [CrossRef]
- Miller, B.R.; Gulick, A.M. Structural biology of nonribosomal peptide synthetases. Methods Mol. Biol. 2016, 1401, 3–29. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.T. Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat. Prod. Rep. 2016, 33, 127–135. [Google Scholar] [CrossRef]
- Marahiel, M.A. A structural model for multimodular NRPS assembly lines. Nat. Prod. Rep. 2016, 33, 136–140. [Google Scholar] [CrossRef] [PubMed]
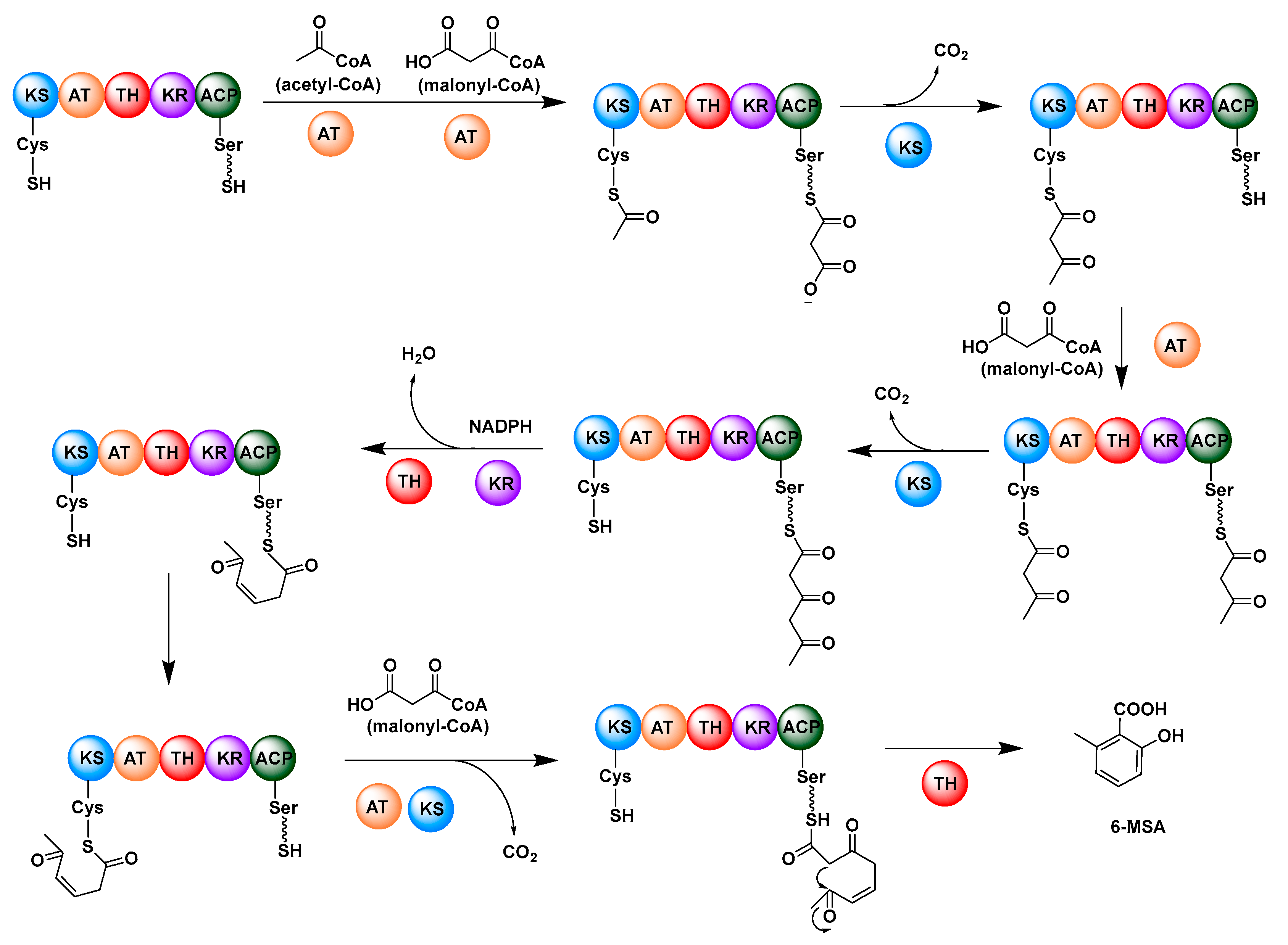
- Spencer, J.B.; Jordan, P.M. Purification and properties of 6-methylsalicylic acid synthase from Penicillium patulum. Biochem. J. 1992, 288, 839–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, J.; Ripka, S.; Siegner, A.; Schiltz, E.; Schweizer, E. The multifunctional 6-methylsalicylic acid synthase gene of Penicillium-patulum-its gene structure relative to that of other polyketide synthases. Eur. J. Biochem. 1990, 192, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Dimroth, P.; Walter, H.; Lynen, F. Biosynthesis of 6-methylsalicylic acid. Eur. J. Biochem. 1970, 13, 98–110. [Google Scholar] [CrossRef]
- Parascandolo, J.S.; Havemann, J.; Potter, H.K.; Huang, F.; Riva, E.; Connolly, J.; Wilkening, I.; Song, L.; Leadlay, P.F.; Tosin, M. Insights into 6-methylsalicylic acid bio-assembly by using chemical probes. Angew. Chem. 2016, 55, 3463–3467. [Google Scholar] [CrossRef] [Green Version]
- Child, C.J.; Spencer, J.B.; Bhogal, P.; ShoolinginJordan, P.M. Structural similarities between 6-methylsalicylic acid synthase from Penicillium patulum and vertebrate type I fatty acid synthase: Evidence from thiol modification studies. Biochemistry 1996, 35, 12267–12274. [Google Scholar] [CrossRef]
- Nicholson, T.P.; Rudd, B.A.M.; Dawson, M.; Lazarus, C.M.; Simpson, T.J.; Cox, R.J. Design and utility of oligonucleotide gene probes for fungal polyketide synthases. CHEM Biol. 2001, 8, 157–178. [Google Scholar] [CrossRef] [Green Version]
- Fujii, I.; Watanabe, A.; Sankawa, U.; Ebizuka, Y. Identification of Claisen cyclase domain in fungal polyketide synthase WA, a naphthopyrone synthase of Aspergillus nidulans. CHEM Biol. 2001, 8, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Bailey, A.M.; Cox, R.J.; Harley, K.; Lazarus, C.M.; Simpson, T.J.; Skellam, E. Characterisation of 3-methylorcinaldehyde synthase (MOS) in Acremonium strictum: First observation of a reductive release mechanism during polyketide biosynthesis. Chem. Commun. 2007, 39, 4053–4055. [Google Scholar] [CrossRef]
- Moriguchi, T.; Kezuka, Y.; Nonaka, T.; Ebizuka, Y.; Fujii, I. Hidden function of catalytic domain in 6-methylsalicylic acid synthase for product release. J. Biol. Chem. 2010, 285, 15637–15643. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, A.; Muntwyler, R.; Kellerschierlein, W. Metabolic products of microorganisms.147. unexpected transformation in chlorothricin series. Helv. Chim. Acta. 1975, 58, 1323–1338. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.Y.; Tian, Z.H.; Shao, L.; Qu, X.D.; Zhao, Q.F.; Tang, J.; Tang, G.L.; Liu, W. Genetic characterization of the chlorothricin gene cluster as a model for spirotetronate antibiotic biosynthesis. Chem. Biol. 2006, 13, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, L.; Qu, X.D.; Jia, X.Y.; Zhao, Q.F.; Tian, Z.H.; Wang, M.; Tang, G.L.; Liu, W. Cloning and characterization of a bacterial iterative type I polyketide synthase gene encoding the 6-methylsalicyclic acid synthase. Biochem. Biophys. Res. Commun. 2006, 345, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Van Lanen, S.G.; Oh, T.J.; Liu, W.; Wendt-Pienkowski, E.; Shen, B. Characterization of the maduropeptin biosynthetic gene cluster from Actinomadura madurae ATCC 39144 supporting a unifying paradigm for enediyne biosynthesis. J. Am. Chem. Soc. 2007, 129, 13082–13094. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Roongsawang, N.; Shirasaka, N.; Lu, W.L.; Flatt, P.M.; Kasanah, N.; Miranda, C.; Mahmud, T. Deciphering pactamycin biosynthesis and engineered production of new pactamycin analogues. ChemBioChem 2009, 10, 2253–2265. [Google Scholar] [CrossRef]
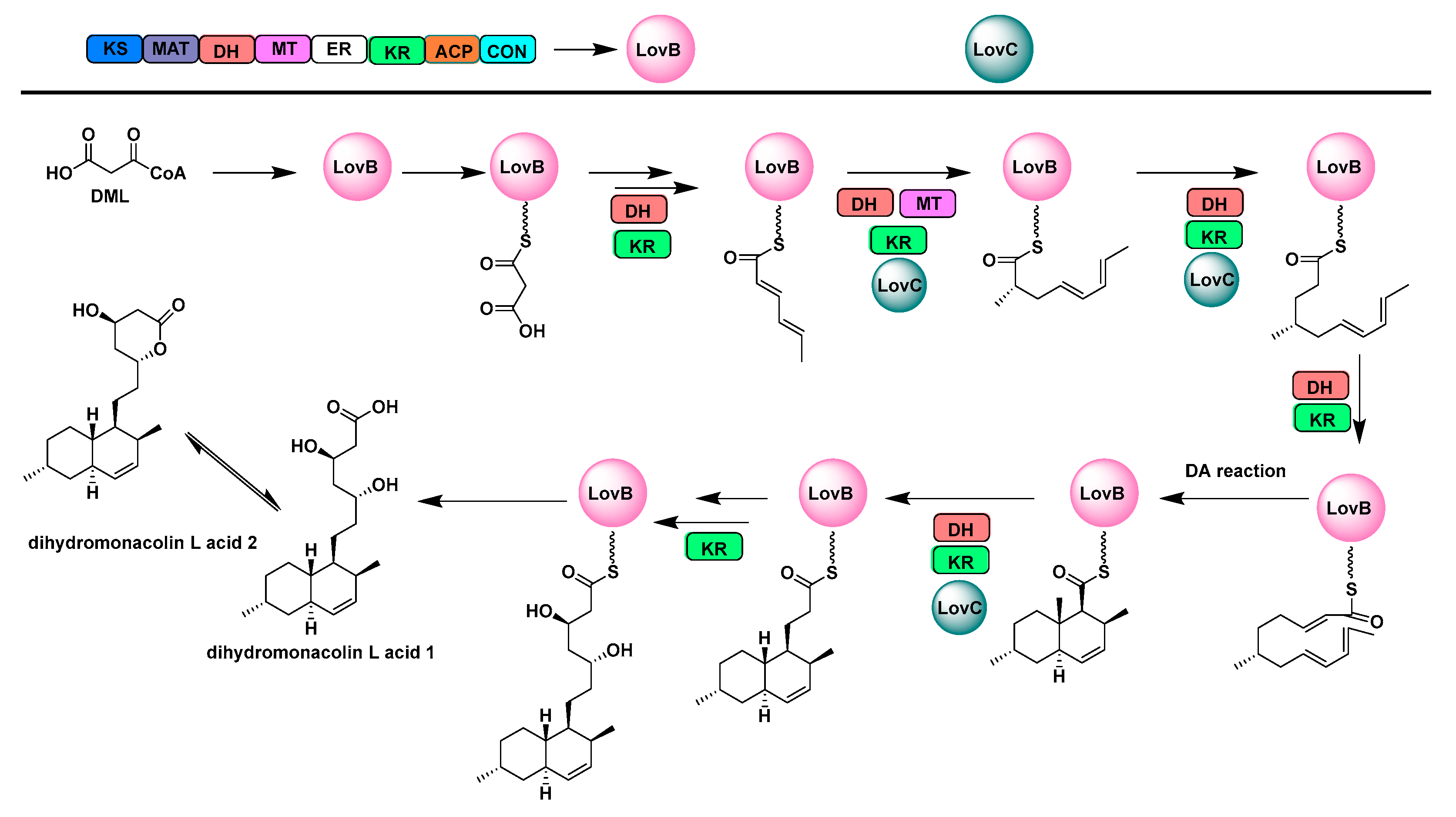
- Wang, J.L.; Liang, J.D.; Chen, L.; Zhang, W.; Kong, L.L.; Peng, C.; Su, C.; Tang, Y.; Deng, Z.X.; Wang, Z.J. Structural basis for the biosynthesis of lovastatin. Nat. Commun. 2021, 12, 867. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Auclair, K.; Kendrew, S.G.; Park, C.; Vederas, J.C.; Hutchinson, C.R. Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 1999, 284, 1368–1372. [Google Scholar] [CrossRef]
