


Methane Reforming Processes: Advances on Mono- and Bimetallic Ni-Based Catalysts Supported on Mg-Al Mixed Oxides

 , and
, and
Abstract
:1. Introduction
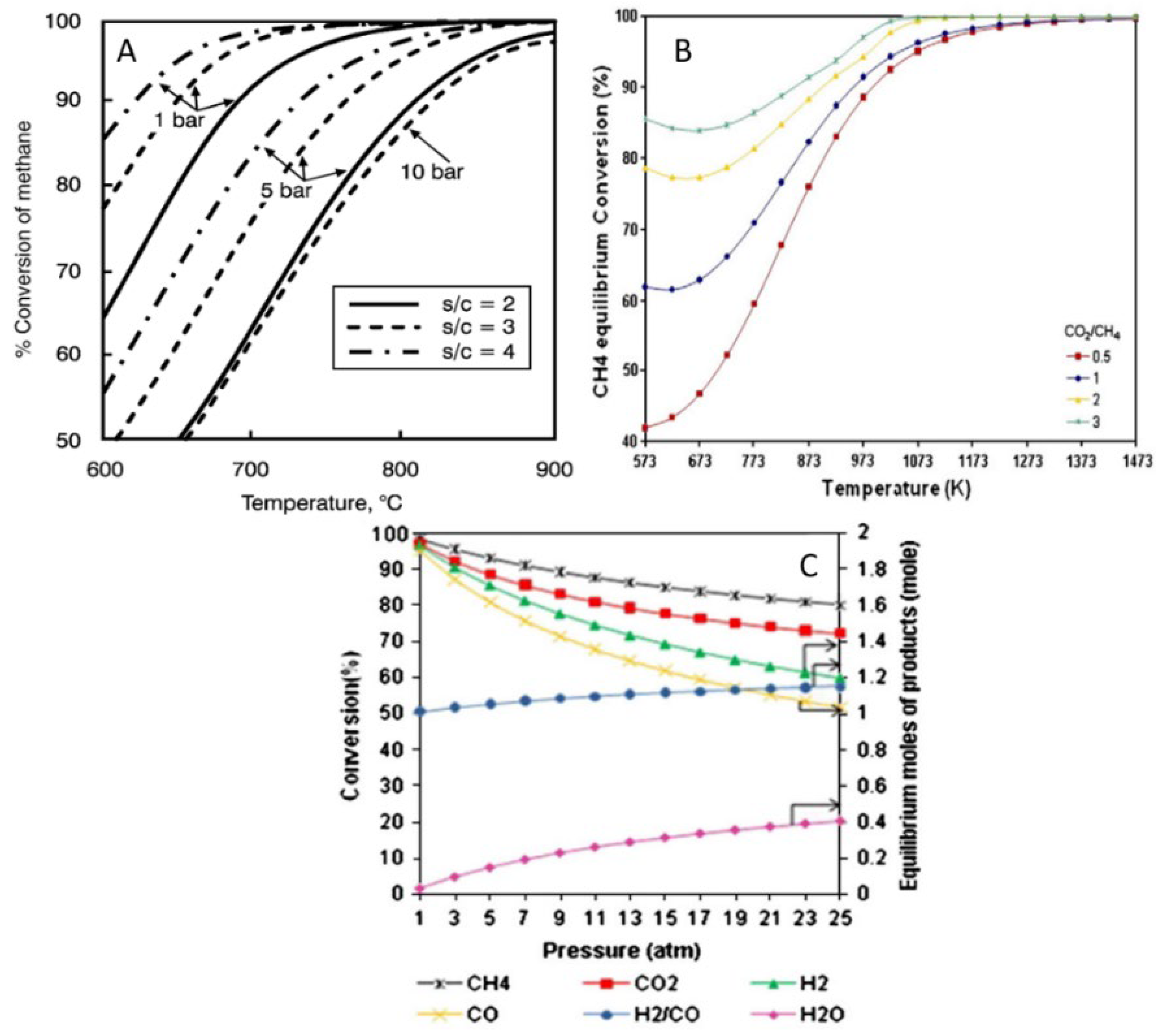
1.1. Steam and Dry Reforming Reactions of CH4: General Aspects
1.2. The Ni(M)/Mg(Al)O Catalysts
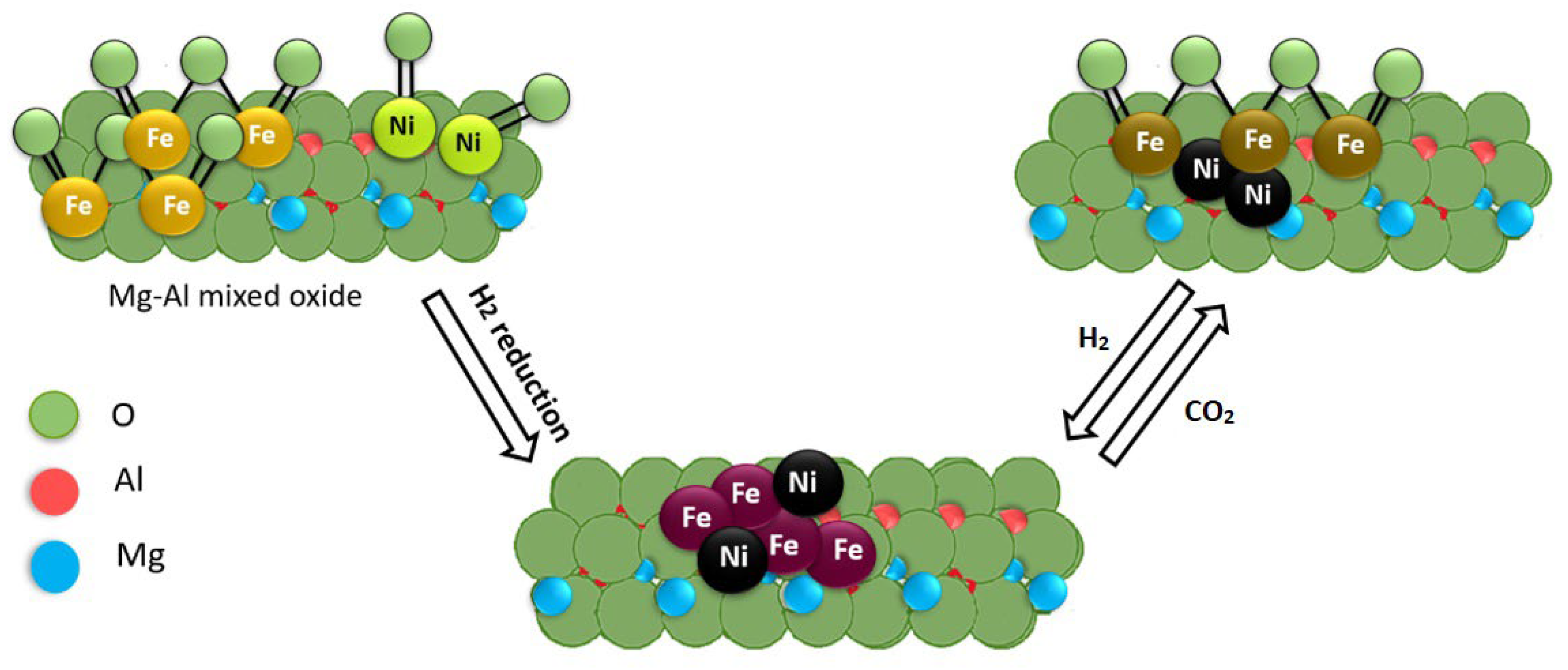
2. Ni-Support Interaction in Ni(M)/Mg(Al)O Catalysts
3. Monometallic Ni/Mg(Al)O Catalysts for SMR and DRM
3.1. Synthesis of Ni/Mg(Al)O Catalysts by Conventional Approaches
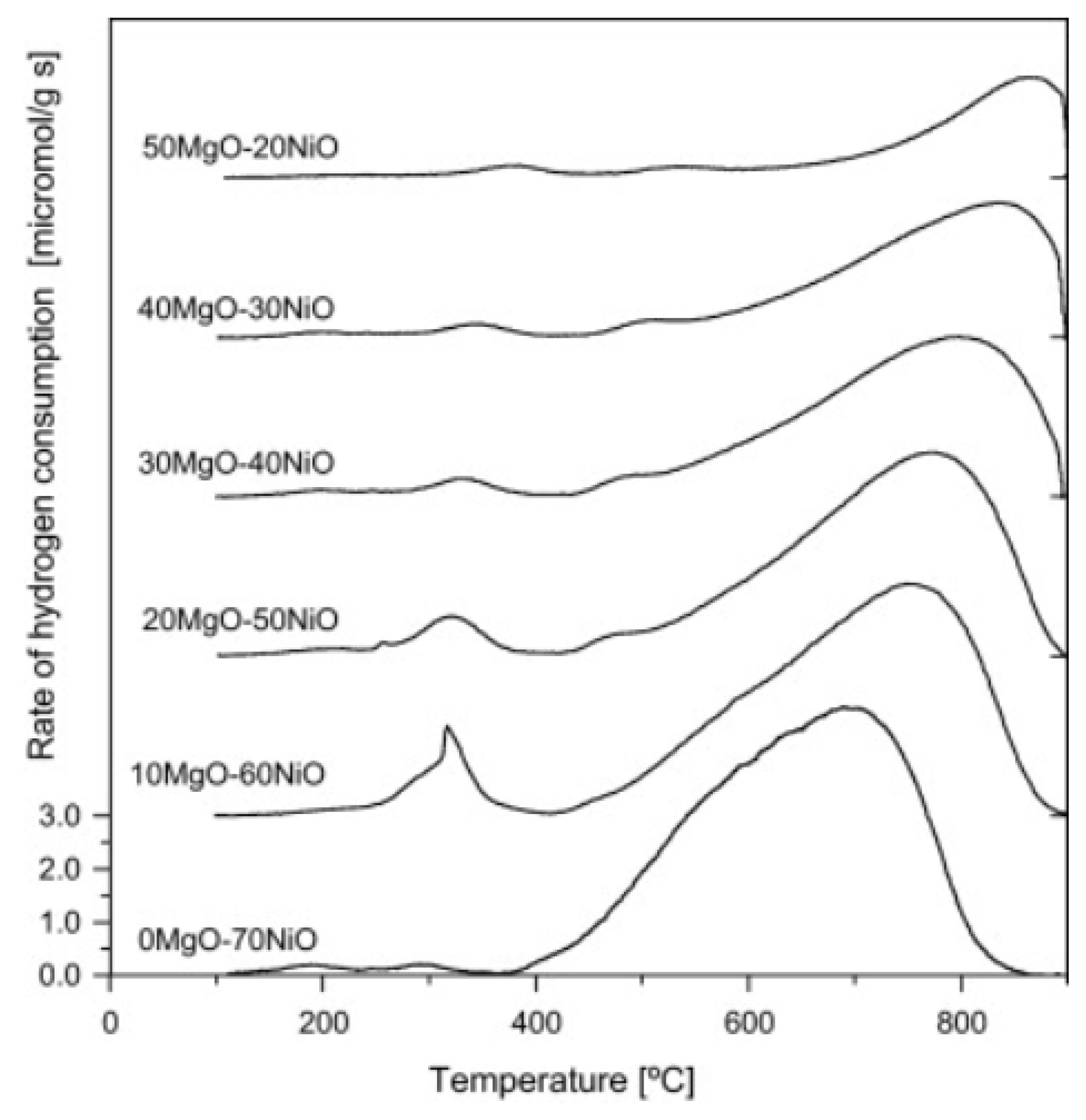
3.1.1. Effect of Ni Loading and Composition Ratio
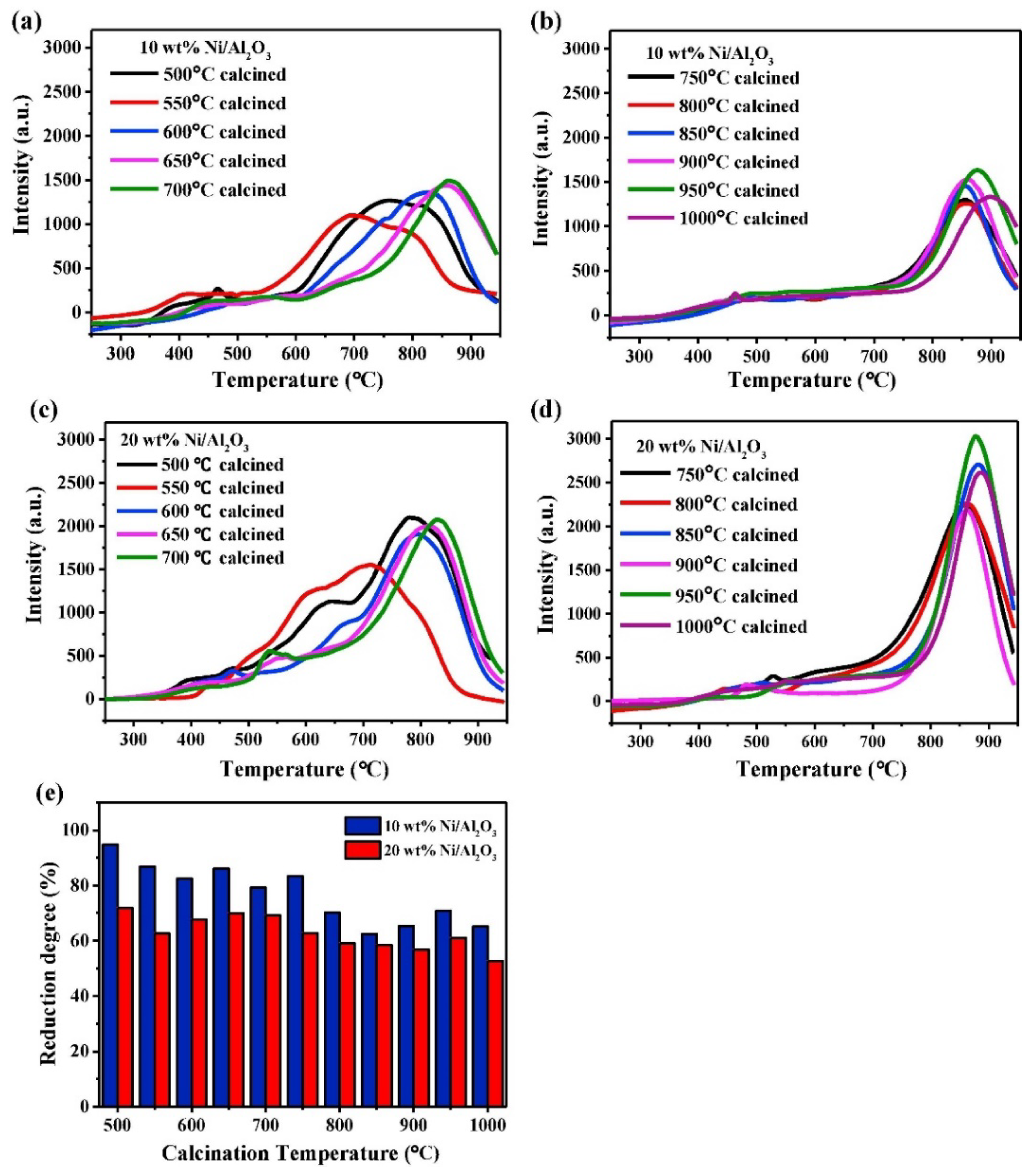
3.1.2. Effect of Heat Treatment
3.2. Synthesis of Ni/Mg(Al)O Catalysts by Advanced Methods
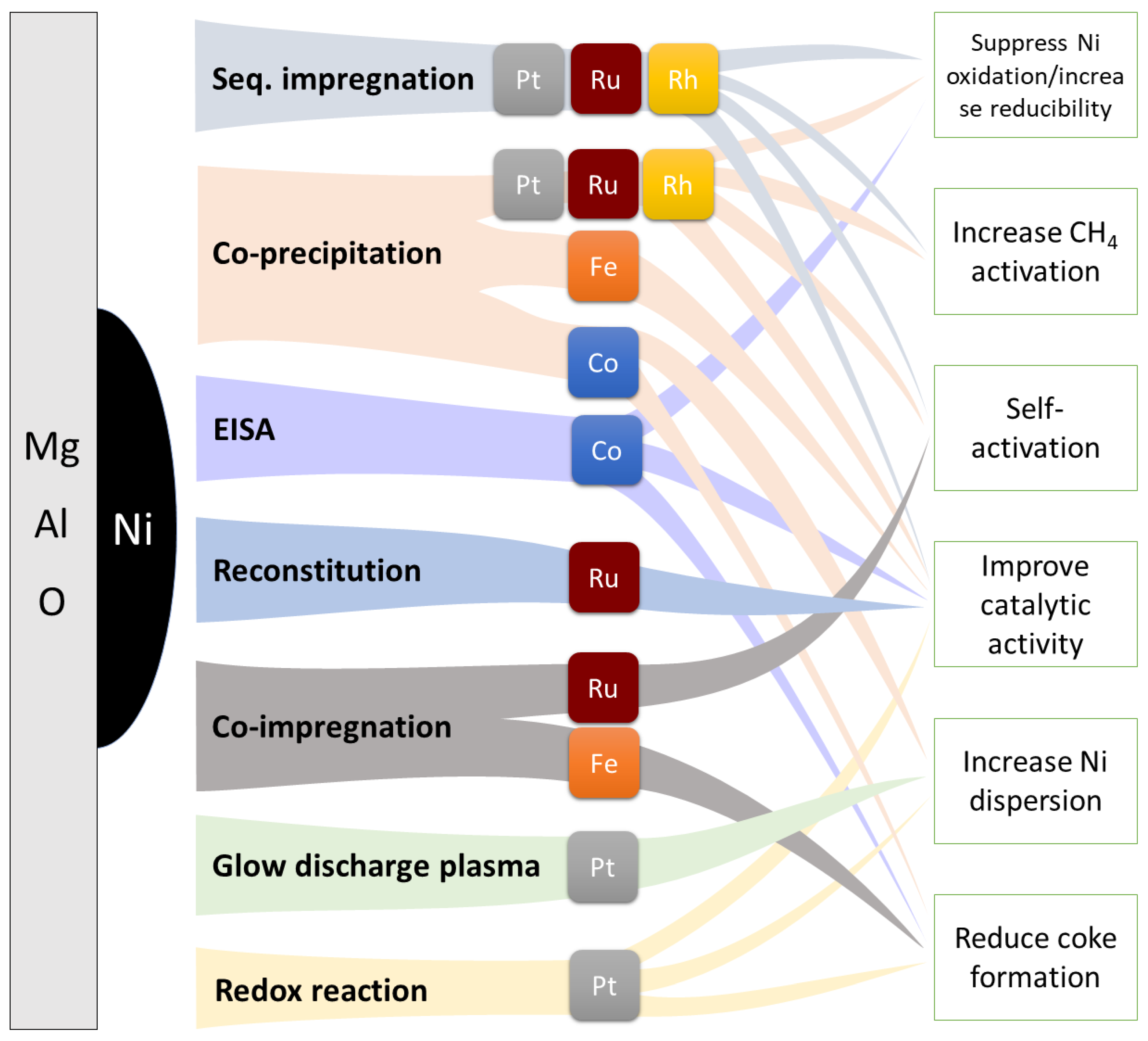
4. Bimetallic Ni-M/Mg(Al)O Catalysts for Reforming Reactions
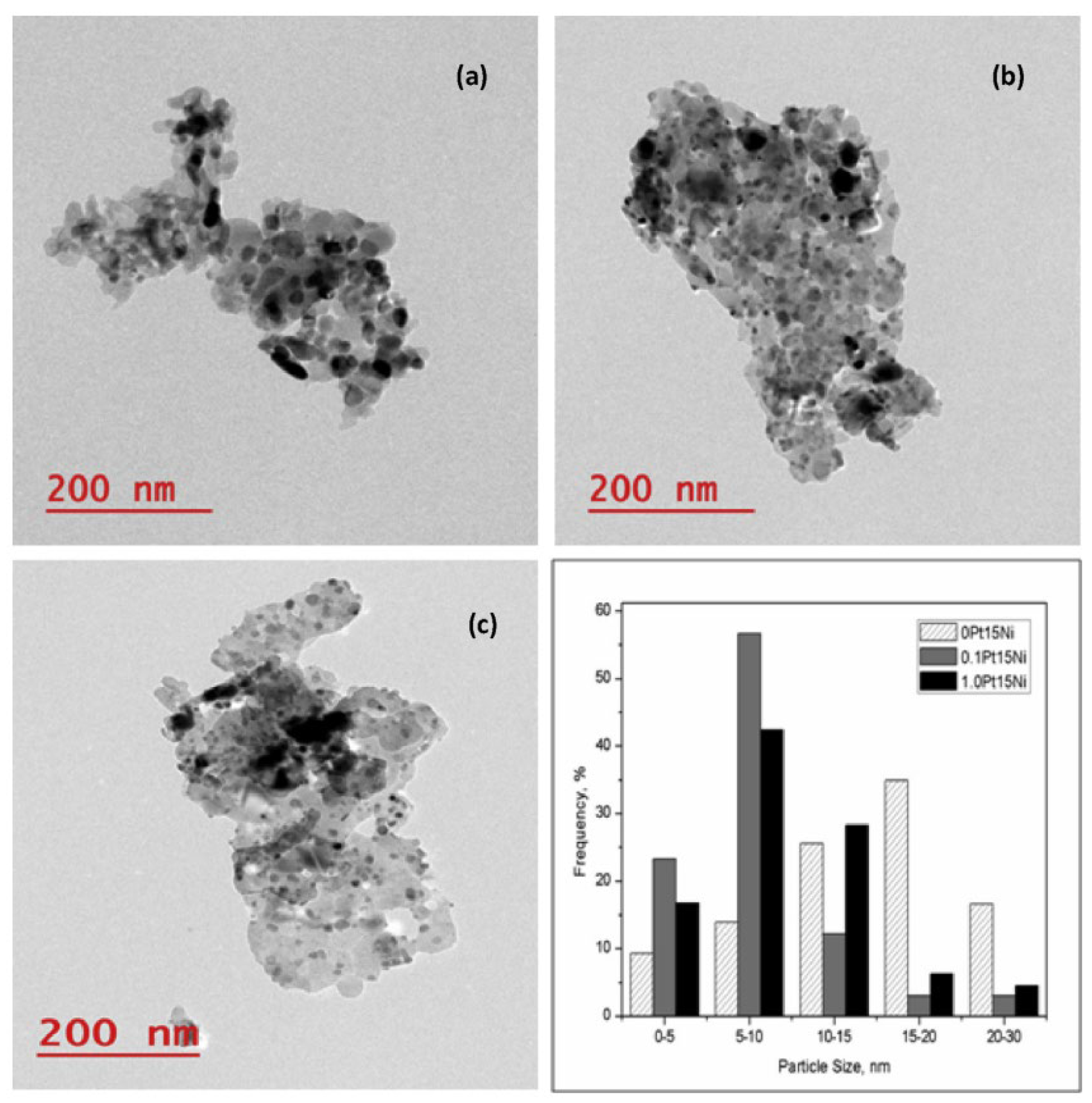
4.1. Bimetallic Ni-M/Mg(Al)O Catalysts for SMR
4.2. Bimetallic Ni-M/Mg(Al)O Catalysts for DRM
5. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- European Commission. Directorate General for Research and Innovation. In Horizon Europe: Strategic Plan 2021–2024; Publications Office: Luxembourg, 2021. [Google Scholar]
- Chen, L.; Qi, Z.; Zhang, S.; Su, J.; Somorjai, G.A. Catalytic Hydrogen Production from Methane: A Review on Recent Progress and Prospect. Catalysts 2020, 10, 858. [Google Scholar] [CrossRef]
- Iulianelli, A.; Liguori, S.; Wilcox, J.; Basile, A. Advances on Methane Steam Reforming to Produce Hydrogen through Membrane Reactors Technology: A Review. Catal. Rev. 2016, 58, 1–35. [Google Scholar] [CrossRef]
- Lavoie, J.M. Review on Dry Reforming of Methane, a Potentially More Environmentally Friendly Approach to the Increasing Natural Gas Exploitation. Front. Chem 2014, 2, 81. [Google Scholar] [CrossRef]
- Jang, W.-J.; Shim, J.-O.; Kim, H.-M.; Yoo, S.-Y.; Roh, H.-S. A Review on Dry Reforming of Methane in Aspect of Catalytic Properties. Catal. Today 2019, 324, 15–26. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A Review of Dry (CO2) Reforming of Methane over Noble Metal Catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- Ismagilov, Z.R.; Matus, E.V.; Ismagilov, I.Z.; Sukhova, O.B.; Yashnik, S.A.; Ushakov, V.A.; Kerzhentsev, M.A. Hydrogen Production through Hydrocarbon Fuel Reforming Processes over Ni Based Catalysts. Catal. Today 2019, 323, 166–182. [Google Scholar] [CrossRef]
- Armor, J.N. The Multiple Roles for Catalysis in the Production of H2. Appl. Catal. A Gen. 1999, 176, 159–176. [Google Scholar] [CrossRef]
- Joensen, F.; Rostrup-Nielsen, J.R. Conversion of Hydrocarbons and Alcohols for Fuel Cells. J. Power Sources 2002, 105, 195–201. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Efstathiou, A.M. Hydrogen Production Technologies: Current State and Future Developments. Conf. Pap. Energy 2013, 2013, 690627. [Google Scholar] [CrossRef]
- Kulkarni, S. A Review on Reforming Reactions with Emphasis on Methane Reforming. Int. J. Res. Rev. 2016, 3, 20–23. [Google Scholar]
- Chai, S.; Zhang, G.; Li, G.; Zhang, Y. Industrial Hydrogen Production Technology and Development Status in China: A Review. Clean Technol. Environ. Policy 2021, 23, 1931–1946. [Google Scholar] [CrossRef]
- Nikoo, M.K.; Amin, N.A.S. Thermodynamic Analysis of Carbon Dioxide Reforming of Methane in View of Solid Carbon Formation. Fuel Process. Technol. 2011, 92, 678–691. [Google Scholar] [CrossRef]
- Özkara-Aydınoğlu, Ş.; Aksoylu, A.E. CO2 Reforming of Methane over Pt–Ni/Al2O3 Catalysts: Effects of Catalyst Composition, and Water and Oxygen Addition to the Feed. Int. J. Hydrogen Energy 2011, 36, 2950–2959. [Google Scholar] [CrossRef]
- Kim, H.Y.; Park, J.-N.; Henkelman, G.; Kim, J.M. Design of a Highly Nanodispersed Pd–MgO/SiO2 Composite Catalyst with Multifunctional Activity for CH4 Reforming. ChemSusChem 2012, 5, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.C.J.; Vannice, M.A. CO2 Reforming of CH4. Catal. Rev. 1999, 41, 1–42. [Google Scholar] [CrossRef]
- Mondal, K.; Sasmal, S.; Badgandi, S.; Chowdhury, D.R.; Nair, V. Dry Reforming of Methane to Syngas: A Potential Alternative Process for Value Added Chemicals—A Techno-Economic Perspective. Environ. Sci. Pollut. Res. 2016, 23, 22267–22273. [Google Scholar] [CrossRef]
- Parsapur, R.K.; Chatterjee, S.; Huang, K.-W. The Insignificant Role of Dry Reforming of Methane in CO2 Emission Relief. ACS Energy Lett. 2020, 5, 2881–2885. [Google Scholar] [CrossRef]
- Noureldin, M.M.B.; Elbashir, N.O.; Gabriel, K.J.; El-Halwagi, M.M. A Process Integration Approach to the Assessment of CO2 Fixation through Dry Reforming. ACS Sustain. Chem. Eng. 2015, 3, 625–636. [Google Scholar] [CrossRef]
- Cai, X.; Hu, Y.H. Advances in Catalytic Conversion of Methane and Carbon Dioxide to Highly Valuable Products. Energy Sci. Eng. 2019, 7, 4–29. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R.; Sehested, J.; Nørskov, J.K. Hydrogen and Synthesis Gas by Steam-and CO2 Reforming. Adv. Catal. 2002, 47, 65–139. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.; Christiansen, L.J. Concepts in Syngas Manufacture. Catal. Sci. Ser. 2011, 10, 392. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R.; Hansen, J.H.B. CO2-Reforming of Methane over Transition Metals. J. Catal. 1993, 144, 38–49. [Google Scholar] [CrossRef]
- Jones, G.; Jakobsen, J.G.; Shim, S.S.; Kleis, J.; Andersson, M.P.; Rossmeisl, J.; Abild-Pedersen, F.; Bligaard, T.; Helveg, S.; Hinnemann, B.; et al. First Principles Calculations and Experimental Insight into Methane Steam Reforming over Transition Metal Catalysts. J. Catal. 2008, 259, 147–160. [Google Scholar] [CrossRef]
- Wu, H.; La Parola, V.; Pantaleo, G.; Puleo, F.; Venezia, A.; Liotta, L. Ni-Based Catalysts for Low Temperature Methane Steam Reforming: Recent Results on Ni-Au and Comparison with Other Bi-Metallic Systems. Catalysts 2013, 3, 563–583. [Google Scholar] [CrossRef]
- De, S.; Zhang, J.; Luque, R.; Yan, N. Ni-Based Bimetallic Heterogeneous Catalysts for Energy and Environmental Applications. Energy Environ. Sci. 2016, 9, 3314–3347. [Google Scholar] [CrossRef]
- Trimm, D.L. Coke Formation and Minimisation during Steam Reforming Reactions. Catal. Today 1997, 37, 233–238. [Google Scholar] [CrossRef]
- Trimm, D.L. Catalysts for the Control of Coking during Steam Reforming. Catal. Today 1999, 49, 3–10. [Google Scholar] [CrossRef]
- Sehested, J. Four Challenges for Nickel Steam-Reforming Catalysts. Catal. Today 2006, 111, 103–110. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Galvita, V.V.; Poelman, H.; Batchu, R.; Buelens, L.C.; Detavernier, C.; Marin, G.B. Mechanism of Carbon Deposits Removal from Supported Ni Catalysts. Appl. Catal. B Environ. 2018, 239, 502–512. [Google Scholar] [CrossRef]
- Li, D.; Nakagawa, Y.; Tomishige, K. Methane Reforming to Synthesis Gas over Ni Catalysts Modified with Noble Metals. Appl. Catal. A Gen. 2011, 408, 1–24. [Google Scholar] [CrossRef]
- Roussière, T.; Schulz, L.; Schelkle, K.M.; Wasserschaff, G.; Milanov, A.; Schwab, E.; Deutschmann, O.; Jentys, A.; Lercher, J.; Schunk, S.A. Structure–Activity Relationships of Nickel–Hexaaluminates in Reforming Reactions Part II: Activity and Stability of Nanostructured Nickel–Hexaaluminate-Based Catalysts in the Dry Reforming of Methane. ChemCatChem 2014, 6, 1447–1452. [Google Scholar] [CrossRef]
- Ginsburg, J.M.; Piña, J.; El Solh, T.; de Lasa, H.I. Coke Formation over a Nickel Catalyst under Methane Dry Reforming Conditions: Thermodynamic and Kinetic Models. Ind. Eng. Chem. Res. 2005, 44, 4846–4854. [Google Scholar] [CrossRef]
- Titus, J.; Roussière, T.; Wasserschaff, G.; Schunk, S.; Milanov, A.; Schwab, E.; Wagner, G.; Oeckler, O.; Gläser, R. Dry Reforming of Methane with Carbon Dioxide over NiO–MgO–ZrO2. Catal. Today 2016, 270, 68–75. [Google Scholar] [CrossRef]
