
Photocatalytic Degradation of Organic and Inorganic Pollutants to Harmless End Products: Assessment of Practical Application Potential for Water and Air Cleaning
 , , and
, , and
Abstract
:
1. Introduction
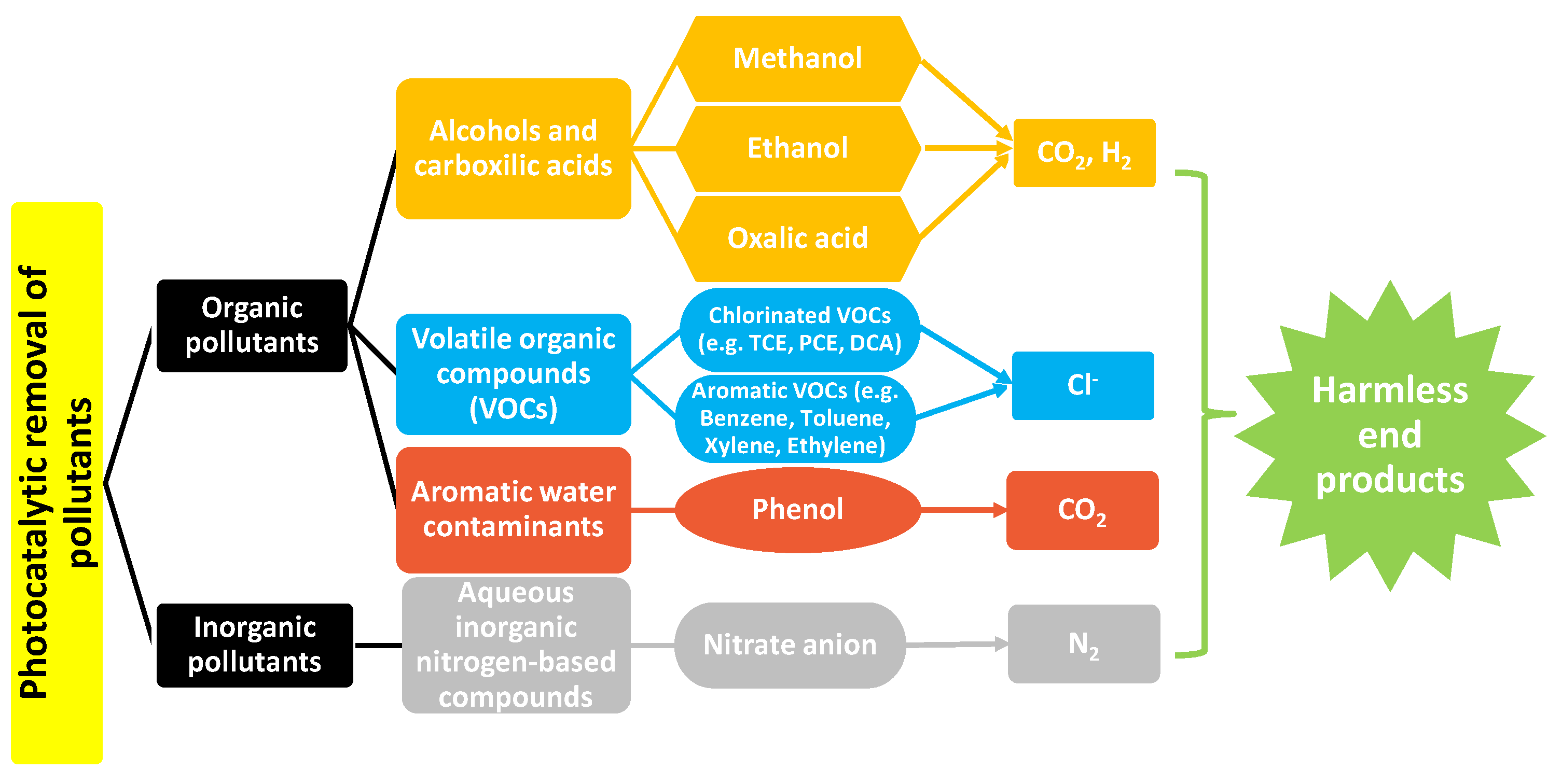
2. Photocatalytic Removal of Organic and Inorganic Pollutants
2.1. Photooxidation of Primary Alcohols and Carboxylic Acids in Gaseous and Liquid Media
2.1.1. Basic Data on Methanol Photodegradation on TiO2
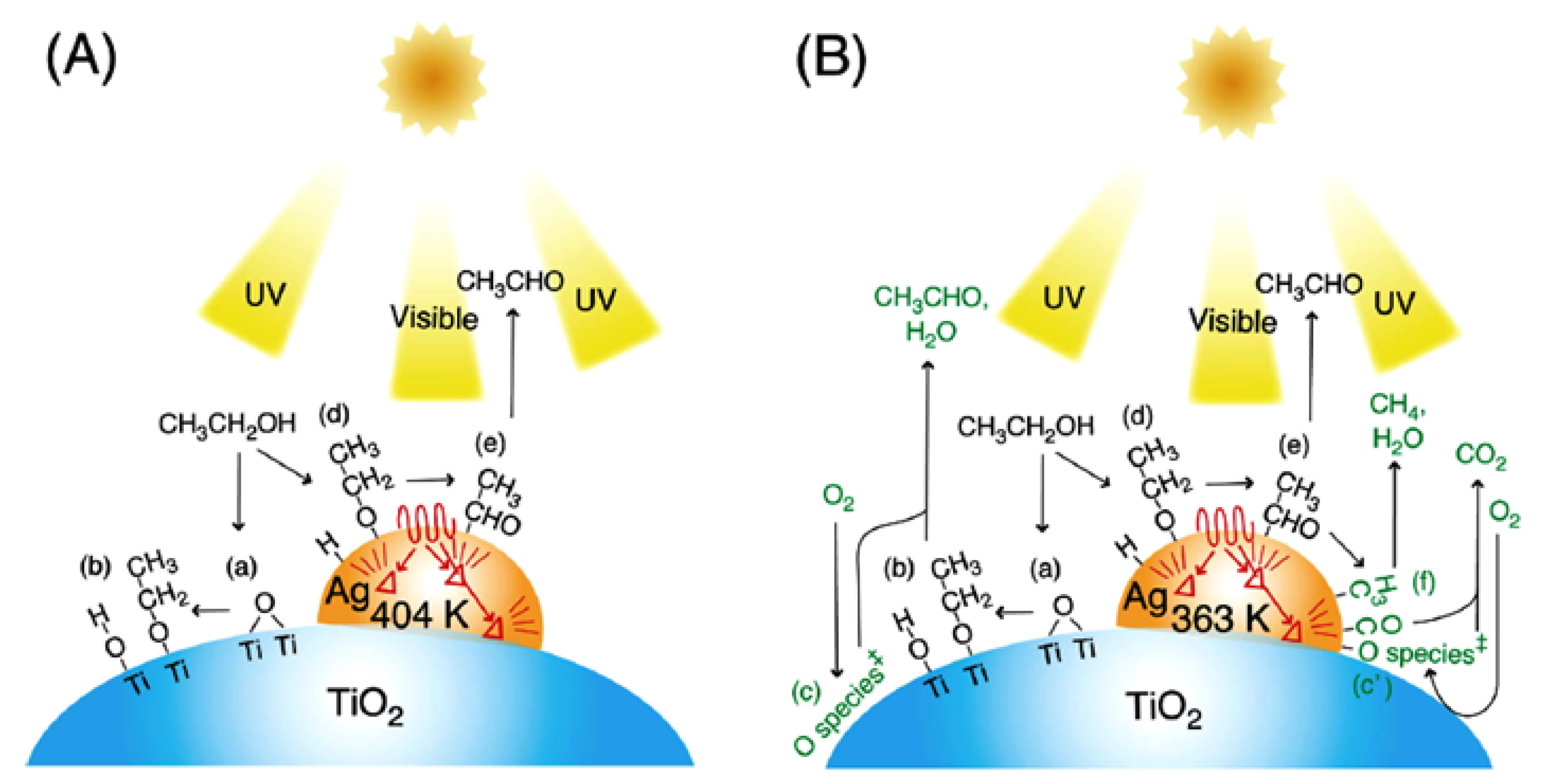
2.1.2. Methanol and Ethanol Oxidation in Gaseous Phase on Bare and Modified Catalysts
2.1.3. Methanol, Ethanol, and Oxalic Acid Oxidation in Liquid Phase on Pristine and Modified Catalysts
2.2. Volatile Organic Compounds (VOCs) from Air and Wastewater
2.2.1. Overview of Volatile Organic Compounds (VOCs)
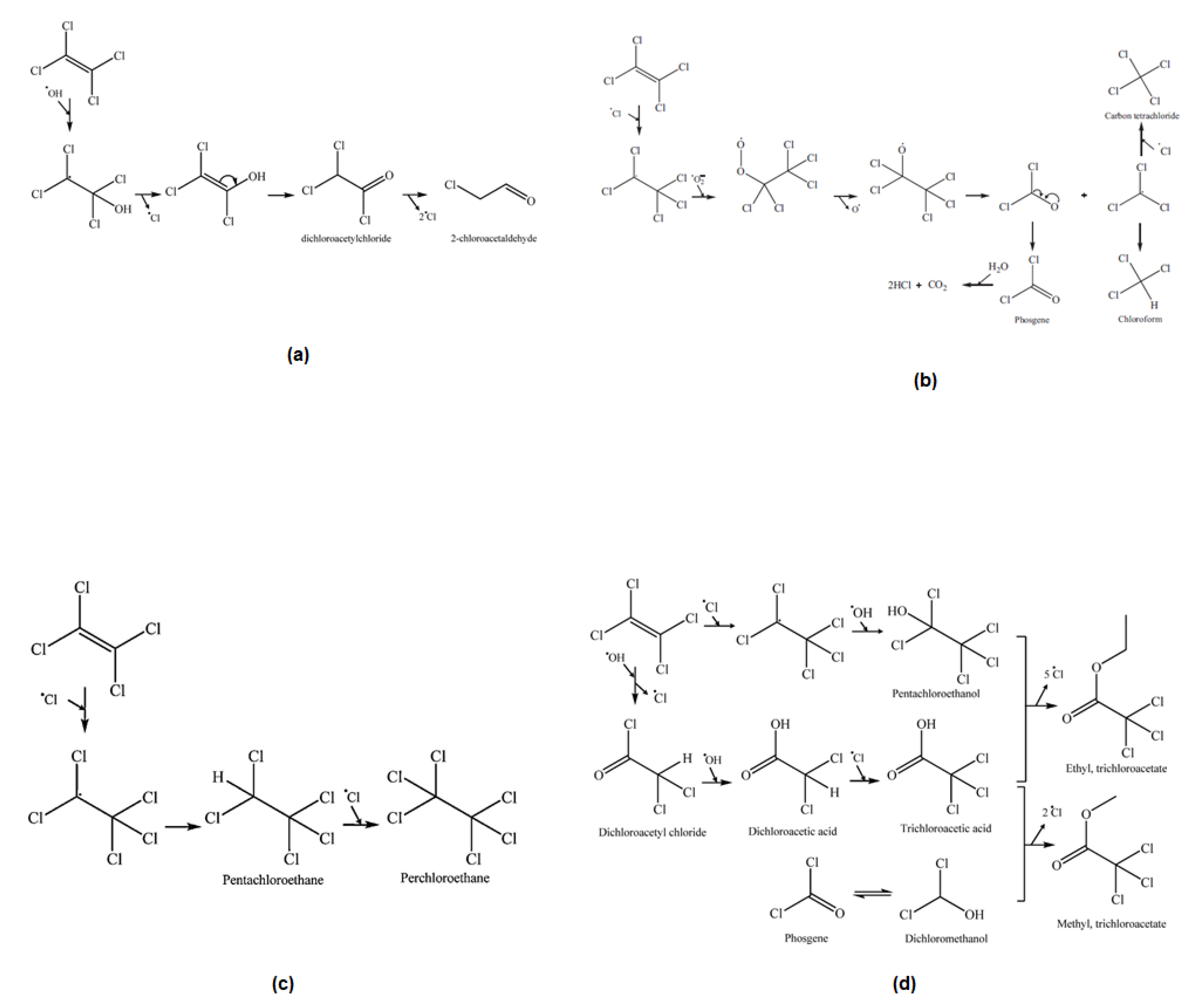
2.2.2. Chlorinated VOCs Abatement from Air and Water
2.2.3. Aromatic VOCs Abatement
2.3. Aromatic Water Contaminants
2.3.1. Phenol: General Properties and Uses
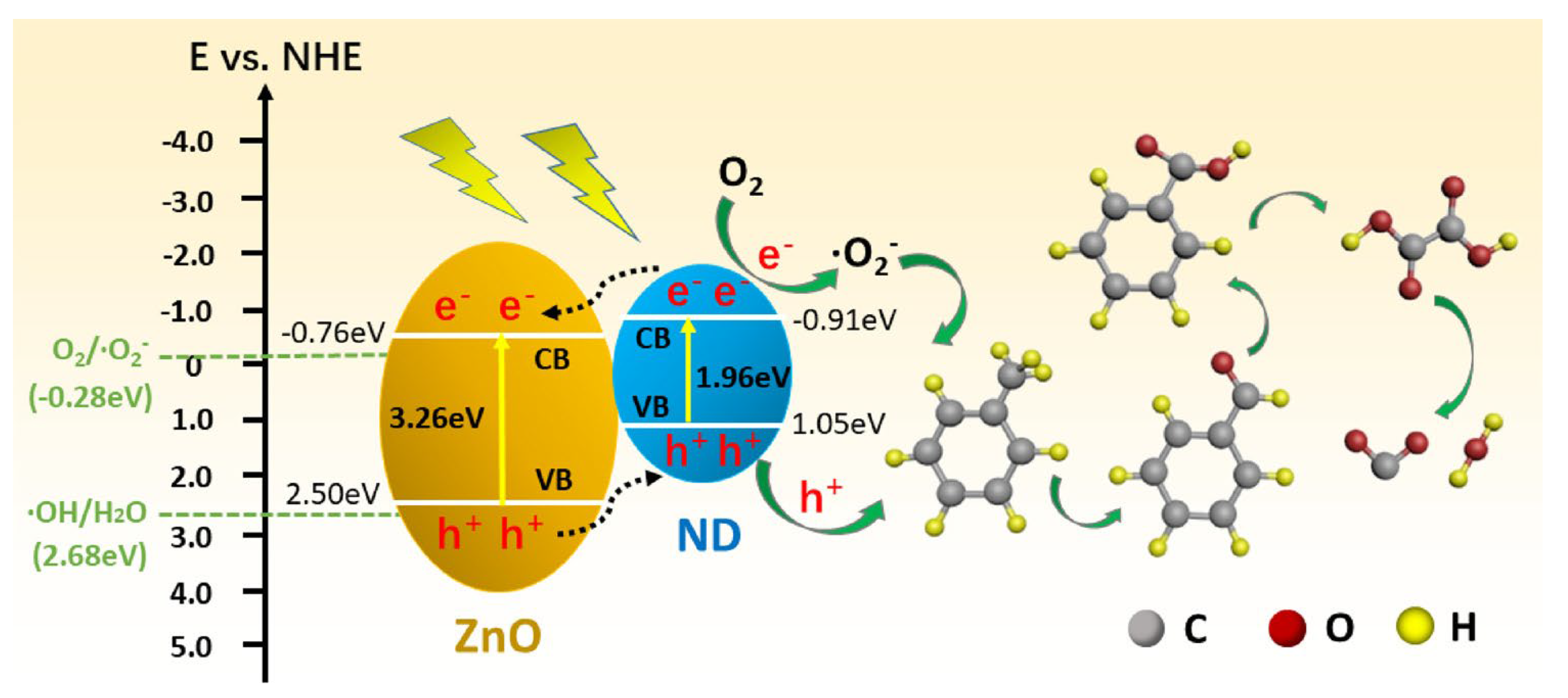
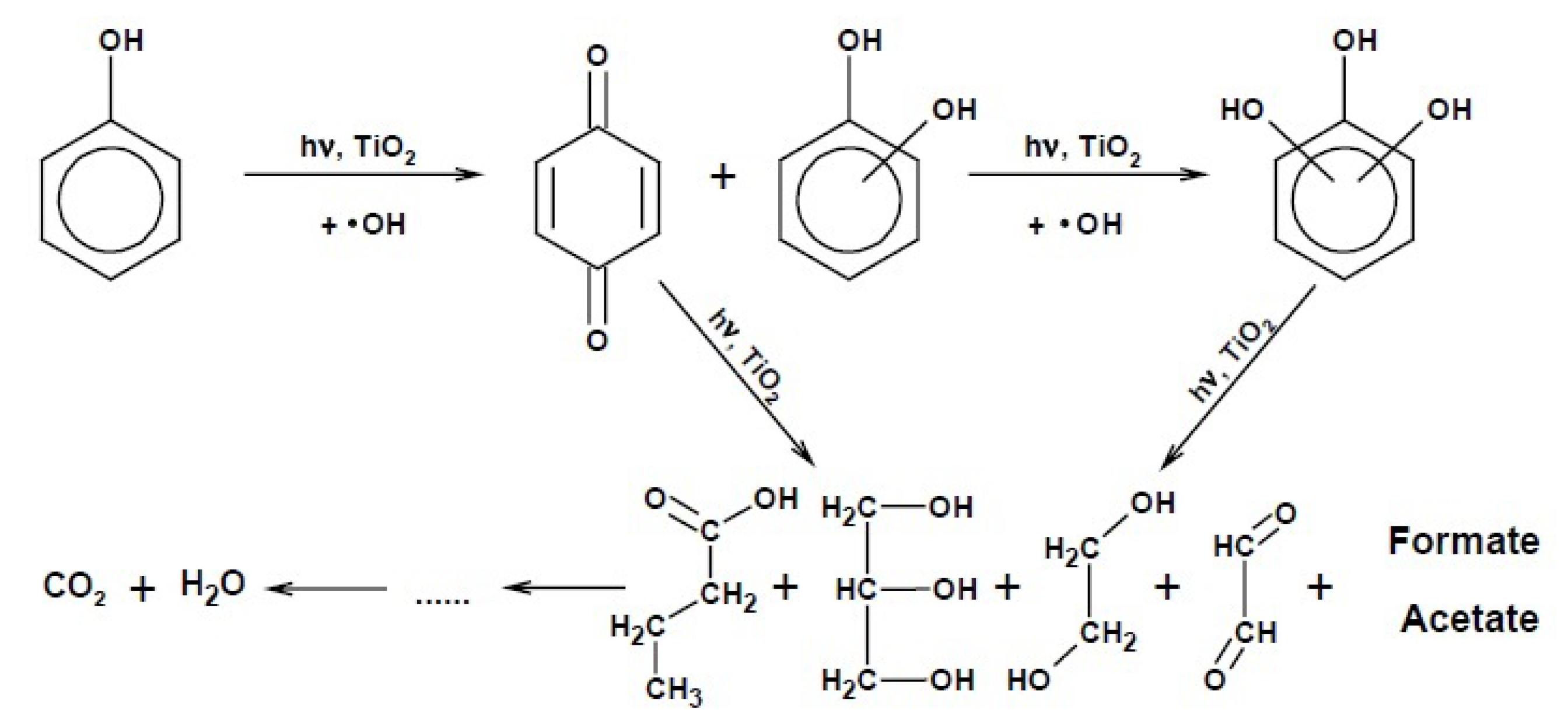
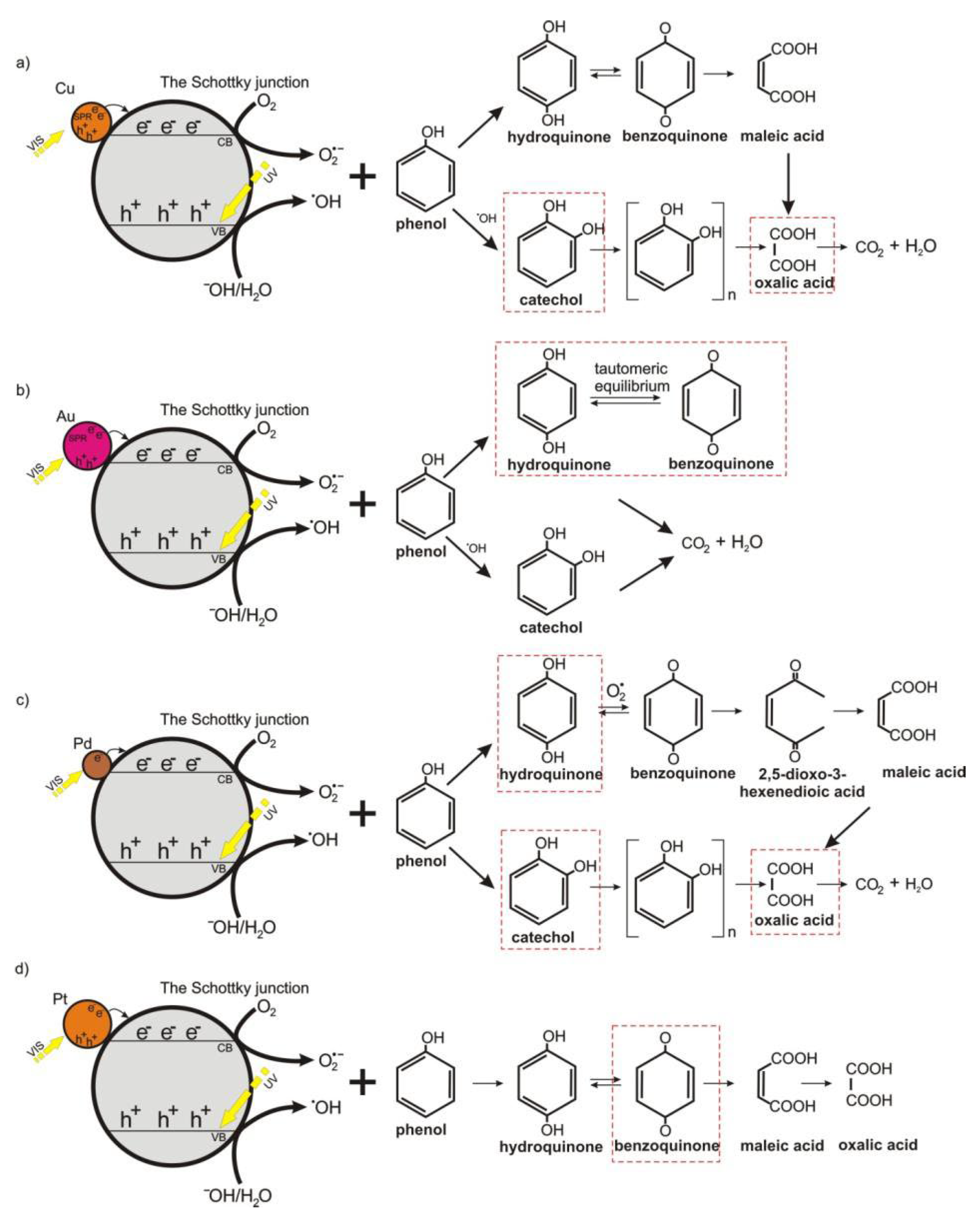
2.3.2. Mechanism of Photocatalytic Removal of Organic Pollutants
2.3.3. Phenol Removal over Various Catalytic Materials
2.3.4. Photocatalysis Coupled with Ozonation Process
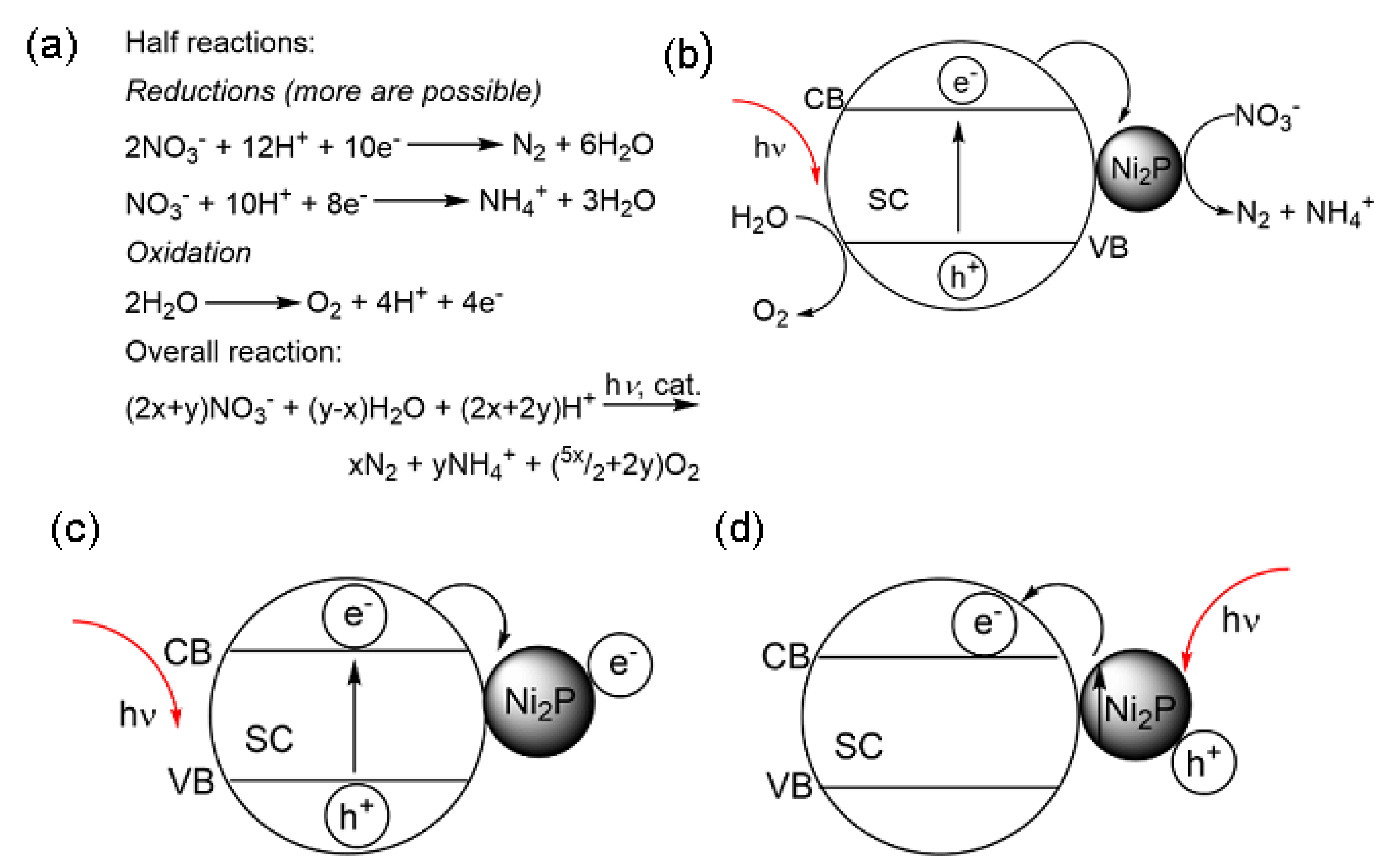
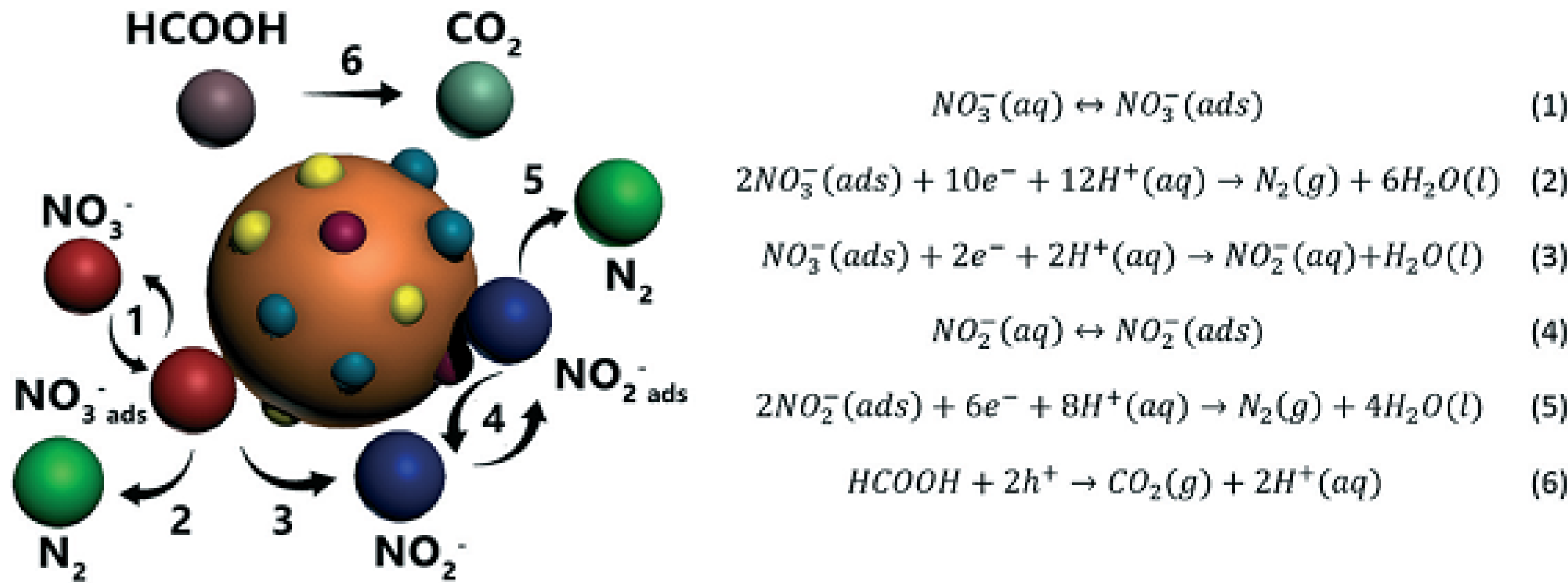
2.4. Aqueous Inorganic Nitrogen-Based Pollutants
2.4.1. Overview of Nitrate Anion and Its Reaction Intermediates (Nitrite and Ammonium Ions)
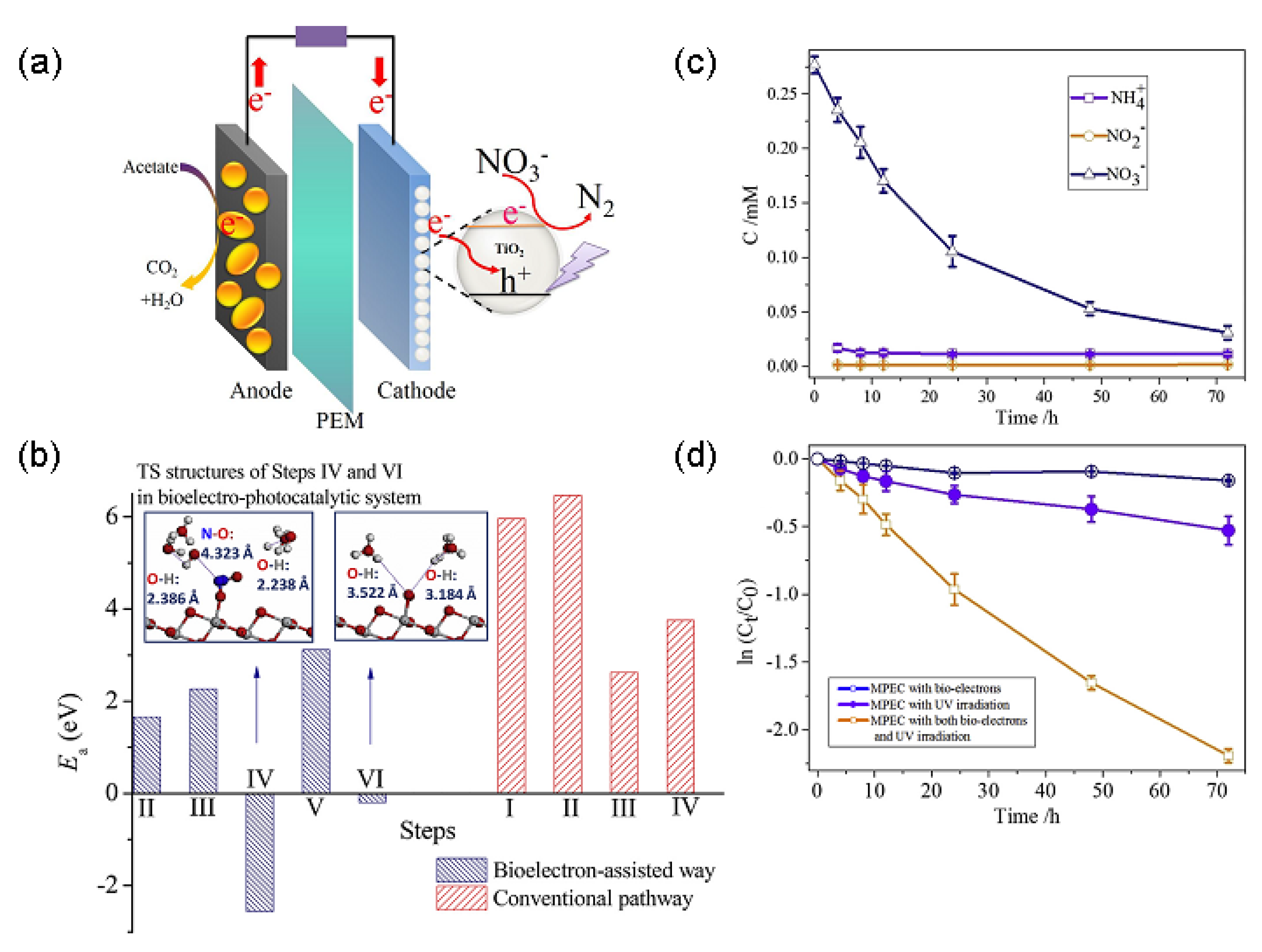
2.4.2. Efficient Nitrate Removal from Wastewater over Different Materials
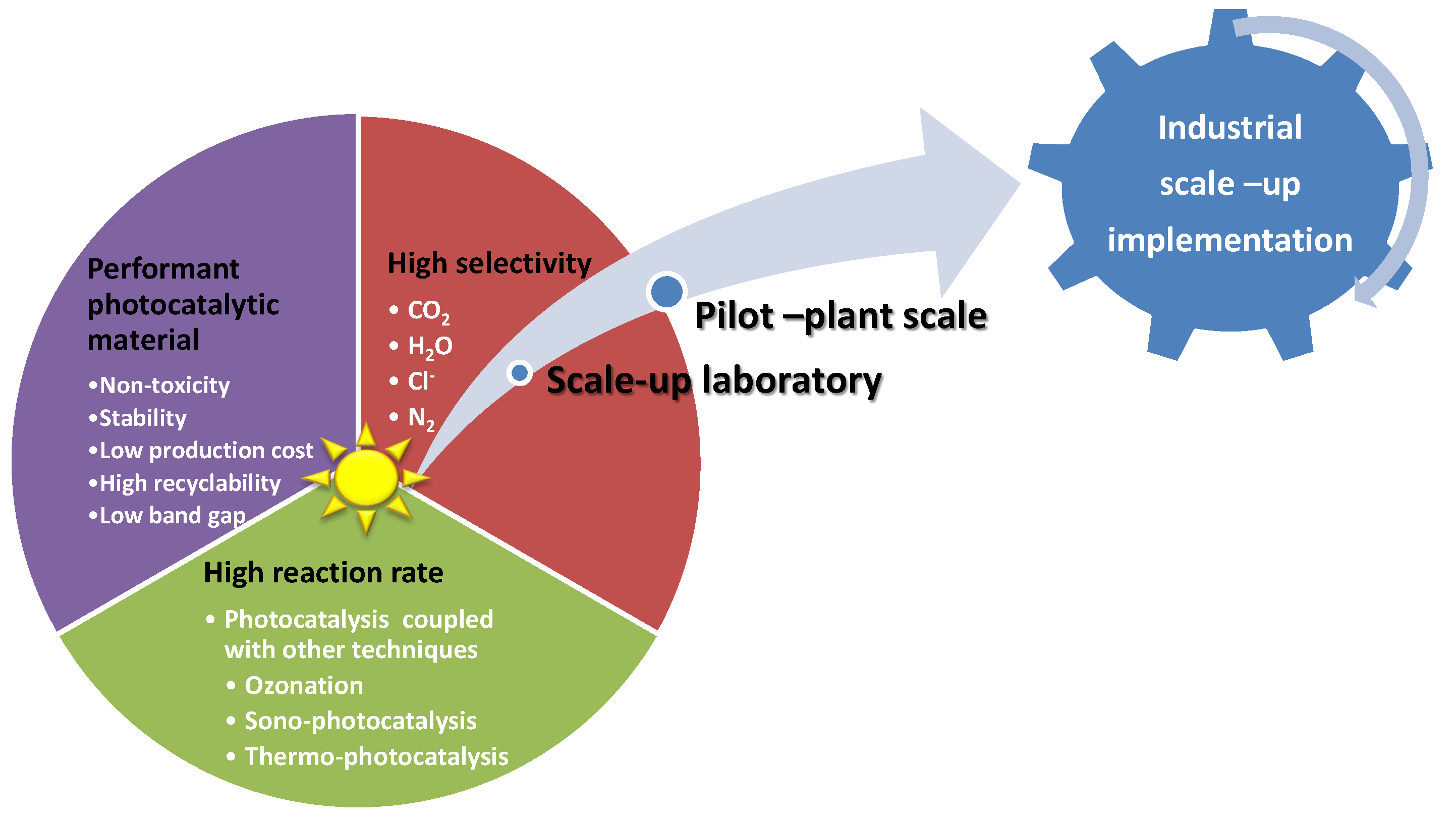
3. Prospectives and Photocatalytic Approaches in Depollution Technologies
3.1. Low Reaction Rates
3.2. Non-Toxicity
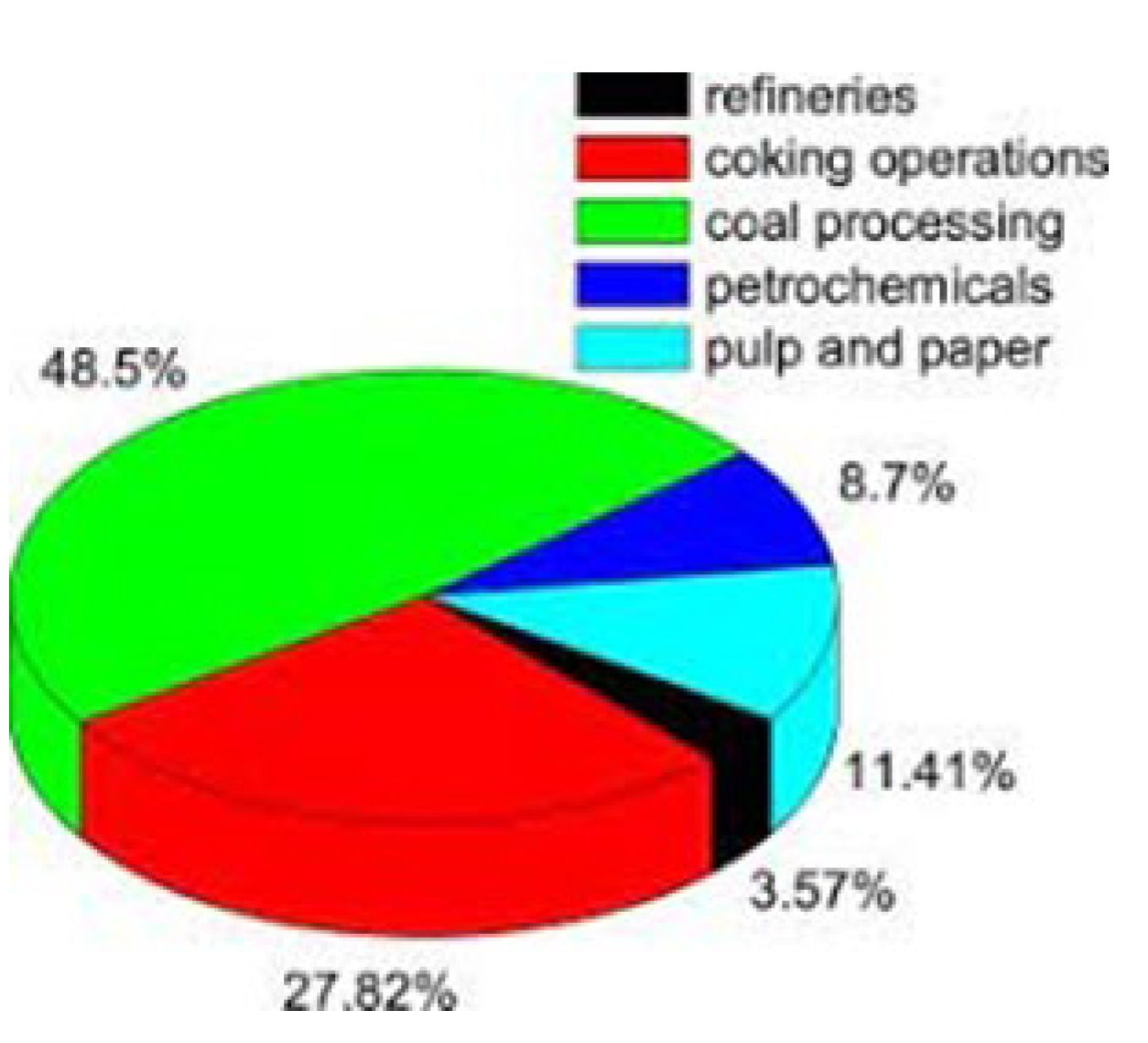
3.3. Industrial Technologies
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ramalingam, G.; Perumal, N.; Priya, A.K.; Rajendran, S. A review of graphene-based semiconductors for photocatalytic degradation of pollutants in wastewater. Chemosphere 2022, 300, 134391. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Chen, S.; Yu, H.; Xu, T.; Wu, S.; Wang, X.; Lu, N.; Quan, X.; Liang, H. Photocatalytic ozonation of organic pollutants in wastewater using a flowing through reactor. J. Hazard. Mater. 2021, 405, 124277. [Google Scholar] [CrossRef] [PubMed]
- Preda, S.; Umek, P.; Zaharescu, M.; Anastasescu, C.; Petrescu, S.V.; Gifu, C.; Eftemie, D.-I.; State, R.; Papa, F.; Balint, I. Iron-modified titanate nanorods for oxidation of aqueous ammonia using combined treatment with ozone and solar light irradiation. Catalysts 2022, 12, 666. [Google Scholar] [CrossRef]
- Mirsadeghi, S.; Zandavar, H.; Rajabi, H.R.; Sajadiasl, F.; Ganjali, M.R.; Pourmortazavi, S.M. Superior degradation of organic pollutants and H2O2 generation ability on environmentally–sound constructed Fe3O4-Cu nanocomposite. J. Mater. Res. Technol. 2021, 14, 808–821. [Google Scholar] [CrossRef]
- Qu, Y.; Chen, Z.; Duan, Y.; Liu, L. H2O2 assisted photocatalysis over Fe-MOF modified BiOBr for degradation of RhB. J. Chem. Technol. Biotechnol. 2022, 97, 2881–2888. [Google Scholar] [CrossRef]
- Binas, V.; Venieri, D.; Kotzias, D.; Kiriakidis, G. Modified TiO2 based photocatalysts for improved air and health quality. J. Materiomics 2017, 3, 3–16. [Google Scholar] [CrossRef]
- Denny, F.; Permana, E.; Scott, J.; Wang, J.; Pui, D.Y.H.; Amal, R. Integrated Photocatalytic Filtration Array for Indoor Air Quality Control. Environ. Sci. Technol. 2010, 44, 5558–5563. [Google Scholar] [CrossRef]
- Shen, M.; Henderson, M.A. Identification of the Active Species in Photochemical Hole Scavenging Reactions of Methanol on TiO2. J. Phys. Chem. Lett. 2011, 2, 2707–2710. [Google Scholar] [CrossRef]
- Tamaki, Y.; Furube, A.; Murai, M.; Hara, K.; Katoh, R.; Tachiya, M. Direct Observation of Reactive Trapped Holes in TiO2 Undergoing Photocatalytic Oxidation of Adsorbed Alcohols: Evaluation of the Reaction Rates and Yields. J. Am. Chem. Soc. 2005, 128, 416–417. [Google Scholar] [CrossRef]
- Mejia, M.I.; Marin, J.M.; Restrepo, G.; Rios, L.A.; Pulgarin, C.; Kiwi, J. Preparation, Testing and Performance of a TiO2/Polyester Photocatalyst for the Degradation of Gaseous Methanol. Appl. Catal. B. 2010, 94, 166–172. [Google Scholar] [CrossRef]
- Setvin, M.; Shi, X.; Hulva, J.; Simschitz, T.; Parkinson, G.S.; Schmid, M.; Valentin, C.D.; Selloni, A.; Diebold, U. Methanol on anatase TiO2 (101): Mechanistic insights into photocatalysis. ACS Catal. 2017, 7, 7081–7091. [Google Scholar] [CrossRef]
- Shen, M.; Acharya, D.P.; Dohnálek, Z.; Henderson, M.A. Importance of diffusion in methanol photochemistry on TiO2 (110). J. Phys. Chem. 2012, 116, 25465–25469. [Google Scholar] [CrossRef]