- Ma, S.M.; Li, J.W.H.; Choi, J.W.; Zhou, H.; Lee, K.K.M.; Moorthie, V.A.; Xie, X.K.; Kealey, J.T.; Da Silva, N.A.; Vederas, J.C.; et al. Complete reconstitution of a highly reducing iterative polyketide synthase. Science 2009, 326, 589–592. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, L.; Davis, C.R.; Roach, C.; Nguyen, D.K.; Aldrich, T.; McAda, P.C.; Reeves, C.D. Lavastatin biosynthesis in Aspergillus terreus: Characterization of blocked mutants, enzyme activities and a multifunctional polyketide synthase gene. CHEM Biol. 1999, 6, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Chooi, Y.H.; Choi, J.W.; Li, S.; Vederas, J.C.; Da Silva, N.A.; Tang, Y. LovG: The thioesterase required for dihydromonacolinl release and lovastatin nonaketide synthase turnover in lovastatin biosynthesis. Angew. Chem. 2013, 52, 6472–6475. [Google Scholar] [CrossRef] [Green Version]
- Ahlert, J.; Shepard, E.; Lomovskaya, N.; Zazopoulos, E.; Staffa, A.; Bachmann, B.O.; Huang, K.X.; Fonstein, L.; Czisny, A.; Whitwam, R.E.; et al. The calicheamicin gene cluster and its iterative type I enediyne PKS. Science 2002, 297, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Christenson, S.D.; Standage, S.; Shen, B. Biosynthesis of the enediyne antitumor antibiotic C-1027. Science 2002, 297, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Sthapit, B.; Oh, T.J.; Lamichhane, R.; Liou, K.; Lee, H.C.; Kim, C.G.; Sohng, J.K. Neocarzinostatin naphthoate synthase: An unique iterative type I PKS from neocarzinostatin producer Streptomyces carzinostaticus. FEBS Lett. 2004, 566, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.M.; Challis, G.L. Recent advances in siderophore biosynthesis. Curr. Opin. Chem. Biol. 2009, 13, 205–215. [Google Scholar] [CrossRef]
- Hai, Y.; Jenner, M.; Tang, Y. Fungal siderophore biosynthesis catalysed by an iterative nonribosomal peptide synthetase. Chem. Sci. 2020, 11, 11525–11530. [Google Scholar] [CrossRef] [PubMed]
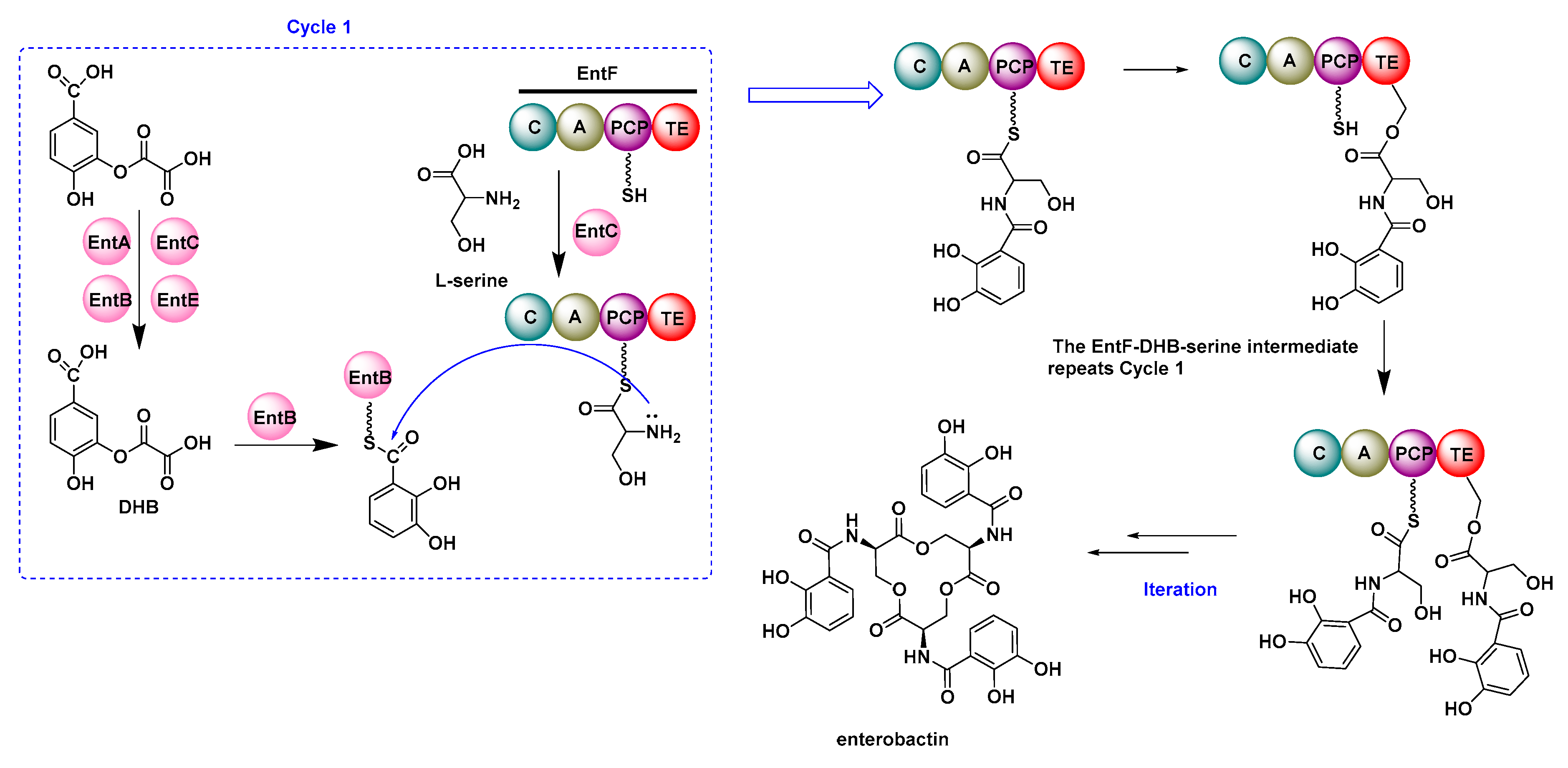
- Raymond, K.N.; Dertz, E.A.; Kim, S.S. Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. USA 2003, 100, 3584–3588. [Google Scholar] [CrossRef] [Green Version]
- Gehring, A.M.; Bradley, K.A.; Walsh, C.T. Enterobactin biosynthesis in Escherichia coli: Isochorismate lyase (EntB) is a bifunctional enzyme that is phosphopantetheinylated by EntD and then acylated by EntE using ATP and 2,3-dihydroxybenzoate. Biochemistry 1997, 36, 8495–8503. [Google Scholar] [CrossRef]
- Reichert, J.; Sakaitani, M.; Walsh, C.T. Characterization of EntF as a serine-activating enzyme. Protein Sci. 1992, 1, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Drake, E.J.; Nicolai, D.A.; Gulick, A.M. Structure of the EntB multidomain nonribosomal peptide synthetase and functional analysis of its interaction with the EntE adenylation domain. CHEM Biol. 2006, 13, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Shaw-Reid, C.A.; Kelleher, N.L.; Losey, H.C.; Gehring, A.M.; Berg, C.; Walsh, C.T. Assembly line enzymology by multimodular nonribosomal peptide synthetases: The thioesterase domain of E-coli EntF catalyzes both elongation and cyclolactonization. CHEM Biol. 1999, 6, 385–400. [Google Scholar] [CrossRef] [Green Version]
- Haese, A.; Schubert, M.; Herrmann, M.; Zocher, R. Molecular characterization of the enniatin synthetase gene encoding a multifunctional enzyme catalyzing n-methyldepsipeptide formation in Fusarium-scirpi. Mol. Microbiol. 1993, 7, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Q. Deciphering the biosynthetic codes for the potent anti-SARS-CoV cyclodepsipeptide valinomycin in Streptomyces tsusimaensis ATCC 15141. ChemBioChem 2006, 7, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Hotta, K.; Praseuth, A.P.; Koketsu, K.; Migita, A.; Boddy, C.N.; Wang, C.C.C.; Oguri, H.; Oikawa, H. Total biosynthesis of antitumor nonribosomal peptides in Escherichia coli. Nat. Chem. Biol. 2006, 2, 423–428. [Google Scholar] [CrossRef]
- Kohli, R.M.; Trauger, J.W.; Schwarzer, D.; Marahiel, M.A.; Walsh, C.T. Generality of peptide cyclization catalyzed by isolated thioesterase domains of nonribosomal peptide synthetases. Biochemistry 2001, 40, 7099–7108. [Google Scholar] [CrossRef]