- Aramouni, N.A.K.; Touma, J.G.; Tarboush, B.A.; Zeaiter, J.; Ahmad, M.N. Catalyst Design for Dry Reforming of Methane: Analysis Review. Renew. Sustain. Energy Rev. 2018, 82, 2570–2585. [Google Scholar] [CrossRef]
- Van Hook, J.P. Methane-Steam Reforming. Catal. Rev. 1980, 21, 1–51. [Google Scholar] [CrossRef]
- Hu, Y.H. Advances in Catalysts for CO2 Reforming of Methane. In Advances in CO2 Conversion and Utilization; American Chemical Society: Washington, DC, USA, 2010; pp. 155–174. [Google Scholar]
- Lee, S. Methane and Its Derivatives; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Hu, Y.H.; Ruckenstein, E. Binary MgO-Based Solid Solution Catalysts for Methane Conversion to Syngas. Catal. Rev. 2002, 44, 423–453. [Google Scholar] [CrossRef]
- Fan, M.-S.; Abdullah, A.Z.; Bhatia, S. Catalytic Technology for Carbon Dioxide Reforming of Methane to Synthesis Gas. ChemCatChem 2009, 1, 192–208. [Google Scholar] [CrossRef]
- Muraza, O.; Galadima, A. A Review on Coke Management during Dry Reforming of Methane. Int. J. Energy Res. 2015, 39, 1196–1216. [Google Scholar] [CrossRef]
- Arora, S.; Prasad, R. An Overview on Dry Reforming of Methane: Strategies to Reduce Carbonaceous Deactivation of Catalysts. RSC Adv. 2016, 6, 108668–108688. [Google Scholar] [CrossRef]
- Guo, J.; Lou, H.; Zhao, H.; Chai, D.; Zheng, X. Dry Reforming of Methane over Nickel Catalysts Supported on Magnesium Aluminate Spinels. Appl. Catal. A Gen. 2004, 273, 75–82. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Bimetallic Catalysts for Upgrading of Biomass to Fuels and Chemicals. Chem. Soc. Rev. 2012, 41, 8075–8098. [Google Scholar] [CrossRef] [PubMed]
- Dal Santo, V.; Gallo, A.; Naldoni, A.; Guidotti, M.; Psaro, R. Bimetallic Heterogeneous Catalysts for Hydrogen Production. Catal. Today 2012, 197, 190–205. [Google Scholar] [CrossRef]
- Usman, M.; Daud, W.W.; Abbas, H.F. Dry Reforming of Methane: Influence of Process Parameters—A Review. Renew. Sustain. Energy Rev. 2015, 45, 710–744. [Google Scholar] [CrossRef]
- Puxley, D.C.; Kitchener, I.J.; Komodromos, C.; Parkyns, N.D. The Effect Of Preparation Method Upon The Structures, Stability And Metal/Support Interactions In Nickel/Alumina Catalysts. In Studies in Surface Science and Catalysis; Poncelet, G., Grange, P., Jacobs, P.A., Eds.; Elsevier: Amsterdam, The Netherlands, 1983; Volume 16, pp. 237–271. [Google Scholar]
- Azancot, L.; Bobadilla, L.F.; Santos, J.L.; Córdoba, J.M.; Centeno, M.A.; Odriozola, J.A. Influence of the Preparation Method in the Metal-Support Interaction and Reducibility of Ni-Mg-Al Based Catalysts for Methane Steam Reforming. Int. J. Hydrogen Energy 2019, 44, 19827–19840. [Google Scholar] [CrossRef]
- Kwon, D.; Kang, J.Y.; An, S.; Yang, I.; Jung, J.C. Tuning the Base Properties of Mg–Al Hydrotalcite Catalysts Using Their Memory Effect. J. Energy Chem. 2020, 46, 229–236. [Google Scholar] [CrossRef]
- Bossola, F.; Evangelisti, C.; Allieta, M.; Psaro, R.; Recchia, S.; Dal Santo, V. Well-Formed, Size-Controlled Ruthenium Nanoparticles Active and Stable for Acetic Acid Steam Reforming. Appl. Catal. B Environ. 2016, 181, 599–611. [Google Scholar] [CrossRef]
- Adamu, S.; Bawah, A.-R.; Muraza, O.; Malaibari, Z.; Hossain, M.M. Effects of Metal Support Interaction on Dry Reforming of Methane over Ni/Ce-Al2O3 Catalysts. Can. J. Chem. Eng. 2020, 98, 2425–2434. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, K.; Pant, K.K.; Choudary, N.V. Tuning the Metal-Support Interaction of Methane Tri-Reforming Catalysts for Industrial Flue Gas Utilization. Int. J. Hydrogen Energy 2020, 45, 1911–1929. [Google Scholar] [CrossRef]
- Özdemir, H.; Öksüzömer, M.A.F.; Gürkaynak, M.A. Effect of the Calcination Temperature on Ni/MgAl2O4 Catalyst Structure and Catalytic Properties for Partial Oxidation of Methane. Fuel 2014, 116, 63–70. [Google Scholar] [CrossRef]
- Li, C.; Chen, Y.-W. Temperature-Programmed-Reduction Studies of Nickel Oxide/Alumina Catalysts: Effects of the Preparation Method. Thermochim. Acta 1995, 256, 457–465. [Google Scholar] [CrossRef]
- Rynkowski, J.M.; Paryjczak, T.; Lenik, M. On the Nature of Oxidic Nickel Phases in NiO/γ-Al2O3 Catalysts. Appl. Catal. A Gen. 1993, 106, 73–82. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, X.; Zhang, Z.; Zhang, L.; Dong, D.; Gao, G.; Westerhof, R.; Syed-Hassan, S.S.A. Steam Reforming of Acetic Acid over Ni/Al2O3 Catalyst: Correlation of Calcination Temperature with the Interaction of Nickel and Alumina. Fuel 2018, 227, 307–324. [Google Scholar] [CrossRef]
- Sahli, N.; Petit, C.; Roger, A.-C.; Kiennemann, A.; Libs, S.; Bettahar, M.M. Ni Catalysts from NiAl2O4 Spinel for CO2 Reforming of Methane. Catal. Today 2006, 113, 187–193. [Google Scholar] [CrossRef]
- Kuzmin, A.; Mironova, N. Composition Dependence of the Lattice Parameter in Solid Solutions. J. Phys. Matter 1998, 10, 7937–7944. [Google Scholar] [CrossRef]
- Parmaliana, A.; Arena, F.; Frusteri, F.; Giordano, N. Temperature-Programmed Reduction Study of NiO–MgO Interactions in Magnesia-Supported Ni Catalysts and NiO–MgO Physical Mixture. J. Chem. Soc. Faraday Trans. 1990, 86, 2663–2669. [Google Scholar] [CrossRef]
- Zanganeh, R.; Rezaei, M.; Zamaniyan, A. Dry Reforming of Methane to Synthesis Gas on NiO–MgO Nanocrystalline Solid Solution Catalysts. Int. J. Hydrogen Energy 2013, 38, 3012–3018. [Google Scholar] [CrossRef]
- Ruckenstein, E.; Hu, Y.H. Carbon Dioxide Reforming of Methane over Nickel/Alkaline Earth Metal Oxide Catalysts. Appl. Catal. A Gen. 1995, 133, 149–161. [Google Scholar] [CrossRef]
- Ou, Z.; Zhang, Z.; Qin, C.; Xia, H.; Deng, T.; Niu, J.; Ran, J.; Wu, C. Highly Active and Stable Ni/Perovskite Catalysts in Steam Methane Reforming for Hydrogen Production. Sustain. Energy Fuels 2021, 5, 1845–1856. [Google Scholar] [CrossRef]
- Hong Phuong, P.; Cam Anh, H.; Tri, N.; Phung Anh, N.; Cam Loc, L. Effect of Support on Stability and Coke Resistance of Ni-Based Catalyst in Combined Steam and CO2 Reforming of CH4. ACS Omega 2022, 7, 20092–20103. [Google Scholar] [CrossRef]
- Gac, W. Acid–Base Properties of Ni–MgO–Al2O3 Materials. Appl. Surf. Sci. 2011, 257, 2875–2880. [Google Scholar] [CrossRef]
- Coleman, L.J.I.; Epling, W.; Hudgins, R.R.; Croiset, E. Ni/Mg–Al Mixed Oxide Catalyst for the Steam Reforming of Ethanol. Appl. Catal. A Gen. 2009, 363, 52–63. [Google Scholar] [CrossRef]
- Damyanova, S.; Pawelec, B.; Arishtirova, K.; Fierro, J.L.G. Ni-Based Catalysts for Reforming of Methane with CO2. Int. J. Hydrogen Energy 2012, 37, 15966–15975. [Google Scholar] [CrossRef]
- Zhang, R.; Xia, G.; Li, M.; Wu, Y.; Nie, H.; Li, D. Effect of Support on the Performance of Ni-Based Catalyst in Methane Dry Reforming. J. Fuel Chem. Technol. 2015, 43, 1359–1365. [Google Scholar] [CrossRef]
- Mehr, J.Y.; Jozani, K.J.; Pour, A.N.; Zamani, Y. Influence of MgO in the CO2–Steam Reforming of Methane to Syngas by NiO/MgO/α-Al2O3 Catalyst. React. Kinet. Catal. Lett. 2002, 75, 267–273. [Google Scholar] [CrossRef]
- Das, S.; Sengupta, M.; Patel, J.; Bordoloi, A. A Study of the Synergy between Support Surface Properties and Catalyst Deactivation for CO2 Reforming over Supported Ni Nanoparticles. Appl. Catal. A Gen. 2017, 545, 113–126. [Google Scholar] [CrossRef]
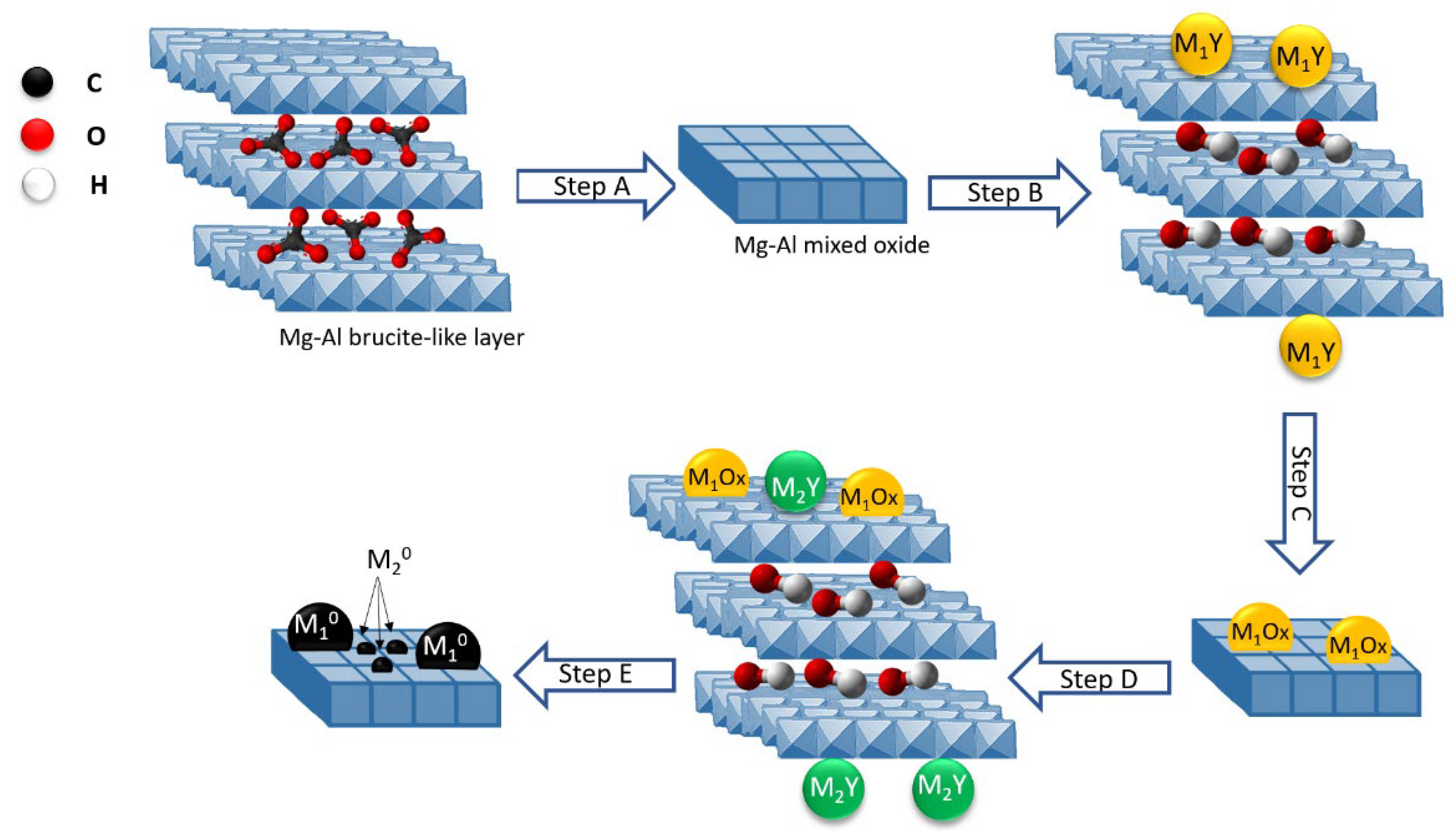
- Williams, G.R.; O’Hare, D. Towards Understanding, Control and Application of Layered Double Hydroxide Chemistry. J. Mater. Chem. 2006, 16, 3065–3074. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-Type Anionic Clays: Preparation, Properties and Applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Sikander, U.; Sufian, S.; Salam, M.A. A Review of Hydrotalcite Based Catalysts for Hydrogen Production Systems. Int. J. Hydrogen Energy 2017, 42, 19851–19868. [Google Scholar] [CrossRef]
