
Applications of Hantzsch Esters in Organocatalytic Enantioselective Synthesis

Abstract
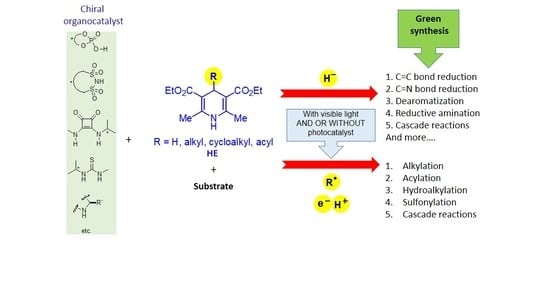
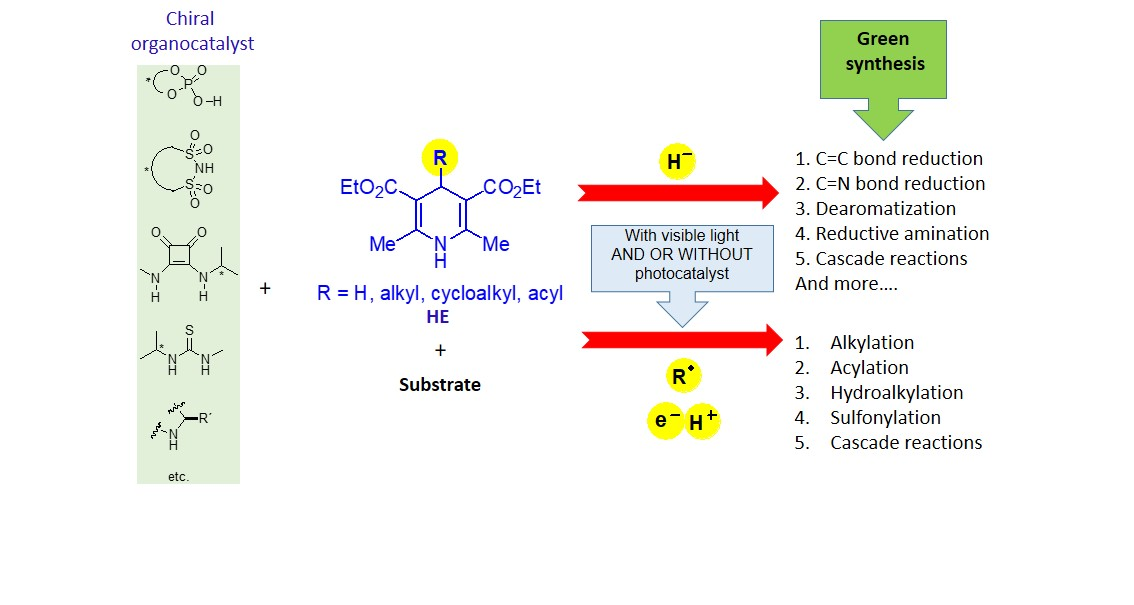
:
{kind=link}
{kind=link}
{kind=link}
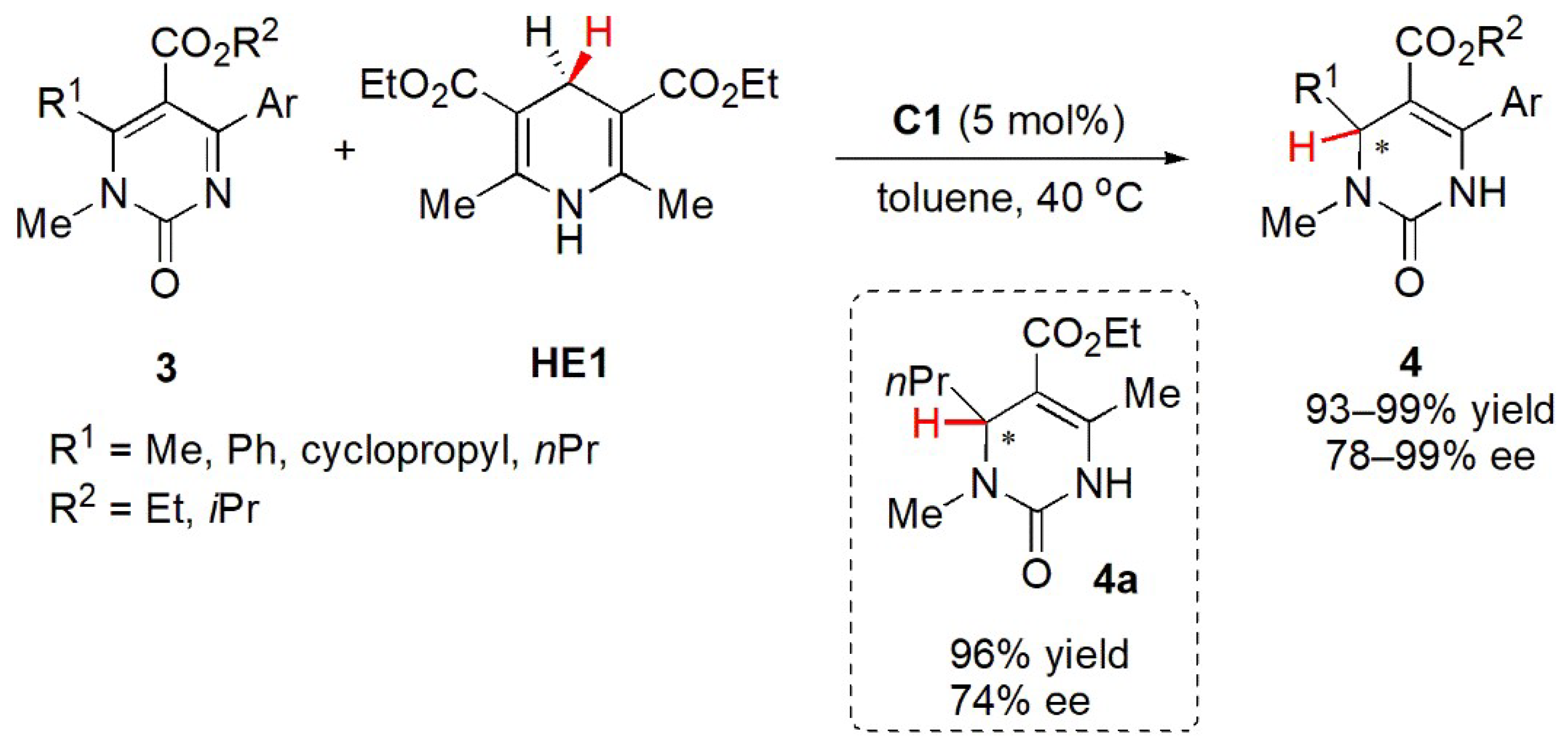{kind=link}
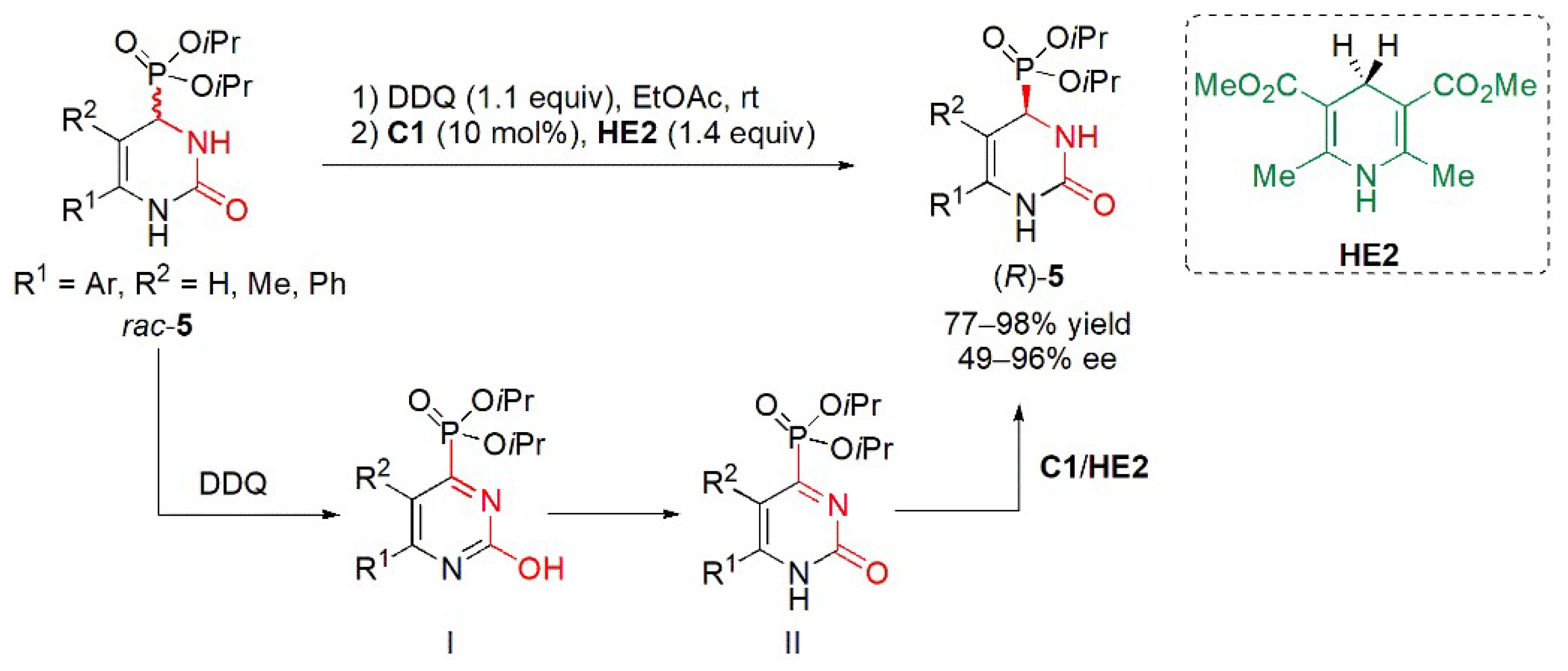{kind=link}
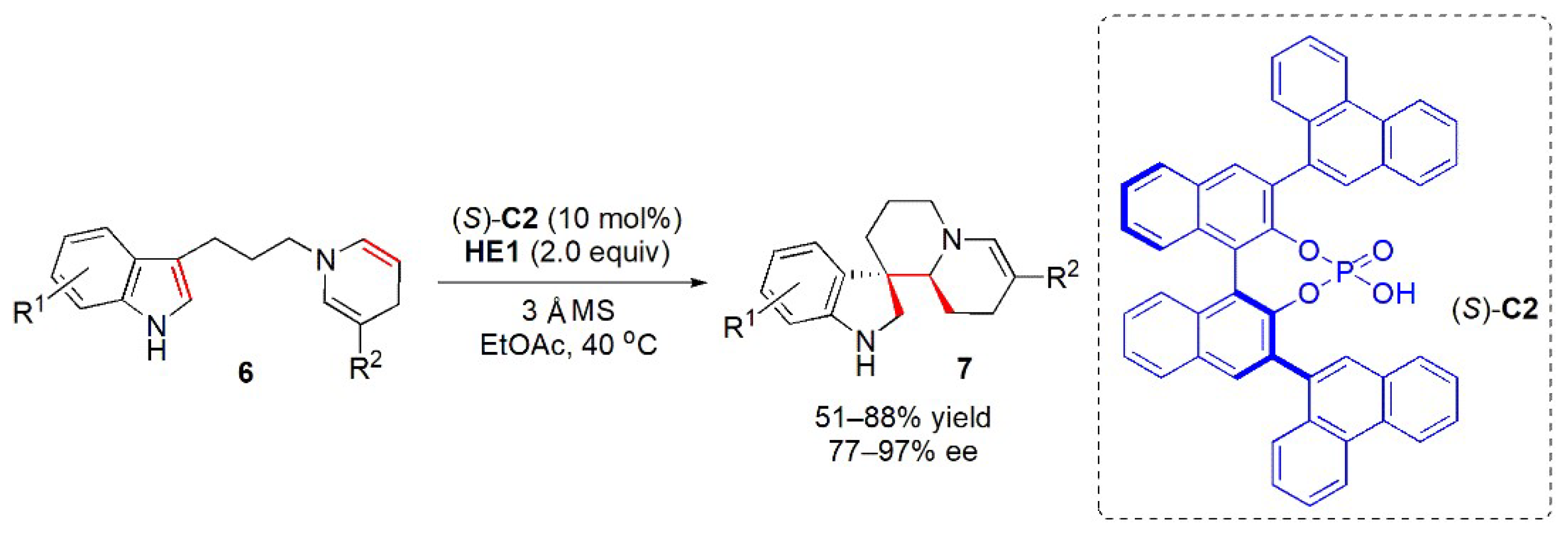{kind=link}