- Hoyer, K.M.; Mahlert, C.; Marahiel, M.A. The iterative gramicidin S thioesterase catalyzes peptide ligation and cyclization. CHEM Biol. 2007, 14, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Koketsu, K.; Minami, A.; Watanabe, K.; Oguri, H.; Oikawa, H. Pictet-Spenglerase involved in tetrahydroisoquinoline antibiotic biosynthesis. Curr. Opin. Chem. Biol. 2012, 16, 142–149. [Google Scholar] [CrossRef]
- Mitsuhashi, T.; Abe, I. Chimeric terpene synthases possessing both terpene cyclization and prenyltransfer activities. ChemBioChem 2018, 19, 1106–1114. [Google Scholar] [CrossRef]
- Toyomasu, T.; Tsukahara, M.; Kaneko, A.; Niida, R.; Mitsuhashi, W.; Dairi, T.; Kato, N.; Sassa, T. Fusicoccins are biosynthesized by an unusual chimera diterpene synthase in fungi. Proc. Natl. Acad. Sci. USA 2007, 104, 3084–3088. [Google Scholar] [CrossRef] [Green Version]
- Srere, P.A. Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 1987, 56, 89–124. [Google Scholar] [CrossRef]
- Miles, E.W.; Rhee, S.; Davies, D.R. The molecular basis of substrate channeling. J. Biol. Chem. 1999, 274, 12193–12196. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.Y.; Holden, H.M.; Raushel, F.M. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu. Rev. Biochem. 2001, 70, 149–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faylo, J.L.; Ronnebaum, T.A.; Christianson, D.W. Assembly-line catalysis in bifunctional terpene synthases. Acc. Chem. Res. 2021, 54, 3780–3791. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.B.; Chou, W.K.W.; Toyomasu, T.; Cane, D.E.; Christianson, D.W. Structure and function of fusicoccadiene synthase, a hexameric bifunctional diterpene synthase. ACS Chem. Biol. 2016, 11, 889–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuhashi, T.; Okada, M.; Abe, I. Identification of chimeric alpha beta gamma diterpene synthases possessing both Type II terpene cyclase and prenyltransferase activities. ChemBioChem 2017, 18, 2104–2109. [Google Scholar] [CrossRef] [PubMed]
- Ronnebaum, T.A.; Eaton, S.A.; Brackhahn, E.A.E.; Christianson, D.W. Engineering the prenyltransferase domain of a bifunctional assembly-line terpene synthase. Biochemistry 2021, 60, 3162–3172. [Google Scholar] [CrossRef]
- Yuan, W.; Lv, S.; Chen, L.Y.; Zhao, Y.; Deng, Z.X.; Hong, K. Production of sesterterpene ophiobolin by a bifunctional terpene synthase in Escherichia coli. Appl. Microbiol. Biotechnol. 2019, 103, 8785–8797. [Google Scholar] [CrossRef]
- Ronnebaum, T.A.; Gupta, K.; Christianson, D.W. Higher-order oligomerization of a chimeric alpha beta gamma bifunctional diterpene synthase with prenyltransferase and class II cyclase activities is concentration-dependent. J. Struct. Biol. 2020, 210, 107463. [Google Scholar] [CrossRef]
- Chang, T.H.; Guo, R.T.; Ko, T.P.; Wang, A.H.J.; Liang, P.H. Crystal structure of type-III geranylgeranyl pyrophosphate synthase from Saccharomyces cerevisiae and the mechanism of product chain length determination. J. Biol. Chem. 2006, 281, 14991–15000. [Google Scholar] [CrossRef] [Green Version]
- Andersen-Ranberg, J.; Kongstad, K.T.; Nielsen, M.T.; Jensen, N.B.; Pateraki, I.; Bach, S.S.; Hamberger, B.; Zerbe, P.; Staerk, D.; Bohlmann, J.; et al. Expanding the landscape of diterpene structural diversity through stereochemically controlled combinatorial biosynthesis. Angew. Chem. 2016, 55, 2142–2146. [Google Scholar] [CrossRef] [Green Version]
- Bian, G.K.; Han, Y.C.; Hou, A.W.; Yuan, Y.J.; Liu, X.H.; Deng, Z.X.; Liu, T.G. Releasing the potential power of terpene synthases by a robust precursor supply platform. Metab. Eng. 2017, 42, 1–8. [Google Scholar] [CrossRef]
- Yuan, Y.J.; Litzenburger, M.; Cheng, S.; Bian, G.K.; Hu, B.; Yan, P.; Cai, Y.S.; Deng, Z.X.; Bernhardt, R.; Liu, T.G. Sesquiterpenoids produced by combining two sesquiterpene cyclases with promiscuous myxobacterial CYP260B1. ChemBioChem 2019, 20, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Minami, A.; Mandi, A.; Liu, C.W.; Taniguchi, T.; Kuzuyama, T.; Monde, K.; Gomi, K.; Oikawa, H. Genome mining for sesterterpenes using bifunctional terpene synthases reveals a unified intermediate of di/sesterterpenes. J. Am. Chem. Soc. 2015, 137, 11846–11853. [Google Scholar] [CrossRef] [PubMed]
- Narita, K.; Sato, H.; Minami, A.; Kudo, K.; Gao, L.; Liu, C.W.; Ozaki, T.; Kodama, M.; Lei, X.G.; Taniguchi, T.; et al. Focused genome mining of structurally related sesterterpenes: Enzymatic formation of enantiomeric and diastereomeric products. Org. Lett. 2017, 19, 6696–6699. [Google Scholar] [CrossRef] [PubMed]