- Dębek, R.; Motak, M.; Grzybek, T.; Galvez, M.E.; Da Costa, P. A Short Review on the Catalytic Activity of Hydrotalcite-Derived Materials for Dry Reforming of Methane. Catalysts 2017, 7, 32. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chang, V.W.; Schumacher, D.J. CO2 Reforming of Methane to Syngas: I: Evaluation of Hydrotalcite Clay-Derived Catalysts. Appl. Clay Sci. 1998, 13, 317–328. [Google Scholar] [CrossRef]
- Fornasari, G.; Gazzano, M.; Matteuzzi, D.; Trifirò, F.; Vaccari, A. Structure and Reactivity of High-Surface-Area Ni/Mg/Al Mixed Oxides. Appl. Clay Sci. 1995, 10, 69–82. [Google Scholar] [CrossRef]
- Shishido, T.; Sukenobu, M.; Morioka, H.; Kondo, M.; Wang, Y.; Takaki, K.; Takehira, K. Partial Oxidation of Methane over Ni/Mg-Al Oxide Catalysts Prepared by Solid Phase Crystallization Method from Mg-Al Hydrotalcite-like Precursors. Appl. Catal. A Gen. 2002, 223, 35–42. [Google Scholar] [CrossRef]
- Shishido, T.; Wang, P.; Kosaka, T.; Takehira, K. Steam Reforming of CH4 over Ni/Mg-Al Catalyst Prepared by Spc-Method from Hydrotalcite. Chem. Lett. 2002, 31, 752–753. [Google Scholar] [CrossRef]
- Di Cosimo, J.I.; Díez, V.K.; Xu, M.; Iglesia, E.; Apesteguía, C.R. Structure and Surface and Catalytic Properties of Mg-Al Basic Oxides. J. Catal. 1998, 178, 499–510. [Google Scholar] [CrossRef]
- Takehira, K.; Shishido, T.; Wang, P.; Kosaka, T.; Takaki, K. Steam Reforming of CH4 over Supported Ni Catalysts Prepared from a Mg–Al Hydrotalcite-like Anionic Clay. Phys. Chem. Chem. Phys. 2003, 5, 3801–3810. [Google Scholar] [CrossRef]
- Shishido, T.; Sukenobu, M.; Morioka, H.; Furukawa, R.; Shirahase, H.; Takehira, K. CO2 Reforming of CH4 over Ni/Mg–Al Oxide Catalysts Prepared by Solid Phase Crystallization Method from Mg–Al Hydrotalcite-like Precursors. Catal. Lett. 2001, 73, 21–26. [Google Scholar] [CrossRef]
- Mehrabadi, B.A.T.; Eskandari, S.; Khan, U.; White, R.D.; Regalbuto, J.R. Chapter One—A Review of Preparation Methods for Supported Metal Catalysts. In Advances in Catalysis; Song, C., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 61, pp. 1–35. [Google Scholar]
- Touahra, F.; Sehailia, M.; Ketir, W.; Bachari, K.; Chebout, R.; Trari, M.; Cherifi, O.; Halliche, D. Effect of the Ni/Al Ratio of Hydrotalcite-Type Catalysts on Their Performance in the Methane Dry Reforming Process. Appl. Petrochem. Res. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Li, L.; Zhang, L.; Zhang, Y.; Li, J. Effect of Ni Loadings on the Catalytic Properties of Ni/MgO(111) Catalyst for the Reforming of Methane with Carbon Dioxide. J. Fuel Chem. Technol. 2015, 43, 315–322. [Google Scholar] [CrossRef]
- Alipour, Z.; Rezaei, M.; Meshkani, F. Effect of Ni Loadings on the Activity and Coke Formation of MgO-Modified Ni/Al2O3 Nanocatalyst in Dry Reforming of Methane. J. Energy Chem. 2014, 23, 633–638. [Google Scholar] [CrossRef]
- Akbari, E.; Alavi, S.M.; Rezaei, M. Synthesis Gas Production over Highly Active and Stable Nanostructured Ni-MgO-Al2O3 Catalysts in Dry Reforming of Methane: Effects of Ni Contents. Fuel 2017, 194, 171–179. [Google Scholar] [CrossRef]
- Dębek, R.; Motak, M.; Duraczyska, D.; Launay, F.; Galvez, M.E.; Grzybek, T.; Da Costa, P. Methane Dry Reforming over Hydrotalcite-Derived Ni–Mg–Al Mixed Oxides: The Influence of Ni Content on Catalytic Activity, Selectivity and Stability. Catal. Sci. Technol. 2016, 6, 6705–6715. [Google Scholar] [CrossRef]
- Lin, X.; Li, R.; Lu, M.; Chen, C.; Li, D.; Zhan, Y.; Jiang, L. Carbon Dioxide Reforming of Methane over Ni Catalysts Prepared from Ni–Mg–Al Layered Double Hydroxides: Influence of Ni Loadings. Fuel 2015, 162, 271–280. [Google Scholar] [CrossRef]
- Qi, Y.; Cheng, Z.; Zhou, Z. Steam Reforming of Methane over Ni Catalysts Prepared from Hydrotalcite-Type Precursors: Catalytic Activity and Reaction Kinetics. Chin. J. Chem. Eng. 2015, 23, 76–85. [Google Scholar] [CrossRef]
- Bian, Z.; Zhong, W.; Yu, Y.; Wang, Z.; Jiang, B.; Kawi, S. Dry Reforming of Methane on Ni/Mesoporous-Al2O3 Catalysts: Effect of Calcination Temperature. Int. J. Hydrogen Energy 2021, 46, 31041–31053. [Google Scholar] [CrossRef]
- Al-Fatesh, A.S.A.; Fakeeha, A.H. Effects of Calcination and Activation Temperature on Dry Reforming Catalysts. J. Saudi Chem. Soc. 2012, 16, 55–61. [Google Scholar] [CrossRef]
- Juan-Juan, J.; Román-Martínez, M.C.; Illán-Gómez, M.J. Nickel Catalyst Activation in the Carbon Dioxide Reforming of Methane: Effect of Pretreatments. Appl. Catal. A Gen. 2009, 355, 27–32. [Google Scholar] [CrossRef]
- Zhou, L.; Li, L.; Wei, N.; Li, J.; Basset, J.-M. Effect of NiAl2O4 Formation on Ni/Al2O3 Stability during Dry Reforming of Methane. ChemCatChem 2015, 7, 2508–2516. [Google Scholar] [CrossRef]
- Feng, J.; Ding, Y.; Guo, Y.; Li, X.; Li, W. Calcination Temperature Effect on the Adsorption and Hydrogenated Dissociation of CO2 over the NiO/MgO Catalyst. Fuel 2013, 109, 110–115. [Google Scholar] [CrossRef]
- Ruckenstein, E.; Hang Hu, Y. The Effect of Precursor and Preparation Conditions of MgO on the CO2 Reforming of CH4 over NiO/MgO Catalysts. Appl. Catal. A Gen. 1997, 154, 185–205. [Google Scholar] [CrossRef]
- Mette, K.; Kühl, S.; Düdder, H.; Kähler, K.; Tarasov, A.; Muhler, M.; Behrens, M. Stable Performance of Ni Catalysts in the Dry Reforming of Methane at High Temperatures for the Efficient Conversion of CO2 into Syngas. ChemCatChem 2014, 6, 100–104. [Google Scholar] [CrossRef]
- Katheria, S.; Gupta, A.; Deo, G.; Kunzru, D. Effect of Calcination Temperature on Stability and Activity of Ni/MgAl2O4 Catalyst for Steam Reforming of Methane at High Pressure Condition. Int. J. Hydrogen Energy 2016, 41, 14123–14132. [Google Scholar] [CrossRef]
- Li, X.; Huang, Y.; Zhang, Q.; Luan, C.; Vinokurov, V.A.; Huang, W. Highly Stable and Anti-Coking Ni/MoCeZr/MgAl2O4-MgO Complex Support Catalysts for CO2 Reforming of CH4: Effect of the Calcination Temperature. Energy Convers. Manag. 2019, 179, 166–177. [Google Scholar] [CrossRef]
- Perez-Lopez, O.W.; Senger, A.; Marcilio, N.R.; Lansarin, M.A. Effect of Composition and Thermal Pretreatment on Properties of Ni–Mg–Al Catalysts for CO2 Reforming of Methane. Appl. Catal. A Gen. 2006, 303, 234–244. [Google Scholar] [CrossRef]
- Djaidja, A.; Libs, S.; Kiennemann, A.; Barama, A. Characterization and Activity in Dry Reforming of Methane on NiMg/Al and Ni/MgO Catalysts. Catal. Today 2006, 113, 194–200. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Dam, A.H.; Xiao, L.; Qi, Y.; Niu, J.; Yang, J.; Zhu, Y.-A.; Holmen, A.; Chen, D. Understanding Effects of Ni Particle Size on Steam Methane Reforming Activity by Combined Experimental and Theoretical Analysis. Catal. Today 2019, 355, 139–147. [Google Scholar] [CrossRef]
- Kim, J.-H.; Suh, D.J.; Park, T.-J.; Kim, K.-L. Effect of Metal Particle Size on Coking during CO2 Reforming of CH4 over Ni–Alumina Aerogel Catalysts. Appl. Catal. A Gen. 2000, 197, 191–200. [Google Scholar] [CrossRef]
- Vogt, C.; Kranenborg, J.; Monai, M.; Weckhuysen, B.M. Structure Sensitivity in Steam and Dry Methane Reforming over Nickel: Activity and Carbon Formation. ACS Catal. 2020, 10, 1428–1438. [Google Scholar] [CrossRef]
- Munnik, P.; de Jongh, P.E.; de Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef]
- Roh, H.-S.; Eum, I.-H.; Jeong, D.-W.; Yi, B.E.; Na, J.-G.; Ko, C.H. The Effect of Calcination Temperature on the Performance of Ni/MgO–Al2O3 Catalysts for Decarboxylation of Oleic Acid. Catal. Today 2011, 164, 457–460. [Google Scholar] [CrossRef]
- Smoláková, L.; Kout, M.; Koudelková, E.; Čapek, L. Effect of Calcination Temperature on the Structure and Catalytic Performance of the Ni/Al2O3 and Ni–Ce/Al2O3 Catalysts in Oxidative Dehydrogenation of Ethane. Ind. Eng. Chem. Res. 2015, 54, 12730–12740. [Google Scholar] [CrossRef]