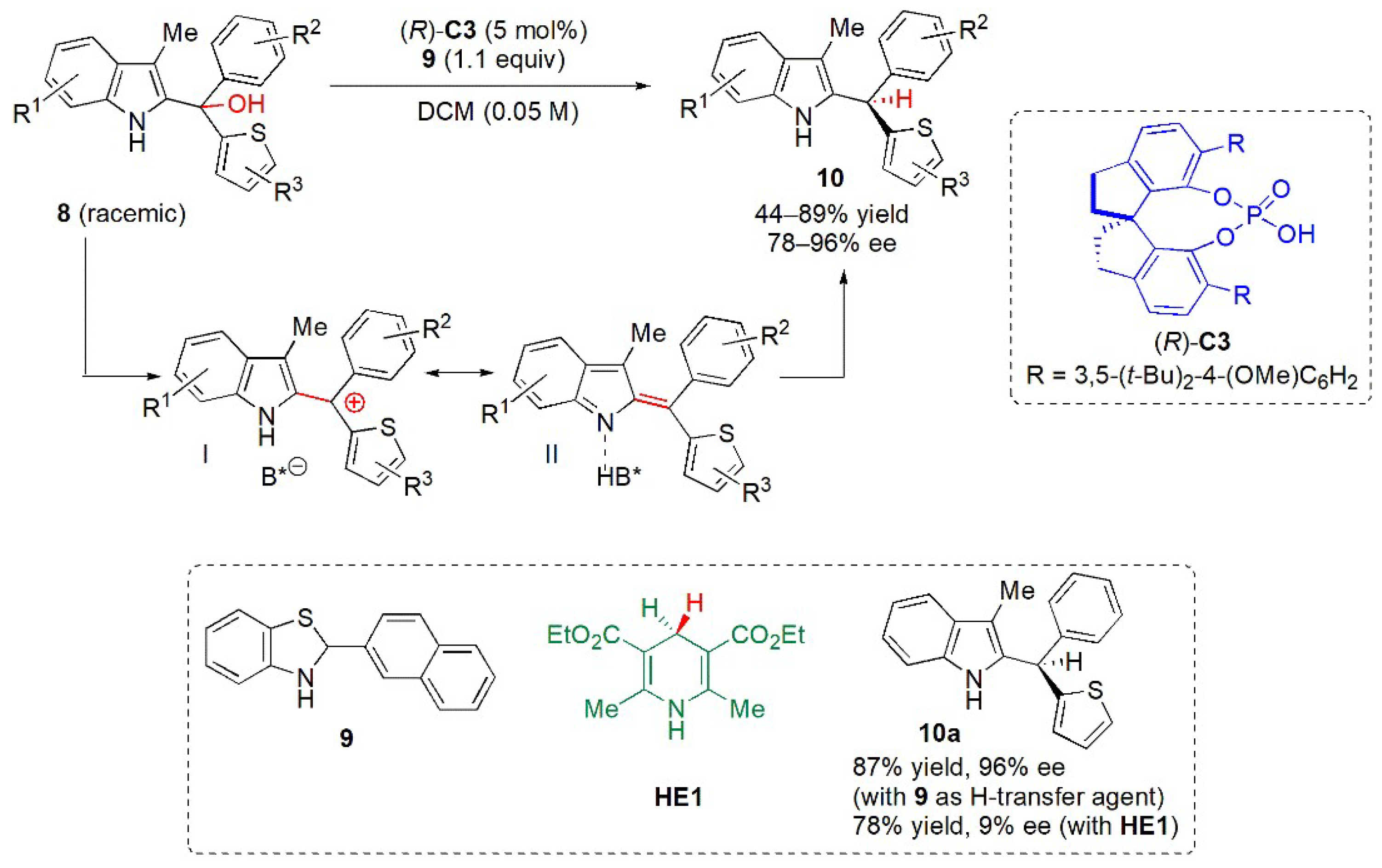{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
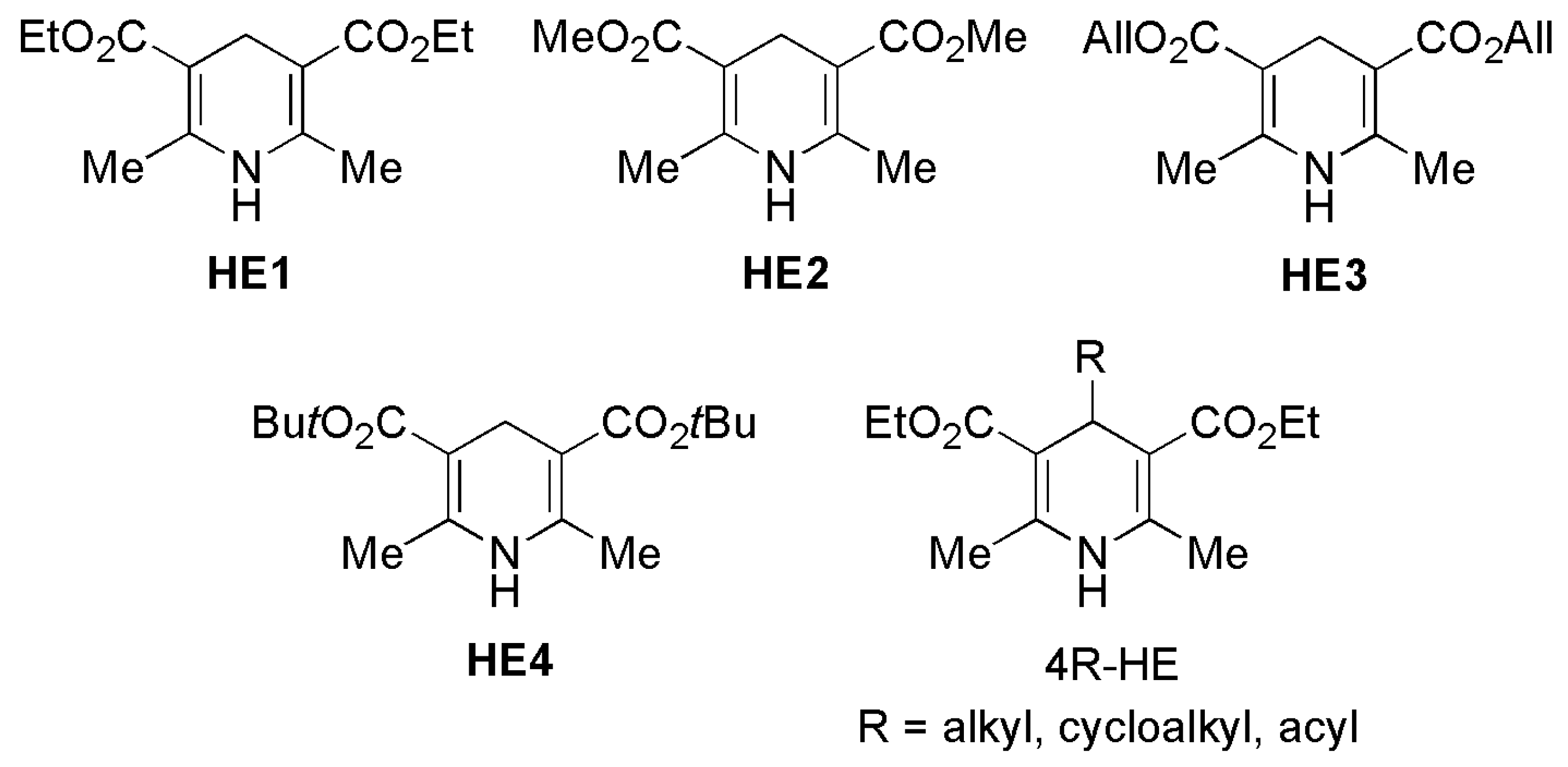
1. Introduction
2. Enantioselective Transfer Hydrogenation
2.1. Brønsted Acids as Catalysts
2.1.1. Phosphoric Acid-Catalyzed Reactions
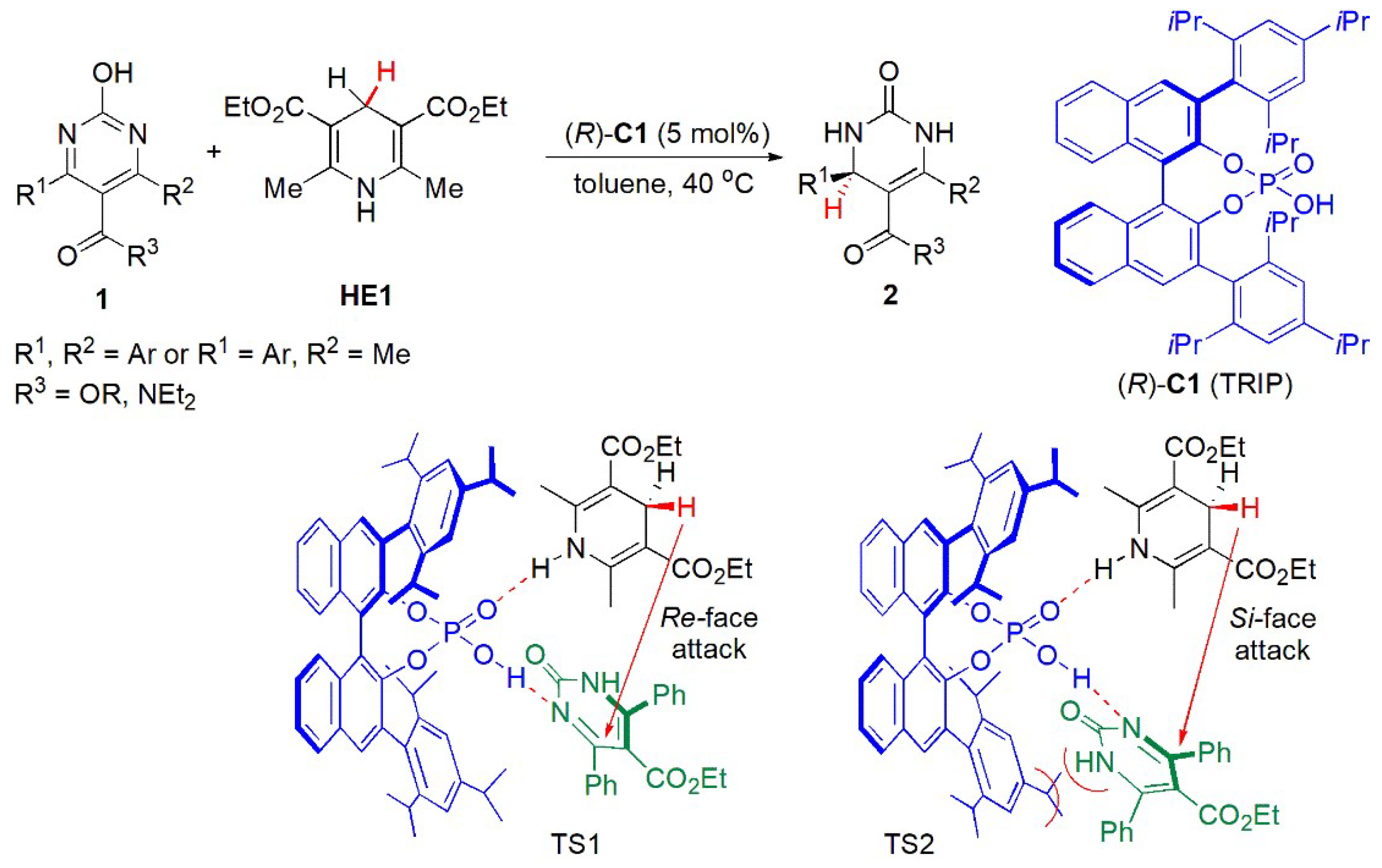
- Synthesis of Dihydropyrimidinones (DHPMs)
- b.
- Reactions of indoles
- c.