- Zhou, C.; Ma, Z.; Ren, Z.; Mao, X.; Dai, D.X.; Yang, X. Effect of defects on photocatalytic dissociation of methanol on TiO2 (110). Chem. Sci. 2011, 2, 1980. [Google Scholar] [CrossRef]
- Panayotov, D.A.; Burrows, S.P.; Morris, J.R. Photooxidation mechanism of methanol on rutile TiO2 nanoparticles. J. Phys. Chem. C. 2012, 116, 6623–6635. [Google Scholar] [CrossRef]
- Humayun, M.; Raziq, F.; Khan, A.; Luo, W. Modification strategies of TiO2 for potential applications in photocatalysis: A critical review. Green Chem. Lett. Rev. 2018, 11, 86–102. [Google Scholar] [CrossRef]
- Misra, M.; Kapur, P.; Singla, M.L. Surface plasmon quenched of near band edge emission and enhanced visible photocatalytic activity of Au@ZnO core-shell nanostructure. Appl. Catal. B Environ. 2014, 150, 605–611. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Y.; Liu, S.; Gao, L.; Mao, L.; Fan, Z.; Shangguan, W.; Jiang, Z. Non-noble metal Cu as a cocatalyst on TiO2 nanorod for highly efficient photocatalytic hydrogen production. Appl. Surf. Sci. 2018, 445, 527–534. [Google Scholar] [CrossRef]
- Rosseler, O.; Shankar, M.V.; Karkmaz-Le Du, M.; Schmidlin, L.; Keller, N.; Keller, V. Solar light photocatalytic hydrogen production from water over Pt and Au/TiO2 (anatase/rutile) photocatalysts: Influence of noble metal and porogen promotion. J. Catal. 2010, 269, 179–190. [Google Scholar] [CrossRef]
- Mizukoshi, Y.; Makise, Y.; Shuto, T.; Hu, J.; Tominaga, A.; Shironita, S.; Tanabe, S. Immobilization of noble metal nanoparticles on the surface of TiO2 by the sonochemical method: Photocatalytic production of hydrogen from an aqueous solution of ethanol. Ult. Sonch. 2007, 14, 387–392. [Google Scholar] [CrossRef]
- Goncearenco, E.; Morjan, I.P.; Dutu, E.; Scarisoreanu, M.; Fleaca, C.; Gavrila-Florescu, L.; Dumitrache, F.; Banici, A.M.; Teodorescu, V.S.; Anastasescu, C.; et al. The effect of noble metal addition on the properties of oxide semiconductors nanoparticles. J. Solid State Chem. 2022, 307, 122817. [Google Scholar] [CrossRef]
- Henderson, M.A.; White, J.M.; Uetsuka, H.; Onishi, H. Selectivity changes during organic photooxidation on TiO2: Role of O2 pressure and organic coverage. J. Catal. 2006, 238, 153–164. [Google Scholar] [CrossRef]
- El-Roz, M.; Bazin, P.; Daturi, M.; Thibault-Starzyk, F. On the mechanism of methanol photooxidation to methylformate and carbon dioxide on TiO2: An operando-FTIR study. Phys. Chem. Chem. Phys. 2015, 17, 11277–11283. [Google Scholar] [CrossRef] [PubMed]
- DePuccio, D.P.; Landry, C.C. Photocatalytic oxidation of methanol using porous Au/WO3 and visible vight. Catal. Sci. Technol. 2016, 6, 7512–7520. [Google Scholar] [CrossRef]
- Muggli, D.S.; Larson, S.A.; Falconer, J.L. Photocatalytic oxidation of ethanol: Isotopic labeling and transient reaction. J. Phys. Chem. 1996, 100, 15886–15889. [Google Scholar] [CrossRef]
- Muggli, D.S.; McCue, J.T.; Falconer, J.L. Mechanism of the photocatalytic oxidation of ethanol on TiO2. J. Catal. 1998, 173, 470–483. [Google Scholar] [CrossRef]
- Yu, Z.; Chuang, S.S.C. In situ IR study of adsorbed species and photogenerated electrons during photocatalytic oxidation of ethanol on TiO2. J. Catal. 2007, 246, 118–126. [Google Scholar] [CrossRef]
- Fukuhara, D.; Joseph, M.T.; Loumissi, T.; Zhang, C.; Itoi, T.; Zhang, H.; Izumi, Y. Local silver site temperature critically reflected partial and complete photooxidation of ethanol using Ag-TiO2 as revealed by extended X-ray absorption fine structure Debye−Waller factor. J. Phys. Chem. C. 2021, 125, 14689–14701. [Google Scholar] [CrossRef]
- Kawai, T.; Sakata, T. Photocatalytic hydrogen production from liquid methanol and water. J. Chem. Soc. Chem. Commun. 1980, 15, 694–695. [Google Scholar] [CrossRef]
- Villarreal, T.L.; Gόmez, R.; Neumann-Spallart, M.; Alonso-Vante, N.; Salvador, P. Semiconductor photooxidation of pollutants dissolved in water: A kinetic model for distinguishing between direct and indirect interfacial hole transfer. I. Photoelectrochemical experiments with polycrystalline anatase electrodes under current doubling and absence of recombination. J. Phys. Chem. B 2004, 108, 15172–15181. [Google Scholar] [CrossRef]
- Haselmann, G.M.; Baumgartner, B.; Wang, J.; Wieland, K.; Gupta, T.; Herzig, C.; Limbeck, A.; Lendl, B.; Eder, D. In situ Pt photodeposition and methanol photooxidation on Pt/TiO2: Pt-loading-dependent photocatalytic reaction pathways studied by liquid-phase infrared spectroscopy. ACS Catal. 2020, 10, 2964–2977. [Google Scholar] [CrossRef]
- Preda, S.; Anastasescu, C.; Balint, I.; Umek, P.; Sluban, M.; Negrila, C.; Angelescu, D.G.; Bratan, V.; Rusu, A.; Zaharescu, M. Charge separation and ROS generation on tubular sodium titanates exposed to simulated solar light. Appl. Surf. Sci. 2019, 470, 1053–1063. [Google Scholar] [CrossRef]