- Khan, A.I.; O’Hare, D. Intercalation Chemistry of Layered Double Hydroxides: Recent Developments and Applications. J. Mater. Chem. 2002, 12, 3191–3198. [Google Scholar] [CrossRef]
- Millet, M.-M.; Tarasov, A.V.; Girgsdies, F.; Algara-Siller, G.; Schlögl, R.; Frei, E. Highly Dispersed Ni0/NixMg1–XO Catalysts Derived from Solid Solutions: How Metal and Support Control the CO2 Hydrogenation. ACS Catal. 2019, 9, 8534–8546. [Google Scholar] [CrossRef]
- Ward, D.A.; Ko, E.I. Preparing Catalytic Materials by the Sol-Gel Method. Ind. Eng. Chem. Res. 1995, 34, 421–433. [Google Scholar] [CrossRef]
- Frenzer, G.; Maier, W.F. Amorphous Porous Mixed Oxides: Sol-Gel Ways to a Highly Versatile Class of Materials and Catalysts. Annu. Rev. Mater. Res. 2006, 36, 281–331. [Google Scholar] [CrossRef]
- Othman, M.R.; Helwani, Z.; Martunus; Fernando, W.J.N. Synthetic Hydrotalcites from Different Routes and Their Application as Catalysts and Gas Adsorbents: A Review. Appl. Organomet. Chem. 2009, 23, 335–346. [Google Scholar] [CrossRef]
- Miller, J.B.; Ko, E.I. Control of Mixed Oxide Textural and Acidic Properties by the Sol-Gel Method. Catal. Today 1997, 35, 269–292. [Google Scholar] [CrossRef]
- Otero Areán, C.; Peñarroya Mentruit, M.; López López, A.J.; Parra, J.B. High Surface Area Nickel Aluminate Spinels Prepared by a Sol–Gel Method. Colloids Surf. A Physicochem. Eng. Asp. 2001, 180, 253–258. [Google Scholar] [CrossRef]
- Li, H.; Xu, H.; Wang, J. Methane Reforming with CO2 to Syngas over CeO2-Promoted Ni/Al2O3-ZrO2 Catalysts Prepared via a Direct Sol-Gel Process. J. Nat. Gas Chem. 2011, 20, 1–8. [Google Scholar] [CrossRef]
- González, A.R.; Asencios, Y.J.O.; Assaf, E.M.; Assaf, J.M. Dry Reforming of Methane on Ni–Mg–Al Nano-Spheroid Oxide Catalysts Prepared by the Sol–Gel Method from Hydrotalcite-like Precursors. Appl. Surf. Sci. 2013, 280, 876–887. [Google Scholar] [CrossRef]
- Salhi, N.; Boulahouache, A.; Petit, C.; Kiennemann, A.; Rabia, C. Steam Reforming of Methane to Syngas over NiAl2O4 Spinel Catalysts. Int. J. Hydrogen Energy 2011, 36, 11433–11439. [Google Scholar] [CrossRef]
- Min, J.-E.; Lee, Y.-J.; Park, H.-G.; Zhang, C.; Jun, K.-W. Carbon Dioxide Reforming of Methane on Ni–MgO–Al2O3 Catalysts Prepared by Sol–Gel Method: Effects of Mg/Al Ratios. J. Ind. Eng. Chem. 2015, 26, 375–383. [Google Scholar] [CrossRef]
- Sunde, T.O.L.; Grande, T.; Einarsrud, M.-A. Modified Pechini Synthesis of Oxide Powders and Thin Films. In Handbook of Sol-Gel Science and Technology; Klein, L., Aparicio, M., Jitianu, A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–30. [Google Scholar]
- Rivas, M.E.; Fierro, J.L.G.; Guil-López, R.; Peña, M.A.; La Parola, V.; Goldwasser, M.R. Preparation and Characterization of Nickel-Based Mixed-Oxides and Their Performance for Catalytic Methane Decomposition. Catal. Today 2008, 133–135, 367–373. [Google Scholar] [CrossRef]
- Rogers, J.L.; Mangarella, M.C.; D’Amico, A.D.; Gallagher, J.R.; Dutzer, M.R.; Stavitski, E.; Miller, J.T.; Sievers, C. Differences in the Nature of Active Sites for Methane Dry Reforming and Methane Steam Reforming over Nickel Aluminate Catalysts. ACS Catal. 2016, 6, 5873–5886. [Google Scholar] [CrossRef]
- Schimmoeller, B.; Pratsinis, S.E.; Baiker, A. Flame Aerosol Synthesis of Metal Oxide Catalysts with Unprecedented Structural and Catalytic Properties. ChemCatChem 2011, 3, 1234–1256. [Google Scholar] [CrossRef]
- Suh, D.J.; Park, T.-J.; Kim, J.-H.; Kim, K.-L. Nickel–Alumina Aerogel Catalysts Prepared by Fast Sol–Gel Synthesis. J. Non-Cryst. Solids 1998, 225, 168–172. [Google Scholar] [CrossRef]
- Sebai, I.; Boulahaouache, A.; Trari, M.; Salhi, N. Preparation and Characterization of 5%Ni/γ-Al2O3 Catalysts by Complexation with NH3 Derivatives Active in Methane Steam Reforming. Int. J. Hydrogen Energy 2019, 44, 9949–9958. [Google Scholar] [CrossRef]
- George, S.M. Atomic Layer Deposition: An Overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef]
- Shang, Z.; Li, S.; Li, L.; Liu, G.; Liang, X. Highly Active and Stable Alumina Supported Nickel Nanoparticle Catalysts for Dry Reforming of Methane. Appl. Catal. B Environ. 2017, 201, 302–309. [Google Scholar] [CrossRef]
- Wang, G.; Luo, F.; Cao, K.; Zhang, Y.; Li, J.; Zhao, F.; Chen, R.; Hong, J. Effect of Ni Content of Ni/γ-Al2O3 Catalysts Prepared by the Atomic Layer Deposition Method on CO2 Reforming of Methane. Energy Technol. 2019, 7, 1800359. [Google Scholar] [CrossRef]
- Jeong, M.-G.; Kim, S.Y.; Kim, D.H.; Han, S.W.; Kim, I.H.; Lee, M.; Hwang, Y.K.; Kim, Y.D. High-Performing and Durable MgO/Ni Catalysts via Atomic Layer Deposition for CO2 Reforming of Methane (CRM). Appl. Catal. A Gen. 2016, 515, 45–50. [Google Scholar] [CrossRef]
- Gould, T.D.; Izar, A.; Weimer, A.W.; Falconer, J.L.; Medlin, J.W. Stabilizing Ni Catalysts by Molecular Layer Deposition for Harsh, Dry Reforming Conditions. ACS Catal. 2014, 4, 2714–2717. [Google Scholar] [CrossRef]
- Littlewood, P.; Liu, S.; Weitz, E.; Marks, T.J.; Stair, P.C. Ni-Alumina Dry Reforming Catalysts: Atomic Layer Deposition and the Issue of Ni Aluminate. Catal. Today 2020, 343, 18–25. [Google Scholar] [CrossRef]
- Kim, H.; Eissa, A.A.-S.; Kim, S.B.; Lee, H.; Kim, W.; Seo, D.J.; Lee, K.; Yoon, W.L. One-Pot Synthesis of a Highly Mesoporous Ni/MgAl2O4 Spinel Catalyst for Efficient Steam Methane Reforming: Influence of Inert Annealing. Catal. Sci. Technol. 2021, 11, 4447–4458. [Google Scholar] [CrossRef]
- Guo, Y.; Li, Y.; Ning, Y.; Liu, Q.; Tian, L.; Zhang, R.; Fu, Q.; Wang, Z. CO2 Reforming of Methane over a Highly Dispersed Ni/Mg–Al–O Catalyst Prepared by a Facile and Green Method. Ind. Eng. Chem. Res. 2020, 59, 15506–15514. [Google Scholar] [CrossRef]
- Huang, J.; Yan, Y.; Saqline, S.; Liu, W.; Liu, B. High Performance Ni Catalysts Prepared by Freeze Drying for Efficient Dry Reforming of Methane. Appl. Catal. B Environ. 2020, 275, 119109. [Google Scholar] [CrossRef]
- Kobayashi, N.; Takahashi, S. Catalyst for Decomposition of Hydrocarbons, Process for Producing the Catalyst, and Process for Producing Hydrogen Using the Catalyst. U.S. Patent 7196036B2, 27 March 2007. [Google Scholar]
- Kwak, G.; Park, H.; Wi, S.; Park, S. High Efficiency Ni-based Catalyst for Steam Methane Reforming and Steam Methane Reforming Reaction using the Same. KR Patent 20220052099A, 27 April 2022. [Google Scholar]
- Hou, Z.; Yashima, T. Small Amounts of Rh-Promoted Ni Catalysts for Methane Reforming with CO2. Catal. Lett. 2003, 89, 193–197. [Google Scholar] [CrossRef]
- Fei, J.; Hou, Z.; Zheng, X.; Yashima, T. Doped Ni Catalysts for Methane Reforming with CO2. Catal. Lett. 2004, 98, 241–246. [Google Scholar] [CrossRef]
- Crisafulli, C.; Scirè, S.; Minicò, S.; Solarino, L. Ni–Ru Bimetallic Catalysts for the CO2 Reforming of Methane. Appl. Catal. A Gen. 2002, 225, 1–9. [Google Scholar] [CrossRef]
- Tomishige, K.; Kanazawa, S.; Sato, M.; Ikushima, K.; Kunimori, K. Catalyst Design of Pt-Modified Ni/Al2O3 Catalyst with Flat Temperature Profile in Methane Reforming with CO2 and O2. Catal. Lett. 2002, 84, 69–74. [Google Scholar] [CrossRef]
- Dias, J.A.C.; Assaf, J.M. Autothermal Reforming of Methane over Ni/γ-Al2O3 Catalysts: The Enhancement Effect of Small Quantities of Noble Metals. J. Power Sources 2004, 130, 106–110. [Google Scholar] [CrossRef]
- Dias, J.A.C.; Assaf, J.M. Autoreduction of Promoted Ni/γ-Al2O3 during Autothermal Reforming of Methane. J. Power Sources 2005, 139, 176–181. [Google Scholar] [CrossRef]