- Chiba, R.; Minami, A.; Gomi, K.; Oikawa, H. Identification of ophiobolin F synthase by a genome mining approach: A sesterterpene synthase from Aspergillus clavatus. Org. Lett. 2013, 15, 594–597. [Google Scholar] [CrossRef]
- Matsuda, Y.; Mitsuhashi, T.; Quan, Z.Y.; Abe, I. Molecular basis for stellatic acid biosynthesis: A genome mining approach for discovery of sesterterpene synthases. Org. Lett. 2015, 17, 4644–4647. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, X.; Sato, Y.; Zhu, G.L.; Minami, A.; Zhang, W.Y.; Ozaki, T.; Zhu, B.; Wang, Z.X.; Wang, X.Y.; et al. Genome-based discovery of enantiomeric pentacyclic sesterterpenes catalyzed by fungal bifunctional terpene synthases. Org. Lett. 2021, 23, 4645–4650. [Google Scholar] [CrossRef]
- Mitsuhashi, T.; Rinkel, J.; Okada, M.; Abe, I.; Dickschat, J.S. Mechanistic characterization of two chimeric sesterterpene synthases from Penicillium. Chemistry 2017, 23, 10053–10057. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.J.; Cai, Y.S.; Cheng, F.C.; Yang, C.J.; Zhang, W.Q.; Yu, W.L.; Yan, J.J.; Deng, Z.X.; Hong, K. Genome mining reveals a multiproduct sesterterpenoid biosynthetic gene cluster in Aspergillus ustus. Org. Lett. 2021, 23, 1525–1529. [Google Scholar] [CrossRef]
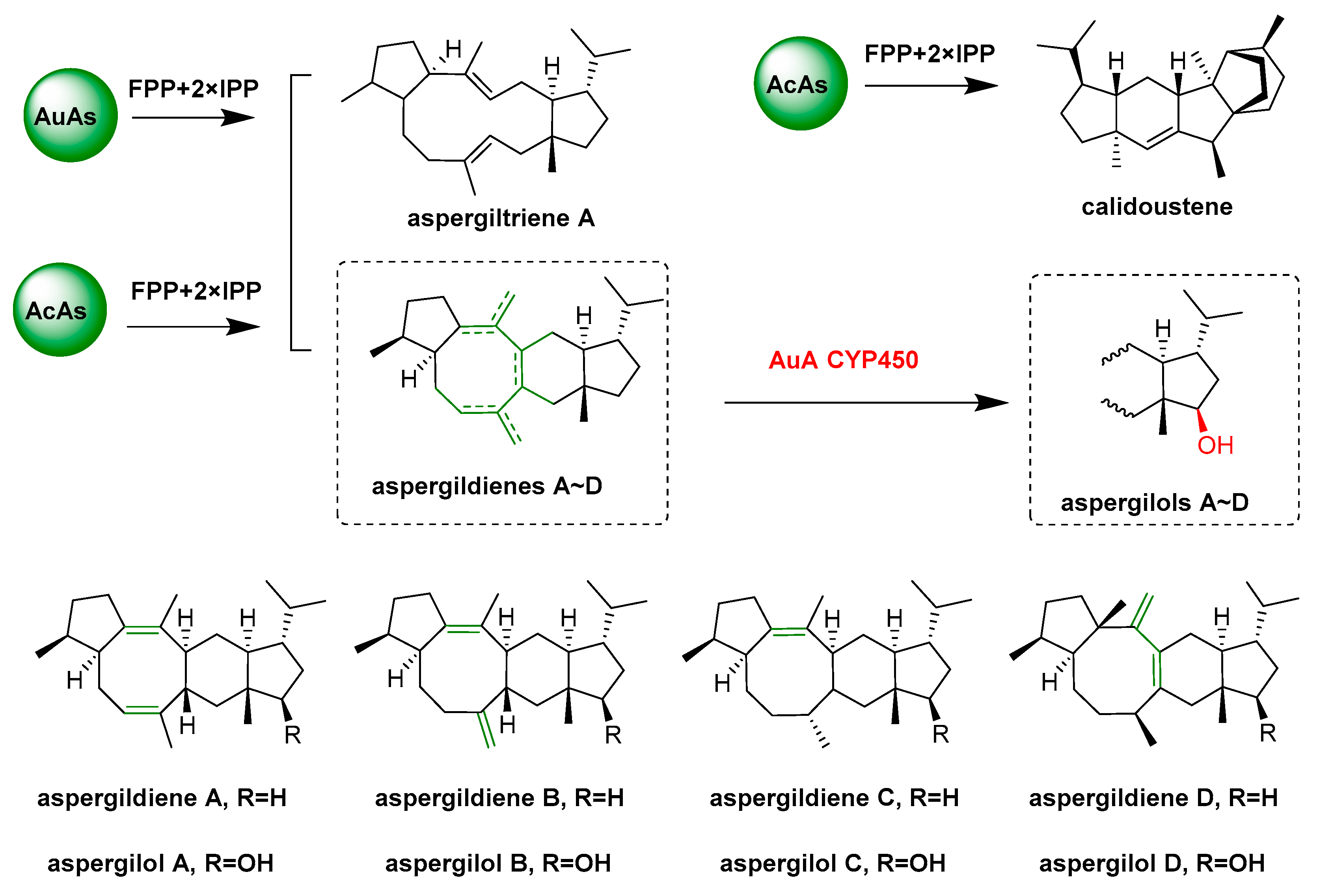
- Quan, Z.Y.; Hou, A.W.; Goldfuss, B.; Dickschat, J.S. Mechanism of the bifunctional multiple product sesterterpene synthase AcAS from Aspergillus calidoustus. Angew. Chem. Int. Ed. 2022, 61, e202117273. [Google Scholar] [CrossRef]
- Chen, R.; Jia, Q.D.; Mu, X.; Hu, B.; Sun, X.; Deng, Z.X.; Chen, F.; Bian, G.K.; Liu, T.G. Systematic mining of fungal chimeric terpene synthases using an efficient precursor-providing yeast chassis. Proc. Natl. Acad. Sci. USA 2021, 118, e2023247118. [Google Scholar] [CrossRef]
- Cane, D.E.; Watt, R.M. Expression and mechanistic analysis of a germacradienol synthase from Streptomyces coelicolor implicated in geosmin biosynthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 1547–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, G.G.; Lombardi, P.M.; Pemberton, T.A.; Matsui, T.; Weiss, T.M.; Cole, K.E.; Koksal, M.; Murphy, F.V.; Vedula, L.S.; Chou, W.K.W.; et al. Structural studies of geosmin synthase, a bifunctional sesquiterpene synthase with alpha alpha domain architecture that catalyzes a unique cyclization-fragmentation reaction sequence. Biochemistry 2015, 54, 7142–7155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liato, V.; Aider, M. Geosmin as a source of the earthy-musty smell in fruits, vegetables and water: Origins, impact on foods and water, and review of the removing techniques. Chemosphere 2017, 181, 9–18. [Google Scholar] [CrossRef]
- Verdoes, J.C.; Krubasik, P.; Sandmann, G.; van Ooyen, A.J.J. Isolation and functional characterisation of a novel type of carotenoid biosynthetic gene from Xanthophyllomyces dendrorhous. Mol. Gen. Genet. 1999, 262, 453–461. [Google Scholar] [CrossRef]
- Lamb, D.C.; Ikeda, H.; Nelson, D.R.; Ishikawa, J.; Skaug, T.; Jackson, C.; Omura, S.; Waterman, M.R.; Kelly, S.L. Cytochrome P450 complement (CYPome) of the avermectin-producer Streptomyces avermitilis and comparison to that of Streptomyces coelicolor A3(2). Biochem. Biophys. Res. Commun. 2003, 307, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, C.J.C.; Bell, S.G.; Wong, L.L. P450(BM3) (CYP102A1): Connecting the dots. Chem. Soc. Rev. 2012, 41, 1218–1260. [Google Scholar] [CrossRef]
- Podust, L.M.; Sherman, D.H. Diversity of P450 enzymes in the biosynthesis of natural products. Nat. Prod. Rep. 2012, 29, 1251–1266. [Google Scholar] [CrossRef] [Green Version]
- Li, S.Y.; Tietz, D.R.; Rutaganira, F.U.; Kells, P.M.; Anzai, Y.; Kato, F.; Pochapsky, T.C.; Sherman, D.H.; Podust, L.M. Substrate recognition by the multifunctional cytochrome P450 MycG in mycinamicin hydroxylation and epoxidation reactions. J. Biol. Chem. 2012, 287, 37880–37890. [Google Scholar] [CrossRef] [Green Version]
- Tudzynski, B.; Kawaide, H.; Kamiya, Y. Gibberellin biosynthesis in Gibberella fujikuroi: Cloning and characterization of the copalyl diphosphate synthase gene. Curr. Genet. 1998, 34, 234–240. [Google Scholar] [CrossRef]
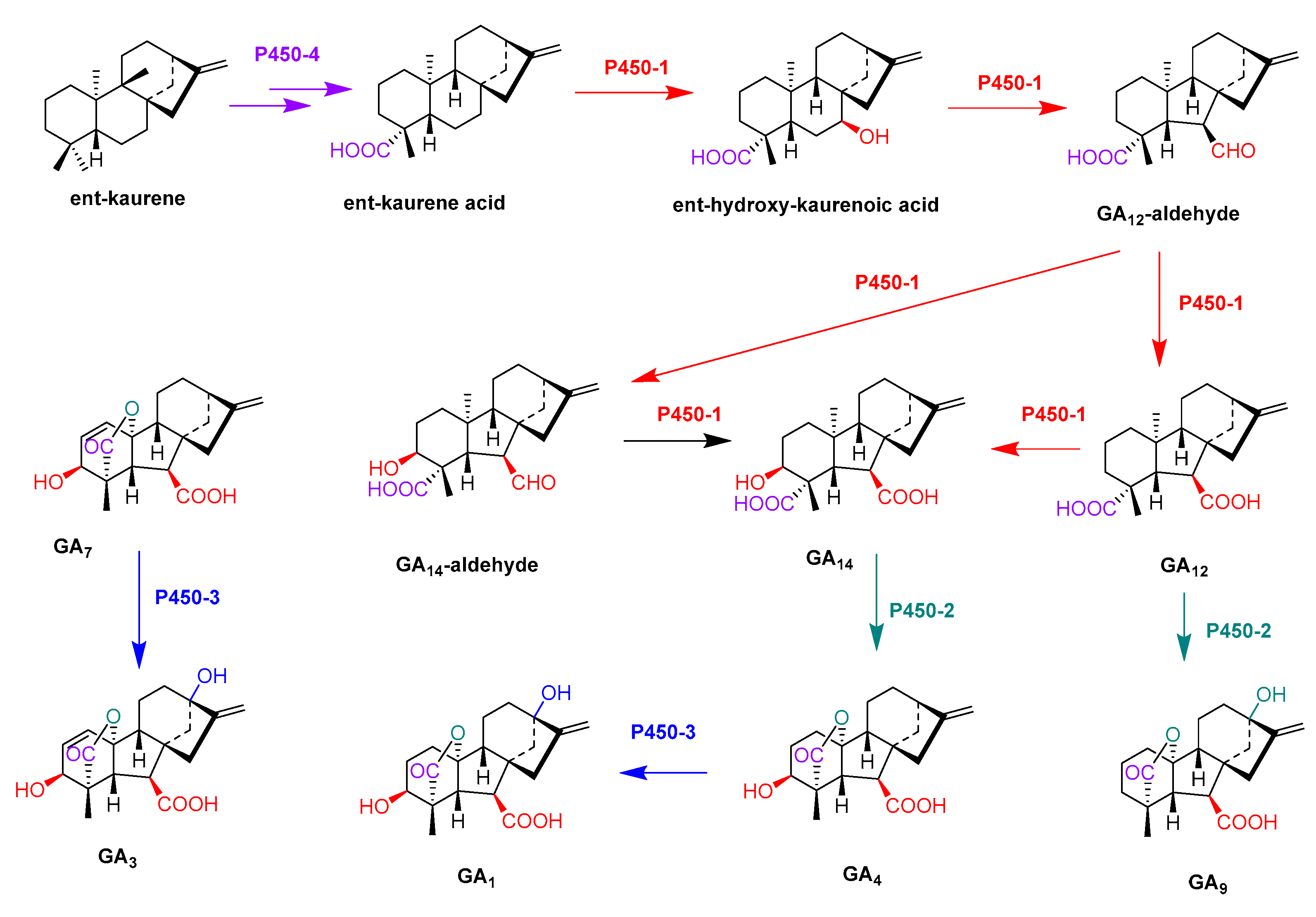
- Rojas, M.C.; Hedden, P.; Gaskin, P.; Tudzynki, B. The P450-1 gene of gibberella fujikuroi encodes a multifunctional enzyme in gibberellin biosynthesis. Proc. Natl. Acad. Sci. USA 2001, 98, 5838–5843. [Google Scholar] [CrossRef] [Green Version]