- Papa, F.; Miyazaki, A.; Scurtu, M.; Ianculescu, A.C.; Balint, I. Morphology, chemical state of nanometric-sized Pt–Cu and Pt–Ag particles, and their photocatalytic activity for mineralization of methanol. J. Nanopart. Res. 2014, 16, 2249. [Google Scholar] [CrossRef]
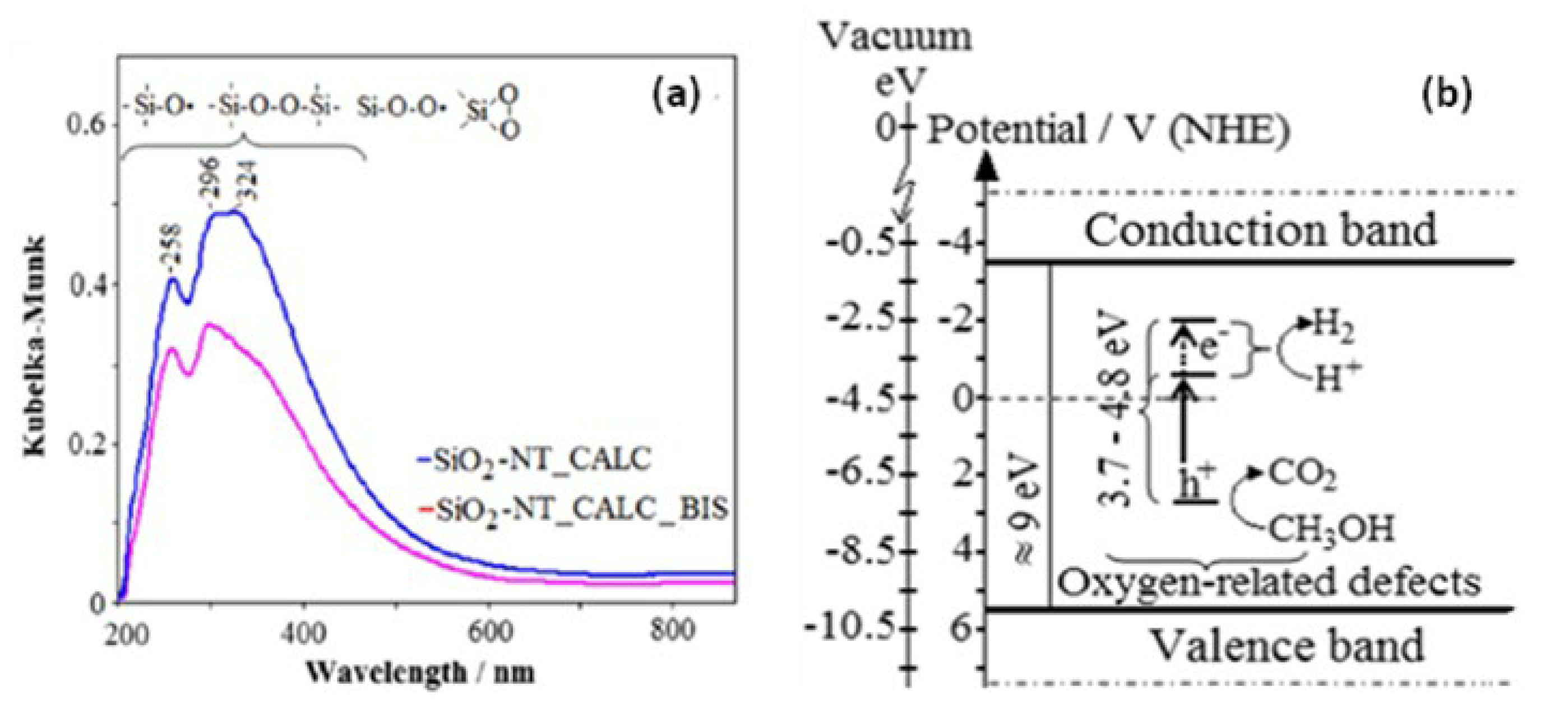
- Anastasescu, C.; Zaharescu, M.; Angelescu, D.; Munteanu, C.; Bratan, V.; Spataru, T.; Negrila, C.; Spataru, N.; Balint, I. Defect-related light absorption, photoluminescence and photocatalytic activity of SiO2 with tubular morphology. Sol. Energ. Mater. Sol. Cells 2017, 159, 325–335. [Google Scholar] [CrossRef]
- Anastasescu, C.; Negrila, C.; Angelescu, D.G.; Atkinson, I.; Anastasescu, M.; Spataru, N.; Zaharescu, M.; Balint, I. Particularities of photocatalysis and formation of reactive oxygen species on insulators and semiconductors: Cases of SiO2, TiO2 and their composite SiO2-TiO2. Catal. Sci. Technol. 2018, 8, 5657–5668. [Google Scholar] [CrossRef]
- Mendive, C.B.; Bredow, T.; Blesabd, M.A.; Bahnemann, D.W. ATR-FTIR measurements and quantum chemical calculations concerning the adsorption and photoreaction of oxalic acid on TiO2. Phys. Chem. Chem. Phys. 2006, 8, 3232–3247. [Google Scholar] [CrossRef]
- Anastasescu, C.; Zaharescu, M.; Balint, I. Unexpected photocatalytic activity of simple and platinum modified tubular SiO2 for the oxidation of oxalic acid to CO2. Catal. Lett. 2009, 132, 81–86. [Google Scholar] [CrossRef]
- Kosanić, M.M. Photocatalytic degradation of oxalic acid over TiO2 powder. J. Photochem. Photobiol. A Chem. 1998, 119, 119–122. [Google Scholar] [CrossRef]
- Craciun, E.; Predoana, L.; Atkinson, I.; Jitaru, I.; Anghel, E.M.; Bratan, V.; Gifu, C.; Anastasescu, C.; Rusu, A.; Raditoiu, V.; et al. Fe3+−doped TiO2 nanopowders for photocatalytic mineralization of oxalic acid under solar light irradiation. J. Photochem. Photobiol. A Chem. 2018, 356, 18–28. [Google Scholar] [CrossRef]
- Kiatkittipong, K.; Iwase, A.; Scott, J.; Amal, R. Photocatalysis of heat treated sodium-and hydrogen-titanate nanoribbons for water splitting, H2/O2 generation and oxalic acid oxidation. Chem. Eng. Sci. 2013, 93, 341–349. [Google Scholar] [CrossRef]
- Cauxa, M.; Fina, F.; Irvine, J.T.S.; Idriss, H.; Howe, R. Impact of the annealing temperature on Pt/g-C3N4 structure, activity and selectivity between photodegradation and water splitting. Catal. Today 2017, 287, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, C.; Dhanalakshmi, R. Photocatalytic performance of particulate semiconductors under natural sunshine—Oxidation of carboxylic acids. Sol. Energy Mater. Sol. Cells 2008, 92, 588–593. [Google Scholar] [CrossRef]
- Wang, Z.; Xie, X.; Wang, X.; Mahmood, A.; Qiu, H.; Sun, J. Difference of photodegradation characteristics between single and mixed VOC pollutants under simulated sunlight irradiation. J Photochem. Photobiol. A Chem. 2019, 384, 112029. [Google Scholar] [CrossRef]
- Lyu, J.; Gao, J.; Zhang, M.; Fu, Q.; Sun, L.; Hu, S.; Zhong, J.; Wang, S. Construction of homojunction-adsorption layer on anatase TiO2 to improve photocatalytic mineralization of volatile organic compounds. Appl. Catal. B Environ. 2017, 202, 664–670. [Google Scholar] [CrossRef]
- Sansotera, M.; Malek Kheylia, S.G.; Baggioli, A.; Bianchi, C.L.; Pedeferri, P.M.; Diamanti, M.V.; Navarrini, W. Absorption and photocatalytic degradation of VOCs by perfluorinated ionomeric coating with TiO2 nanopowders for air purification. Chem. Eng. J. 2019, 361, 885–896. [Google Scholar] [CrossRef]
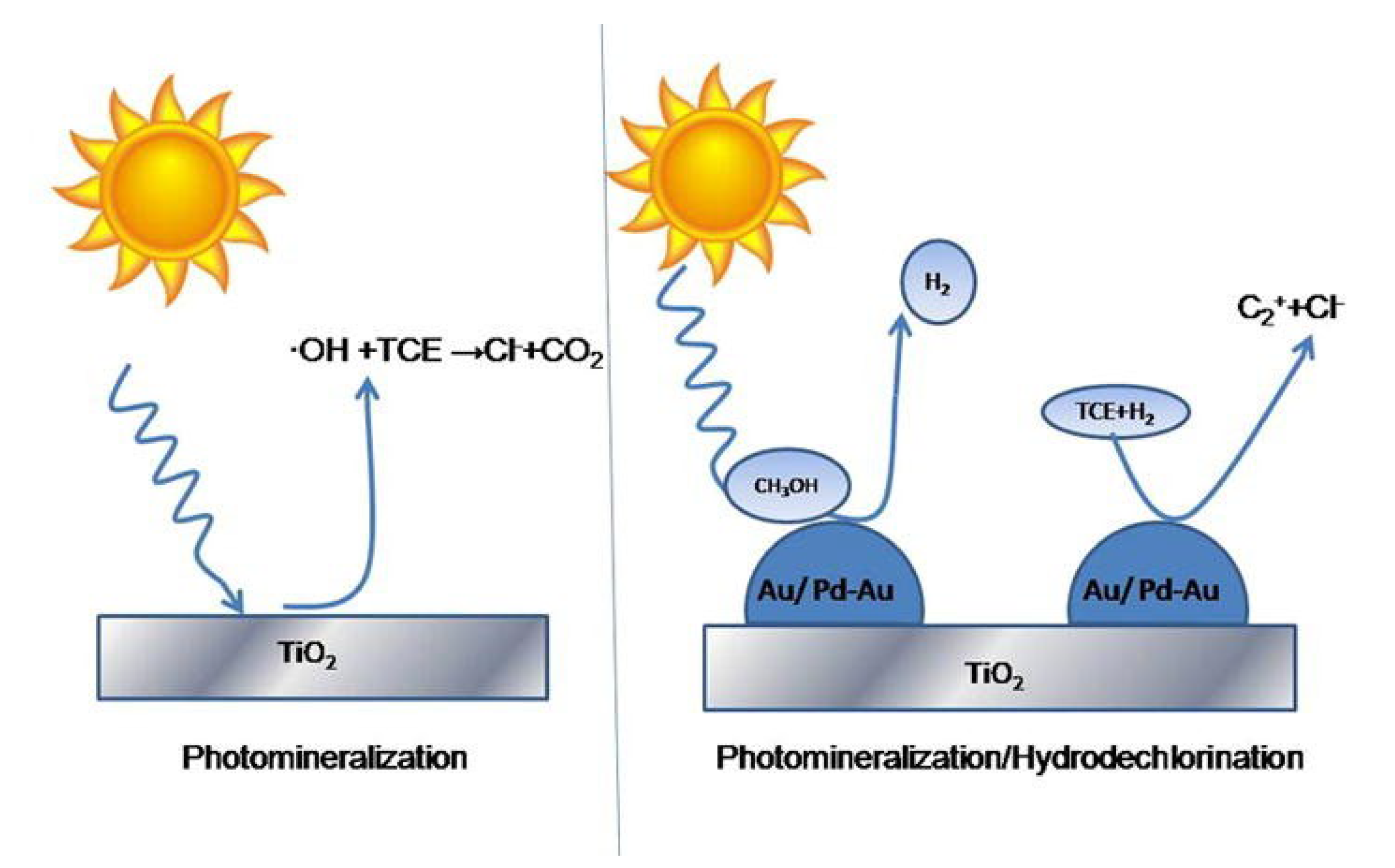
- State, R.; Papa, F.; Tabakova, T.; Atkinson, I.; Negrila, C.; Balint, I. Photocatalytic abatement of trichlorethylene over Au and Pd–Au supported on TiO2 by combined photomineralization/hydrodechlorination reactions under simulated solar irradiation. J. Catal. 2017, 346, 101–108. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Chang, S.-H.; Chung, W.-C.; Chang, M.-B. Photocatalytic removal of trichloroethylene from water with LaFeO3. Environ. Sci. Pollut. Res. 2019, 26, 26276–26285. [Google Scholar] [CrossRef]
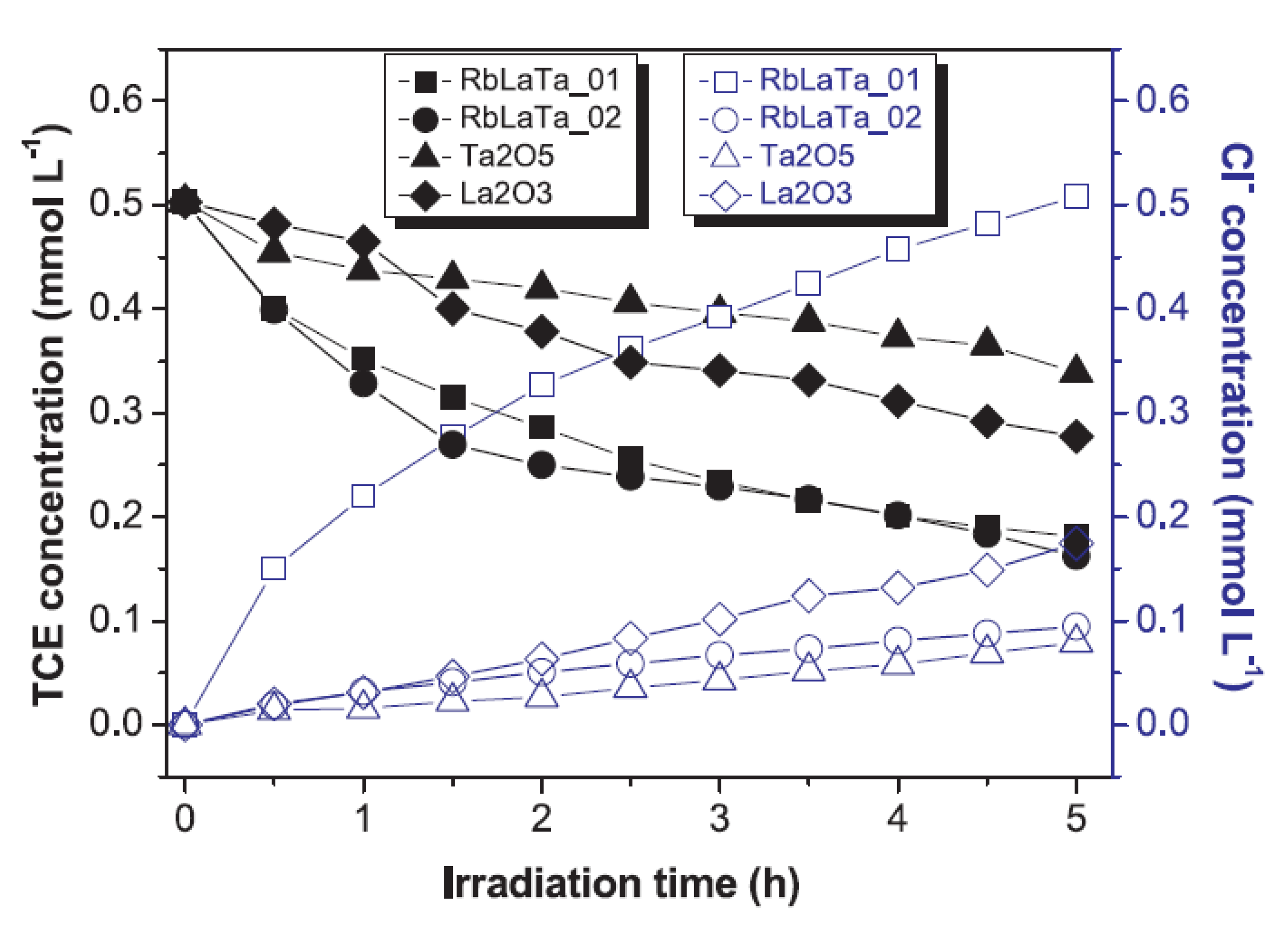
- Raciulete, M.; Papa, F.; Negrila, C.; Bratan, V.; Munteanu, C.; Pandele-Cusu, J.; Culita, D.C.; Alkinson, I.; Balint, I. Strategy for modifying layered perovskites toward efficient solar light-driven photocatalysts for removal of chlorinated pollutants. Catalysts 2020, 10, 637. [Google Scholar] [CrossRef]
- Raciulete, M.; Papa, F.; Kawamoto, D.; Munteanu, C.; Culita, D.C.; Negrila, C.; Atkinson, I.; Bratan, V.; Pandele-Cusu, J.; Balint, I. Particularities of trichloroethylene photoatalytic degradation over crystalline RbLaTa2O7 nanowire bundles grown by solid-state synthesis route. J. Environ. Chem. Eng. 2019, 7, 102789. [Google Scholar] [CrossRef]