- Daza, C.E.; Gallego, J.; Mondragón, F.; Moreno, S.; Molina, R. High Stability of Ce-Promoted Ni/Mg–Al Catalysts Derived from Hydrotalcites in Dry Reforming of Methane. Fuel 2010, 89, 592–603. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Sutthisripok, W.; Assabumrungrat, S. Synthesis Gas Production from Dry Reforming of Methane over CeO2 Doped Ni/Al2O3: Influence of the Doping Ceria on the Resistance toward Carbon Formation. Chem. Eng. J. 2005, 112, 13–22. [Google Scholar] [CrossRef]
- Abd Ghani, N.A.; Azapour, A.; Syed Muhammad, A.F.; Abdullah, B. Dry Reforming of Methane for Hydrogen Production over NiCo Catalysts: Effect of NbZr Promoters. Int. J. Hydrogen Energy 2019, 44, 20881–20888. [Google Scholar] [CrossRef]
- Navarro, M.V.; Plou, J.; López, J.M.; Grasa, G.; Murillo, R. Effect of Oxidation-Reduction Cycles on Steam-Methane Reforming Kinetics over a Nickel-Based Catalyst. Int. J. Hydrogen Energy 2019, 44, 12617–12627. [Google Scholar] [CrossRef]
- Álvarez, M.A.; Bobadilla, L.F.; Garcilaso, V.; Centeno, M.A.; Odriozola, J.A. CO2 Reforming of Methane over Ni-Ru Supported Catalysts: On the Nature of Active Sites by Operando DRIFTS Study. J. CO2 Util. 2018, 24, 509–515. [Google Scholar] [CrossRef]
- Foletto, E.L.; Alves, R.W.; Jahn, S.L. Preparation of Ni/Pt Catalysts Supported on Spinel (MgAl2O4) for Methane Reforming. J. Power Sources 2006, 161, 531–534. [Google Scholar] [CrossRef]
- Jaiswar, V.K.; Katheria, S.; Deo, G.; Kunzru, D. Effect of Pt Doping on Activity and Stability of Ni/MgAl2O4 Catalyst for Steam Reforming of Methane at Ambient and High Pressure Condition. Int. J. Hydrogen Energy 2017, 42, 18968–18976. [Google Scholar] [CrossRef]
- Yoshida, K.; Begum, N.; Ito, S.; Tomishige, K. Oxidative Steam Reforming of Methane over Ni/α-Al2O3 Modified with Trace Noble Metals. Appl. Catal. A Gen. 2009, 358, 186–192. [Google Scholar] [CrossRef]
- Li, B.; Kado, S.; Mukainakano, Y.; Miyazawa, T.; Miyao, T.; Naito, S.; Okumura, K.; Kunimori, K.; Tomishige, K. Surface Modification of Ni Catalysts with Trace Pt for Oxidative Steam Reforming of Methane. J. Catal. 2007, 245, 144–155. [Google Scholar] [CrossRef]
- Arena, F.; Horrell, B.A.; Cocke, D.L.; Parmaliana, A.; Giordano, N. Magnesia-Supported Nickel Catalysts I. Factors Affecting the Structure and Morphological Properties. J. Catal. 1991, 132, 58–67. [Google Scholar] [CrossRef]
- Nurunnabi, M.; Mukainakano, Y.; Kado, S.; Miyazawa, T.; Okumura, K.; Miyao, T.; Naito, S.; Suzuki, K.; Fujimoto, K.-I.; Kunimori, K.; et al. Oxidative Steam Reforming of Methane under Atmospheric and Pressurized Conditions over Pd/NiO–MgO Solid Solution Catalysts. Appl. Catal. A Gen. 2006, 308, 1–12. [Google Scholar] [CrossRef]
- Katheria, S.; Deo, G.; Kunzru, D. Rh-Ni/MgAl2O4 Catalyst for Steam Reforming of Methane: Effect of Rh Doping, Calcination Temperature and Its Application on Metal Monoliths. Appl. Catal. A Gen. 2019, 570, 308–318. [Google Scholar] [CrossRef]
- Prins, R. Hydrogen Spillover. Facts and Fiction. Chem. Rev. 2012, 112, 2714–2738. [Google Scholar] [CrossRef] [PubMed]
- Takehira, K. “Intelligent” Reforming Catalysts: Trace Noble Metal-Doped Ni/Mg(Al)O Derived from Hydrotalcites. J. Nat. Gas Chem. 2009, 18, 237–259. [Google Scholar] [CrossRef]
- Li, D.; Zhan, Y.; Nishida, K.; Oumi, Y.; Sano, T.; Shishido, T.; Takehira, K. “Green” Preparation of “Intelligent” Pt-Doped Ni/Mg(Al)O Catalysts for Daily Start-up and Shut-down CH4 Steam Reforming. Appl. Catal. A Gen. 2009, 363, 169–179. [Google Scholar] [CrossRef]
- Miyata, T.; Li, D.; Shiraga, M.; Shishido, T.; Oumi, Y.; Sano, T.; Takehira, K. Promoting Effect of Rh, Pd and Pt Noble Metals to the Ni/Mg(Al)O Catalysts for the DSS-like Operation in CH4 Steam Reforming. Appl. Catal. A Gen. 2006, 310, 97–104. [Google Scholar] [CrossRef]
- Katheria, S.; Kunzru, D.; Deo, G. Kinetics of Steam Reforming of Methane on Rh–Ni/MgAl2O4 Catalyst. React. Kinet. Mech. Catal. 2020, 130, 91–101. [Google Scholar] [CrossRef]
- Jeong, J.H.; Lee, J.W.; Seo, D.J.; Seo, Y.; Yoon, W.L.; Lee, D.K.; Kim, D.H. Ru-Doped Ni Catalysts Effective for the Steam Reforming of Methane without the Pre-Reduction Treatment with H2. Appl. Catal. A Gen. 2006, 302, 151–156. [Google Scholar] [CrossRef]
- Baek, S.-C.; Jun, K.-W.; Lee, Y.-J.; Kim, J.D.; Park, D.Y.; Lee, K.-Y. Ru/Ni/MgAl2O4 Catalysts for Steam Reforming of Methane: Effects of Ru Content on Self-Activation Property. Res. Chem. Intermed. 2012, 38, 1225–1236. [Google Scholar] [CrossRef]
- Nawfal, M.; Gennequin, C.; Labaki, M.; Nsouli, B.; Aboukaïs, A.; Abi-Aad, E. Hydrogen Production by Methane Steam Reforming over Ru Supported on Ni–Mg–Al Mixed Oxides Prepared via Hydrotalcite Route. Int. J. Hydrogen Energy 2015, 40, 1269–1277. [Google Scholar] [CrossRef]
- Kim, N.Y.; Yang, E.-H.; Lim, S.-S.; Jung, J.S.; Lee, J.-S.; Hong, G.H.; Noh, Y.-S.; Lee, K.Y.; Moon, D.J. Hydrogen Production by Steam Reforming of Methane over Mixed Ni/MgAl + CrFe3O4 Catalysts. Int. J. Hydrogen Energy 2015, 40, 11848–11854. [Google Scholar] [CrossRef]
- Kim, D.H.; Youn, J.-R.; Seo, J.-C.; Kim, S.B.; Kim, M.-J.; Lee, K. One-Pot Synthesis of NiCo/MgAl2O4 Catalyst for High Coke-Resistance in Steam Methane Reforming: Optimization of Ni/Co Ratio. Catal. Today 2022, in press. [Google Scholar] [CrossRef]
- Bossola, F.; Roongcharoen, T.; Coduri, M.; Evangelisti, C.; Somodi, F.; Sementa, L.; Fortunelli, A.; Dal Santo, V. Discovering Indium as Hydrogen Production Booster for a Cu/SiO2 Catalyst in Steam Reforming of Methanol. Appl. Catal. B Environ. 2021, 297, 120398. [Google Scholar] [CrossRef]
- Etminan, A.; Sadrnezhaad, S.K. A Two Step Microwave-Assisted Coke Resistant Mesoporous Ni-Co Catalyst for Methane Steam Reforming. Fuel 2022, 317, 122411. [Google Scholar] [CrossRef]
- Moon, D.J.; Lee, Y.J.; Jung, J.S.; Lee, J.H.; Lee, S.H.; Kim, B.H.; Kim, H.J.; Yang, E.H. Alkaline Earth Metal Co-Precipitated Nickel-Based Catalyst for Steam Carbon Dioxide Reforming of Natural Gas. US Patent 20140339475A, 3 March 2015. [Google Scholar]
- Du, J.; Liu, P.; Gao, Z.; Zhang, W.; Zhang, L.; Li, X. Magnesium Aluminate Spinel Type Composite Oxide Carrier, Preparation Method Thereof and Steam Reforming Catalyst. CN Patent 112844388A, 29 November 2022. [Google Scholar]
- Chin, Y.-H.; King, D.L.; Roh, H.-S.; Wang, Y.; Heald, S.M. Structure and Reactivity Investigations on Supported Bimetallic AuNi Catalysts Used for Hydrocarbon Steam Reforming. J. Catal. 2006, 244, 153–162. [Google Scholar] [CrossRef]
- Binary Alloy Phase Diagrams, 2nd ed; Massalski, T.B.; Okamoto, H.; Subramanian, P.R.; Kacprzak, L. (Eds.) ASM International: Materials Park, OH, USA, 1990. [Google Scholar]
- Bengaard, H.S.; Nørskov, J.K.; Sehested, J.; Clausen, B.S.; Nielsen, L.P.; Molenbroek, A.M.; Rostrup-Nielsen, J.R. Steam Reforming and Graphite Formation on Ni Catalysts. J. Catal. 2002, 209, 365–384. [Google Scholar] [CrossRef]
- Zhan, Y.; Li, D.; Nishida, K.; Shishido, T.; Oumi, Y.; Sano, T.; Takehira, K. Preparation of “Intelligent” Pt/Ni/Mg(Al)O Catalysts Starting from Commercial Mg–Al LDHs for Daily Start-up and Shut-down Steam Reforming of Methane. Appl. Clay Sci. 2009, 45, 147–154. [Google Scholar] [CrossRef]
- Ko, J.; Kim, B.-K.; Han, J.W. Density Functional Theory Study for Catalytic Activation and Dissociation of CO2 on Bimetallic Alloy Surfaces. J. Phys. Chem. C 2016, 120, 3438–3447. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, H.; Dalai, A.K. Effects of Metal Content on Activity and Stability of Ni-Co Bimetallic Catalysts for CO2 Reforming of CH4. Appl. Catal. A Gen. 2008, 339, 121–129. [Google Scholar] [CrossRef]
- Niu, J.; Wang, Y.; Liland, E.S.; Regli, K.S.; Yang, J.; Rout, K.R.; Luo, J.; Rønning, M.; Ran, J.; Chen, D. Unraveling Enhanced Activity, Selectivity, and Coke Resistance of Pt–Ni Bimetallic Clusters in Dry Reforming. ACS Catal. 2021, 11, 2398–2411. [Google Scholar] [CrossRef]