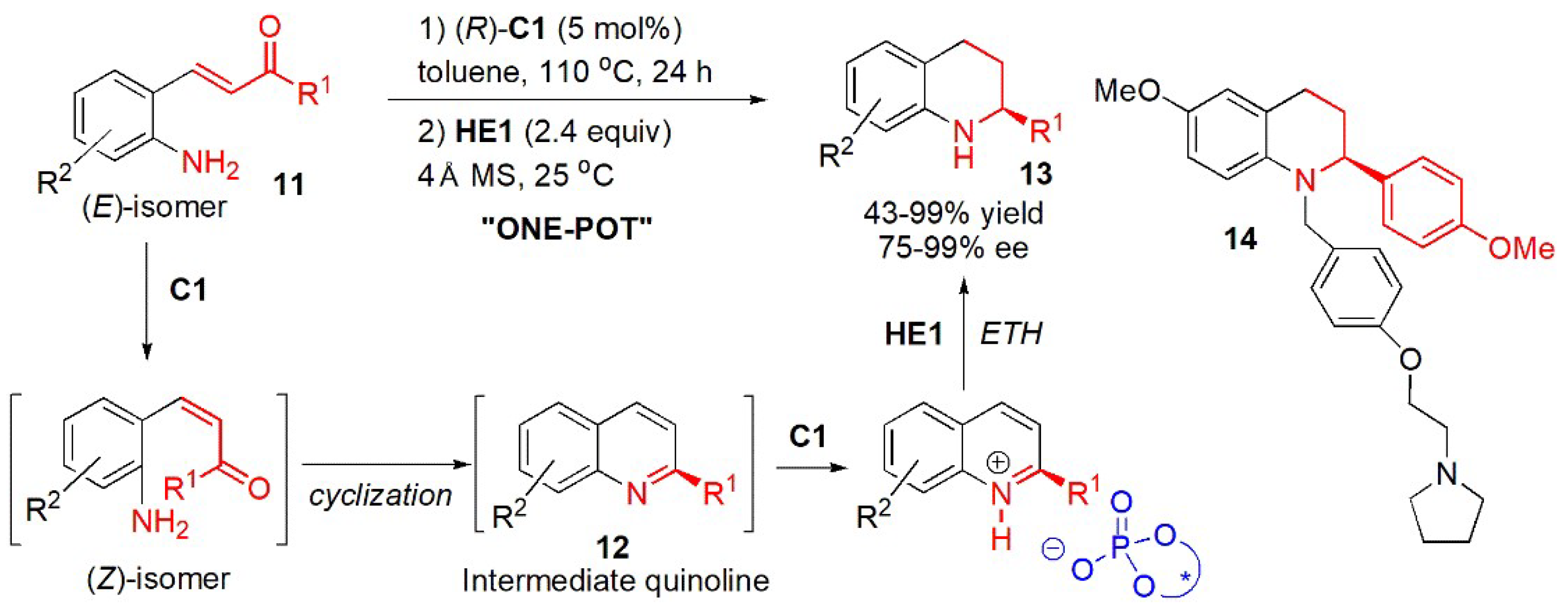
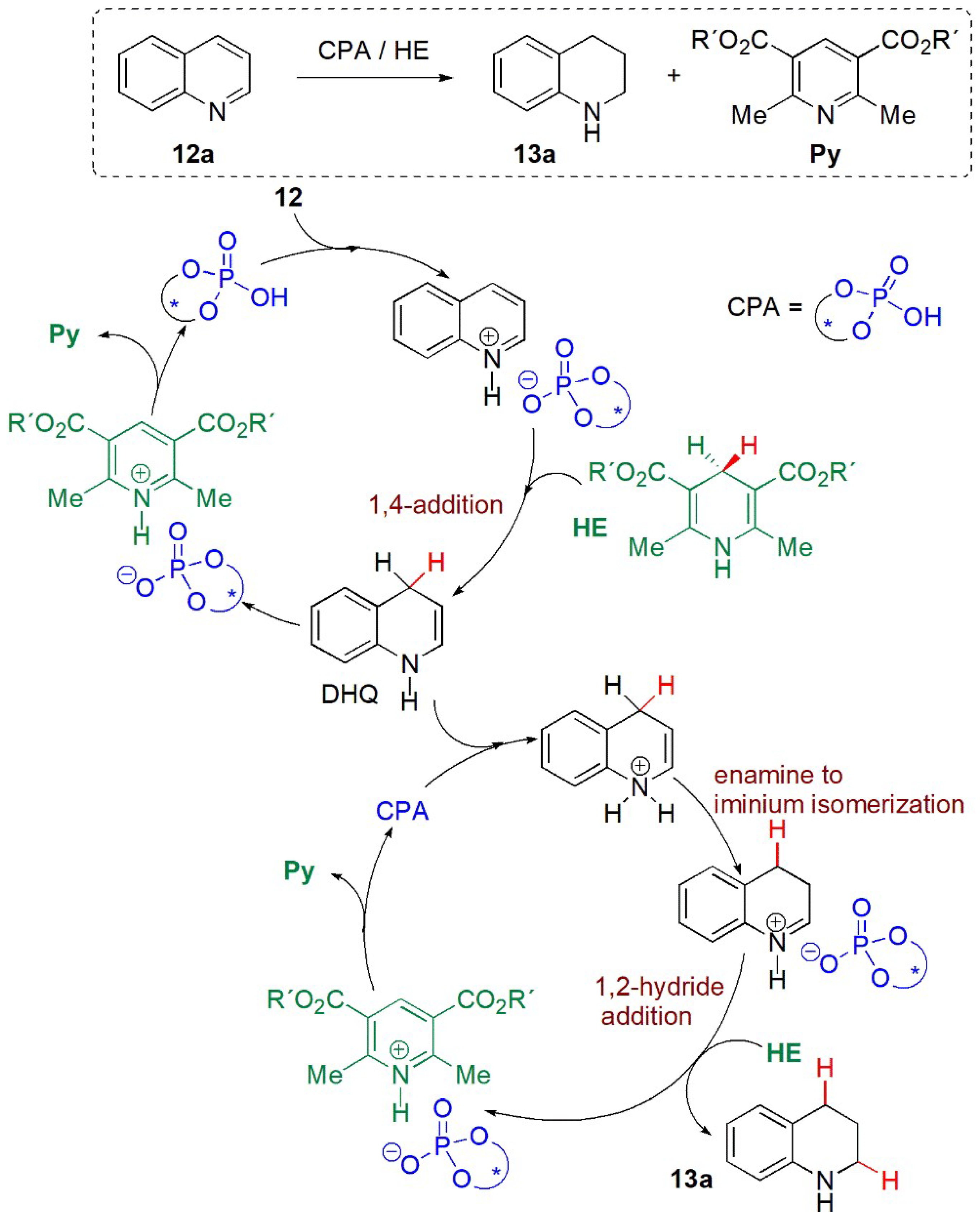
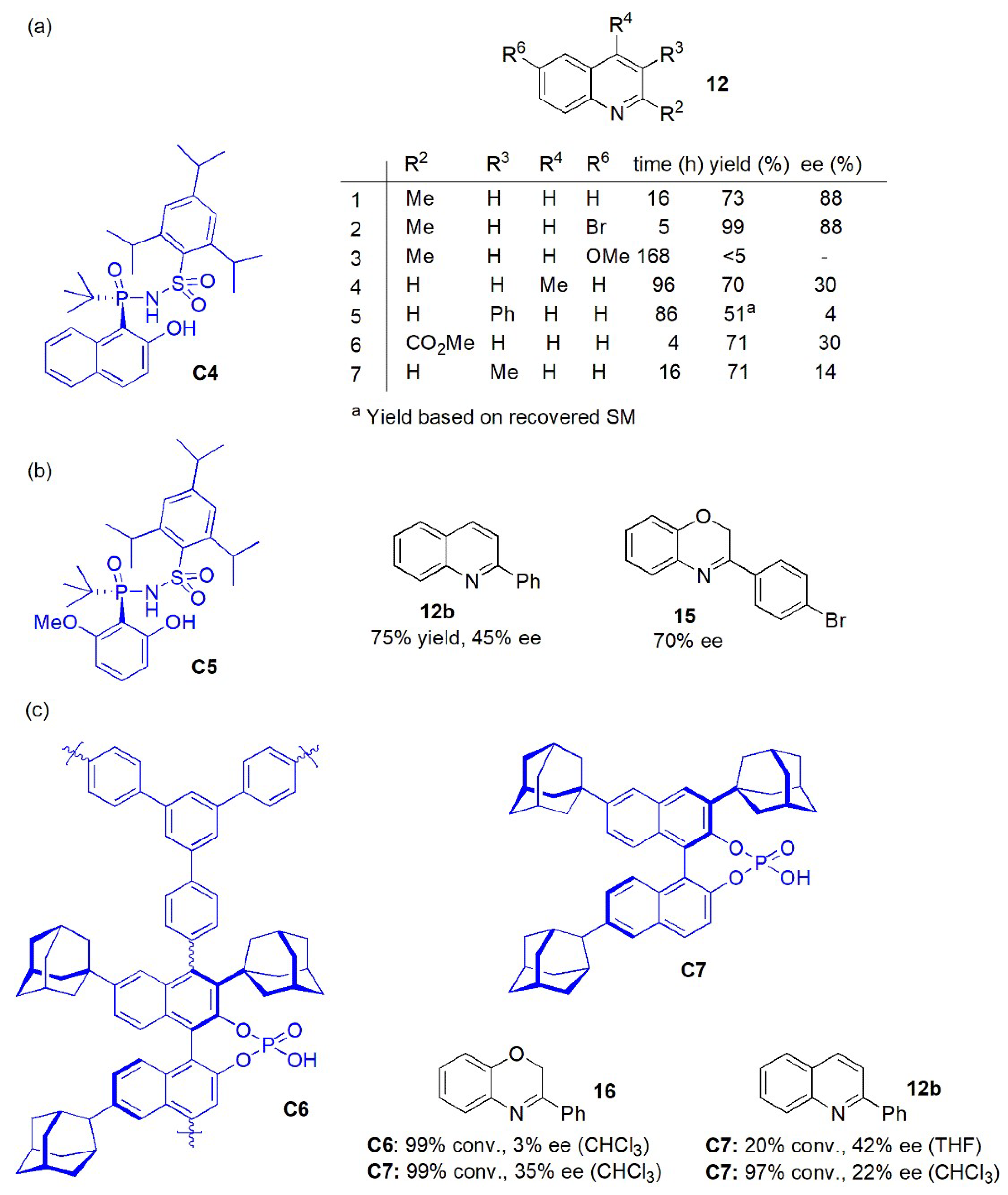
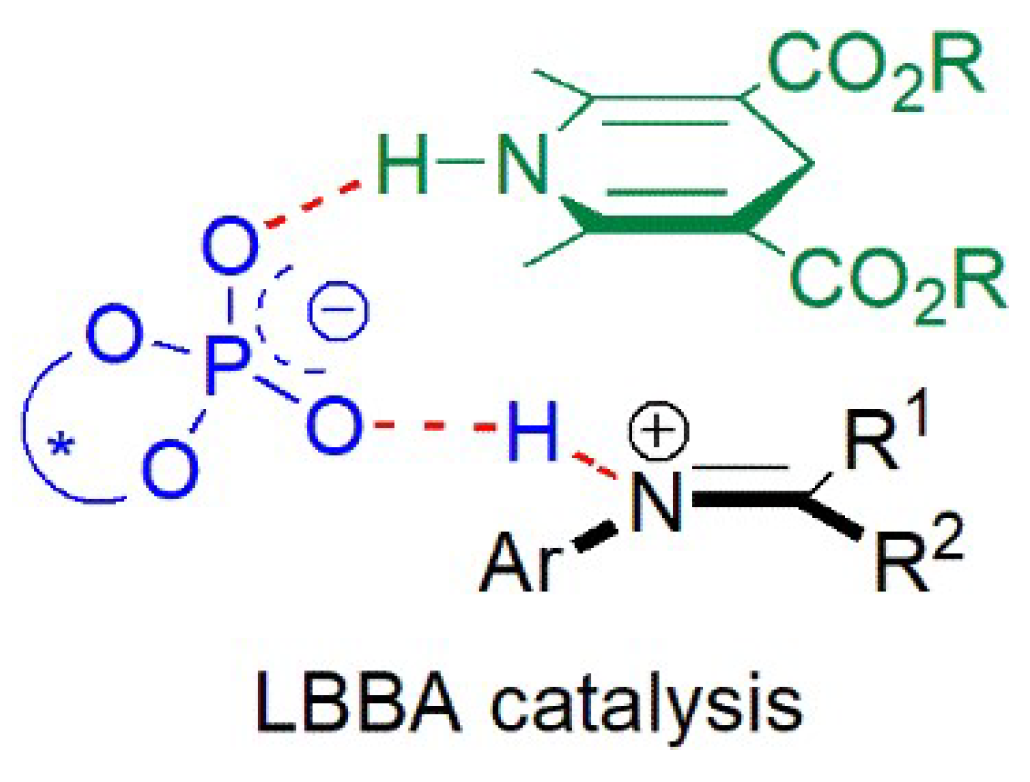
- Reactions of quinolines

- d.