- Raciulete, M.; Papa, F.; Culita, D.C.; Munteanu, C.; Atkinson, I.; Bratan, V.; Pandele-Cusu, J.; State, R.; Balint, I. Inpact of RbLaTa2O7 layered perovskite synthesis conditions on their activity for photocatalytic abatement of trichloroethylene. Rev. Roum. Chim. 2018, 63, 821–828. [Google Scholar]
- Joo, J.C.; Ahn, C.H.; Jang, D.G.; Yoon, Y.H.; Kim, J.K.; Campos, L.; Ahn, H. Photocatalytic degradation of trichloroethylene in aqueous phase using nano-ZNO/Laponite composites. J. Hazard. Mater. 2013, 263, 569–574. [Google Scholar] [CrossRef]
- Friedmann, D.A. General overview of heterogeneous photocatalysis as a remediation technology for wastewaters containing pharmaceutical compounds. Water 2022, 14, 3588. [Google Scholar] [CrossRef]
- Monteiro, R.A.R.; Silva, A.M.T.; Ângelo, J.R.M.; Silva, G.V.; Mendes, A.M.; Boaventura, R.A.R.; Vilar, V.J.P. Photocatalytic oxidation of gaseous perchloroethylene over TiO2 based paint. J. Photochem. Photobiol. A Chem. 2015, 311, 41–52. [Google Scholar] [CrossRef]
- Egerton, T.A.; Mattinson, J.A. The influence of platinum on UV and ‘visible’ photocatalysis by rutile and Degussa P25. J. Photochemi. Photobiol. A Chem. 2008, 194, 283–289. [Google Scholar] [CrossRef]
- Grzechulska–Damszel, J.; Grześkowiak, M.; Przepiórski, J.; Morawski, A.W. Photocatalytic decomposition of low-concentrated trichloroethylene and tetrachloroethylene in water. Int. J. Environ. Res. 2014, 8, 347–352. [Google Scholar] [CrossRef]
- Suárez, S.; Jansson, I.; Ohtani, B.; Sánchez, B. From titania nanoparticles to decahedral anatase particles: Photocatalytic activity of TiO2/zeolite hybrids for VOCs oxidation. Catal. Today 2019, 326, 2–7. [Google Scholar] [CrossRef]
- Chen, C.-J.; Wu, C.-C.; Hsieh, L.-T.; Chen, K.-C. Treatment of trichloroethylene with photocatalyst-coated optical fiber. Water 2019, 11, 2391. [Google Scholar] [CrossRef]
- Dutschke, M.; Schnabel, T.; Schütz, F.; Springer, C. Degradation of chlorinated volatile organic compounds from contaminated ground water using a carrier-bound TiO2/UV/O3-system. J. Environ. Manag. 2022, 304, 114236. [Google Scholar] [CrossRef]
- Ji, J.; Xu, Y.; Huang, H.; He, M.; Liu, S.; Liu, G.; Xie, R.; Feng, Q.; Shu, Y.; Zhan, Y.; et al. Mesoporous TiO2 under VUV irradiation: Enhanced photocatalytic oxidation for VOCs degradation at room temperature. Chem. Eng. J. 2017, 327, 490–499. [Google Scholar] [CrossRef]
- Fiorenza, R.; Spitaleria, L.; Perricelli, F.; Nicotra, G.; Fragalà, M.E.; Scirè, S.; Gulino, A. Efficient photocatalytic oxidation of VOCs using ZnO@Au nanoparticles. J. Photochem. Photobiol. A Chem. 2023, 434, 114232. [Google Scholar] [CrossRef]
- Liu, J.; Wang, P.; Qu, W.; Li, H.; Shi, L.; Zhang, D. Nanodiamond-decorated ZnO catalysts with enhanced photocorrosion-resistance for photocatalytic degradation of gaseous toluene. Appl. Catal. B Environ. 2019, 257, 117880. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, G.; Chen, Z.; Lian, H.; Gan, L.; Pan, M. Hierarchically nanostructured Ag/ZnO/nBC for VOC photocatalytic degradation: Dynamic adsorption and enhanced charge transfer. J. Environ. Chem. Eng. 2022, 10, 108690. [Google Scholar] [CrossRef]
- Ouachtak, H.; Akhouairi, S.; Haounatic, R.; Addic, A.A.; Jadad, A.; Tahaa, M.L.; Douch, J. 3,4-Dihydroxybenzoic acid removal from water by goethite modified natural sand column fixed-bed: Experimental study and mathematical modeling. Desalin. Water. Treatm. 2020, 194, 439–449. Available online: https://www.deswater.com/DWT_abstracts/vol_194/194_2020_439.pdf (accessed on 20 January 2023). [CrossRef]
- Haounati, R.; Alakhras, F.; Ouachtak, H.; Saleh, T.A.; Al-Mazaideh, G.; Alhajri, E.; Jada, A.; Hafid, N.; Addi, A.A. Synthesized of zeolite@Ag2O nanocomposite as superb stability photocatalysis toward hazardous rhodamine B dye from water. Arab. J. Sci. Eng. 2023, 48, 169–179. [Google Scholar] [CrossRef]
- Raza, W.; Lee, J.; Raza, N.; Luo, Y.; Kim, K.H.; Yang, J. Removal of phenolic compounds from industrial waste water based on membrane-based technologies. J. Ind. Eng. Chem. 2019, 71, 1–18. [Google Scholar] [CrossRef]
- Min, K.; Freeman, C.; Kang, H.; Choi, S.U. The regulation by phenolic compounds of soil organic matter dynamics under a changing environment. Biomed. Res. Int. 2015, 2015, 825098. [Google Scholar] [CrossRef]
- Weber, M.; Weber, M.; Kleine-Boymann, M. Phenol. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2004; Volume 26, pp. 503–519. [Google Scholar] [CrossRef]
- Michałowicz, J.; Duda, W. Phenols—Sources and toxicity. Pol. J. Environ. Stud. 2007, 16, 347–362. [Google Scholar]
- ATSDR—Agency for Toxic Substances and Disease Registry. Medical Management Guidelines for Phenol. 2014. Available online: https://wwwn.cdc.gov/TSP/MMG/MMGDetails.aspx?mmgid=144&toxid=27 (accessed on 14 December 2022).