- Álvarez Moreno, A.; Ramirez-Reina, T.; Ivanova, S.; Roger, A.-C.; Centeno, M.Á.; Odriozola, J.A. Bimetallic Ni–Ru and Ni–Re Catalysts for Dry Reforming of Methane: Understanding the Synergies of the Selected Promoters. Front. Chem. 2021, 9, 694976. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Das, S.; Wai, M.H.; Hongmanorom, P.; Kawi, S. A Review on Bimetallic Nickel-Based Catalysts for CO2 Reforming of Methane. ChemPhysChem 2017, 18, 3117–3134. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Aparicio, P.; Fernandez-Garcia, M.; Guerrero-Ruiz, A.; Rodriguez-Ramos, I. Evaluation of the Role of the Metal–Support Interfacial Centers in the Dry Reforming of Methane on Alumina-Supported Rhodium Catalysts. J. Catal. 2000, 190, 296–308. [Google Scholar] [CrossRef]
- Nematollahi, B.; Rezaei, M.; Khajenoori, M. Combined Dry Reforming and Partial Oxidation of Methane to Synthesis Gas on Noble Metal Catalysts. Int. J. Hydrogen Energy 2011, 36, 2969–2978. [Google Scholar] [CrossRef]
- Wang, H.Y.; Au, C.T. Carbon Dioxide Reforming of Methane to Syngas over SiO2-Supported Rhodium Catalysts. Appl. Catal. A Gen. 1997, 155, 239–252. [Google Scholar] [CrossRef]
- Wu, H.; Pantaleo, G.; La Parola, V.; Venezia, A.M.; Collard, X.; Aprile, C.; Liotta, L.F. Bi- and Trimetallic Ni Catalysts over Al2O3 and Al2O3-MOx (M=Ce or Mg) Oxides for Methane Dry Reforming: Au and Pt Additive Effects. Appl. Catal. B Environ. 2014, 156–157, 350–361. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, F.; Wang, N.; Hao, S.; Chu, W. Plasma-Treated Bimetallic Ni–Pt Catalysts Derived from Hydrotalcites for the Carbon Dioxide Reforming of Methane. Catal. Lett. 2014, 144, 293–300. [Google Scholar] [CrossRef]
- Osaki, T.; Mori, T. Role of Potassium in Carbon-Free CO2 Reforming of Methane on K-Promoted Ni/Al2O3 Catalysts. J. Catal. 2001, 204, 89–97. [Google Scholar] [CrossRef]
- García-Diéguez, M.; Pieta, I.S.; Herrera, M.C.; Larrubia, M.A.; Alemany, L.J. Nanostructured Pt- and Ni-Based Catalysts for CO2-Reforming of Methane. J. Catal. 2010, 270, 136–145. [Google Scholar] [CrossRef]
- Li, L.; Anjum, D.H.; Zhu, H.; Saih, Y.; Laveille, P.V.; D’Souza, L.; Basset, J.-M. Synergetic Effects Leading to Coke-Resistant NiCo Bimetallic Catalysts for Dry Reforming of Methane. ChemCatChem 2015, 7, 427–433. [Google Scholar] [CrossRef]
- Hu, Y.H. Solid-Solution Catalysts for CO2 Reforming of Methane. Catal. Today 2009, 148, 206–211. [Google Scholar] [CrossRef]
- Duan, X.; Pan, J.; Yang, X.; Wan, C.; Lin, X.; Li, D.; Jiang, L. Nickel-cobalt Bimetallic Catalysts Prepared from Hydrotalcite-like Compounds for Dry Reforming of Methane. Int. J. Hydrogen Energy 2022, 47, 24358–24373. [Google Scholar] [CrossRef]
- Torimoto, M.; Sekine, Y. Effects of Alloying for Steam or Dry Reforming of Methane: A Review of Recent Studies. Catal. Sci. Technol. 2022, 12, 3387–3411. [Google Scholar] [CrossRef]
- Tsyganok, A.I.; Inaba, M.; Tsunoda, T.; Uchida, K.; Suzuki, K.; Takehira, K.; Hayakawa, T. Rational Design of Mg–Al Mixed Oxide-Supported Bimetallic Catalysts for Dry Reforming of Methane. Appl. Catal. A Gen. 2005, 292, 328–343. [Google Scholar] [CrossRef]
- Miyata, S. Physico-Chemical Properties of Synthetic Hydrotalcites in Relation to Composition. Clays Clay Miner. 1980, 28, 50–56. [Google Scholar] [CrossRef]
- Tsyganok, A.I.; Inaba, M.; Tsunoda, T.; Suzuki, K.; Takehira, K.; Hayakawa, T. Combined Partial Oxidation and Dry Reforming of Methane to Synthesis Gas over Noble Metals Supported on Mg–Al Mixed Oxide. Appl. Catal. A Gen. 2004, 275, 149–155. [Google Scholar] [CrossRef]
- Wysocka, I.; Hupka, J.; Rogala, A. Catalytic Activity of Nickel and Ruthenium–Nickel Catalysts Supported on SiO2, ZrO2, Al2O3, and MgAl2O4 in a Dry Reforming Process. Catalysts 2019, 9, 540. [Google Scholar] [CrossRef]
- Lucrédio, A.F.; Assaf, J.M.; Assaf, E.M. Methane Conversion Reactions on Ni Catalysts Promoted with Rh: Influence of Support. Appl. Catal. A Gen. 2011, 400, 156–165. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Galvita, V.V.; Poelman, H.; Marin, G.B. Enhanced Carbon-Resistant Dry Reforming Fe-Ni Catalyst: Role of Fe. ACS Catal. 2015, 5, 3028–3039. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Batchu, R.; Galvita, V.V.; Poelman, H.; Marin, G.B. Carbon Gasification from Fe–Ni Catalysts after Methane Dry Reforming. Appl. Catal. B Environ. 2016, 185, 42–55. [Google Scholar] [CrossRef]
- Wan, C.; Song, K.; Pan, J.; Huang, M.; Luo, R.; Li, D.; Jiang, L. Ni–Fe/Mg(Al)O Alloy Catalyst for Carbon Dioxide Reforming of Methane: Influence of Reduction Temperature and Ni–Fe Alloying on Coking. Int. J. Hydrogen Energy 2020, 45, 33574–33585. [Google Scholar] [CrossRef]
- Wang, L.; Li, D.; Koike, M.; Koso, S.; Nakagawa, Y.; Xu, Y.; Tomishige, K. Catalytic Performance and Characterization of Ni-Fe Catalysts for the Steam Reforming of Tar from Biomass Pyrolysis to Synthesis Gas. Appl. Catal. A Gen. 2011, 392, 248–255. [Google Scholar] [CrossRef]
- Ray, K.; Sengupta, S.; Deo, G. Reforming and Cracking of CH4 over Al2O3 Supported Ni, Ni-Fe and Ni-Co Catalysts. Fuel Process. Technol. 2017, 156, 195–203. [Google Scholar] [CrossRef]
- Boudjeloud, M.; Boulahouache, A.; Rabia, C.; Salhi, N. La-Doped Supported Ni Catalysts for Steam Reforming of Methane. Int. J. Hydrogen Energy 2019, 44, 9906–9913. [Google Scholar] [CrossRef]
- Ohi, T.; Miyata, T.; Li, D.; Shishido, T.; Kawabata, T.; Sano, T.; Takehira, K. Sustainability of Ni Loaded Mg–Al Mixed Oxide Catalyst in Daily Startup and Shutdown Operations of CH4 Steam Reforming. Appl. Catal. A Gen. 2006, 308, 194–203. [Google Scholar] [CrossRef]
- Benito, P.; Dal Santo, V.; De Grandi, V.; Marelli, M.; Fornasari, G.; Psaro, R.; Vaccari, A. Coprecipitation versus Chemical Vapour Deposition to Prepare Rh/Ni Bimetallic Catalysts. Appl. Catal. B Environ. 2015, 179, 150–159. [Google Scholar] [CrossRef]
- Huang, Y.; Du, J.; Ling, C.; Zhou, T.; Wang, S. Methane Dehydrogenation on Au/Ni Surface Alloys—A First-Principles Study. Catal. Sci. Technol. 2013, 3, 1343–1354. [Google Scholar] [CrossRef]
- Niu, J.; Wang, Y.; Qi, Y.; Dam, A.H.; Wang, H.; Zhu, Y.-A.; Holmen, A.; Ran, J.; Chen, D. New Mechanism Insights into Methane Steam Reforming on Pt/Ni from DFT and Experimental Kinetic Study. Fuel 2020, 266, 117143. [Google Scholar] [CrossRef]
- De Coster, V.; Srinath, N.V.; Theofanidis, S.A.; Pirro, L.; Van Alboom, A.; Poelman, H.; Sabbe, M.K.; Marin, G.B.; Galvita, V.V. Looking inside a Ni-Fe/MgAl2O4 Catalyst for Methane Dry Reforming via Mössbauer Spectroscopy and in Situ QXAS. Appl. Catal. B Environ. 2022, 300, 120720. [Google Scholar] [CrossRef]
- Meloni, E.; Martino, M.; Palma, V. A Short Review on Ni Based Catalysts and Related Engineering Issues for Methane Steam Reforming. Catalysts 2020, 10, 352. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon Type | Effect on Catalysts | Effect on Reactor | Favored Conditions | Preventive Measures |
|---|---|---|---|---|
| Whisker carbon | Catalyst pellet break-up | Increase on pressure build-up | Feed of olefins and aromatics, low H2O/C ratio, high operating temperature | Increase H2O/C ratio |
| Gum | Deactivation by blocking active site | Feed of aromatics, low H2O/C ratio, absence of H2, low operating temperature | Increase H2O/C ratio Increase operating temperature H2 co-feed | |
| Pyrolytic coke | Deactivation by blocking active site | Coke accumulation in reactor wall, increase on pressure build up | Feed of olefins, long residence time, high operating temperature, feed containing sulfur | Reduce residence time Remove sulfur from feed |
| Catalyst (Ni Loading wt.%) | Synthesis Method | Catalytic Process | Parameter Investigated (1) | Reaction Conditions | Main Findings (3) | Ref. |