- Tudzynski, B.; Rojas, M.C.; Gaskin, P.; Hedden, P. The gibberellin 20-oxidase of Gibberella fujikuroi is a multifunctional monooxygenase. J. Biol. Chem. 2002, 277, 21246–21253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tudzynski, B.; Mihlan, M.; Rojas, M.C.; Linnemannstons, P.; Gaskin, P.; Hedden, P. Characterization of the final two genes of the gibberellin biosynthesis gene cluster of Gibberella fujikuroi-des and P450-3 encode GA(4) desaturase and the 13-hydroxylase, respectively. J. Biol. Chem. 2003, 278, 28635–28643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Kourmpetli, S.; Ward, D.A.; Thomas, S.G.; Gong, F.; Powers, S.J.; Carrera, E.; Taylor, B.; Gonzalez, F.N.D.; Tudzynski, B.; et al. Characterization of the fungal gibberellin desaturase as a 2-Oxoglutarate-dependent dioxygenase and its utilization for enhancing plant growth. Plant Physiol. 2012, 160, 837–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inouye, M.; Takada, Y.; Muto, N.; Beppu, T.; Horinouchi, S. Characterization and expression of a P-450-like mycinamicin biosynthesis gene using a novel Micromonospora escherichia-coli shuttle cosmid vector. Mol. Gen. Genet. 1994, 245, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Anzai, Y.; Li, S.Y.; Chaulagain, M.R.; Kinoshita, K.; Kato, F.; Montgomery, J.; Sherman, D.H. Functional analysis of MycCI and MycG, cytochrome P450 enzymes involved in biosynthesis of mycinamicin macrolide antibiotics. CHEM Biol. 2008, 15, 950–959. [Google Scholar] [CrossRef] [Green Version]
- Anzai, Y.; Tsukada, S.; Sakai, A.; Masuda, R.; Harada, C.; Domeki, A.; Li, S.Y.; Kinoshita, K.; Sherman, D.H.; Kato, F. Function of cytochrome P450 enzymes MycCI and MycG in Micromonospora griseorubida, a producer of the macrolide antibiotic mycinamicin. Antimicrob. Agents Chemother. 2012, 56, 3648–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.E.A.; Traitcheva, N.; Knupfer, U.; Hertweck, C. Sequential asymmetric polyketide heterocyclization catalyzed by a single cytochrome P450 monooxygenase (AurH). Angew. Chem. Int. Ed. 2008, 47, 8872–8875. [Google Scholar] [CrossRef]
- Muller, M.; He, J.; Hertweck, C. Dissection of the late steps in aureothin biosynthesis. ChemBioChem 2006, 7, 37–39. [Google Scholar] [CrossRef]
- Zocher, G.; Richter, M.E.A.; Mueller, U.; Hertweck, C. Structural fine-tuning of a multifunctional cytochrome P450 monooxygenase. J. Am. Chem. Soc. 2011, 133, 2292–2302. [Google Scholar] [CrossRef]
- Lin, H.C.; Tsunematsu, Y.; Dhingra, S.; Xu, W.; Fukutomi, M.; Chooi, Y.H.; Cane, D.E.; Calvo, A.M.; Watanabe, K.; Tang, Y. Generation of complexity in fungal terpene biosynthesis: Discovery of a multifunctional cytochrome P450 in the fumagillin pathway. J. Am. Chem. Soc. 2014, 136, 4426–4436. [Google Scholar] [CrossRef]
- Iizaka, Y.; Takeda, R.; Senzaki, Y.; Fukumoto, A.; Anzai, Y. Cytochrome P450 enzyme RosC catalyzes a multistep oxidation reaction to form the non-active compound 20-carboxyrosamicin. FEMS Microbiol. Lett. 2017, 364, fnx110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iizaka, Y.; Higashi, N.; Ishida, M.; Oiwa, R.; Ichikawa, Y.; Takeda, M.; Anzai, Y.; Kato, F. Function of cytochrome P450 enzymes RosC and RosD in the biosynthesis of rosamicin macrolide antibiotic produced by Micromonospora rosaria. Antimicrob. Agents Chemother. 2013, 57, 1529–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.; Kang, Q.J.; Jiang, C.Y.; Han, M.; Bai, L.Q. Engineered biosynthesis of pimaricin derivatives with improved antifungal activity and reduced cytotoxicity. Appl. Microbiol. Biotechnol. 2015, 99, 6745–6752. [Google Scholar] [CrossRef] [PubMed]
- Proctor, R.H.; Plattner, R.D.; Desjardins, A.E.; Busman, M.; Butchko, R.A.E. Fumonisin production in the maize pathogen Fusarium verticillioides: Genetic basis of naturally occurring chemical variation. J. Agric. Food Chem. 2006, 54, 2424–2430. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Iwabuchi, T.; Wakimoto, T.; Awakawa, T.; Abe, I. Uncovering the unusual D-ring construction in terretonin biosynthesis by collaboration of a multifunctional cytochrome P450 and a unique isomerase. J. Am. Chem. Soc. 2015, 137, 3393–3401. [Google Scholar] [CrossRef]
- Barriuso, J.; Nguyen, D.T.; Li, J.W.H.; Roberts, J.N.; MacNevin, G.; Chaytor, J.L.; Marcus, S.L.; Vederas, J.C.; Ro, D.K. Double oxidation of the cyclic nonaketide dihydromonacolin l to Monacolin j by a single cytochrome P450 monooxygenase, LovA. J. Am. Chem. Soc. 2011, 133, 8078–8081. [Google Scholar] [CrossRef]