- World Health Organization. Phenol Health and Safety Guide. 1994. Available online: http://apps.who.int/iris/bitstream/handle/10665/39958/9241510889-eng.pdf;jsessionid=A1A871567C5E2FEBB6594D0839772049?sequence=1 (accessed on 14 December 2022).
- Matos, J.; Laine, J.; Herrmann, J.-M. Effect of the type of activated carbons on the photocatalytic degradation of aqueous organic pollutants by UV-irradiated titania. J. Catal. 2001, 200, 10–20. [Google Scholar] [CrossRef]
- Gaya, U.I.; Abdullah, A.H. Heterogeneous photocatalytic degradation of organic contaminants over titanium dioxide: A review of fundamentals, progress and problems. J. Photochem. Photobiol. C Photochem. Rev. 2008, 9, 1–12. [Google Scholar] [CrossRef]
- Dang, T.T.T.; Le, S.T.T.; Channei, D.; Khanitchaidecha, W.; Nakaruk, A. Photodegradation mechanisms of phenol in the photocatalytic process. Res. Chem. Intermed. 2016, 42, 5961–5974. [Google Scholar] [CrossRef]
- Sobczynski, A.; Duczmal, L.; Zmudzinski, W. Phenol destruction by photocatalysis on TiO2: An attempt to solve the reaction mechanism. J. Mol. Catal. A Chem. 2004, 213, 225–230. [Google Scholar] [CrossRef]
- Guo, Z.; Ma, R.; Li, G. Degradation of phenol by nanomaterial TiO2 in wastewater. Chem. Eng. J. 2006, 119, 55–59. [Google Scholar] [CrossRef]
- Wysocka, I.; Kowalska, E.; Trzcinski, K.; Łapinski, M.; Nowaczyk, G.; Zielinska-Jurek, A. UV-Vis-induced degradation of phenol over magnetic photocatalysts modified with Pt, Pd, Cu and Au nanoparticles. Nanomater. 2018, 8, 28. [Google Scholar] [CrossRef]
- Chatterjee, D.; Dasgupta, S. Visible light induced photocatalytic degradation of organic pollutants. J. Photochem. Photobiol. C Photochem. Rev. 2005, 6, 186–205. [Google Scholar] [CrossRef]
- Fu, F.; Shen, H.; Xue, W.; Zhen, Y.; Soomro, A.R.; Yang, X.; Wang, D.; Xu, B.; Chi, R. Alkali-assisted synthesis of direct Z-scheme based Bi2O3/Bi2MoO6 photocatalyst for highly efficient photocatalytic degradation of phenol and hydrogen evolution reaction. J. Catal. 2019, 375, 399–409. [Google Scholar] [CrossRef]
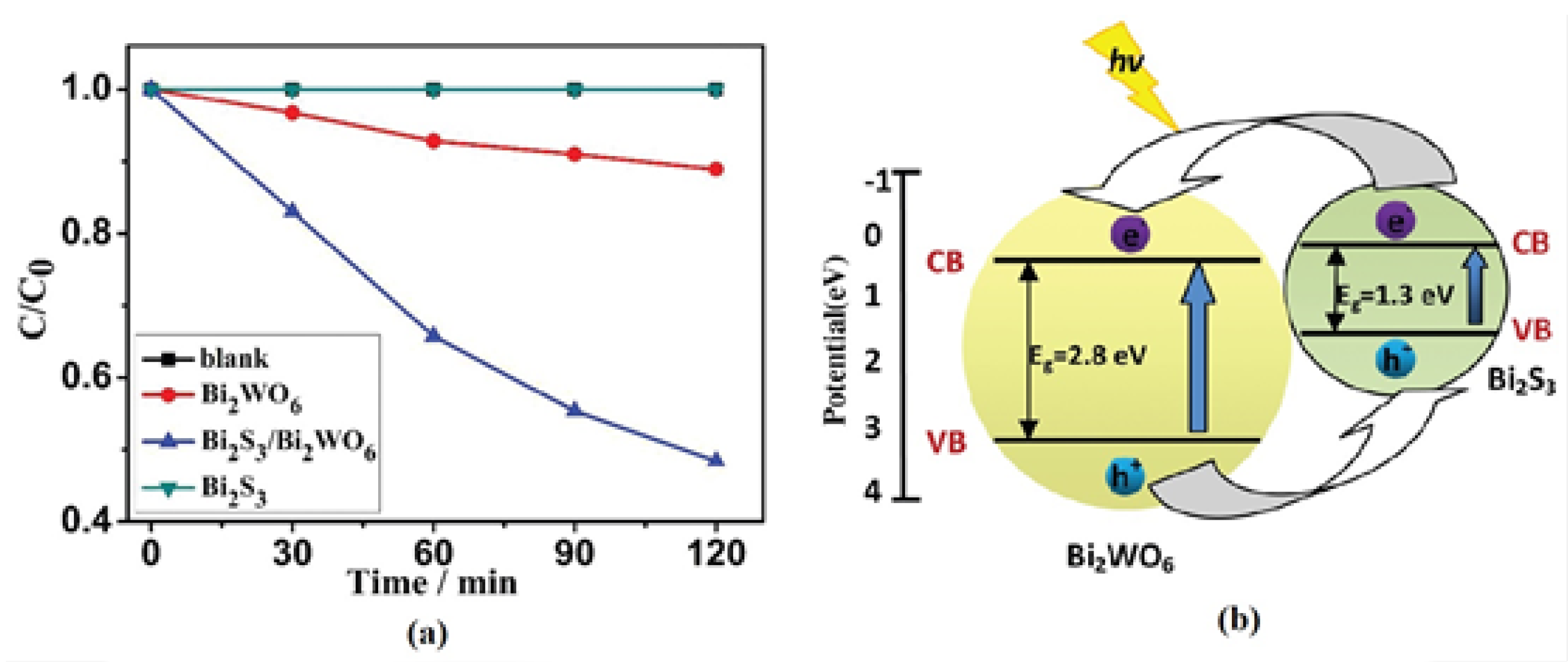
- Zhang, Z.; Wang, W.; Wang, L.; Sun, S. Enhancement of visible-light photocatalysis coupling with narrow-band-bap semiconductor: A case study on Bi2S3/Bi2WO6. ACS. Appl. Mater. Interfaces 2012, 4, 593–597. [Google Scholar] [CrossRef]
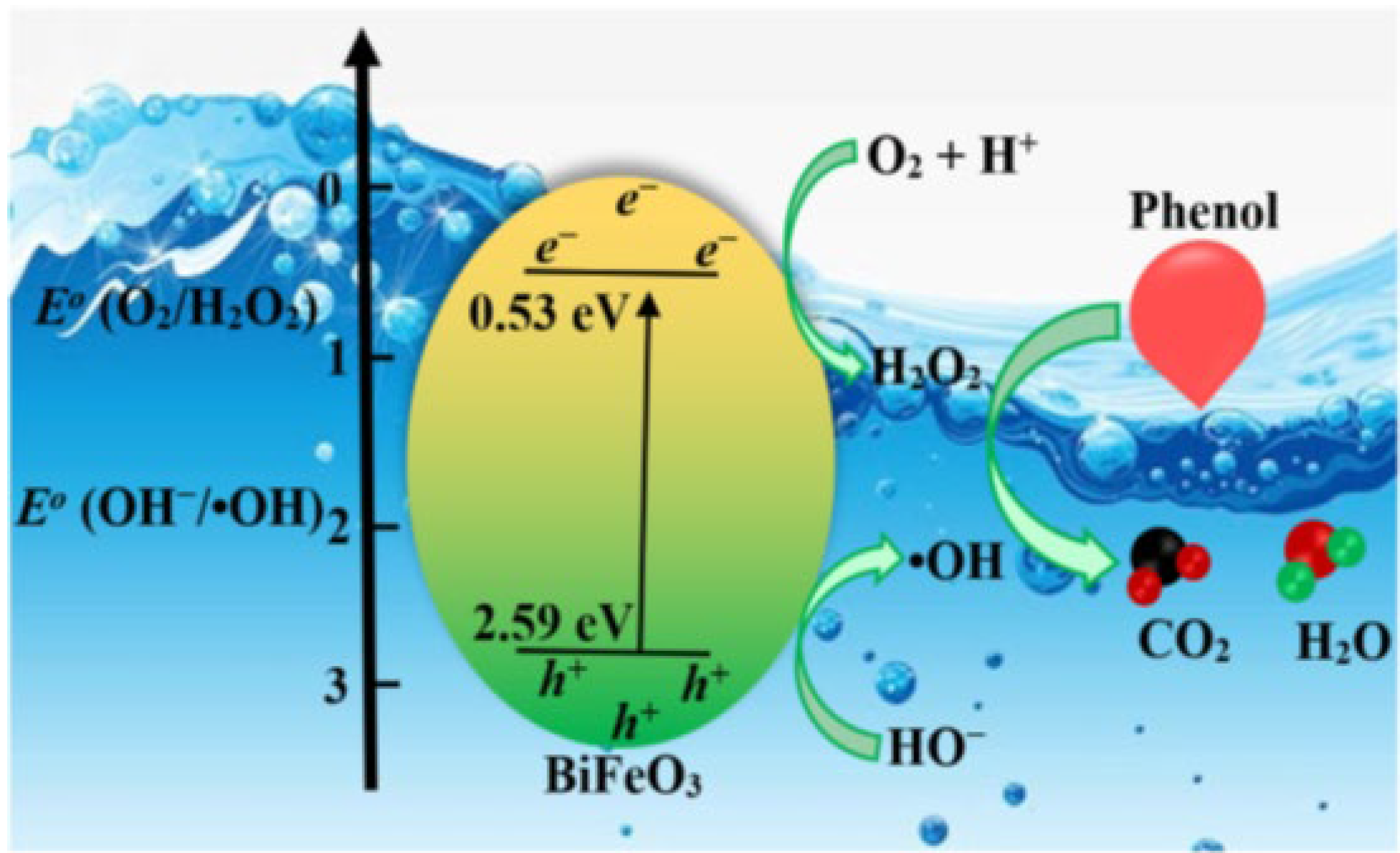
- Chien, S.W.; Ng, D.Q.; Kumar, D.; Lam, S.M.; Jaffari, Z.H. Investigating the effects of various synthesis routes on morphological, optical, photoelectrochemical and photocatalytic properties of single-phase perovskite BiFeO3. J. Phys. Chem. Solid. 2022, 160, 110342. [Google Scholar] [CrossRef]
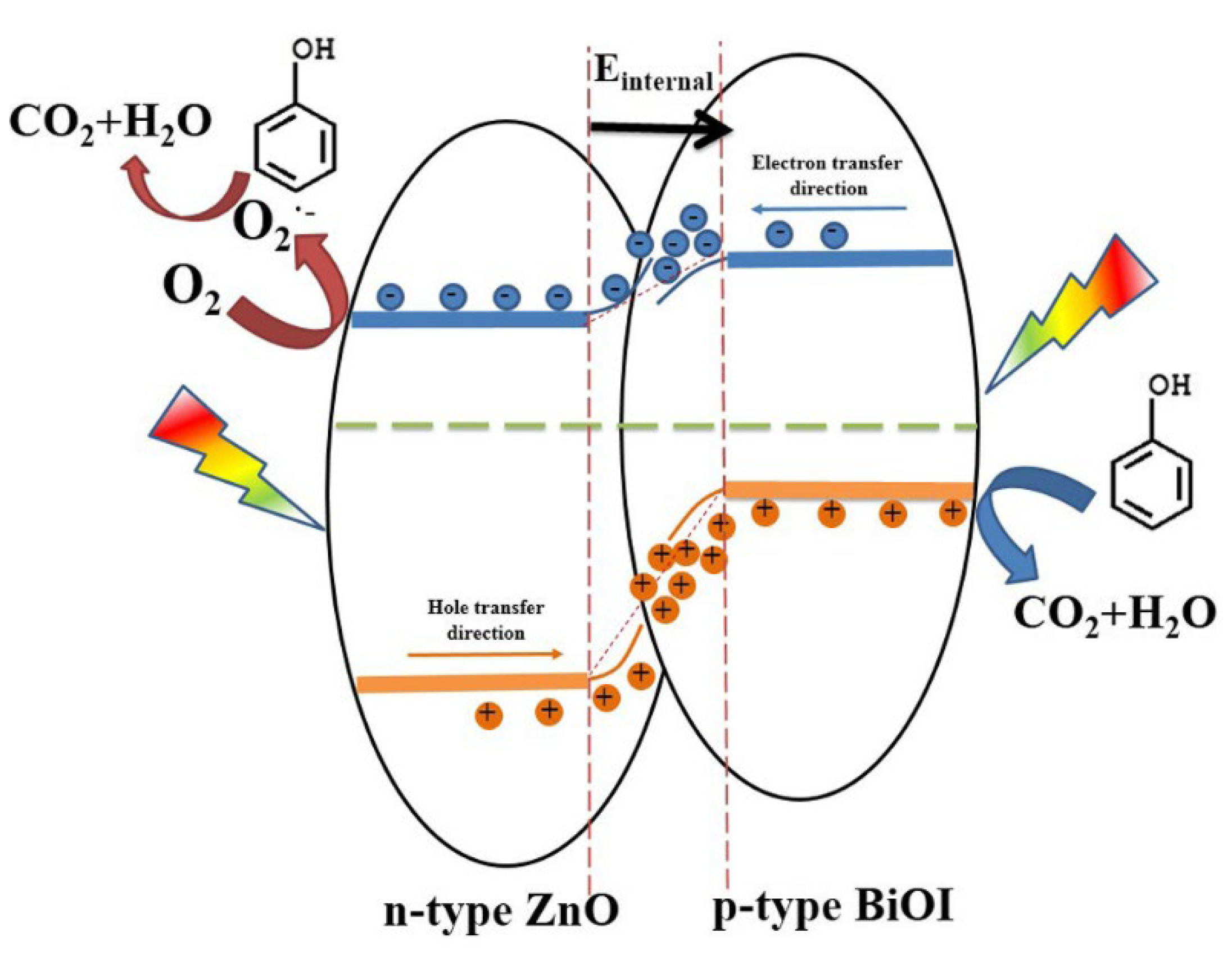
- Jiang, J.; Wang, H.; Chen, X.; Li, S.; Xie, T.; Wang, D.; Lin, Y. Enhanced photocatalytic degradation of phenol and photogenerated charges transfer property over BiOI-loaded ZnO composites. J. Colloid Interface Sci. 2017, 494, 130–138. [Google Scholar] [CrossRef]
- Zhang, Y.; Selvara, R.; Sillanpää, M.; Kim, Y.; Tai, C.-W. The influence of operating parameters on heterogeneous photocatalytic mineralization of phenol over BiPO4. Chem. Eng. J. 2014, 245, 117–123. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, X.; Dong, W.; Zhang, Q.; Liu, J.; Li, R.; Wang, Y.; Zhang, X.; Fan, C. CeO2 nanoparticles decorated Bi4O7 nanosheets for enhanced photodegradation performance of phenol. Mater. Lett. 2022, 322, 132465. [Google Scholar] [CrossRef]
- Yuan, Y.; Guo, R.-T.; Hong, L.-F.; Lin, Z.-D.; Ji, X.-Y.; Pan, W.-G. Fabrication of a dual S-scheme Bi7O9I3/g-C3N4/Bi3O4Cl heterojunction with enhanced visible-light-driven performance for phenol degradation. Chemosphere 2022, 287, 132241. [Google Scholar] [CrossRef]
- Jiang, C.; Ge, Y.; Chen, W.; Hua, L.; Li, H.; Zhang, Y.; Cao, S. Hierarchically-structured TiO2/MnO2 hollow spheres exhibiting the complete mineralization of phenol. Catalysts 2019, 9, 390. [Google Scholar] [CrossRef] [Green Version]
- Sonawane, R.S.; Dongare, M.K. Sol-gel synthesis of Au/TiO2 thin films for photocatalytic degradation of phenol in sunlight. J. Mol. Catal. A Chem. 2006, 243, 68–76. [Google Scholar] [CrossRef]
- Chiou, C.H.; Juang, R.S. Photocatalytic degradation of phenol in aqueous solutions by Pr-doped TiO2 nanoparticles. J. Hazard. Mater. 2007, 149, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Moreira, J.; Gomaa, H.; Ray, A.K. Visible-solar-light-driven photocatalytic degradation of phenol with dye-sensitized TiO2: Parametric and kinetic study. Ind. Eng. Chem. Res. 2012, 51, 4523–4532. [Google Scholar] [CrossRef]
- Dlamini, M.C.; Dlamini, M.L.; Mente, P.; Tlhaole, B.; Erasmus, R.; Maubane-Nkadimeng, M.S.; Moma, J.A. Photocatalytic abatement of phenol on amorphous TiO2-BiOBr-bentonite heterostructures under visible light irradiation. J. Ind. Eng. Chem. 2022, 111, 419–436. [Google Scholar] [CrossRef]
- Rehman, G.U.; Tahir, M.; Goh, P.S.; Ismail, A.F.; Khan, I.U. Controlled synthesis of reduced graphene oxide supported magnetically separable Fe3O4@rGO@AgI ternary nanocomposite for enhanced photocatalytic degradation of phenol. Powder Technol. 2019, 356, 547–558. [Google Scholar] [CrossRef]
- Boukhatem, H.; Khalaf, H.; Djouadi, L.; Gonzalez, F.V.; Navarro, R.M.; Santaballa, J.A.; Canle, M. Photocatalytic activity of mont-La (6%)-Cu0.6Cd0.4S catalyst for phenol degradation under near UV visible light irradiation. Appl. Catal. B Environm. 2018, 211, 114–125. [Google Scholar] [CrossRef]
- Fan, H.; Yi, G.; Zhang, Z.; Zhang, X.; Li, P.; Zhang, C.; Chen, L.; Zhang, Y.; Sun, Q. Binary TiO2/RGO photocatalyst for enhanced degradation of phenol and its application in underground coal gasification wastewater treatment. Opt. Mater. 2021, 120, 111482. [Google Scholar] [CrossRef]
- Othman, I.; Haija, M.A.; Ismail, I.; Zain, J.H.; Banat, F. Preparation and catalytic performance of CuFe2O4 nanoparticles supported on reduced graphene oxide CuFe2O4/rGO) for phenol degradation. Mater. Chem. Phys. 2019, 238, 121931. [Google Scholar] [CrossRef]
- Mohamed, A.; Yousef, S.; Nasser, W.S.; Osman, T.A.; Knebel, A.; Sanchez, E.P.V.; Hashem, T. Rapid photocatalytic degradation of phenol from water using composite nanofibers under UV. Environ. Sci. Eur. 2020, 32, 160. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, L.; Lei, J.; Liu, Y.; Zhang, J. Photo-Fenton degradation of phenol by CdS/rGO/Fe2+ at natural pH with in situ-generated H2O2. Appl. Catal. B 2019, 241, 367–374. [Google Scholar] [CrossRef]
- Yang, X.; Jia, Q.; Pang, J.; Yang, Y.; Zheng, S.; Jia, J.; Qin, Z. Hierarchical porous N-TiO2/carbon foam composite for enhancement of photodegradation activity under simulated sunlight. Diam. Relat. Mater. 2022, 128, 109234. [Google Scholar] [CrossRef]
- Yuan, Y.; Pan, W.-G.; Guo, R.-T.; Hong, L.-F.; Lin, Z.-D.; Ji, X.-Y. Flower spherical-like Bi7O9I3/AgI S-scheme heterojunction for phenol photodegradation: The synergetic effect of dual surface plasmon resonance and photothermal property. Sep. Purif. Technol. 2022, 297, 121538. [Google Scholar] [CrossRef]
- Jing, Y.; Yin, H.; Li, C.; Chen, J.; Wu, S.; Liu, H.; Xie, L.; Lei, Q.; Sun, M.; Yu, S. Fabrication of Pt doped TiO2–ZnO@ ZIF-8 core@shell photocatalyst with enhanced activity for phenol degradation. Environ. Res. 2022, 203, 111819. [Google Scholar] [CrossRef]
- Wang, X.; Xu, H.; Luo, X.; Li, M.; Dai, M.; Chen, Q.; Song, H. Enhanced photocatalytic properties of CeO2/TiO2 heterostructures for phenol degradation. Colloid Interface Sci. Commun. 2021, 44, 100476. [Google Scholar] [CrossRef]
- Sharma, N.; Pap, Z.; Baán, K.; Gyulavari, T.; Karacs, G.; Nemeth, Z.; Garg, S.; Hernadi, K. Effective removal of phenol by activated charcoal/BiOCl composite under UV light irradiation. J. Mol. Struct. 2022, 1254, 132344. [Google Scholar] [CrossRef]
- Al-Hamdia, A.M.; Sillanpää, M.; Bora, T.; Duttac, J. Efficient photocatalytic degradation of phenol in aqueous solution bySnO2:Sb nanoparticles. Appl. Surf. Sci. 2016, 370, 229–236. [Google Scholar] [CrossRef]
- Sandulescu, A.; Anastasescu, C.; Papa, F.; Raciulete, M.; Vasile, A.; Spataru, T.; Scarisoreanu, M.; Fleaca, C.; Mihailescu, C.N.; Teodorescu, V.S.; et al. Advancements on basic working principles of photo-driven oxidative degradation of organic substrates over pristine and noble metal-modified TiO2. Model case of phenol photo oxidation. Catalysts 2021, 11, 487. [Google Scholar] [CrossRef]
- Mendoza-Damian, G.; Tzompantzi, F.; Mantilla, A.; Pérez-Hernández, R.; Hernández-Gordillo, A. Improved photocatalytic activity of SnO2–ZnAl LDH prepared by one step Sn4+ incorporation. Appl. Clay Sci. 2016, 121, 127–136. [Google Scholar] [CrossRef]
- Raciulete, M.; Anastasescu, C.; Papa, F.; Atkinson, I.; Bradu, C.; Negrila, C.; Eftemie, D.-I.; Culita, D.C.; Miyazaki, A.; Bratan, V.; et al. Band-gap engineering of layered perovskites by Cu spacer insertion as photocatalysts for depollution reaction. Catalysts 2022, 12, 1529. [Google Scholar] [CrossRef]
- Hilsabeck, K.I.; Meiser, J.L.; Sneha, M.; Harrison, J.A.; Zare, R.N. Nonresonant photons catalyze photodissociation of phenol. J. Am. Chem. Soc. 2019, 141, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Messele, S.A.; Bengoa, C.; Stüber, F.E.; Giralt, J.; Fortuny, A.; Fabregat, A.; Font, J. Enhanced degradation of phenol by a Fenton-like system (Fe/EDTA/H2O2) at circumneutral pH. Catalysts 2019, 9, 474. [Google Scholar] [CrossRef]
- Wei, X.Y.; Shao, S.J.; Ding, X.; Jiao, W.Z.; Liu, Y.Z. Degradation of phenol with heterogeneous catalytic ozonation enhanced by high gravity technology. J. Clean. Prod. 2019, 119, 179–189. [Google Scholar] [CrossRef]
- Nawrocki, J.; Kasprzyk-Hordern, B. The efficiency and mechanisms of catalytic ozonation. Appl. Catal. B Environ. 2010, 99, 27–42. [Google Scholar] [CrossRef]