- Miscellaneous
2.1.2. Disulfonimide-Catalyzed Reactions
2.2. Bifunctional Catalysts Based on Thioureas, Squaramides and Related Substances
2.3. Amines as Catalysts
3. Hantzsch Esters in “Transfer-Alkylation” and Other Photoredox Radical Reactions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arthur Rudolph Hantzsch (1857–1935) and the synthesis of nitrogen heterocycles. Synform 2021, 12, A201–A210.
- Faisca Phillips, A.M.; Pombeiro, A.J. Recent advances in organocatalytic enantioselective transfer hydrogenation. Org. Biomol. Chem. 2017, 15, 2307–2340. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; You, S.L. Transfer hydrogenation with Hantzsch esters and related organic hydride donors. Chem. Soc. Rev. 2012, 41, 2498–2518. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J.D. Molecular Biology of the Cell; Garland: New York, NY, USA, 2002. [Google Scholar]
- Yang, J.W.; Fonseca, M.T.H.; List, B. A metal-free transfer hydrogenation: Organocatalytic conjugate reduction of α,β-unsaturated aldehydes. Angew. Chem. Int. Ed. 2004, 43, 6660–6662. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, S.G.; Tuttle, J.B.; MacMillan, D.W.C. Enantioselective Organocatalytic Hydride Reduction. J. Am. Chem. Soc. 2005, 127, 32–33. [Google Scholar] [CrossRef]
- Wang, C.; Wu, X.; Xiao, J. Broader, greener, and more efficient: Recent advances in asymmetric transfer hydrogenation. Asian Chem. 2008, 3, 1750–1770. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The golden age of transfer hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Gladiali, S.; Alberico, E. Asymmetric transfer hydrogenation: Chiral ligands and applications. Chem. Soc. Rev. 2006, 35, 226–236. [Google Scholar] [CrossRef]
- Zehani, S.; Gelbard, G. Asymmetric reductions catalysed by chiral shift reagents. J. Chem. Soc. Chem. Commun. 1985, 1162–1163. [Google Scholar] [CrossRef]
- Yang, J.W.; List, B. Catalytic asymmetric transfer hydrogenation of α-ketoesters with Hantzsch esters. Org. Lett. 2006, 8, 5653–5655. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J. Hantzsch Dihydropyridine Synthesis. In Name Reactions; Springer: Cham, Switzerland, 2021. [Google Scholar]
- Cheng, X.; Huang, W. Hantzsch esters as multifunctional reagents in visible-light photoredox catalysis. Synlett 2016, 28, 148–158. [Google Scholar] [CrossRef]
- Bhunia, A.; Studer, A. Recent advances in radical chemistry proceeding through pro-aromatic radicals. Chem 2021, 7, 2060–2100. [Google Scholar] [CrossRef]
- Hedstrand, D.M.; Kruizinga, W.H.; Kellogg, R.M. Light induced and dye accelerated reductions of phenacyl onium salts by 1,4-dihydropyridines. Tetrahedron Lett. 1978, 19, 1255–1258. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [Green Version]
- Narayanam, M.R.; Tucker, J.; Stephenson, C.R.J. Electron-transfer photoredox catalysis: Development of a tin-free reductive dehalogenation reaction. J. Am. Chem. Soc. 2009, 131, 8756–8757. [Google Scholar] [CrossRef]
- Nguyen, J.D.; D’Amato, E.M.; Narayanam, M.R.; Stephenson, C.R. Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions. J. Nat. Chem. 2012, 4, 854–859. [Google Scholar] [CrossRef]
- Jung, J.; Kim, J.; Park, G.; You, Y.; Cho, E.J. Selective debromination and α-hydroxylation of α-bromo ketones using Hantzsch esters as photoreductants. Adv. Synth. Catal. 2016, 358, 74–80. [Google Scholar] [CrossRef]
- Li, G.; Chen, R.; Wu, L.; Fu, Q.; Zhang, X.; Tang, Z. Alkyl transfer from C-C cleavage. Angew. Chem. Int. Ed. 2013, 52, 8432–8436. [Google Scholar] [CrossRef]
- Wang, P.-Z.; Chen, J.-R.; Xiao, W.-J. Hantzsch esters: An emerging versatile class of reagents in photoredox catalyzed organic synthesis. Org. Biomol. Chem. 2019, 17, 6936–6951. [Google Scholar] [CrossRef]
- Ye, S.; Wu, J. 4-Substituted Hantzsch esters as alkylation reagents in organic synthesis. Acta Chim. Sin. 2019, 77, 814–831. [Google Scholar] [CrossRef]
- Shen, G.B.; Xie, L.; Yu, H.Y.; Liu, J.; Fu, Y.H.; Yan, M. Theoretical investigation on the nature of 4-substituted Hantzsch esters as alkylation agents. RSC Adv. 2020, 10, 31425–31434. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.L.; Xu-Xu, Q.F.; Zheng, C.; You, S.L. Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions. Chem. Soc. Rev. 2020, 49, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Pálvölgyi, Á.M.; Scharinger, F.; Schnürch, M.; Bica-Schröder, K. Chiral phosphoric acids as versatile tools for organocatalytic asymmetric transfer hydrogenations. Eur. J. Org. Chem. 2021, 2021, 5367–5381. [Google Scholar] [CrossRef]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef]