- Cochrane, R.V.K.; Vederas, J.C. Highly selective but multifunctional oxygenases in secondary metabolism. Acc. Chem. Res. 2014, 47, 3148–3161. [Google Scholar] [CrossRef]
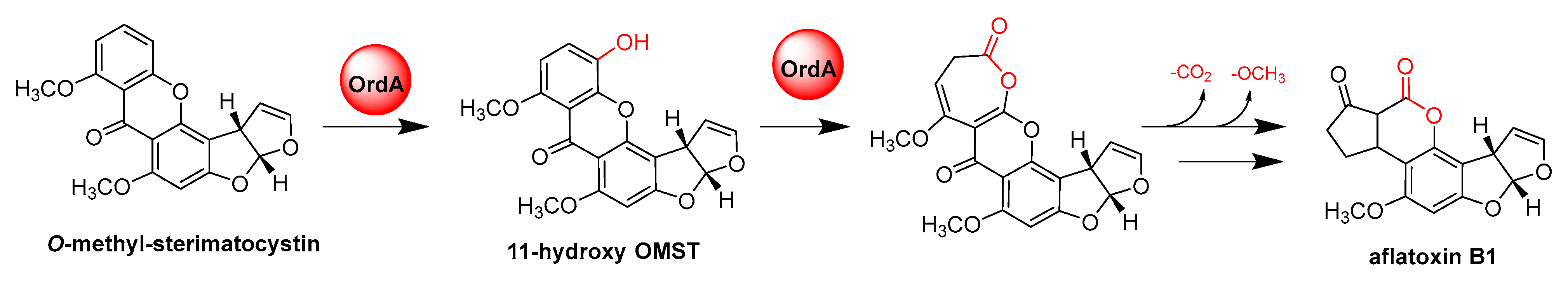
- Udwary, D.W.; Casillas, L.K.; Townsend, C.A. Synthesis of 11-hydroxyl O-methylsterigmatocystin and the role of a cytochrome P-450 in the final step of aflatoxin biosynthesis. J. Am. Chem. Soc. 2002, 124, 5294–5303. [Google Scholar] [CrossRef]
- Yu, J.; Chang, P.K.; Ehrlich, K.C.; Cary, J.W.; Montalbano, B.; Dyer, J.M.; Bhatnagar, D.; Cleveland, T.E. Characterization of the critical amino acids of an Aspergillus parasiticus cytochrome P-450 monooxygenase encoded by ordA that is involved in the biosynthesis of aflatoxins B1, G1, B2, and G2. Appl. Environ. Microbiol. 1998, 64, 4834–4841. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.Q.; Wilson, D.; Zhao, L.S.; Liu, H.W.; Sherman, D.H. Hydroxylation of macrolactones YC-17 and narbomycin is mediated by the pikC-encoded cytochrome P450 in Streptomyces venezuelae. CHEM Biol. 1998, 5, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Lin, X.; Lei, L.; Lamb, D.C.; Kelly, S.L.; Waterman, M.R.; Cane, D.E. Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor A3(2). J. Biol. Chem. 2008, 283, 8183–8189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Lei, L.; Vassylyev, D.G.; Lin, X.; Cane, D.E.; Kelly, S.L.; Yuan, H.; Lamb, D.C.; Waterman, M.R. Crystal structure of albaflavenone monooxygenase containing a moonlighting terpene synthase active site. J. Biol. Chem. 2009, 284, 36711–36719. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Waterman, M.R. Moonlighting cytochrome P450 monooxygenases. IUBMB Life 2011, 63, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Huijbers, M.M.E.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J.H. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef]
- Walsh, C.T.; Wencewicz, T.A. Flavoenzymes: Versatile catalysts in biosynthetic pathways. Nat. Prod. Rep. 2013, 30, 175–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebehmed, J.; Alphand, V.; de Berardinis, V.; de Brevern, A.G. Evolution study of the Baeyer-Villiger monooxygenases enzyme family: Functional importance of the highly conserved residues. Biochimie 2013, 95, 1394–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.X.; Zhang, W.K.; Chooi, Y.H.; Wang, L.; Cao, B.; Deng, Z.X.; Chu, Y.W.; You, D.L. A multifunctional monooxygenase XanO4 catalyzes xanthone formation in xantholipin biosynthesis via a cryptic demethoxylation. Cell Chem. Biol. 2016, 23, 508–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.C.; Dietrich, D.; Xu, W.; Patel, A.; Thuss, J.A.J.; Wang, J.J.; Yin, W.B.; Qiao, K.J.; Houk, K.N.; Vederas, J.C.; et al. A carbonate-forming Baeyer-Villiger monooxygenase. Nat. Chem. Biol. 2014, 10, 552–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Xiang, L.; Izumikawa, M.; Meluzzi, D.; Moore, B.S. Enzymatic total synthesis of enterocin polyketides. Nat. Chem. Biol. 2007, 3, 557–558. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Kalaitzis, J.A.; Moore, B.S. EncM, a versatile enterocin biosynthetic enzyme involved in Favorskii oxidative rearrangement, aldol condensation, and heterocycle-forming reactions. Proc. Natl. Acad. Sci. USA 2004, 101, 15609–15614. [Google Scholar] [CrossRef] [Green Version]
- Aik, W.; McDonough, M.A.; Thalhammer, A.; Chowdhury, R.; Schofield, C.J. Role of the jelly-roll fold in substrate binding by 2-oxoglutarate oxygenases. Curr. Opin. Struct. Biol. 2012, 22, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Clifton, I.J.; McDonough, M.A.; Ehrismann, D.; Kershaw, N.J.; Granatino, N.; Schofield, C.J. Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins. J. Inorg. Biochem. 2006, 100, 644–669. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, K.S.; Granatino, N.; Welford, R.W.D.; McDonough, M.A.; Schofield, C.J. Oxidation by 2-oxoglutarate oxygenases: Non-haem iron systems in catalysis and signalling. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2005, 363, 807–828. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Awakawa, T.; Wakimoto, T.; Abe, I. Spiro-ring formation is catalyzed by a multifunctional dioxygenase in austinol biosynthesis. J. Am. Chem. Soc. 2013, 135, 10962–10965. [Google Scholar] [CrossRef]