- Lincho, J.; Zaleska-Medynska, A.; Martins, R.C.; Gomes, J. Nanostructured photocatalysts for the abatement of contaminants by photocatalysis and photocatalytic ozonation: An overview. Sci. Total Environ. 2022, 837, 155776. [Google Scholar] [CrossRef]
- Xiao, J.; Xie, Y.; Cao, H. Organic pollutants removal in wastewater by heterogeneous photocatalytic ozonation. Chemosphere 2015, 121, 1–17. [Google Scholar] [CrossRef]
- Xie, Y.; Peng, S.; Feng, Y.; Wu, D. Enhanced mineralization of oxalate by highly active and stable Ce(III)-doped g-C3N4 catalyzed ozonation. Chemosphere 2020, 239, 124612. [Google Scholar] [CrossRef]
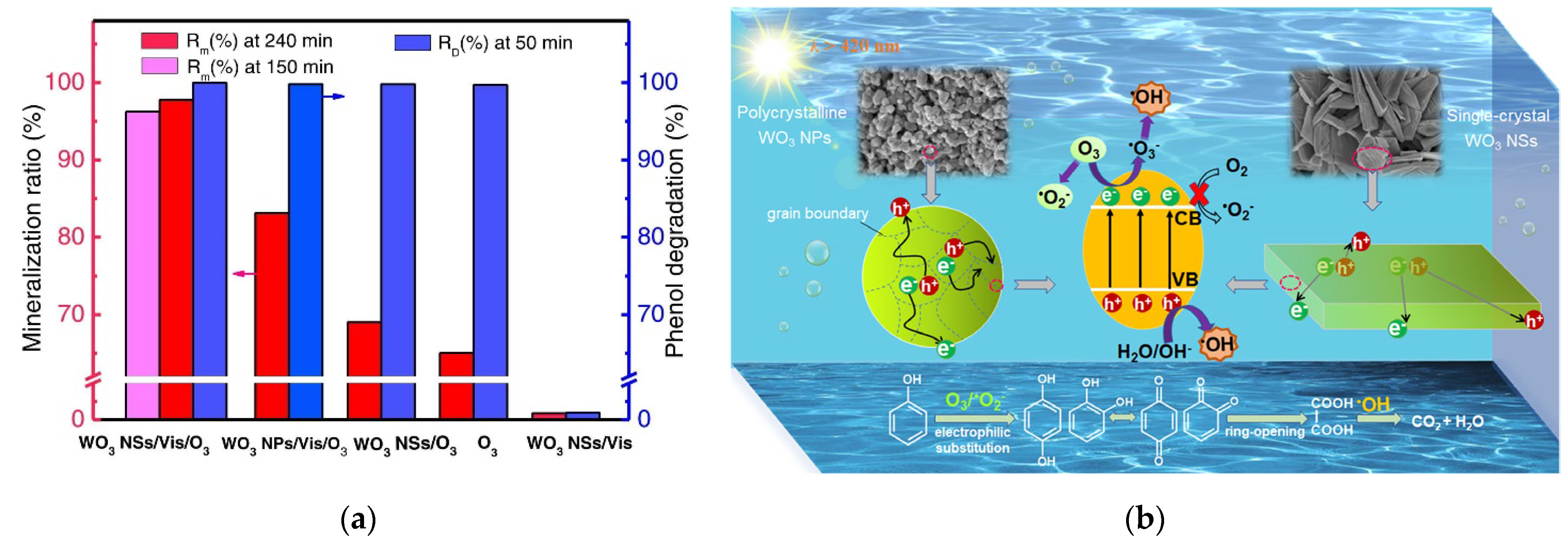
- Yu, H.; Wang, M.; Yan, J.; Dang, H.; Zhu, H.; Liu, Y.; Wen, M.; Li, G.; Wu, L. Complete mineralization of phenolic compounds in visible-light-driven photocatalytic ozonation with single-crystal WO3 nanosheets: Performance and mechanism investigation. J. Hazard. Mater. 2022, 433, 128811. [Google Scholar] [CrossRef]
- Nishimoto, S.; Mano, T.; Kameshima, Y.; Miyake, M. Photocatalytic water treatment over WO3 under visible light irradiation combined with ozonation. Chem. Phys. Lett. 2012, 500, 86–89. [Google Scholar] [CrossRef]
- Tawabini, B.; Zubair, A. Bromate control in phenol-contaminated water treated by UV and ozone processes. Desalination 2011, 267, 16–19. [Google Scholar] [CrossRef]
- An, W.; Tian, L.; Hu, J.; Liu, L.; Cui, W.; Liang, Y. Efficient degradation of organic pollutants by catalytic ozonation and photocatalysis synergy system using double-functional MgO/g-C3N4 catalyst. Appl. Surf. Sci. 2020, 534, 147518. [Google Scholar] [CrossRef]
- Bhatnagar, A.; Sillanpää, M. A review of emerging adsorbents for nitrate removal from water. Chem. Eng. J. 2011, 168, 493–504. [Google Scholar] [CrossRef]
- Loganathan, P.; Vigneswaran, S.; Kandasamy, J. Enhanced removal of nitrate from water using surface modification of adsorbents—A review. J. Environ. Manag. 2013, 131, 363–374. [Google Scholar] [CrossRef]
- Tugaoen, H.O.N.; Garcia-Segura, S.; Hristovski, K.; Westerhoff, P. Challenges in photocatalytic reduction of nitrate as a water treatment technology. Sci. Total Environ. 2017, 599, 1524–1551. [Google Scholar] [CrossRef]
- Anderson, J.A. Photocatalytic nitrate reduction over Au/TiO2. Catal. Today 2011, 175, 316–321. [Google Scholar] [CrossRef]
- Lin, Z.-Q.; Yuan, S.-J.; Li, W.-W.; Chen, J.-J.; Sheng, G.-P.; Yu, H.-Q. Denitrification in an integrated bioelectro-photocatalytic system. Water Res. 2017, 109, 88–93. [Google Scholar] [CrossRef]
- Yue, M.; Wang, R.; Ma, B.; Cong, R.; Gao, W.; Yang, T. Superior performance of CuInS2 for photocatalytic water treatment: Full conversion of highly stable nitrate ions into harmless N2 under visible light. Catal. Sci. Technol. 2016, 6, 8300–8308. [Google Scholar] [CrossRef]
- Anderson, J.A. Simultaneous photocatalytic degradation of nitrate and oxalic acid over gold promoted titania. Catal. Today 2012, 181, 171–176. [Google Scholar] [CrossRef]
- Soares, O.S.G.P.; Pereira, M.F.R.; Órfão, J.J.M.; Faria, J.L.; Silva, C.G. Photocatalytic nitrate reduction over Pd–Cu/TiO2. Chem. Eng. J. 2014, 251, 123–130. [Google Scholar] [CrossRef]
- Hirayama, J.; Kamiya, Y. Combining the photocatalyst Pt/TiO2 and the nonphotocatalyst SnPd/Al2O3 for effective photocatalytic purification of groundwater polluted with nitrate. ACS Catal. 2014, 4, 2207–2215. [Google Scholar] [CrossRef]
- Hirayama, J.; Abe, R.; Kamiya, Y. Combinational effect of Pt/SrTiO3:Rh photocatalyst and SnPd/Al2O3 non-photocatalyst for photocatalytic reduction of nitrate to nitrogen in water under visible light irradiation. Appl. Catal. B Environ. 2014, 144, 721–729. [Google Scholar] [CrossRef]
- Ren, H.T.; Jia, S.Y.; Zou, J.J.; Wu, S.H.; Han, X. A Facile Preparation of Ag2O/P25 Photocatalyst for Selective Reduction of Nitrate. Appl. Catal. B Environ. 2015, 176, 53–61. [Google Scholar] [CrossRef]
- Oka, M.; Miseki, Y.; Saito, K.; Kudo, A. Photocatalytic reduction of nitrate ions to dinitrogen over layered perovskite BaLa4Ti4O15 using water as an electron donor. Appl. Catal. B Environ. 2015, 179, 407–411. [Google Scholar] [CrossRef]
- Sá, J.; Alcaraz Agüera, C.; Gross, S.; Anderson, J.A. Photocatalytic nitrate reduction over metal modified TiO2. Appl. Catal. B Environ. 2009, 85, 192–200. [Google Scholar] [CrossRef]
- Doudrick, K.; Yanga, T.; Hristovski, K.; Westerhoff, P. Photocatalytic nitrate reduction in water: Managing the hole scavenger and reaction by-product selectivity. Appl. Catal. B Environ. 2013, 136, 40–47. [Google Scholar] [CrossRef]
- Gekko, H.; Hashimotoa, K.; Kominami, H. Photocatalytic reduction of nitrite to dinitrogen in aqueous suspensions of metal-loaded titanium (IV) oxide in the presence of a hole scavenger: An ensemble effect of silver and palladium co-catalysts. Phys. Chem. Chem. Phys. 2012, 14, 7965–7970. [Google Scholar] [CrossRef]
- Adachi, M.; Kudo, A. Effect of Surface Modification with layered double hydroxide on reduction of nitrate to nitrogen over BaLa4Ti4O15 photocatalyst. Chem. Lett. 2012, 41, 1007–1008. [Google Scholar] [CrossRef]
- Shi, H.; Li, C.; Wang, L.; Wang, W.; Meng, X. Selective reduction of nitrate into N2 by novel Z-scheme NH2-MIL-101(Fe)/BiVO4 heterojunction with enhanced photocatalytic activity. J. Hazard. Mater. 2022, 424, 127711. [Google Scholar] [CrossRef]
- Ruiz-Beviá, F.; Fernández-Torres, M.J. Effective catalytic removal of nitrates from drinking water: An unresolved problem? J. Clean. Prod. 2019, 217, 398–408. [Google Scholar] [CrossRef]
- Zazo, J.A.; García-Muñoz, P.; Pliego, G.; Silveira, J.E.; Jaffe, P.; Casas, J.A. Selective reduction of nitrate to N2 using ilmenite as a low cost photo-catalyst. Appl. Catal. B Environ. 2020, 273, 118930. [Google Scholar] [CrossRef]
- Silveira, J.E.; Ribeiro, A.R.; Carbajo, J.; Pliego, G.; Zazo, J.A.; Casas, J.A. The photocatalytic reduction of NO3− to N2 with ilmenite (FeTiO3): Effects of groundwater matrix. Water Res. 2021, 200, 117250. [Google Scholar] [CrossRef]
- Gomathi Devi, L.; Kavitha, R. A review on non metal ion doped titania for the photocatalytic degradation of organic pollutants under UV/solar light: Role of photogenerated charge carrier dynamics in enhancing the activity. Appli. Catal. B Environ. 2013, 140, 559–587. [Google Scholar] [CrossRef]
- Girish Kumar, S.; Koteswara Rao, K.S.R. Comparison of modification strategies towards enhanced charge carrier separation and photocatalytic degradation activity of metal oxide semiconductors (TiO2, WO3 and ZnO). Appl. Surf. Sci. 2017, 391, 124–148. [Google Scholar] [CrossRef]
- Lazar, M.A.; Varghese, S.; Nair, S.S. Photocatalytic water treatment by titanium dioxide: Recent updates. Catalysts 2012, 2, 572–601. [Google Scholar] [CrossRef]
- Zhang, L.; Mohamed, H.H.; Dillert, R.; Bahnemann, D. Kinetics and mechanisms of charge transfer processes in photocatalytic systems: A review. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 263–276. [Google Scholar] [CrossRef]
- Luiz, D.B.; Andersen, S.L.F.; Berger, C.; José, H.J.; Moreira, R.F.P.M. Photocatalytic reduction of nitrate ions in water over metal-modified TiO2. J. Photochem. Photobiol. A Chem. 2012, 246, 36–44. [Google Scholar] [CrossRef]
- Doudrick, K.; Monzón, O.; Mangonon, A.; Hristovski, K.; Westerhoff, P. Nitrate reduction in water using commercial titanium dioxide photocatalysts (P25, P90, and Hombikat UV100). J. Environ. Eng. 2012, 138, 852–861. [Google Scholar] [CrossRef]
- Kominami, H.; Nakaseko, T.; Shimada, Y.; Furusho, A.; Inoue, H.; Murakami, S.Y.; Kera, Y.; Ohtani, B. Selective photocatalytic reduction of nitrate to nitrogen molecules in an aqueous suspension of metal-loaded titanium (IV) oxide particles. Chem. Commun. 2005, 23, 2933–2935. [Google Scholar] [CrossRef]
- Li, L.; Xu, Z.; Liu, F.; Shao, Y.; Wang, J.; Wan, H.; Zheng, S. Photocatalytic nitrate reduction over Pt–Cu/TiO2 catalysts with benzene as hole scavenger. J. Photochem. Photobio. A Chem. 2010, 212, 113–121. [Google Scholar] [CrossRef]
- Li, Y.; Wasgestian, F. Photocatalytic reduction of nitrate ions on TiO2 by oxalic acid. J. Photochem. Photobiol. A Chem. 1998, 112, 255–259. [Google Scholar] [CrossRef]
- Zhang, F.; Jin, R.; Chen, J.; Shao, C.; Gao, W.; Li, L.; Guan, N. High photocatalytic activity and selectivity for nitrogen in nitrate reduction on Ag/TiO2 catalyst with fine silver clusters. J. Catal. 2005, 232, 424–431. [Google Scholar] [CrossRef]
- Nakamura, K.; Yoshida, Y.; Mikami, I.; Okuhara, T. Selective hydrogenation of nitrate in water over Cu–Pd/mordenite. Appl. Catal. B Environ. 2006, 65, 31–36. [Google Scholar] [CrossRef]
- Prüsse, U.; Hähnlein, M.; Daum, J.; Vorlop, K.D. Improving the catalytic nitrate reduction. Catal. Today 2000, 55, 79–90. [Google Scholar] [CrossRef]
- Prüsse, U.; Vorlop, K.D. Supported bimetallic palladium catalysts for water-phase nitrate reduction. J. Molec. Catal. A Chem. 2001, 173, 313–328. [Google Scholar] [CrossRef]
- Compton, J.E.; Harrison, J.A.; Dennis, R.L.; Greaver, T.L.; Hill, B.H.; Jordan, S.J.; Walker, H.; Campbell, H.V. Ecosystem services altered by human changes in the nitrogen cycle: A new perspective for US decision making. Ecol. Lett. 2011, 14, 804–815. [Google Scholar] [CrossRef]
- Garcia-Segura, S.; Mostafa, E.; Baltruschat, H. Could NOx be released during mineralization of pollutants containing nitrogen by hydroxyl radical? Ascertaining the release of N-volatile species. Appl. Catal. B Environ. 2017, 207, 376–384. [Google Scholar] [CrossRef]
- Kumar, S.G.; Rao, K.K. Tungsten-based nanomaterials (WO3 & Bi2WO6): Modifications related to charge carrier transfer mechanisms and photocatalytic applications. Appl. Surf. Sci. 2015, 355, 939–958. [Google Scholar] [CrossRef]
- Ranjit, K.T.; Viswanathan, B. Photocatalytic reduction of nitrite and nitrate ions to ammonia on M/TiO2 catalysts. J. Photochem. Photobiol. A Chem. 1997, 108, 73–78. [Google Scholar] [CrossRef]