- Akiyama, T.; Mori, K. Stronger Brϕnsted acids: Recent progress. Chem. Rev. 2015, 115, 9277–9306. [Google Scholar] [CrossRef]
- Faisca Phillips, A.M. Non-covalent interactions in asymmetric reactions catalyzed by chiral phosphoric acids. In Noncovalent Interactions in Catalysis; Mahmudov, K.T., Kopylovich, M.N., Guedes da Silva, M.F.C., Pombeiro, A.J.L., Eds.; Royal Society of Chemistry: Cambridge, UK, 2019; pp. 253–281. [Google Scholar]
- Sorgenfrei, N.; Hioe, J.; Greindl, J.; Rothermel, K.; Morana, F.; Lokesh, N.; Gschwind, R.M. NMR spectroscopic characterization of charge assisted strong hydrogen bonds in Brønsted acid catalysis. J. Am. Chem. Soc. 2016, 138, 16345–16354. [Google Scholar] [CrossRef] [Green Version]
- Rothermel, K.; Melikian, M.; Hioe, J.; Greindl, J.; Gramüller, J.; Zabka, M.; Sorgenfrei, N.; Hausler, T.; Morana, F.; Gschwind, R.M. Internal acidity scale and reactivity evaluation of chiral phosphoric acids with different 3,3′- substituents in Brønsted acid catalysis. Chem. Sci. 2019, 10, 10025–10034. [Google Scholar] [CrossRef] [Green Version]
- Knowles, R.R.; Jacobsen, E.N. Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. USA 2010, 107, 20678–20685. [Google Scholar] [CrossRef] [Green Version]
- Maji, R.; Mallojjala, S.C.; Wheeler, S.E. Chiral phosphoric acid catalysis: From numbers to insights. Chem. Soc. Rev. 2018, 47, 1142–1158. [Google Scholar] [CrossRef]
- Simón, L.; Goodman, J.M. Theoretical study of the mechanism of Hantzsch ester hydrogenation of imines catalyzed by chiral BINOL-phosphoric acids. J. Am. Chem. Soc. 2008, 130, 8741–8747. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Chaudhary, S.; Kumar, K.; Gupta, M.K.; Rawal, R.K. Recent synthetic and medicinal perspectives of dihydropyrimidinones: A review. Eur. J. Med. Chem. 2017, 132, 108–134. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.H.S.; Masson, F.T.; Simeoni, L.A.; Homem-de-Mello, M. Biological activity of dihydropyrimidinone (DHPM) derivatives: A systematic review. Eur. J. Med. Chem. 2018, 143, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Debonis, S.; Simorre, J.P.; Crevel, I.; Lebeau, L.; Skoufias, D.A.; Blangy, A.; Ebel, C.; Gans, P.; Cross, R.; Hackney, D.D.; et al. Interaction of the Mitotic Inhibitor Monastrol with Human Kinesin Eg5. Biochemistry 2003, 42, 338–349. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.J.; Shi, L.; Feng, G.S.; Sun, L.; Zhou, Y.G. Enantioselective synthesis of 3,4-dihydropyrimidin-2(1 H)-ones through organocatalytic transfer hydrogenation of 2-hydroxypyrimidines. J. Org. Chem. 2019, 84, 4435–4442. [Google Scholar] [CrossRef]
- Meng, F.-J.; Shi, L.; Jiang, W.-F.; Lu, X.-B. Enantioselective 1,4-reduction of pyrimidin-2-ones to synthesize novel 3,4-dihydropyrimidin-2(1H)-ones containing an alkyl-substituted stereogenic center. Asian J. Org. Chem. 2020, 9, 778–781. [Google Scholar] [CrossRef]
- Meng, F.J.; Shao, B.R.; Velopolcek, M.K.; Guo, X.; Feng, G.S.; Shi, L. Redox deracemization of phosphonate-substituted dihydropyrimidines. Org. Biomol. Chem. 2021, 19, 10570–10574. [Google Scholar] [CrossRef]
- Stçckigt, J.; Antonchick, A.P.; Wu, F.; Waldmann, H. The Pictet–Spengler reaction in nature and in organic chemistry. Angew. Chem. Int. Ed. 2011, 50, 8538–8564. [Google Scholar] [CrossRef]
- Shiri, M. Indoles in multicomponent processes (MCPs). Chem. Rev. 2012, 112, 3508–3549. [Google Scholar] [CrossRef]
- Faisca Phillips, A.M.M.M. Organocatalytic enantioselective dearomatization reactions for the synthesis of nitrogen heterocycles. In Synthetic Approaches to Nonaromatic Nitrogen Heterocycles; Faisca Phillips, A.M.M.M., Ed.; John Wiley & Sons: Chichester, UK, 2020; pp. 273–323. [Google Scholar]
- Wang, S.-G.; Xia, Z.-L.; Xu, R.-Q.; Liu, X.-J.; Zheng, C.; You, S.-L. Construction of chiral tetrahydro-β-carbolines: Asymmetric Pictet-Spengler reaction of indolyl dihydropyridines. Angew. Chem. Int. Ed. 2017, 56, 7440–7443. [Google Scholar] [CrossRef]
- Bariwal, J.; Voskressensky, L.G.; Van der Eycken, E.V. Recent advances in spirocyclization of indole derivatives. Chem. Soc. Rev. 2018, 47, 3831–3848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.-L.; Zheng, C.; Wang, S.-G.; You, S.-L. Catalytic asymmetric dearomatization of indolyl dihydropyridines through an enamine isomerization/spirocyclization/transfer hydrogenation sequence. Angew. Chem. Int. Ed. 2018, 57, 2653–2656. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Roy, D.; Panda, D. Overview on the recent strategies for the enantioselective synthesis of 1, 1-diarylalkanes, triarylmethanes and related molecules containing the diarylmethine stereocenter. ChemCatChem 2018, 10, 1941–1967. [Google Scholar] [CrossRef]