- Lo, H.C.; Entwistle, R.; Guo, C.J.; Ahuja, M.; Szewczyk, E.; Hung, J.H.; Chiang, Y.M.; Oakley, B.R.; Wang, C.C.C. Two separate gene clusters encode the biosynthetic pathway for the meroterpenoids austinol and dehydroaustinol in Aspergillus nidulans. J. Am. Chem. Soc. 2012, 134, 4709–4720. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Iwabuchi, T.; Fujimoto, T.; Awakawa, T.; Nakashima, Y.; Mori, T.; Zhang, H.P.; Hayash, F.; Abe, I. Discovery of key dioxygenases that diverged the paraherquonin and acetoxydehydroaustin pathways in Penicillium brasilianum. J. Am. Chem. Soc. 2016, 138, 12671–12677. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Mori, T.; Nakamura, H.; Awakawa, T.; Hoshino, S.; Senda, M.; Senda, T.; Abe, I. Structure function and engineering of multifunctional non-heme iron dependent oxygenases in fungal meroterpenoid biosynthesis. Nat. Commun. 2018, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Hamed, R.B.; Gomez-Castellanos, J.R.; Henry, L.; Ducho, C.; McDonough, M.A.; Schofield, C.J. The enzymes of beta-lactam biosynthesis. Nat. Prod. Rep. 2013, 30, 21–107. [Google Scholar] [CrossRef]
- Townsend, C.A. New reactions in clavulanic acid biosynthesis. Curr. Opin. Chem. Biol. 2002, 6, 583–589. [Google Scholar] [CrossRef]
- Lloyd, M.D.; Lipscomb, S.J.; Hewitson, K.S.; Hensgens, C.M.H.; Baldwin, J.E.; Schofield, C.J. Controlling the substrate selectivity of deacetoxycephalosporin/deacetylcephalosporin C synthase. J. Biol. Chem. 2004, 279, 15420–15426. [Google Scholar] [CrossRef] [Green Version]
- Tarhonskaya, H.; Szollossi, A.; Leung, I.K.H.; Bush, J.T.; Henry, L.; Chowdhury, R.; Iqbal, A.; Claridge, T.D.W.; Schofield, C.J.; Flashman, E. Studies on deacetoxycephalosporin c synthase support a consensus mechanism for 2-oxoglutarate dependent oxygenases. Biochemistry 2014, 53, 2483–2493. [Google Scholar] [CrossRef] [PubMed]
- Phelan, R.M.; Townsend, C.A. Mechanistic insights into the bifunctional non-heme iron oxygenase carbapenem synthase by active site saturation mutagenesis. J. Am. Chem. Soc. 2013, 135, 7496–7502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasquez, J.E.; van der Donk, W.A. Genome mining for ribosomally synthesized natural products. Curr. Opin. Chem. Biol. 2011, 15, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Oman, T.J.; van der Donk, W.A. Follow the leader: The use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol. 2010, 6, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Knerr, P.J.; van der Donk, W.A. Discovery, biosynthesis, and engineering of lantipeptides. Annu. Rev. Biochem. 2012, 81, 479–505. [Google Scholar] [CrossRef]
- Krawczyk, B.; Voller, G.H.; Voller, J.; Ensle, P.; Sussmuth, R.D. Curvopeptin: A new lanthionine-containing class III lantibiotic and its co-substrate promiscuous synthetase. ChemBioChem 2012, 13, 2065–2071. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, Y.; Velasquez, J.E.; van der Donk, W.A. Evolution of lanthipeptide synthetases. Proc. Natl. Acad. Sci. USA 2012, 109, 18361–18366. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; van der Donk, W.A. Biosynthesis of the class III lantipeptide catenulipeptin. ACS Chem. Biol. 2012, 7, 1529–1535. [Google Scholar] [CrossRef]
- van der Donk, W.A.; Nair, S.K. Structure and mechanism of lanthipeptide biosynthetic enzymes. Curr. Opin. Struct. Biol. 2014, 29, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Lopez, F.J.; Carretero-Molina, D.; Sanchez-Hidalgo, M.; Martin, J.; Gonzalez, I.; Roman-Hurtado, F.; de la Cruz, M.; Garcia-Fernandez, S.; Reyes, F.; Deisinger, J.P.; et al. Cacaoidin, first member of the new lanthidin RiPP family. Angew. Chem. 2020, 59, 12654–12658. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, F.; Cheng, Z.; Bashiri, G.; Wang, J.; Hong, J.L.; Wang, Y.M.; Xu, L.J.; Chen, X.F.; Huang, S.X.; et al. Functional genome mining reveals a class v lanthipeptide containing ad-amino acid introduced by an F420H2-dependent reductase. Angew. Chem. 2020, 59, 18029–18035. [Google Scholar] [CrossRef] [PubMed]
- Repka, L.M.; Chekan, J.R.; Nair, S.K.; van der Donka, W.A. Mechanistic understanding of lanthipeptide biosynthetic enzymes. Chem. Rev. 2017, 117, 5457–5520. [Google Scholar] [CrossRef] [PubMed]
- Lagedroste, M.; Reiners, J.; Knospe, C.V.; Smits, S.H.J.; Schmitt, L. A structural view on the maturation of lanthipeptides. Front. Microbiol. 2020, 11, 1183. [Google Scholar] [CrossRef]
- Kodani, S.; Hudson, M.E.; Durrant, M.C.; Buttner, M.J.; Nodwell, J.R.; Willey, J.M. The SapB morphogen is a lantibiotic-like peptide derived from the product of the developmental gene ramS in Streptomyces coelicolor. Proc. Natl. Acad. Sci. USA 2004, 101, 11448–11453. [Google Scholar] [CrossRef] [Green Version]