|---|---|---|---|---|---|---|
| Ni/Al2O3 (46.6–73.5 wt.%) | Co-precipitation | DRM | Ni/Al ratio | GHSV = 12 L g−1 h−1 CO2/CH4 = 1 T = 400–700 °C (2) Max CH4 conv. = 90% (700 °C) | High Ni/Al ratio resulted in high reducibility and large particles Molar ratio Ni/Al = 2 showed the best result Higher calcination temperature resulted in higher activity | [82] |
| Ni/MgO (2–20 wt.%) | Impregnation on MgO nanosheets | DRM | Ni loading; (2–20 wt.%) | GHSV = 36 L g−1 h−1 CO2/CH4 = 1 T = 450–650 °C Max CH4 conv. = 63% (650 °C) | High Ni loading resulted in larger particles Activity and stability increased with Ni loading from 2 to 10 wt.% Activity and stability decreased with Ni loading from 10 to 20 wt.% | [83] |
| Ni/3 wt.%MgO-Al2O3 (5–20 wt.%) | Ni, Mg impregnation on sol-gel Al2O3 | DRM | Ni loading (5–20 wt.%) | GHSV = 12 L g−1 h−1 CO2/CH4 = 1 T = 550–700 °C Max CH4 conv. = 76% (700 °C) | High Ni loading led to segregation of NiO phase Coke formation was higher for higher Ni loadings | [84] |
| Ni/Mg(Al)O (2.5–15 wt.%–) | Co-precipitation | DRM | Ni loading (2.5–15 wt.%) | GHSV = 18 Lg−1 h−1 CO2/CH4 = 1 T = 550–700 °C Max CH4 conv. = 74% (700 °C) | High Ni loading promoted NiO phase formation, improving reducibility Activity and coke formation proportionated to Ni content | [85] |
| Ni/Mg(Al)O (1–15 wt.%) | Impregnation on co-precipitated MgAl2O4 | DRM | Ni loading (1–15 wt.%) | GHSV = 90 L g−1 h−1 CO2/CH4 = 1 T = 750 °C Max CH4 conv. = 93% | High Ni loading improved reducibility, activity, and coke generation. Too low Ni loading (i.e., 1 wt.%) showed deactivation overtime | [43] |
| Ni/Mg(Al)O (4–60 wt.%) | Co-precipitation (LDH) | DRM | Ni loading (4–60 wt.%) | GHSV = 20,000 h−1 CO2/CH4 = 1 T = 550–750 °C Max CH4 conv. = 90% (750 °C) | High Ni loading increased particles size, reducibility, and basicity No obvious sintering is presented after reaction High Ni loading promotes carbon formation. | [86] |
| Ni/Mg(Al)O (3–18 wt.%) | Co-precipitation | DRM | Ni loading (3–18 wt.%) | GHSV = 60 L g−1 h−1 CO2/CH4 = 1 T = 500–800 °C Max CH4 conv. = 95% (800 °C) | Active site dispersion is optimal at Ni loading of 15 wt.% High CH4 conversion and CO disproportionation using high Ni loadings At high Treaction, high Ni loading catalyst was more stable At low Treaction, high Ni loading catalyst was less stable | [87] |
| Ni/Mg(Al)O (15 wt.%) | Co-precipitation (LDH) | SMR | Molar Ni/Mg-Al (2, 3, 4) | GHSV = 900 L g−1 h−1 H2O/CH4 = 4 T = 650 °C Max CH4 conv. = 42% | Highest surface area, Ni dispersion was obtained with molar Ni/Mg-Al = 3 Molar Ni/Mg-Al = 3 showed highest activity in SMR | [88] |
| Ni/Mg(Al)O (6.3–12.6 wt.%) | Co-precipitation (spc) or impregnation | SMR | Synthesis method, molar Ni/Mg ratio | GHSV = 120 L g−1 h−1 H2O/CH4 = 2 T = 800 °C Max CH4 conv. = 95% | Highest dispersion, best activity, and stability were observed with co-precipitated catalyst with a Ni/Mg molar ratio = 0.5/2.5 | [77] |
| Ni/Mg(Al)O (16.3 wt.%) | Co-precipitation (spc) | SMR | Molar ratio Mg/Al | GHSV = 180–900 L g−1 h−1 H2O/CH4 = 2 T = 800 °C Max CH4 conv. = 98% | Low molar Mg/Al ratio resulted in Al(OH)3 and amorphous solid NiO-MgO and stable MgAl2O4 Co-precipitated catalyst particles size remained after reaction Co-precipitated Ni0.5/Mg2.5-Al showed high activity, stable for 600 h | [79] |
| Ni/Al2O3 (5 wt.%) | Impregnation in mesoporous Al2O3 | DRM | Tcal (500–800 °C) | GHSV = 120 L g−1 h−1 CO2/CH4 = 1 T = 650–700 °C Max CH4 conv. = 78% (700 °C) | Calcination at 700 °C promoted spinel and limitedNiO-Al2O3 phase formation Too high calcination temperature (800 °C) reduced catalyst activity | [89] |
| Ni/Al2O3 (3 wt.%) | Impregnation | DRM | Tcal (500–900 °C)Tred (500–800 °C) | GHSV = 2.6 L g−1 h−1 CO2/CH4 = 1 T = 500–800 °C Max CH4 conv. = 85% (800 °C) | High Tcal reduced surface area but promoted basicity No difference in activity was observed with Tcal Reduction at lower than 700 °C showed no or little activity | [90] |
| Ni/Al2O3 (10 wt.%) | Impregnation | DRM | Tcal, Tred | GHSV = 20 L g−1 h−1 CO2/CH4 = 1 T = 500, 700 °C Max CH4 conv. = 87% (700 °C) | High Tcal promoted formation of spinel phase Calcination pretreatment did not affect activity and coke deposition Reduced catalyst minimized the coke deposition | [91] |
| Ni/Al2O3 (16 wt.%) | Impregnation | DRM | Tcal (350–900 °C) | GHSV = 480 L g−1 h−1 CO2/CH4 = 1 T = 500–800 °C Max CH4 conv. = NR | Catalyst calcined at 900 °C forms NiAl2O4. The catalyst was stable for 100 h TOS Strong Ni-Al interaction prevented particles being pushed by coke formed, reducing catalyst breakage. | [92] |
| Ni/Mg (20 wt.%) | Impregnation | DRM | Tcal (600–800 °C) | GHSV = 20 L g−1 h−1 CO2/CH4 = 1 T = 800 °C Max CH4 conv. = 93% | High Tcal reduced interaction of Ni-MgO and reducibility No weakly interacting NiO when Tcalc = 800 °C More CO2 adsorption site with increased Tcalc Catalyst calcined at higher temperature is more active | [93] |
| Ni/MgO (13.1 wt.%) | Impregnation | DRM | Mg precursor Tcal of support | GHSV = 60 L g−1 h−1 CO2/CH4 = 1 T = 790 °C Max CH4 conv. = 75% | [MgCO3]4Mg(OH)2 precursor formed stable MgO at high Tcal Ni impregnated on [MgCO3]4Mg(OH)2 showed highest CO yield Induction time to reach stable conversion depended on precursor and Tcal of support | [94] |
| Ni/Mg(Al)O (55 wt.%) | Co-precipitation | DRM | Tcal, Tred | GHSV = 1000 L g−1 h−1 CO2/CH4 = 1.25 T = 800–900 °C Max CH4 conv. = 74% (900 °C) | Low Tcalc resulted in rock-salt type with amorphous phase High Tcalc resulted in spinel type with high crystallinity Reduction at 800 °C and 900 °C resulted in small nanoparticle (9 nm) Reduction at 1000 °C promoted sintering to large particles (19 nm) High reaction temperature resulted in lower coke formation | [95] |
| Ni/Mg(Al)O (15 wt.%) | Impregnation on LDH (MG30) | SMR | Tcal (350–1000 °C) | GHSV = 65 L g−1 h−1 P = 1–10 bar T = 600 °C H2O/CH4 = 5 Max CH4 conv. = 50% | High Tcal reduced crystallite size Catalysts with Tcal lower than 650 °C deactivated with increased reaction pressure Optimal catalyst calcined at 850 °C | [96] |
| Ni/MoCeZr/MgAl2O4-MgO (10 wt.%) | Co-precipitation | DRM | Tcal | GHSV = 60 L g−1 h−1 CO2/CH4 = 1 T = 900 °C Max CH4 conv. = 95% | Higher Tcal (900 °C) promoted MgAl2O4 phase but decreased the surface area. NiAl2O4 phase was not observed. At Tcal = 800 °C the catalyst showed the highest activity and stability | [97] |
| Ni/Mg(Al)O (22.3–50.3 wt.%) | Co-precipitation | DRM | Molar Mg/Al, Ni/Mg ratio Tcal (550–700 °C) Tred (550–700 °C) | GHSV = 45 L g−1 h−1 CO2/CH4 = 2 T = 500–700 °C Max CH4 conv. = 92% (700 °C) | Surface area was independent from Ni content but decreased with low molar Mg/Al ratio Reduction, but not calcination, affected the activity Mg/Al affected activity more than Ni/Mg Reduction temperature enhances activity and H2 selectivity Low Mg/Al molar ratio negatively affected the activity | [98] |
| Ni/Mg(Al)O (2.5–10 wt.%) | Co-precipitation or Impregnation | DRM | Tcal, Tred, preparation method | GHSV = 15 L g−1 h−1 CO2/CH4 = 1 T = 700–800 °C Max CH4 conv. = 97% (700 °C) | High Tcal reduced specific surface area and dampened reducibility Activity decreased when catalyst was treated at high Tcal Ni-Mg/Al by co-precipitation showed good resistance to coke Ni/MgO by impregnation showed higher activity than co-precipitation | [99] |
| Synthesis conditions | Advantages | Disadvantages |
|---|---|---|
| High calcination temperature |
|
|
| High reduction temperature |
|
|
| High Ni loading |
|
|
| Catalyst (Ni Loading) | Synthesis Method | Catalytic Process | Reaction Conditions (1) | Main Findings | Ref. |
|---|---|---|---|---|---|