- Yan, Q.; Duan, M.; Chen, C.; Deng, Z.; Wu, M.; Yu, P.; He, M.-L.; Zhu, G.; Houk, K.N.; Sun, J. Organocatalytic discrimination of non-directing aryl and heteroaryl groups: Enantioselective synthesis of bioactive indole-containing triarylmethanes. Chem. Sci. 2022, 13, 5767–5773. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Antonchick, A.P.; Theissmann, T. A highly enantioselective Brønsted acid catalyzed cascade reaction: Organocatalytic transfer hydrogenation of quinolines and their application in the synthesis of alkaloids. Angew. Chem. Int. Ed. 2006, 45, 3683–3686. [Google Scholar] [CrossRef] [PubMed]
- Park, D.Y.; Lee, S.Y.; Jeon, J.; Cheon, C.-H. Enantioselective synthesis of tetrahydroquinolines from 2-aminochalcones via a consecutive one-pot reaction catalyzed by chiral phosphoric acid. J. Org. Chem. 2018, 83, 12486–12495. [Google Scholar] [CrossRef]
- Yuan, M.; Mbaezue, I.I.; Zhou, Z.; Topic, F.; Tsantrizos, Y.S. P-Chiral, N-phosphoryl sulfonamide Brønsted acids with an intramolecular hydrogen bond interaction that modulates organocatalysis. Org. Biomol Chem. 2019, 17, 8690–8694. [Google Scholar] [CrossRef]
- Han, Z.S.; Wu, H.; Qu, B.; Wang, Y.; Wu, L.; Zhang, L.; Xu, Y.; Wu, L.; Zhang, Y.; Lee, H.; et al. New class of P-stereogenic chiral Brønsted acid catalysts derived from chiral phosphinamides. Tetrahedron Lett. 2019, 60, 1834–1837. [Google Scholar] [CrossRef]
- Rueping, M.; Sugiono, E.; Steck, A.; Theissmann, T. Synthesis and application of polymer-supported chiral Brønsted acid organocatalysts. Adv. Synth. Catal. 2010, 352, 281–287. [Google Scholar] [CrossRef]
- Kundu, D.S.; Schmidt, J.; Bleschke, C.; Thomas, A.; Blechert, S. A microporous binol-derived phosphoric acid. Angew. Chem. Int. Ed. 2012, 51, 5456–5459. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Noto, R. Supported proline and proline derivatives as recyclable organocatalysts. Chem. Soc. Rev. 2008, 37, 1666–1688. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, G.; Ding, K. Self-supported catalysts. Chem. Rev. 2008, 109, 322–359. [Google Scholar] [CrossRef] [PubMed]
- Monterde, C.; Navarro, R.; Iglesias, M.; Sánchez, F. Adamantyl-BINOL as platform for chiral porous polymer aromatic frameworks. Multiple applications as recyclable catalysts. J. Catal. 2019, 377, 609–618. [Google Scholar] [CrossRef]
- Marcelli, T.; Hammar, P.; Himo, F. Phosphoric acid catalyzed enantioselective transfer hydrogenation of imines: A density functional theory study of reaction mechanism and the origins of enantioselectivity. Chem.-Eur. J. 2008, 14, 8562–8571. [Google Scholar] [CrossRef] [PubMed]
- Pastor, J.; Rezabal, E.; Voituriez, A.; Betzer, J.F.; Marinetti, A.; Frison, G. Revised theoretical model on enantiocontrol in phosphoric acid catalyzed H-transfer hydrogenation of quinoline. J. Org. Chem. 2018, 83, 2779–2787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storer, R.I.; Carrera, D.E.; Ni, Y.; MacMillan, D.W. Enantioselective organocatalytic reductive amination. J. Am. Chem. Soc. 2006, 128, 84–86. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Nicoletti, M.; List, B. Catalytic asymmetric reductive amination of aldehydes via dynamic kinetic resolution. J. Am. Chem. Soc. 2006, 128, 13074–13075. [Google Scholar] [CrossRef]
- Wakchaure, V.N.; Zhou, J.; Hoffmann, S.; List, B. Catalytic asymmetric reductive amination of alpha-branched ketones. Angew. Chem. Int. Ed. Engl. 2010, 49, 4612–4614. [Google Scholar] [CrossRef]
- Khan, I.A.; Saxena, A.K. Metal-Free, mild, nonepimerizing, chemo- and enantio- or diastereoselective N-alkylation of amines by alcohols via oxidation/imine–iminium formation/reductive amination: A pragmatic synthesis of octahydropyrazinopyridoindoles and higher ring analogues. J. Org. Chem. 2013, 78, 11656–11669. [Google Scholar] [CrossRef]
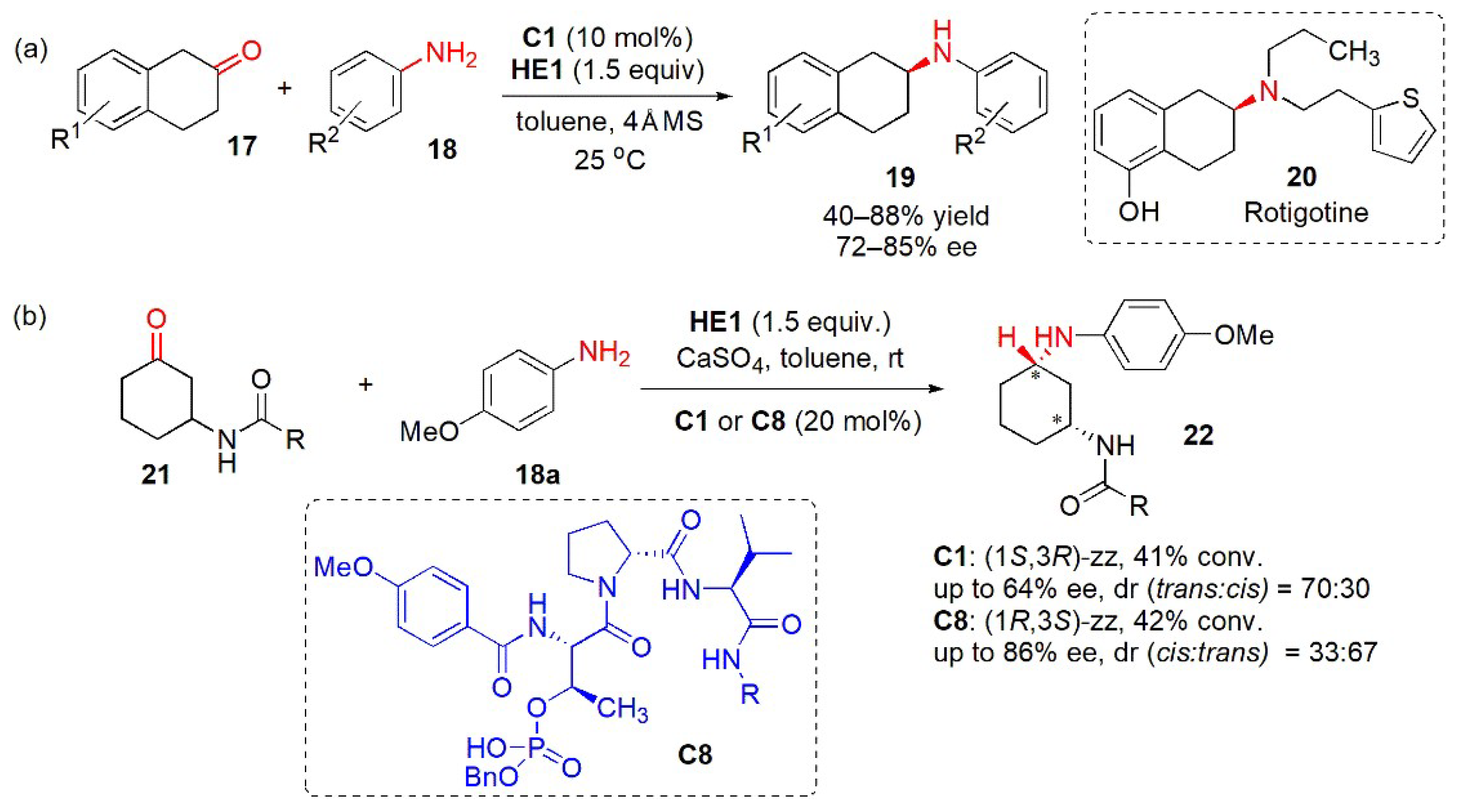
- Park, D.Y.; Kim, K.-H.; Cheon, C.-H. Enantioselective synthesis of β-aminotetralins via chiral phosphoric acid-catalyzed reductive amination of β-tetralones. Adv. Synth. Catal. 2018, 360, 462. [Google Scholar] [CrossRef]
- Zareba, G. Rotigotine: A novel dopamine agonist for the transdermal treatment of Parkinson’s disease. Drugs Today (Barc) 2006, 42, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Shugrue, C.R.; Featherston, A.L.; Lackner, R.M.; Lin, A.; Miller, S.J. Divergent stereoselectivity in phosphothreonine (pThr)-catalyzed reductive aminations of 3-amidocyclohexanones. J. Org. Chem. 2018, 83, 4491–4504. [Google Scholar] [CrossRef] [PubMed]