- Muller, W.M.; Schmiederer, T.; Ensle, P.; Sussmuth, R.D. In vitro biosynthesis of the prepeptide of Type-III lantibiotic labyrinthopeptin A2 including formation of a c-c bond as a post-translational modification. Angew. Chem. 2010, 49, 2436–2440. [Google Scholar] [CrossRef]
- Goto, Y.; Li, B.; Claesen, J.; Shi, Y.X.; Bibb, M.J.; van der Donk, W.A. Discovery of unique lanthionine synthetases reveals new mechanistic and evolutionary Insights. PLoS Biol. 2010, 8, e1000339. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of palladium-catalyzed c-n cross-coupling reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Gkotsi, D.S.; Dhaliwal, J.; McLachlan, M.M.W.; Mulholand, K.R.; Goss, R.J.M. Halogenases: Powerful tools for biocatalysis (mechanisms applications and scope). Curr. Opin. Chem. Biol. 2018, 43, 119–126. [Google Scholar] [CrossRef]
- Gribble, G.W. Naturally occurring organohalogen compounds. Acc. Chem. Res. 1998, 31, 141–152. [Google Scholar] [CrossRef]
- Chankhamjon, P.; Tsunematsu, Y.; Ishida-Ito, M.; Sasa, Y.; Meyer, F.; Boettger-Schmidt, D.; Urbansky, B.; Menzel, K.D.; Scherlach, K.; Watanabe, K.; et al. Regioselective dichlorination of a non-activated aliphatic carbon atom and phenolic bismethylation by a multifunctional fungal flavoenzyme. Angew. Chem. 2016, 55, 11955–11959. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ohashi, M.; Hung, Y.-S.; Scherlach, K.; Watanabe, K.; Hertweck, C.; Tang, Y. AoiQ catalyzes geminal dichlorination of 1,3-diketone natural products. J. Am. Chem. Soc. 2021, 143, 7267–7271. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.X.; Wang, Q.; Deng, Z.X.; You, D.L. Flavin adenine dinucleotide-dependent halogenase XanH and engineering of multifunctional fusion halogenases. Appl. Environ. Microbiol. 2020, 86, e01225-20. [Google Scholar] [CrossRef]
- Andorfer, M.C.; Belsare, K.D.; Girlich, A.M.; Lewis, J.C. Aromatic halogenation by using bifunctional flavin reductase-halogenase fusion enzymes. ChemBioChem 2017, 18, 2099–2103. [Google Scholar] [CrossRef] [PubMed]
- Higashide, E.; Asai, M.; Ootsu, K.; Tanida, S.; Kozai, Y.; Hasegawa, T.; Kishi, T.; Sugino, Y.; Yoneda, M. Ansamitocin, a group of novel maytansinoid antibiotics with antitumour properties from Nocardia. Nature 1977, 270, 721–722. [Google Scholar] [CrossRef] [PubMed]
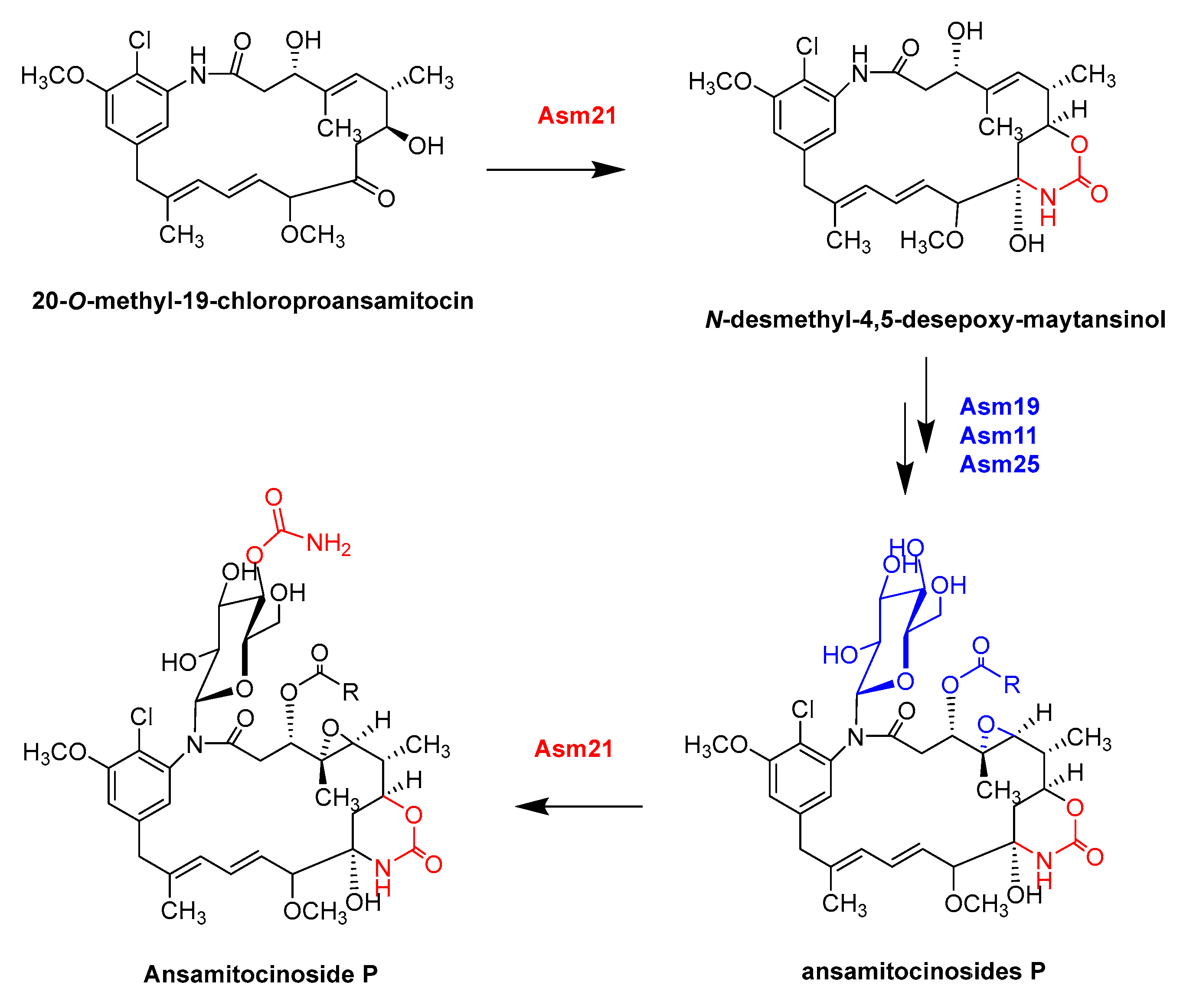
- Yu, T.W.; Bai, L.Q.; Clade, D.; Hoffmann, D.; Toelzer, S.; Trinh, K.Q.; Xu, J.; Moss, S.J.; Leistner, E.; Floss, H.G. The biosynthetic gene cluster of the maytansinoid antitumor agent ansamitocin from Actinosynnema pretiosum. Proc. Natl. Acad. Sci. USA 2002, 99, 7968–7973. [Google Scholar] [CrossRef] [Green Version]
- Moss, S.J.; Bai, L.Q.; Toelzer, S.; Carroll, B.J.; Mahmud, T.; Yu, T.W.; Floss, H.G. Identification of Asm19 as an acyltransferase attaching the biologically essential ester side chain of ansamitocins using N-desmethyl-4,5-desepoxymaytansinol, not maytansinol, as its substrate. J. Am. Chem. Soc. 2002, 124, 6544–6545. [Google Scholar] [CrossRef]
- Spiteller, P.; Bai, L.Q.; Shang, G.D.; Carroll, B.J.; Yu, T.W.; Floss, H.G. The post-polyketide synthase modification steps in the biosynthesis of the antitumor agent ansamitocin by Actinosynnema pretiosum. J. Am. Chem. Soc. 2003, 125, 14236–14237. [Google Scholar] [CrossRef]
- Zhao, P.J.; Bai, L.Q.; Ma, J.; Zeng, Y.; Li, L.; Zhang, Y.R.; Lu, C.H.; Dai, H.Q.; Wu, Z.X.; Li, Y.Y.; et al. Amide N-glycosylation by Asm25, an N-glycosyltransferase of ansamitocins. CHEM Biol. 2008, 15, 863–874. [Google Scholar] [CrossRef]
- Sun, P.; Zhao, Q.F.; Yu, F.T.; Zhang, H.; Wu, Z.H.; Wang, Y.Y.; Wang, Y.; Zhang, Q.L.; Liu, W. Spiroketal Formation and modification in avermectin biosynthesis involves a dual activity of AveC. J. Am. Chem. Soc. 2013, 135, 1540–1548. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.-T.; Shi, T.-T.; Ding, L.; Xie, J.; Zhao, P.-J. Multifunctional Enzymes in Microbial Secondary Metabolic Processes. Catalysts 2023, 13, 581. https://doi.org/10.3390/catal13030581
Wang J-T, Shi T-T, Ding L, Xie J, Zhao P-J. Multifunctional Enzymes in Microbial Secondary Metabolic Processes. Catalysts. 2023; 13(3):581. https://doi.org/10.3390/catal13030581
Chicago/Turabian StyleWang, Jun-Tao, Ting-Ting Shi, Lin Ding, Juan Xie, and Pei-Ji Zhao. 2023. "Multifunctional Enzymes in Microbial Secondary Metabolic Processes" Catalysts 13, no. 3: 581. https://doi.org/10.3390/catal13030581