- Park, H.; Park, Y.; Kim, W.; Choi, W. Surface modification of TiO2 photocatalyst for environmental applications. J. Photochem. Photobiol. C Photochem. Rev. 2013, 15, 1–20. [Google Scholar] [CrossRef]
- Yang, J.; Dai, J.; Li, J. Visible-light-induced photocatalytic removal of aqueous nitrate with Nd, N-codoped titania nanoparticles. Sci. Adv. Mater. 2013, 5, 1013–1023. [Google Scholar] [CrossRef]
- Hamanoi, O.; Kudo, A. Reduction of nitrate and nitrite ions over Ni-ZnS photocatalyst under visible light irradiation in the presence of a sacrificial reagent. Chem. Lett. 2002, 31, 838–839. [Google Scholar] [CrossRef]
- Miyazaki, A.; Matsuda, K.; Papa, F.; Scurtu, M.; Negrila, C.; Dobrescu, G.; Balint, I. Impact of particle size and metal–support interaction on denitration behavior of well-defined Pt–Cu nanoparticles. Catal. Sci. Technol. 2015, 5, 492–503. [Google Scholar] [CrossRef]
- State, R.; Scurtu, M.; Miyazaki, A.; Papa, F.; Atkinson, I.; Munteanu, C.; Balint, I. Influence of metal-support interaction on nitrate hydrogenation over Rh and Rh-Cu nanoparticles dispersed on Al2O3 and TiO2 supports. Arab. J. Chem. 2017, 10, 975–984. [Google Scholar] [CrossRef]
- Vasile, A.; Papa, F.; Bratan, V.; Munteanu, C.; Teodorescu, M.; Atkinson, I.; Anastasescu, M.; Kawamoto, D.; Negrila, C.; Ene, C.D.; et al. Water denitration over titania-supported Pt and Cu by combined photocatalytic and catalytic processes: Implications for hydrogen generation properties in a photocatalytic system. J. Environ. Chem. Eng. 2022, 10, 107129. [Google Scholar] [CrossRef]
- Chelu, M.; State, R.; Munteanu, C.; Atkinson, I.; Rusu, A.; Bratan, V.; Musuc, A.; Balint, I.; Papa, F. Enhanced photocatalytic activity of ZnO nanoparticles obtained by “green” synthesis with well dispersed Pd-Au bimetallic nanoparticles. Rev. Roum. Chim. 2018, 63, 837–845. Available online: https://revroum.lew.ro/wp-content/uploads/2018/09/Art%2007.pdf (accessed on 19 December 2022).
- Park, C.; Kwak, H.; Moon, G.H.; Kim, W. Biomimetic photocatalysts for the conversion of aqueous-and gas-phase nitrogen species to molecular nitrogen via denitrification and ammonia oxidation. J. Mater. Chem. A 2021, 9, 19179–19205. [Google Scholar] [CrossRef]
- Bems, B.; Jentoft, F.; Schlögl, C.R. Photoinduced decomposition of nitrate in drinking water in the presence of titania and humic acids. Appl. Catal. B Environ. 1999, 20, 155–163. [Google Scholar] [CrossRef]
- Orth, W.S.; Gillham, R.W. Dechlorination of trichloroethene in aqueous solution using Fe0. Environ. Sci. Technol. 1995, 30, 66–71. [Google Scholar] [CrossRef]
- Alowitz, M.J.; Scherer, M.M. Kinetic of nitrate, nitrite and Cr(VI) reduction by iron metal. Environ. Sci. Technol. 2002, 36, 299–306. [Google Scholar] [CrossRef]
- Hou, Z.; Chen, F.; Wang, J.; François-Xavier, C.P.; Wintgens, T. Novel Pd/GdCrO3 composite for photo-catalytic reduction of nitrate to N2 with high selectivity and activity. Appl. Catal. B Environ. 2018, 232, 124–134. [Google Scholar] [CrossRef]
- Wehbe, N.; Jaafar, M.; Guillard, C.; Herrmann, J.-M.; Miachon, S.; Puzenat, E.; Guilhaume, N. Comparative study of photocatalytic and non-photocatalytic reduction of nitrates in water. Appl. Catal. A Gen. 2009, 368, 1–8. [Google Scholar] [CrossRef]
- Gao, W.; Jin, R.; Chen, J.; Guan, X.; Zeng, H.; Zhang, F. Titania-supported bimetallic catalysts for photocatalytic reduction of nitrate. Catal. Today 2004, 90, 331–336. [Google Scholar] [CrossRef]
- Mikami, I.; Sakamoto, Y.; Yoshinaga, Y.; Okuhara, T. Kinetic and adsorption studies on the hydrogenation of nitrate and nitrite in water using Pd-Cu on active carbon support. Appl. Catal. B Environ. 2003, 44, 79–86. [Google Scholar] [CrossRef]
- Devadas, A.; Vasudevan, S.; Epron, F. Nitrate reduction in water: Influence of the addition of a second metal on the performances of the Pd/CeO2 catalyst. J. Hazard. Mater. 2012, 669, 1412–1417. [Google Scholar] [CrossRef]
- Hirayama, J.; Kondo, H.; Miura, Y.K.; Abe, R.; Kamiya, Y. Highly effective photocatalytic system comprising semiconductor photocatalyst and supported bimetallic non-photocatalyst for selective reduction of nitrate to nitrogen in water. Catal. Commun. 2012, 20, 99–102. [Google Scholar] [CrossRef]
- Liu, G.; You, S.; Ma, M.; Huang, H.; Ren, N. Removal of nitrate by photocatalytic denitrification using nonlinear optical material. Environ. Sci. Techn. 2016, 50, 11218–11225. [Google Scholar] [CrossRef]
- Sato, T.; Sato, K.; Fujishiro, Y.; Yoshioka, T.; Okuwaki, A. Photochemical reduction of nitrate to ammonia using layered hydrous titanate/cadmium sulphide nanocomposites. J. Chem. Technol. Biotechnol. 1996, 67, 345–349. [Google Scholar] [CrossRef]
- Ketir, W.; Bouguelia, A.; Trari, M. NO3− removal with a new delafossite CuCrO2 photocatalyst. Desalination 2009, 244, 144–152. [Google Scholar] [CrossRef]
- Ranjit, K.T.; Viswanathan, B. Photocatalytic reduction of nitrite and nitrate ions over doped TiO2 catalysts. J. Photochem. Photobiol. A Chem. 1997, 107, 215–220. [Google Scholar] [CrossRef]
- Kato, H.; Kudo, A. Photocatalytic reduction of nitrate ions over tantalate photocatalysts. Phys. Chem. Chem. Phys. 2002, 4, 2833–2838. [Google Scholar] [CrossRef]
- Kudo, A.; Domen, K.; Maruya, K.; Onishi, T. Photocatalytic reduction of NO3− to form NH3 over Pt–TiO2. Chem. Lett. 1987, 6, 1019–1022. [Google Scholar] [CrossRef]
- Bahadori, E.; Tripodi, A.; Ramis, G.; Rossetti, I. Semi-batch photocatalytic reduction of nitrates: Role of process conditions and co-catalysts. ChemCatChem 2019, 11, 4642–4652. [Google Scholar] [CrossRef]
- Osterloh, F.E. Inorganic nanostructures for photoelectrochemical and photocatalytic water splitting. Chem. Soc. Rev. 2013, 42, 2294–2320. [Google Scholar] [CrossRef]
- Wang, Z.; Li, C.; Domen, K. Recent developments in heterogeneous photocatalysts for solar-driven overall water splitting. Chem. Soc. Rev. 2019, 48, 2109–2125. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Kamat, P.V. Quantum dot solar cells. Semiconductor nanocrystals as light harvesters. J. Phys. Chem. C 2008, 112, 18737–18753. [Google Scholar] [CrossRef]
- Linic, S.; Aslam, U.; Boerigter, C.; Morabito, M. Photochemical transformations on plasmonic metal nanoparticles. Nature Mater. 2015, 14, 567–576. [Google Scholar] [CrossRef]
- Wei, L.; Adamson, M.A.; Vela, J. Ni2P-modified Ta3N5 and TaON for photocatalytic nitrate reduction. Chem. Nano Mat. 2020, 6, 1179–1185. [Google Scholar] [CrossRef]
- Wei, L.; Liu, D.J.; Rosales, B.A.; Evans, J.W.; Vela, J. Mild and selective hydrogenation of nitrate to ammonia in the absence of noble metals. ACS Catal. 2020, 10, 3618–3628. [Google Scholar] [CrossRef]
- Hou, Z.; Chu, J.; Liu, C.; Wang, J.; Li, A.; Lin, T.; François-Xavier, C.P. High efficient photocatalytic reduction of nitrate to N2 by Core-shell Ag/SiO2@ cTiO2 with synergistic effect of light scattering and surface plasmon resonance. Chem. Eng. J. 2021, 415, 128863. [Google Scholar] [CrossRef]
- Liu, Y.; Gong, X.; Yang, W.; Wang, B.; Yang, Z.; Liu, Y. Selective reduction of nitrate into nitrogen using Cu/Fe bimetal combined with sodium sulfite. Ind. Eng. Chem. Res. 2019, 58, 5175–5185. [Google Scholar] [CrossRef]
- Shang, Z.; An, X.; Liu, H.; Hu, C.; Qu, J. Silvered TiO2 for facet-dependent photocatalytic denitrification. ACS Appl. Nano Mater. 2021, 4, 13534–13542. [Google Scholar] [CrossRef]
- Silveira, J.E.; De Souza, A.S.; Pansini, F.N.; Ribeiro, A.R.; Scopel, W.L.; Zazo, J.A.; Casas, J.A.; Paz, W.S. A comprehensive study of the reduction of nitrate on natural FeTiO3: Photocatalysis and DFT calculations. Sep. Purif. Technol. 2023, 306, 122570. [Google Scholar] [CrossRef]
- Silveira, J.E.; Garcia-Costa, A.L.; Carbajo, J.; Ribeiro, A.R.; Pliego, G.; Paz, W.S.; Zazo, J.A.; Casas, J.A. Nitrate removal in saline water by photo-reduction using natural FeTiO3 as catalyst. Chem. Eng. J. Adv. 2022, 12, 100387. [Google Scholar] [CrossRef]
- Wang, L.; Fu, W.; Zhuge, Y.; Wang, J.; Yao, F.; Zhong, W.; Ge, X. Synthesis of polyoxometalates (POM)/TiO2/Cu and removal of nitrate nitrogen in water by photocatalysis. Chemosphere 2021, 278, 130298. [Google Scholar] [CrossRef]
- Liu, X.; Liu, H.; Wang, Y.; Yang, W.; Yu, Y. Nitrogen-rich g-C3N4@ AgPd Mott-Schottky heterojunction boosts photocatalytic hydrogen production from water and tandem reduction of NO3− and NO2−. J. Colloid Interface Sci. 2021, 581, 619–626. [Google Scholar] [CrossRef]
- Soliman, A.M.; Alshamsi, D.; Murad, A.A.; Aldahan, A.; Ali, I.M.; Ayesh, A.I.; Elhaty, I.A. Photocatalytic removal of nitrate from water using activated carbon-loaded with bimetallic Pd-Ag nanoparticles under natural solar radiation. J. Photochem. Photobiol. A Chem. 2022, 433, 114175. [Google Scholar] [CrossRef]
- Liu, Y.; Lee, J.; Zhao, Y.; Zhang, M.; Wang, L.; Duan, Q. A novel preparation approach and denitrification performance of TiO2/Fe0 photocatalysts. Desalination Water Treat. 2016, 57, 3125–3131. [Google Scholar] [CrossRef]
- Ahmed, M.J.; Hameed, B.H.; Khan, M.A. Recent advances on activated carbon-based materials for nitrate adsorption: A review. J. Anal. Appl. Pyrolysis 2022, 169, 105856. [Google Scholar] [CrossRef]
- Cheng, Z.W.; Jiang, Y.F.; Zhang, L.L.; Chen, J.M.; Wei, Y.Y. Conversion characteristics and kinetic analysis of gaseous α-pinene degraded by a VUV light in various reaction media. Sep. Purif. Technol. 2011, 77, 26–32. [Google Scholar] [CrossRef]
- Geng, Q.; Wang, H.; Chen, R.; Chen, L.; Li, K.; Dong, F. Advances and challenges of photocatalytic technology for air purification. Natl. Sci. Open 2022, 1, 20220025. [Google Scholar] [CrossRef]
- Younis, S.A.; Kim, K.H. Heterogeneous photocatalysis scalability for environmental remediation: Opportunities and challenges. Catalysts 2020, 10, 1109. [Google Scholar] [CrossRef]
- Figueredo, M.; Rodríguez, E.M.; Fernando, J.R.; Beltrán, J. Photocatalytic ozonation in water treatment: Is there really a synergy between systems? Water Res. 2021, 206, 117727. [Google Scholar] [CrossRef]