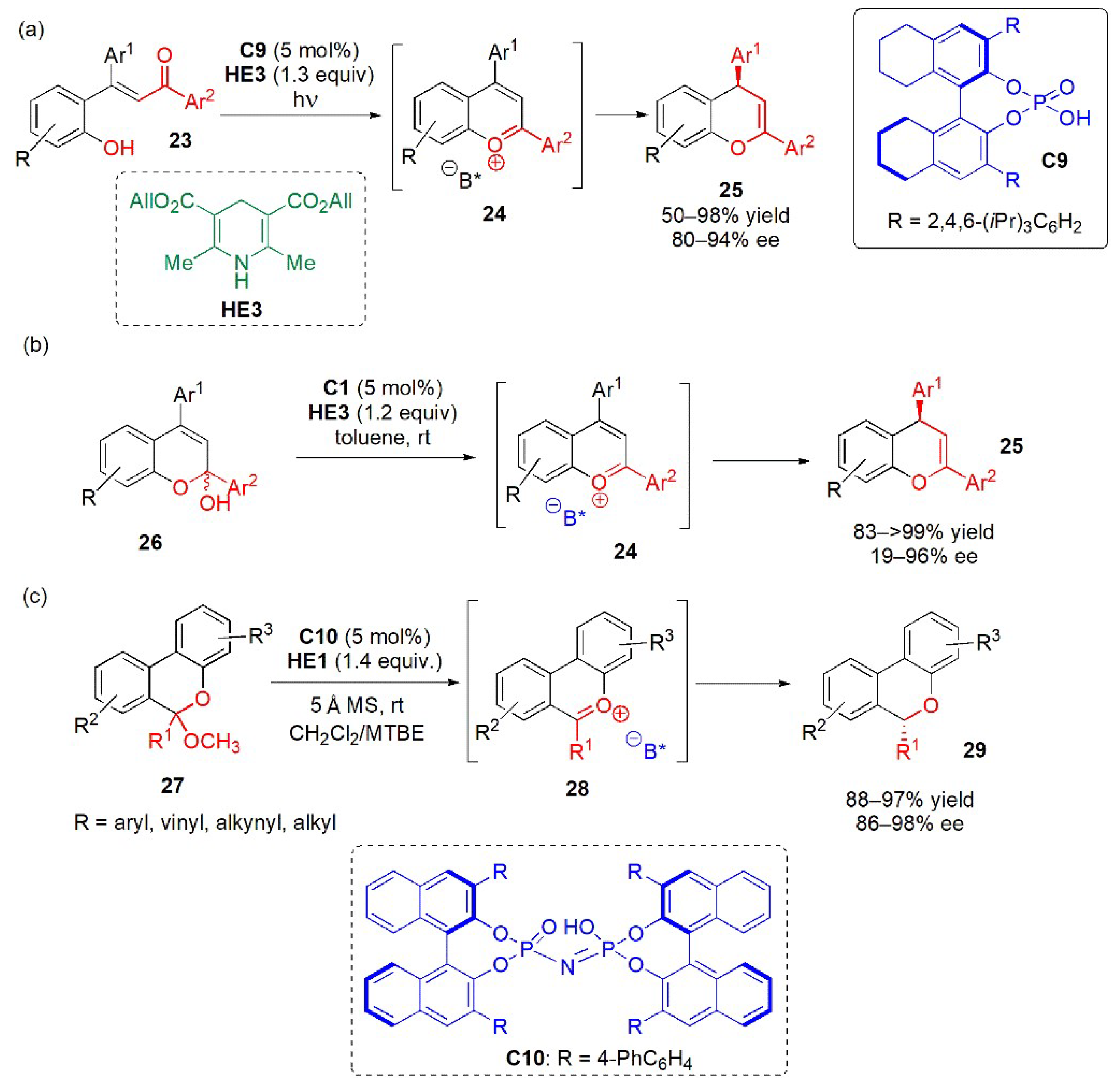
- Hsiao, C.C.; Liao, H.H.; Sugiono, E.; Atodiresei, I.; Rueping, M. Shedding light on organocatalysis-light-assisted asymmetric ion-pair catalysis for the enantioselective hydrogenation of pyrylium ions. Chem.-A Eur. J. 2013, 19, 9775–9779. [Google Scholar] [CrossRef]
- Terada, M.; Yamanaka, T.; Toda, Y. Chiral anion catalysis in the enantioselective 1,4-reduction of the 1-benzopyrylium ion as a reactive intermediate. Chem. Eur. J. 2013, 19, 13658–13662. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wan, M.; Sun, S.; Fu, Z.; Huang, H.; Liu, L. Efficient access to chiral benzo[c]chromenes via asymmetric transfer hydrogenation of ketals. Org. Chem. Front. 2018, 5, 1280–1283. [Google Scholar] [CrossRef]
- Touge, T.; Nara, H.; Fujiwhara, M.; Kayaki, Y.; Ikariya, T. Efficient access to chiral benzhydrols via asymmetric transfer hydrogenation of unsymmetrical benzophenones with bifunctional oxo-tethered ruthenium catalysts. J. Am. Chem. Soc. 2016, 138, 10084–10087. [Google Scholar] [CrossRef] [PubMed]
- Zhi, L.; Tegley, C.M.; Pio, B.; Edwards, J.P.; Motamedi, M.; Jones, T.K.; Marschke, K.B.; Mais, D.E.; Risek, B.; Schrader, W.T. 5-Benzylidene-1,2-dihydrochromeno[3,4-f]quinolines as selective progesterone receptor modulators. J. Med. Chem. 2003, 46, 4104–4112. [Google Scholar] [CrossRef]
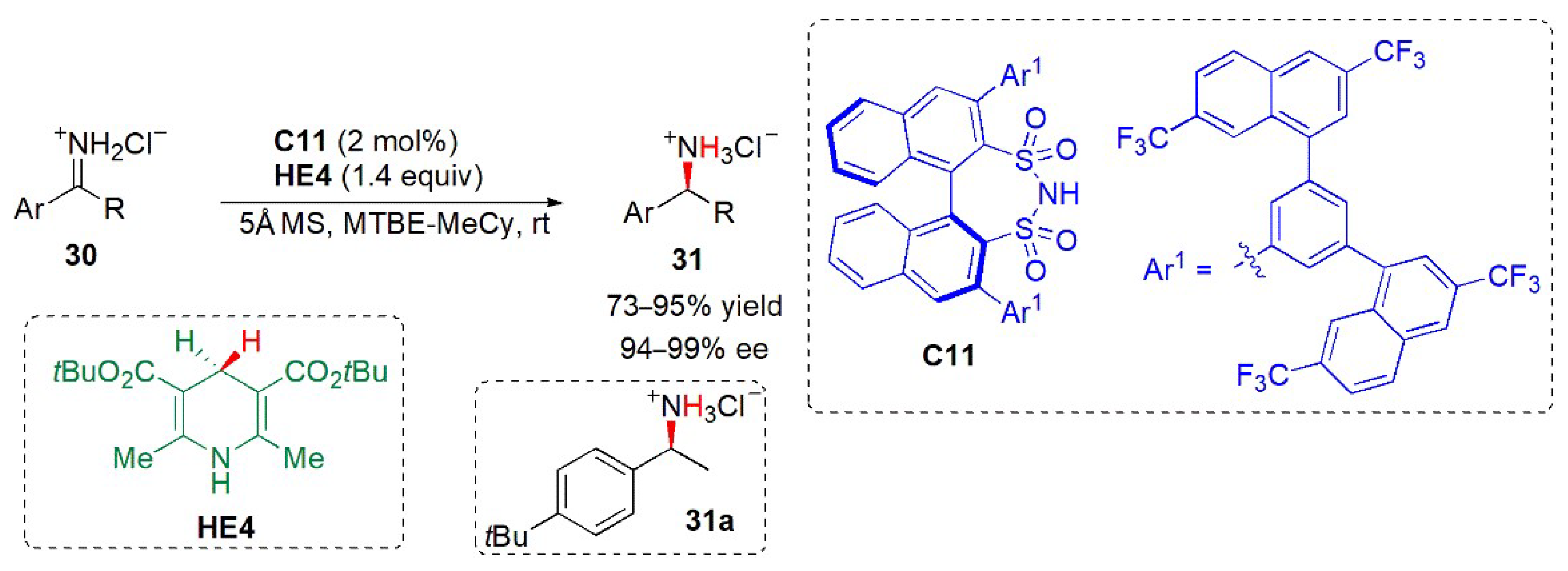
- García-García, P.; Lay, F.; García-García, P.; Rabalakos, C.; List, B. A powerful chiral counteranion motif for asymmetric catalysis. Angew. Chem. Int. Ed. 2009, 48, 4363–4366. [Google Scholar] [CrossRef]
- Wakchaure, V.N.; Kaib, P.S.J.; Leutzsch, M.; List, B. Disulfonimide-catalyzed asymmetric reduction of N-alkyl imines. Angew. Chem. Int. Ed. 2015, 54, 1852–11856. [Google Scholar] [CrossRef]
- Rothermel, K.; Žabka, M.; Hioe, J.; Gschwind, R.M. Disulfonimides versus phosphoric acids in Brønsted acid catalysis: The effect of weak hydrogen bonds and multiple acceptors on complex structures and reactivity. J. Org. Chem. 2019, 84, 13221–13231. [Google Scholar] [CrossRef]
- Wakchaure, V.N.; Obradors, C.; List, B. Chiral Brønsted acids catalyze asymmetric additions to substrates that are already protonated: Highly enantioselective disulfonimide-catalyzed Hantzsch ester reductions of NH–imine hydrochloride salts. Synlett 2020, 31, 1707–1712. [Google Scholar]
- Zhao, Q.; Wen, J.; Tan, R.; Huang, K.; Metola, P.; Wang, R.; Anslyn, E.V.; Zhang, X. Rhodium-catalyzed asymmetric hydrogenation of unprotected NH imines assisted by a thiourea. Angew. Chem. Int. Ed. 2014, 53, 8467–8470. [Google Scholar] [CrossRef] [PubMed]
- Žabka, M.; Gschwind, R.M. Ternary complexes of chiral disulfonimides in transfer-hydrogenation of imines: The relevance of late intermediates in ion pair catalysis. Chem. Sci. 2021, 12, 15263–15272. [Google Scholar] [CrossRef] [PubMed]
- Faisca Phillips, A.M.; Prechtl, M.G.H.; Pombeiro, A.J.L. Non-covalent interactions in enantioselective organocatalysis: Theoretical and mechanistic studies of reactions mediated by dual H-bond donors, bifunctional squaramides, thioureas and related catalysts. Catalysts 2021, 11, 569. [Google Scholar] [CrossRef]
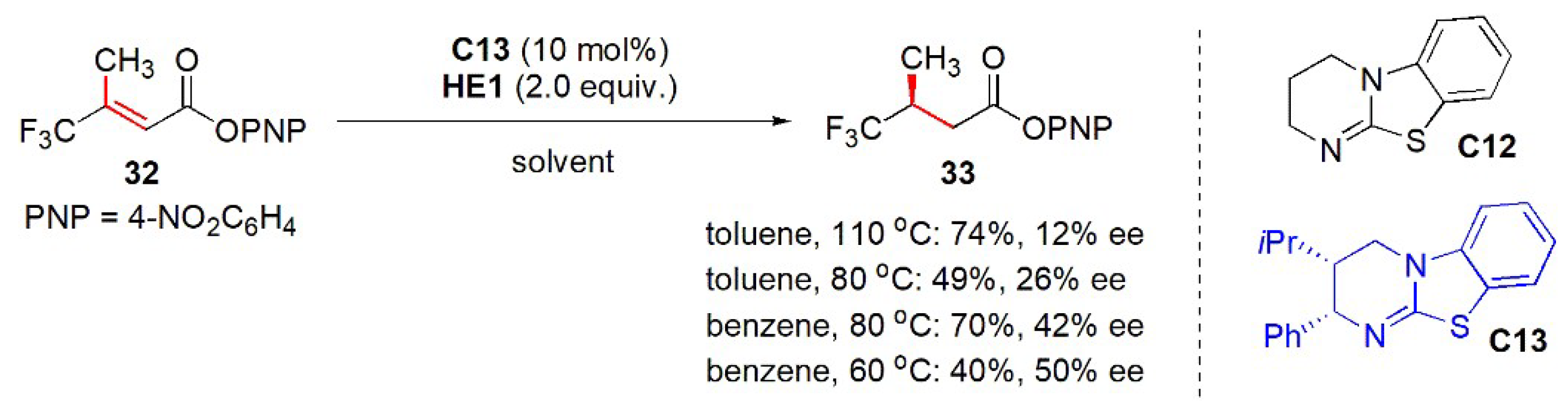
- Wu, J.; Young, C.M.; Smith, A.D. Isothiourea-catalysed transfer hydrogenation of α,β-unsaturated para-nitrophenyl esters. Tetrahedron 2021, 78, 131758. [Google Scholar] [CrossRef]
- Corti, V.; Riccioli, R.; Martinelli, A.; Sandri, S.; Fochi, M.; Bernardi, L. Stereodivergent entry to β-branched β-trifluoromethyl α-amino acid derivatives by sequential catalytic asymmetric reactions. Chem. Sci. 2021, 12, 10233–10241. [Google Scholar] [CrossRef]
- Yang, J.W.; Fonseca, M.T.H.; Vignola, N.; List, B. Metal-free, organocatalytic asymmetric transfer hydrogenation of alpha,beta-unsaturated aldehydes. Angew. Chem. Int. Ed. 2005, 44, 108–110. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Li, D.-H.; Liu, Y.-K. Diversified synthesis of chiral chromane-containing polyheterocyclic compounds via asymmetric organocatalytic cascade reactions. ACS Omega 2018, 3, 16615–16625. [Google Scholar] [CrossRef]