- Adewui, Y.G. Sonochemistry in environmental remediation. 2. Heterogeneous sonophotocatalytic oxidation processes for the treatment of pollutants in water. Environ. Sci. Technol. 2005, 39, 8557–8570. [Google Scholar] [CrossRef]
- Khan, M.F.; Bakhtiar, S.H.; Zada, A.; Raziq, F.; Saleemi, H.A.; Khan, M.S.; Ismail, P.M.; Alguno, A.C.; Capangpangan, R.Y.; Ali, A.; et al. Ag modified ZnO microsphere synthesis for efficient sonophotocatalytic degradation of organic pollutants and CO2 conversion. Environ. Nanotechnol. Monit. Manag. 2022, 18, 100711. [Google Scholar] [CrossRef]
- Paustian, D.; Franke, M.; Stelter, M.; Braeutigam, P. Sonophotocatalysis—Limits and possibilities for synergistic effects. Catalysts 2022, 12, 754. [Google Scholar] [CrossRef]
- Nair, V.; Muñoz-Batista, M.J.; Fernández-García, M.; Luque, R.; Colmenares, J.C. Thermo-photocatalysis: Environmental and energy applications. ChemSusChem 2019, 12, 2098–2116. [Google Scholar] [CrossRef]
- Muñoz-Batista, M.J.; Eslava-Castillo, A.M.; Kubacka, A.; Fernández-García, M. Thermo-photo degradation of 2-propanol using a composite ceria-titania catalyst: Physico-chemical interpretation from a kinetic model. Appl. Catal. B Environ. 2018, 225, 298–306. [Google Scholar] [CrossRef]
- Wang, G.; Huang, B.; Lou, Z.; Wang, Z.; Qin, X.; Zhang, X.; Dai, Y. Valence state heterojunction Mn3O4/MnCO3: Photo and thermal synergistic catalyst. Appl. Catal. B Environ. 2016, 180, 6–12. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Y.; Shen, H.; Shuai, D. Simultaneous coupling of photocatalytic and biological processes: A promising synergistic alternative for enhancing decontamination of recalcitrant compounds in water. Chem. Eng. J. 2021, 403, 126365. [Google Scholar] [CrossRef]
- Tyagi, S.; Rawtani, D.; Khatri, N.; Tharmavaram, M. Strategies for nitrate removal from aqueous environment using nanotechnology: A review. J. Water Process. Eng. 2018, 21, 84–95. [Google Scholar] [CrossRef]
- Andronic, L.; Isac, L.; Miralles-Cuevas, S.; Visa, M.; Oller, I.; Duta, A.; Malato, S. Pilot-plant evaluation of TiO2 and TiO2-based hybrid photocatalysts for solar treatment of polluted water. J. Hazard. Mater. 2016, 320, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Salmerón, I.; Rivas, G.; Oller, I.; Martínez-Piernas, A.; Agüera, A.; Malato, S. Nanofiltration retentate treatment from urban wastewater secondary effluent by solar electrochemical oxidation processes. Sep. Purif. Techn. 2021, 254, 117614. [Google Scholar] [CrossRef]
- Gomes, J.; Roccamante, M.; Contreras, S.; Medina, F.; Oller, I.; Martin, R.C. Scale-up impact over solar photocatalytic ozonation with benchmark-P25 and N-TiO2 for insecticides abatement in water. J. Environm. Chem. Eng. 2021, 9, 104915. [Google Scholar] [CrossRef]
- Augugliaro, V.; Litter, M.; Palmisano, L.; Soria, J. The combination of heterogeneous photocatalysis with chemical and physical operations: A tool for improving the photoprocess performance. J. Photochem. Photobiol. C Photochem. Rev. 2006, 7, 127–144. [Google Scholar] [CrossRef]
- Ajmal, Z.; Naciri, Y.; Hsini, A.; Bresolin, B.M.; Qadeer, A.; Nauman, M.; Arif, M.; Irshad, M.K.; Khan, K.A.; Djellabi, R.; et al. Prospects of Photocatalysis in the Management of Nitrate Contamination in Potable Water. In Progress and Prospects in the Management of Oxyanion Polluted Aqua Systems. Environmental Contamination Remediation and Management; Oladoja, N.A., Unuabonah, E.I., Eds.; Springer: Cham, Switzerland, 2021; Chapter 7; pp. 185–217. [Google Scholar] [CrossRef]
- Sacco, O.; Vaiano, V.; Sannino, D. Main parameters influencing the design of photocatalytic reactors for wastewater treatment: A mini review. J. Chem. Technol. Biotechnol. 2020, 95, 2608–2618. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photocatalyst | Reactivity Sequence |
|---|---|
| ZnO | oxalic acid > acetic acid > citric acid The catalyst is not stable in formic acid solution |
| CuO | formic acid > oxalic acid > acetic acid > citric acid |
| TiO2 | formic acid > oxalic acid > acetic acid > citric acid |
| Bi2O3 | formic acid > oxalic acid = acetic acid > citric acid |
| Pb2O3 | formic acid > oxalic acid = acetic acid > citric acid |
| PbO2 | formic acid > oxalic acid = acetic acid > citric acid |
| Parameter | Value | ||
|---|---|---|---|
| Flow rate Ozone concentration Irradiance | 30 1289 33.6 | L h−1 mg m−3 W m−2 | |
| VOC | Degradation (SE) in % | ||
| DCE | C2H2Cl2 | 99.4 | (±0.3) |
| TCE | C2HCl3 | 99.3 | (±0.2) |
| PCE | C2Cl4 | 98.0 | (±0.2) |
| TCM | CHCl3 | 84.8 | (±7.8) |
| PCM | CCl4 | 14.0 | (±11.4) |
| Type of Photocatalyst | Experimental Conditions | Light Source | Reaction Time | Degradation Efficiency | TOC Removal Efficiency | Main Active Species | Ref. |
|---|---|---|---|---|---|---|---|
| TiO2/MnO2 follow sphere | [phenol] = 5 mg L−1 [photocatalyst] = 20 mg | Simulated solar light (XHA 300 W Xe lamp AM 1.5 G filter | 180 min | 100% | 91% | superoxide radical | [84] |
| 2% Au/TiO2 | [phenol] = 30 mg L−1 | Solar light | 3.5 h | 100% | - | - | [85] |
| Pr(0.072%)-TiO2 | [phenol] = 50 ppm, [photocatalyst] = 1 g L−1, pH = 6.58 | UV light | 2 h | 94.4% | - | - | [86] |
| EY-TiO2/Pt(0.5%) | [phenol] = 40 ppm, [photocatalyst] = 0.8 g L−1, 0.2 M TEOA, neutral pH | Visible solar light | 2 h | 100% | - | superoxyde radical | [87] |
| TiO2-BiOBr-Bentonite | [phenol] = 20 ppm, [photocatalyst] = 150 mg/100 mL solution | Visible light | 70 min | 100% | 83% | superoxide radical and e− (major role) h+, •OH (appreciable role) | [88] |
| Fe3O4@rGO@AgI | [phenol] = 50 ppm [photocatalyst] = 0.2 g/350 mL solution | UV-C light, λ = 254 nm | 9 h | 99% | - | mainly •OH | [89] |
| mont-La(6%)-Cu0.6Cd0.4S nanocomposite | [phenol] = 20 mg L−1 [photocatalyst] = 60 mg/L | Medium-pressure Hg-vapor lamp, near UV-Vis irradiation | 4 h | 86% | 77.8% | •OH and h+ radicals | [90] |
| TiO2/rGO | [phenol] = 20 ppm | UV light | 180 min | 97.87% | - | •OH, superoxide radical, and h+ | [91] |
| CuFe2O4/rGO | [phenol] = 200 ppm [photocatalyst] = 60 mg/L | Visible light | 15 min | 100% | - | - | [92] |
| PAN-CNT/TiO2-NH2 | [phenol] = 10 ppm, pH = 5 [photocatalyst] =20 mg | UV light | 7 min | 99.2% | - | •OH and superoxide radical | [93] |
| CdS/rGO/Fe2+ | [phenol] = 10 ppm [photocatalyst] = 20 mg/20 mL | 300 W Xe lamp irradiation (λ > 420 nm) | 60 min | 100% | 43.66% | •OH | [94] |
| N-TiO2/HPCF (Hierarchical Porous Carbon Foam) | [phenol] = 30 mg L−1 [photocatalyst] = 50 mg/50 mL aq. Ph | 500 W Xe lamp, simulated sunlight irradiation | 2 h | 97% | 78 % after 6 h of illumination | - | [95] |
| (2:1) Bi-Bi7O9I3/Ag-AgI | [phenol] = 10 ppm [photocatalyst] = 50 mg/100 mL aq. Ph | 300 Xe lamp, λ > 420 nm | 120 min | 100% | 95.38% | •OH (more significant role than h+) | [96] |
| Pt/TiO2-ZnO@ZIF-8 | [phenol] = 5 ppm | UV light | 24 min | 99.7% | - | - | [97] |
| CeO2/TiO2 | [phenol] = 38 ppm [photocatalyst] = 20 mg/40 mL aq. Ph | 300 W high-pressure UV mercury lamp, UV irradiation | 120 min | 99.1% | - | •OH, h+ | [98] |
| Photocatalyst | Light Source | Sacrificial Agent | NO3− Conversion (%) | NO2− Selectivity (%) | NH4+ Selectivity (%) | N2 Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|
| Fresh Ag/P25 | High-pressure Hg lamp 300 W | Formic acid 0.04 M | 99.6 | 2.3 (yield of NO2−, mgN L−1) | 9.3 (yield of NH4+, mgN L−1) | 88.4 | [125] |
| 5% Ag2O/P25 | High-pressure Hg lamp 300 W | Formic acid 0.04 M | 97.2 | 2.4 (yield of NO2−, mgN L−1 | 14.0 (yield of NH4+, mgN L−1) | 83.1 | [125] |
| Fe/TiO2 | High-pressure Hg lamp 110 W | Formic acid 40 mM | 100 | 0 | 13.0 | 87.0 | [127] |
| Cu/TiO2 | High-pressure Hg lamp 110 W | Formic acid 40 mM | 100 | 0 | 37.0 | 63.0 | [127] |
| Cr-TiO2 | Low-pressure Hg lamp 17 W | Formic acid 450 mg L−1 (~9.8 mM) | 56.29 | - | - | 98.53 | [139] |
| Zn-TiO2 | Low-pressure Hg lamp 17 W | Formic acid 450 mg L−1 (~9.8 mM) | 91.67 | - | - | 95.45 | [139] |
| Ag/TiO2 | High-pressure Hg lamp 125 W | Formic acid 0.04 mol/L | 98.4 | 0 | 0 | 100 | [144] |
| Ag/TiO2 | High-pressure Hg lamp 125 W | Oxalic acid | 16.7 | 2.20 | 0.37 | 84.6 | [144] |
| Pd-Cu/TiO2 | - | Formic acid | 62 | 0 | 6.0 | 94.0 | [164] |
| LiNbO3 | High-pressure Hg lamp 110 W | Humic acid 1.0 mmol/L | 90.1 | - | - | 86.2 | [169] |
| LiNbO3 | High-pressure Hg lamp 110 W | Formic acid 1.0 mmol/L | 98.4 | 0.13 | 1.2 | 95.8 | [169] |
| Ag2O/Ag/101-TiO2 | - | - | 98.57 | 3.55 | 2.42 | 94.03 | [185] |
| Ag2O/Ag/101-TiO2 | - | - | 99.10 | 4.56 | 14.34 | 81.10 | [185] |
| nzv Fe/TiO2 | UV-A lamp 20 W nzv Fe/TiO2 | Formic acid 27 mM | 80.0 | 0 | 39.1 | 60.9 | [191] |
| SiW9/TiO2/Cu | High-pressure mercury lamp 125 W | Formic acid 30 mmol/L | 76.53 | - | - | 82.09 | [188] |
| Ag3Pd7/g-C1.95N4 | Two light bulbs 40W | - | 87.4 | 61.8 | - | ≈100 | [189] |
| Ag-Pd NPs/activated carbon | Natural solar radiation | Formic acid 0.05 M | 85 | traces | traces | High selectivity to N2 | [190] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavel, M.; Anastasescu, C.; State, R.-N.; Vasile, A.; Papa, F.; Balint, I. Photocatalytic Degradation of Organic and Inorganic Pollutants to Harmless End Products: Assessment of Practical Application Potential for Water and Air Cleaning. Catalysts 2023, 13, 380. https://doi.org/10.3390/catal13020380
Pavel M, Anastasescu C, State R-N, Vasile A, Papa F, Balint I. Photocatalytic Degradation of Organic and Inorganic Pollutants to Harmless End Products: Assessment of Practical Application Potential for Water and Air Cleaning. Catalysts. 2023; 13(2):380. https://doi.org/10.3390/catal13020380
Chicago/Turabian StylePavel, Monica, Crina Anastasescu, Razvan-Nicolae State, Anca Vasile, Florica Papa, and Ioan Balint. 2023. "Photocatalytic Degradation of Organic and Inorganic Pollutants to Harmless End Products: Assessment of Practical Application Potential for Water and Air Cleaning" Catalysts 13, no. 2: 380. https://doi.org/10.3390/catal13020380