- Wei, Y.; Zhou, Q.-Q.; Tan, F.; Lu, L.-Q.; Xiao, W.-J. Visible-light-driven organic photochemical reactions in the absence of external photocatalysts. Synthesis 2019, 51, 3021–3054. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Mazzarella, D.; Melchiorre, P. Synthetic methods driven by the photoactivity of electron donor− acceptor complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. [Google Scholar] [CrossRef] [Green Version]
- Arceo, E.; Jurberg, I.D.; Álvarez-Fernández, A.; Melchiorre, P. Photochemical activity of a key donor–acceptor complex can drive stereoselective catalytic α-alkylation of aldehydes. Nature Chem. 2019, 5, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Goti, G.; Bieszczad, B.; Vega-Peñaloza, A.; Melchiorre, P. Stereocontrolled synthesis of 1,4-dicarbonyl compounds by photochemical organocatalytic acyl radical addition to enals. Angew. Chem. Int. Ed. Engl. 2019, 58, 1213–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, K.; Tan, S.M.; Lee, R.; Yang, S.; Jia, H.; Zhao, X.; Qiao, B.; Jiang, Z. Catalytic enantioselective addition of prochiral radicals to vinylpyridines. J. Am. Chem. Soc. 2019, 141, 5437–5443. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Dai, Y.; Jia, H.; Li, J.; Bu, L.; Qiao, B.; Zhao, X.; Jiang, Z. Conjugate addition−enantioselective protonation of N-aryl glycines to α-branched 2-vinylazaarenes via cooperative photoredox and asymmetric catalysis. J. Am. Chem. Soc. 2018, 140, 6083–6087. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Muir, C.W.; Leach, A.G.; Kennedy, A.R.; Watson, A.J.B. Catalytic enantioselective synthesis of α-chiral azaheteroaryl ethylamines by asymmetric protonation. Angew. Chem. Int. Ed. 2018, 57, 11374–11377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepburn, H.B.; Melchiorre, P. Brønsted acid-catalysed conjugate addition of photochemically generated α-amino radicals to alkenylpyridines. Chem. Commun. 2016, 52, 3520–3523. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, X.; Liu, H.; Xu, L.; Zhuang, J.; Li, J.; Li, H.; Wang, W. Organocatalytic enantioselective direct additions of aldehydes to 4-vinylpyridines and electron-deficient vinylarenes and their synthetic applications. J. Am. Chem. Soc. 2015, 137, 2303–2310. [Google Scholar] [CrossRef]
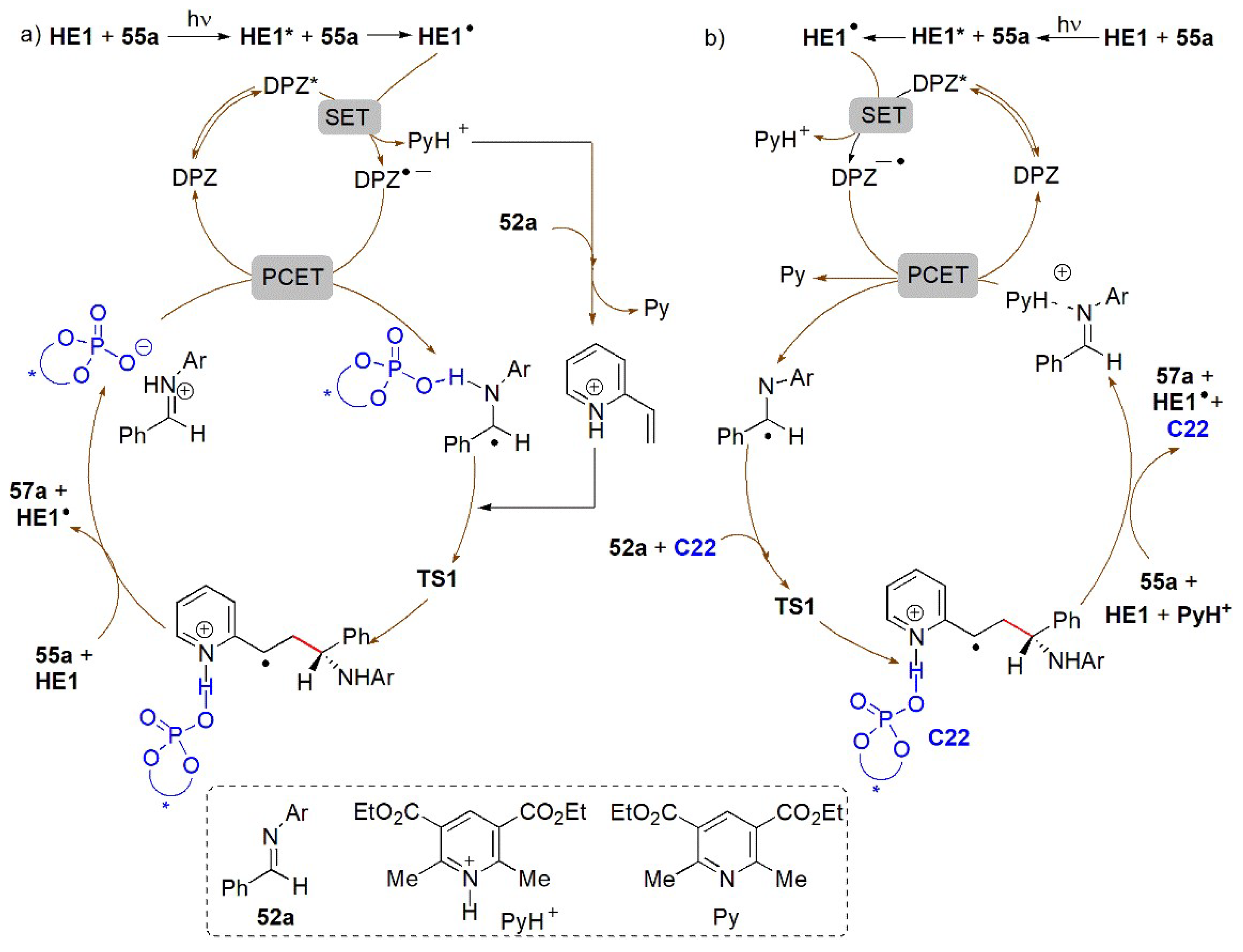
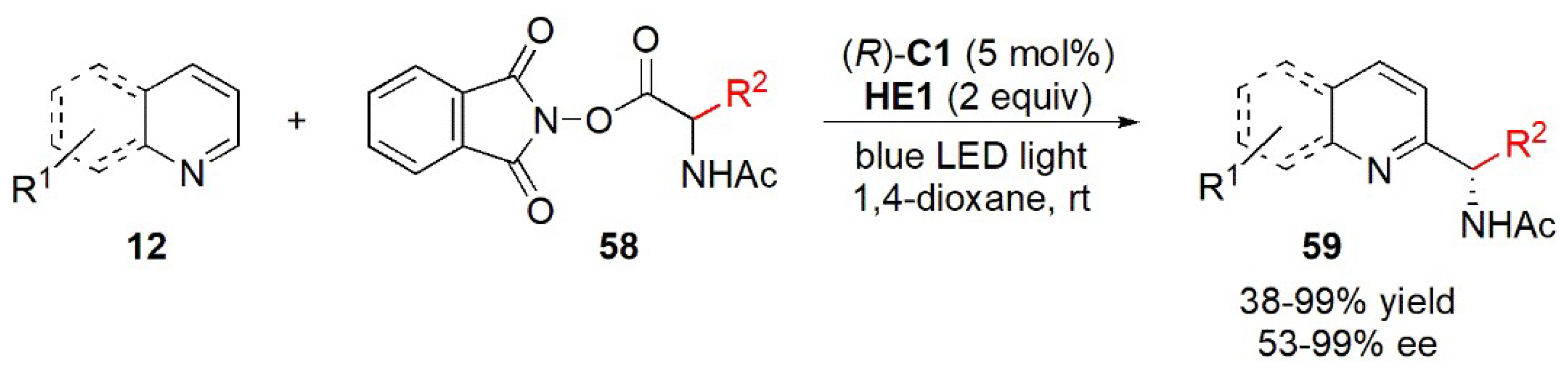
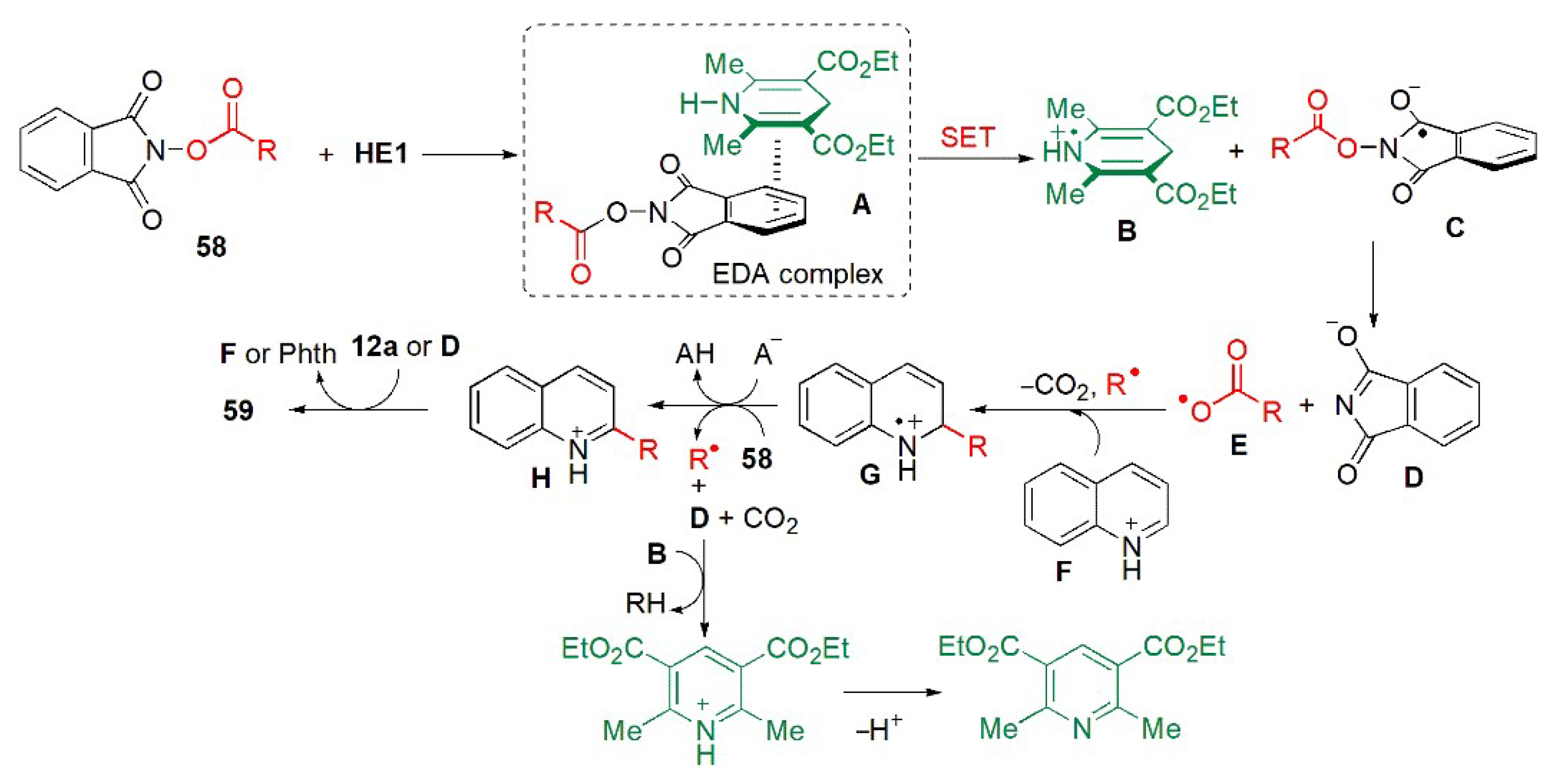
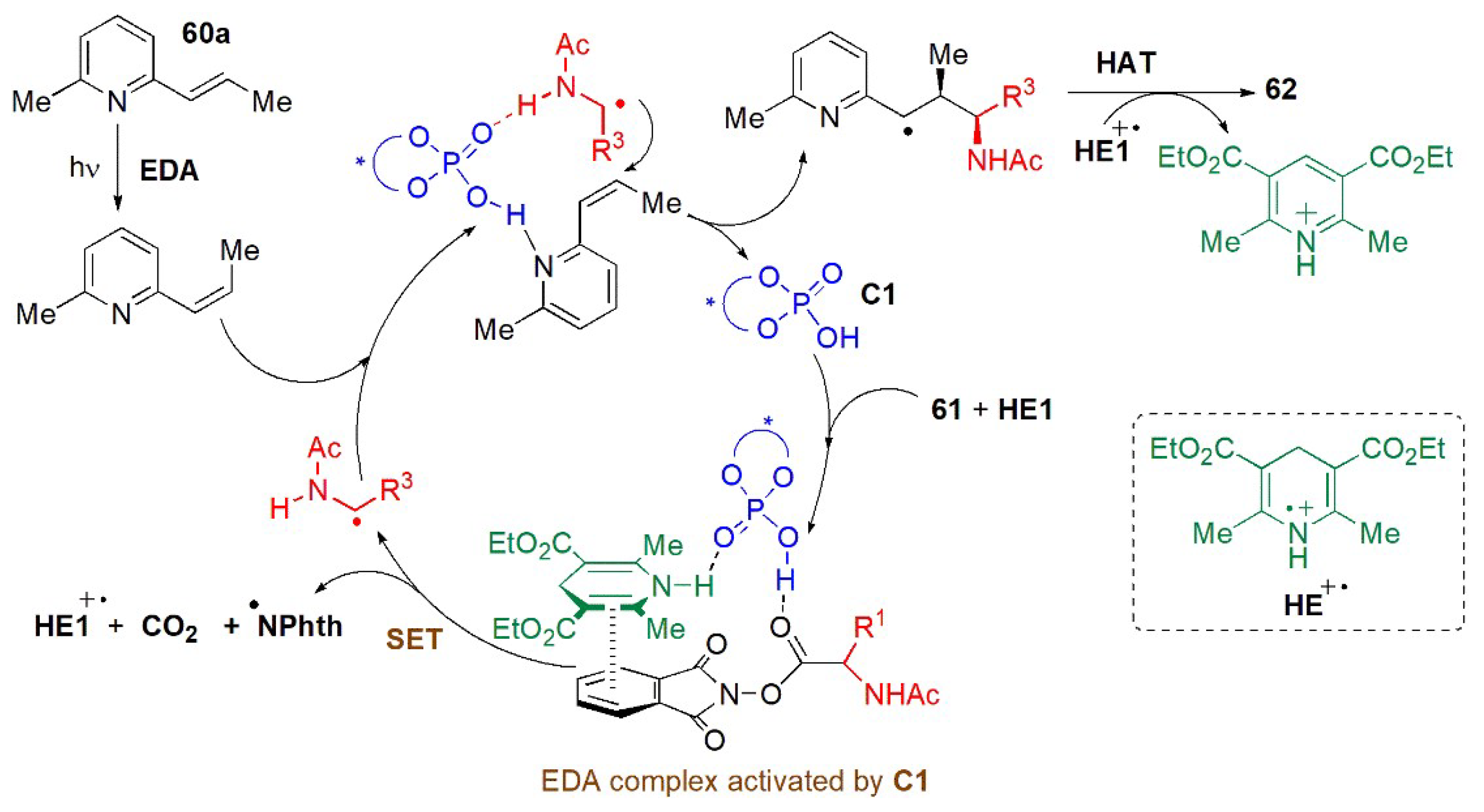
- Li, J.; Tan, S.S.; Kyne, S.H.; Chana, P.W.H. Minisci-type alkylation of N-heteroarenes by N-acyloxy)phthalimide esters mediated by a Hantzsch ester and blue LED light. Adv. Synth. Catal. 2022, 364, 802–810. [Google Scholar] [CrossRef]
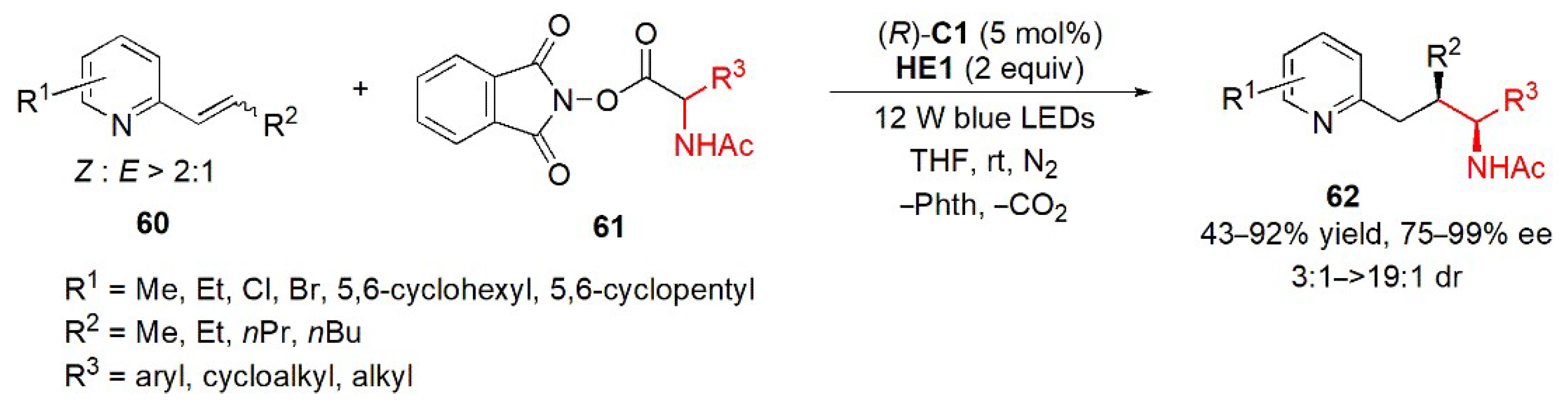
- Guo, J.; Xie, Y.; Lai, Z.-M.; Weng, J.; Chan, A.S.C.; Lu, G. Enantioselective hydroalkylation of alkenylpyridines enabled by merging photoactive electron donor−acceptor complexes with chiral bifunctional organocatalysis. ACS Catal. 2022, 12, 13065–13074. [Google Scholar] [CrossRef]
- Xiong, T.; Zhang, Q. Recent Advances in the direct construction of enantioenriched stereocenters through addition of radicals to internal alkenes. Chem. Soc. Rev. 2021, 50, 8857–8873. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
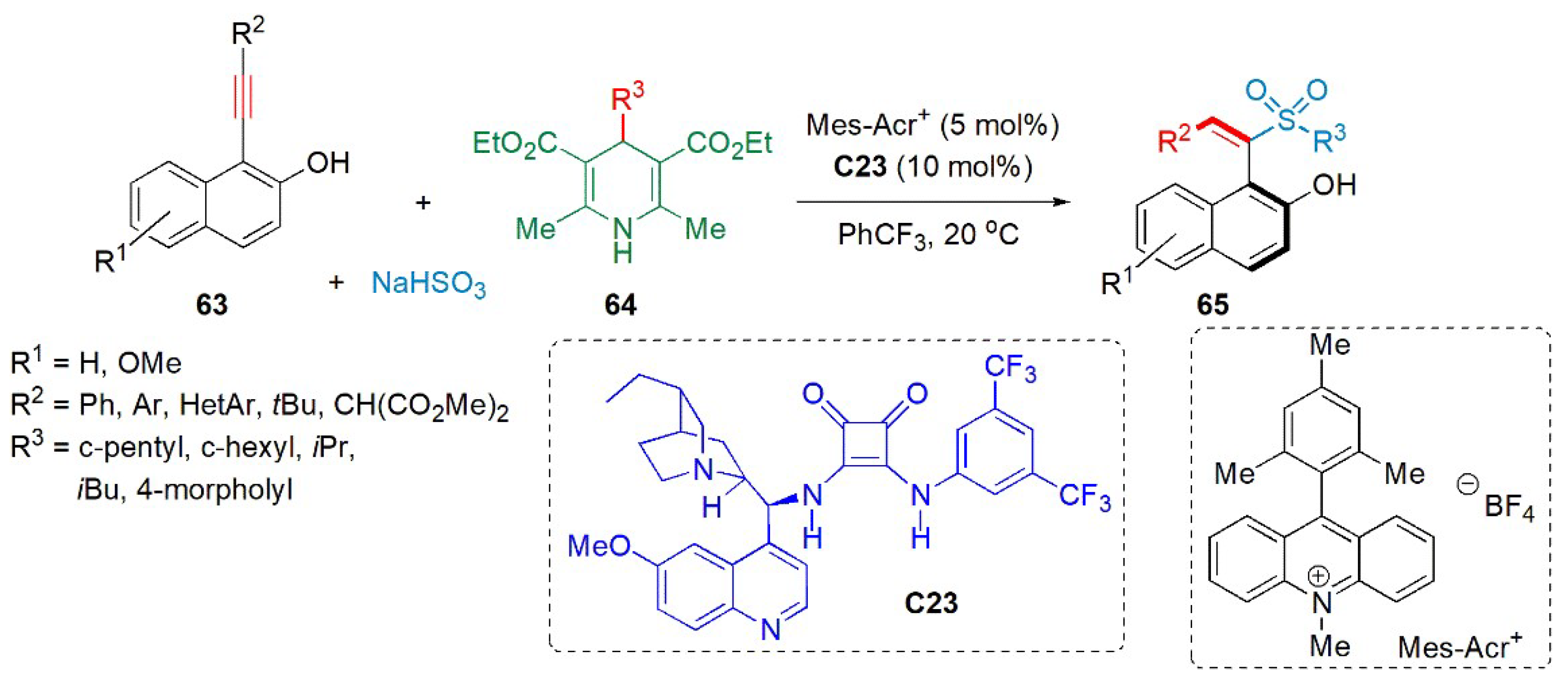
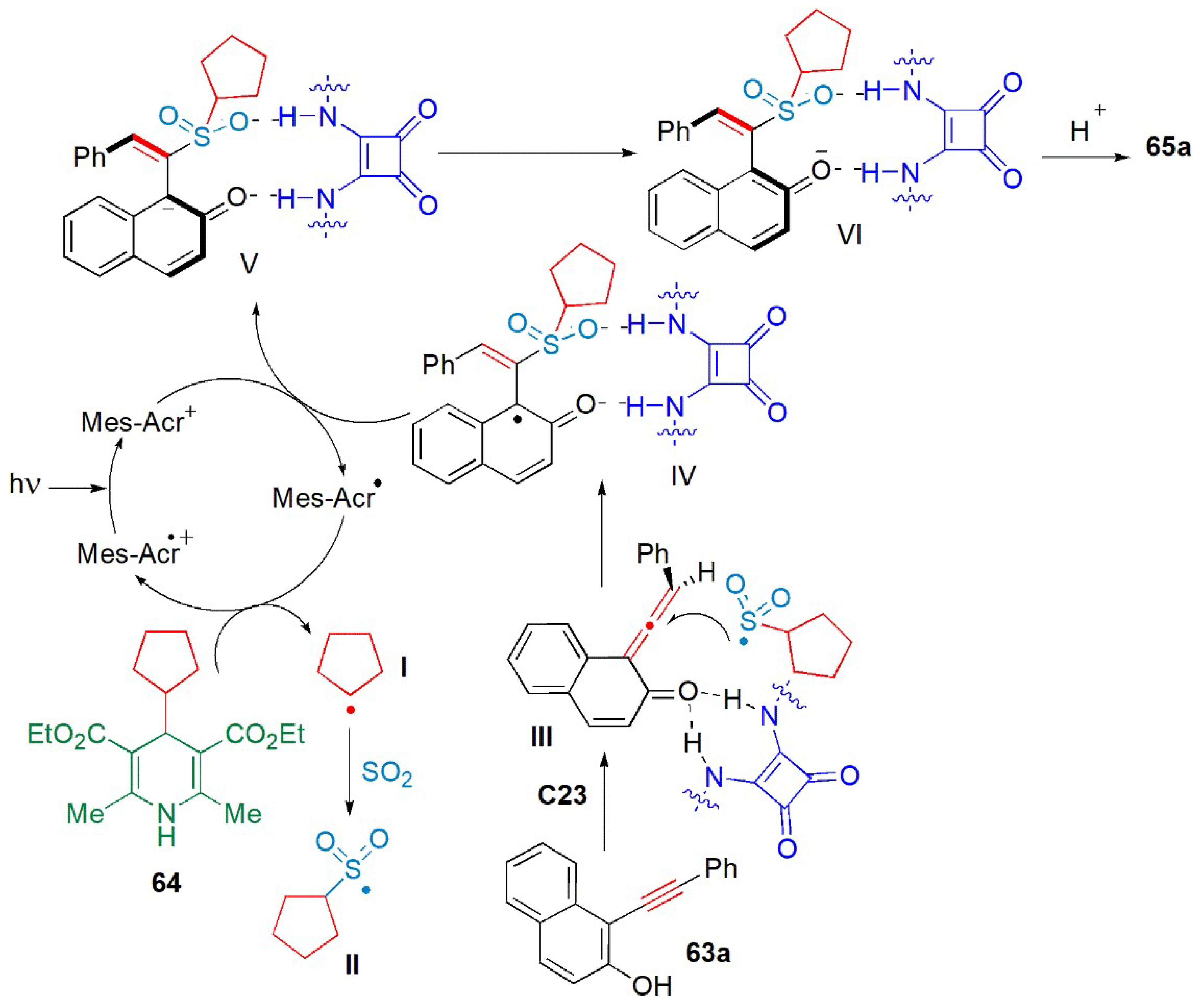
- Wang, X.; Ding, Q.; Yang, C.; Yang, J.; Wu, J. Enantioselective sulfonylation using sodium hydrogen sulfite, 4-substituted Hantzsch esters and 1-(arylethynyl)naphthalen-2-ols. Org. Chem. Front. 2023, 10, 92–98. [Google Scholar] [CrossRef]
- Wang, X.; Liu, R.; Ding, Q.; Xiao, W.; Wu, J. Synergistic photoredox and tertiary amine catalysis: Generation of allylic sulfones from Morita-Baylis-Hillman acetates and sulfur dioxide. Org. Chem. Front. 2021, 8, 3308–3313. [Google Scholar] [CrossRef]
- Zhang, C.; Tang, Z.; Qiu, Y.; Tang, J.; Ye, S.; Li, Z.; Wu, J. Access to axially chiral styrenes via a photoinduced asymmetric radical reaction involving a sulfur dioxide insertion. Chem. Catal. 2022, 2, 164–177. [Google Scholar] [CrossRef]
























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faisca Phillips, A.M.; Pombeiro, A.J.L. Applications of Hantzsch Esters in Organocatalytic Enantioselective Synthesis. Catalysts 2023, 13, 419. https://doi.org/10.3390/catal13020419
Faisca Phillips AM, Pombeiro AJL. Applications of Hantzsch Esters in Organocatalytic Enantioselective Synthesis. Catalysts. 2023; 13(2):419. https://doi.org/10.3390/catal13020419
Chicago/Turabian StyleFaisca Phillips, Ana Maria, and Armando J. L. Pombeiro. 2023. "Applications of Hantzsch Esters in Organocatalytic Enantioselective Synthesis" Catalysts 13, no. 2: 419. https://doi.org/10.3390/catal13020419