| Ni/Mg(Al)O (4.3–19.0 wt.%) | Sol-gel | DRM | GHSV = 120 L g−1 h−1 CO2/CH4 = 1 T = 800 °C (2) Max CH4 conv. = 98% | Sol-gel hydrotalcite formed periclase instead of spinel phase upon calcination Catalysts from sol-gel precursor calcined at 650 °C possessed the best activity and low carbon formation. | [114] |
| Ni/Al2O3 (30.4–52 wt.%) | Sol-gel | DRM | GHSV = 30 L g−1 h−1 CO2/CH4 = 1 T = 700–800 °C Max CH4 conv. = NR | Sol-gel method enabled homogeneous and stochiometric spinel formation. Ni-rich catalysts contained separated Ni particles, promoting carbon formation | [115] |
| Ni/Mg(Al)O (15 wt.%) | Sol-gel | DRM | GHSV = 72 L g−1 h−1 CO2/CH4 = 1 T = 800 °C Max CH4 conv. = 90% | Sol-gel Ni/Al2O3 showed poor activity and high coke formation Sol-gel Ni/MgO showed high activity despite low reducibility Sol-gel Ni/Mg(Al)O obtained the highest activity and steady performance | [116] |
| Ni/Al2O3 (20–45 wt.%) | Pechini method | DRM | GHSV = 52,400 h−1 CO2/CH4 = 1 T = 700 °C Max CH4 conv. = 43% | Highly uniform, highly crystalline, and grain-sized NiAl2O4 were formed Unreduced stochiometric spinel NiAl2O4 was active for DRM | [119] |
| Ni/Al2O3 (20–45 wt.%) | Pechini method | SMR | GHSV = 65,500 h−1 H2O/CH4 = 2.4 T = 700 °C Max CH4 conv. = 82% | Highly uniform, highly crystalline, and grain-sized NiAl2O4 were formed Reduced Ni particles were active for SMR | [119] |
| Ni/Al2O3 (10 wt.%) | Aerogel | DRM | GHSV = 24 L g−1 h−1 CO2/CH4 = 1 T = 700 °C Max CO2 conv. = 60% | High surface area support and uniformed active sites were prepared by supercritical CO2 drying Higher loading of Ni in aerosol catalysts led to higher coke formation | [121] |
| Ni/Al2O3 (6 wt.%) | Impregnation of Ni-complex | SMR | GHSV = 24 L g−1 h−1 H2O/CH4 = 3 T = 500–800 °C Max CH4 conv. = 99 % (800 °C) | Complexing Ni before impregnation hindered particle growth Ni-triethylamine performed well in SMR | [122] |
| Ni/Al2O3 (1 wt.%) | Atomic layered deposition (ALD) | DRM | GHSV = 34 L g−1 h−1 CO2/CH4 = 1 T = 700–850 °C Max CH4 conv. = 99% (850 °C) | ALD catalysts showed higher activity than impregnated ones. ALD catalysts required a longer induction period | [124] |
| Ni/Al2O3 (0.8–2 wt.%) | ALD | DRM | GHSV = 7.2 L g−1 h−1 CO2/CH4 = 1 T = 400–800 °C Max CH4 conv. = 80% (800 °C) | ALD catalysts contained more NiO, resulting in higher activity compared to impregnated catalysts | [125] |
| Ni/MgO (NR) | ALD | DRM | GHSV = 12 L g−1 h−1 CO2/CH4 = 1 T = 800 °C Max CH4 conv. = 73% | MgO-deposited catalysts obtained higher basicity The activity and stability of the catalysts were greatly enhanced with ALD | [126] |
| Ni/Al2O3 (0.8 wt.%) | Molecular layered deposition | DRM | GHSV = 30 L g−1 h−1 CO2/CH4 = 1 T = 700 °C Max CH4 conv. = 38% | MLD catalysts exhibited sintering resistance 10 MLD cycled catalyst was stable up to 108 h time of stream | [127] |
| Ni/Al2O3 (2 wt.%) | ALD | DRM | GHSV = 72 L g−1 h−1 CO2/CH4 = 1 T = 700 °C Max CH4 conv. = 39% | ALD catalysts show higher stability compared to spinel NiAl2O4 Induction time was required for ALD to restructure the overlayer to active Ni species | [128] |
| Ni/Mg(Al)O (not found) | EISA | SMR | GHSV = 10 L g−1 h−1 H2O/CH4 = 3 T = 800 °C Max CH4 conv. = 97.6% | The most active catalyst prepared via sequential inert-air thermal treatments | [129] |
| Ni/Mg(Al)O | CAHD | DRM | GHSV = 96 L g−1 h−1 CO2/CH4 = 1 T = 700 °C Max CH4 conv. = 79% | CAHD catalysts had higher surface area, uniform pore size, high basicity, and small Ni nanoparticle (2.1 nm) The activity and stability of the catalysts were higher than the co-impregnated Ni/MgO-Al2O3 catalyst. | [130] |
| Ni/Mg(Al)O (LDH) | Freeze drying | DRM | GHSV = 480 L g−1 h−1 CO2/CH4 = 1 T = 800 °C Max CH4 conv. = 93% | Freeze drying preserved LDH-like platelet structure and resulted in higher Ni dispersion. Freeze dried catalysts showed higher activity (rate 8.3 mmol CH4 s−1 g-cat−1 at 900 °C) and higher coke resistance | [131] |
| Ni/Mg(Al)O (12 wt.%) | Impregnation + 3D printing | SMR | GHSV = 6 L g−1 h−1 H2O/CH4 = - T = 800 °C Max CH4 conv. = 75% | Ni impregnated on a 3D printed support with a binder for large-sized reactors | [132] |
| Ni/Mg(Al)O (8 wt.%) | Co-precipitation | SMR | GHSV = 2.5 L g−1 h−1 H2O/CH4 = 1.8 T = 700 °C Max CH4 conv. = 93.2% | Coke deposition after 350 h on stream of 0.32 wt.% Catalyst molded into 2.5 mm diameter spherical beads | [133] |
| Catalyst (M1/M2) | Synthesis Method | Reaction Conditions (1) | Main Finding | Ref |
|---|---|---|---|---|
| Pt-Ni/Mg(Al)O (0.1 wt.%/15 wt.%) | Sequential impregnation; Support: obtained by sol–gel method, calcined at 1100 °C | GHSV = 9.6 L g−1 h−1 H2O/CH4 = 4 T = 600 °C (2) Max CH4 Conv. = 80% |
| [145] |
| Pt-Ni/Mg(Al)O (0.1 wt.%/15 wt.%) | Sequential impregnation; Support: Commercial MG30, calcined at 900 °C | GHSV = 83 L g−1 h−1 H2O/CH4 = 5 T = 600 °C Max CH4 Conv. = 65% |
| [146] |
| Rh-Ni/Mg(Al)O (0.5 wt.%/15 wt.%) | Sequential impregnation; Support: Commercial MG30, calcined at 900 °C | GHSV = 83 L g−1 h−1 H2O/CH4 = 5 T = 600 °C Max CH4 Conv. = 59.2% |
| [151] |
| Ru-Ni/Mg(Al)O (0.5 wt.%/20 wt.%) | Sequential impregnation; Support: co-precipitated, calcined at 900 °C | GHSV = 160 L g−1 h−1 H2O/CH4 = 1.2 T = 650 °C Max CH4 Conv. = NR |
| [156] |
| Ru-Ni/Mg(Al)O (0.1 wt.%/12 wt.%) | Stepwise impregnation; Co-impregnation; Support: commercial MG30 | GHSV = 50 L g−1 h−1 H2O/CH4 = 3 T = 700 °C Max CH4 Conv. = 89% |
| [158] |
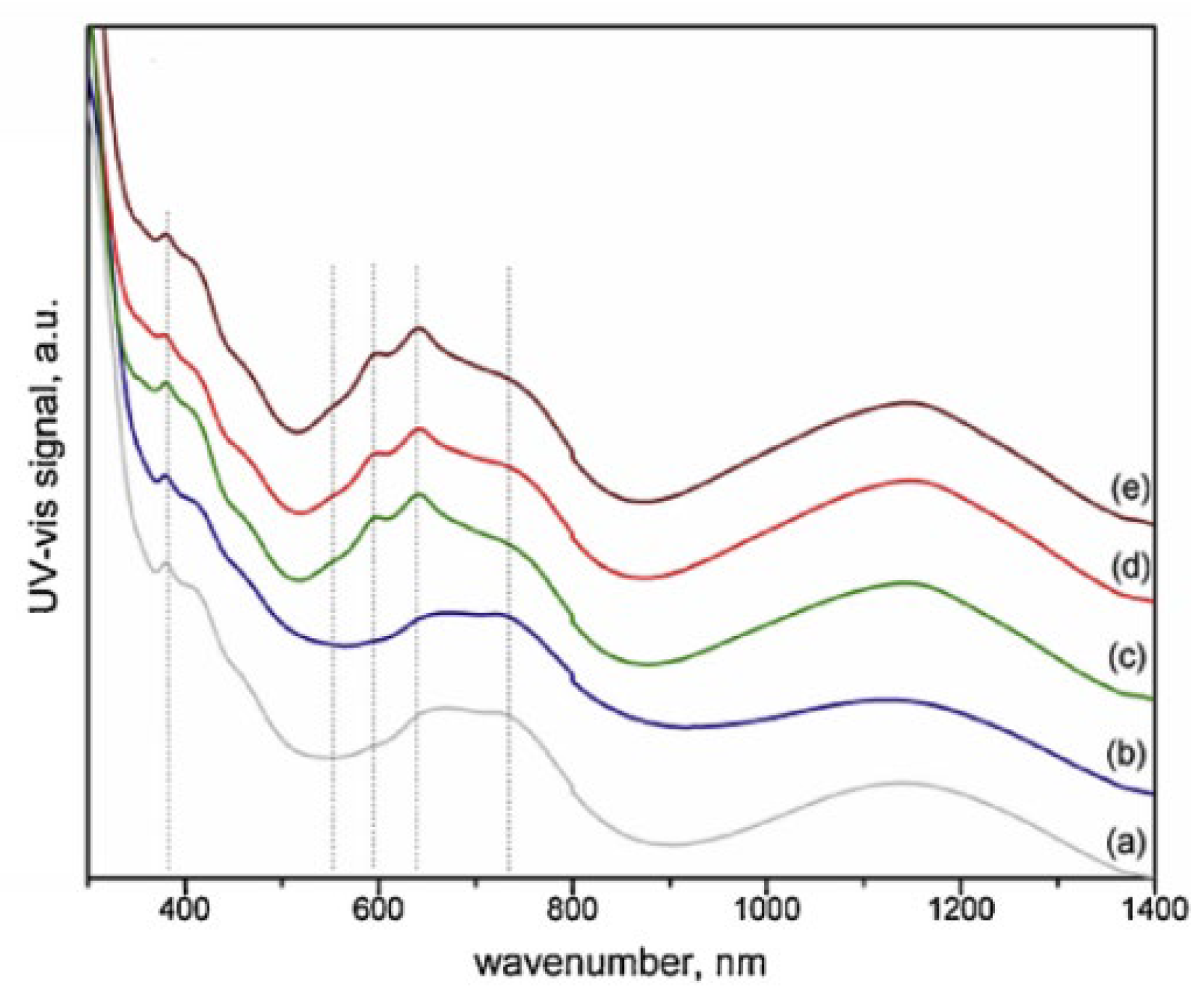
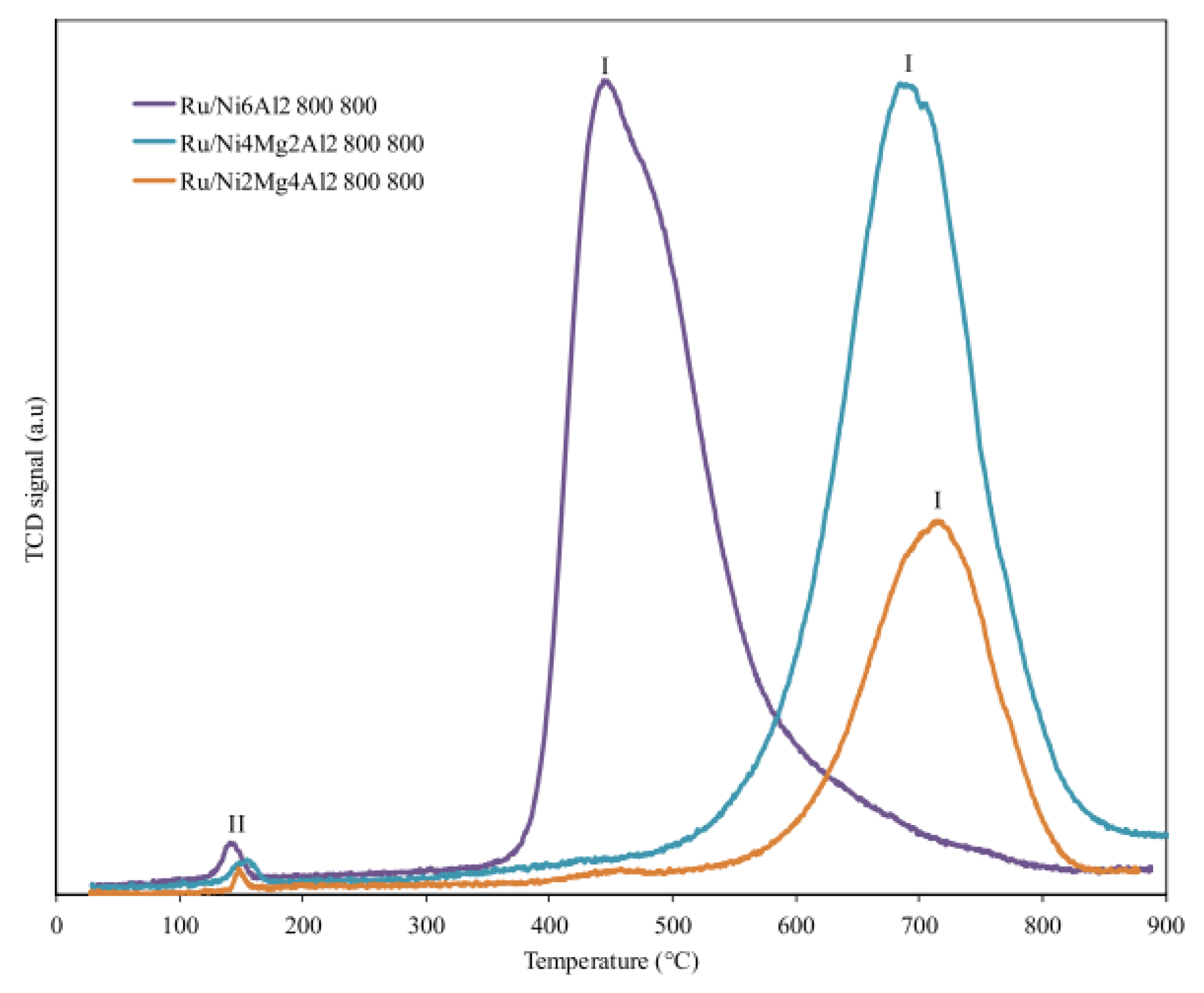
| Ru/Ni4Mg2Al2 (0.5 wt.%) Ru/Ni2Mg4Al2 (0.5 wt.%) | Co-precipitation followed by Impregnation; Support: Coprecipitation, calcined at 800 °C | GHSV = 6 L g−1 h−1 H2O/CH4 = 1 T = 650 °C Max CH4 Conv. = 76% |
| [159] |
| Fe-Ni/Mg(Al)O (1 wt.%/15 wt.%) Cr-Ni/Mg(Al)O (1 wt.%/15 wt.%) Ni/Mg(Al)O + CrFe3O4 (15 wt.%/10 wt.%) | Sequential dry impregnation; Support: Commercial MG70 | GHSV = 3000 h−1 H2O/CH4 = 2 T = 700 °C Max CH4 Conv. = 84% |
| [160] |
| NiCo/MgAl2O4 (15 wt.%/5 wt.%)) | EISA | H2O/CH4 = 1 T = 700 °C GHSV = 10 L g−1 h−1 Max CH4 Conv. = ~84% |
| [161] |
| Ni-Ca/Mg(Al)O (20 wt.%/0.35 wt.%) | Co-precipitation and impregnation | H2O/CH4/CO2 = 1.63/1/0.6 T = 900 °C GHSV = 3000 h−1 Max CH4 Conv. = 83.2% |
| [164] |
| Ni-Sr/Mg(Al)O (16 wt.%/0.8 wt.%) | Impregnation | GHSV = 1200 h−1 H2O/CH4 = 2.5 T = 750 °C Max CH4 conv. = 69.3% |
| [165] |
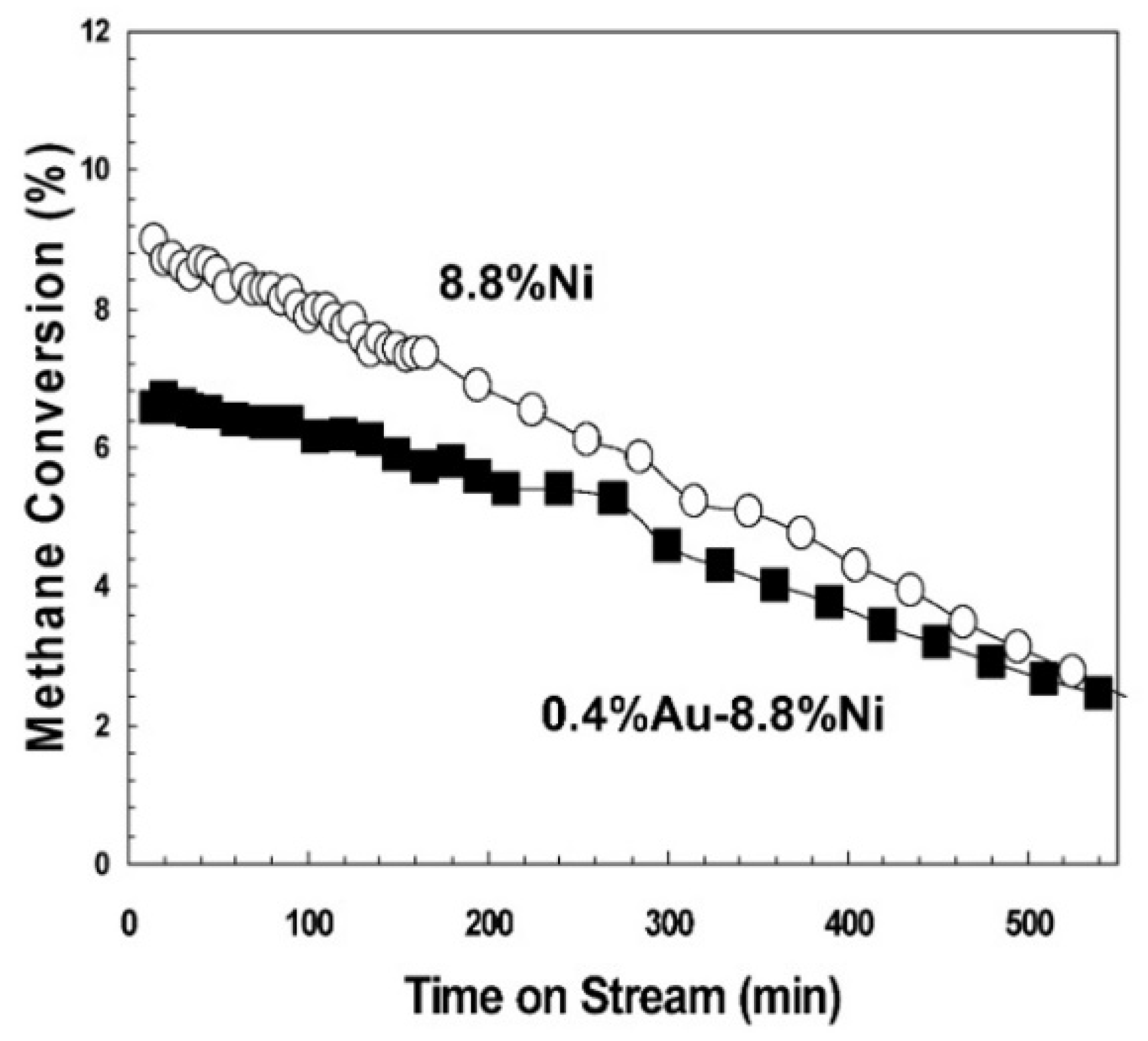
| Au–Ni/Mg(Al)O (0.4 wt.%/8.8 wt.%) | Wet Impregnation; Support: Commercial MG30, calcined at 700 °C | GHSV = 3300 L g−1 h−1 H2O/CH4= 1 T = 550 °C Max CH4 Conv. = 6.4% |
| [166] |
| Pt-Ni/Mg3.5Al2O4 (0.05 wt.%/10 wt.%) | sequential impregnation; Support: commercial Mg-Al with Mg/Al molar ratios of 3.5, 1.3 and 0.5 | GHSV = 1600 L g−1 h−1 H2O/CH4 = 2 T = 600 °C Max CH4 Conv. = 42.6% |
| [169] |
| Catalyst (M1/M2 wt.%) | Synthesis Method | Reaction Conditions(1) | Main Finding | Ref. |
|---|---|---|---|---|
| Pt-Ni/Mg(Al)O (0.5 wt.%/12 wt.%) Pt-Ni/Mg(Al)O (1 wt.%/12 wt.%) | Redox reaction (Pt2+ + Ni → Pt + Ni2+); Support: Ni/Mg/Al obtained by co-precipitation, calcined at 600 °C | GHSV = 360 L g−1 h−1 CO2/CH4 = 1 T = 700 °C Max CH4 Conv. = 65% |
| [172] |
| Ni-Pt/Al2O3 (4 wt.%/0.2 wt.%) Ni-Au/Al2O3 (4 wt.%/0.2 wt.%) Ni-Au-Pt/Al2O3 (4 wt.%/0.2 wt.%) Ni-Au-Pt/Mg(Al)O (4 wt.%/0.2 wt.%) | Sequential impregnation; Support: Sol–gel | GHSV = 60 L g−1 h−1 CO2/CH4 = 1 T = 700 °C (2) Max CH4 Conv. = 86.8% |
| [178] |
| Pt-Ni/Mg(Al)O (0.1 wt.%/8 wt.%) P-Pt-Ni/Mg(Al)O (0.1 wt.%/8 wt.%) | Impregnation and glow discharge plasma pretreatment; Support: co-precipitation | GHSV = 72 L g−1 h−1 CH4/CO2 = 1 T = 550 °C Max CH4 Conv. = 31.7% |
| [179] |
| NiCo2/Mg0.9(Al)O (1.7 wt.%/2.92 wt.%) NiCo2/Mg0.9(Al)O (2.79 wt.%/5.17 wt.%) | Co-precipitation; Support: co-precipitation | GHSV = 120 L g−1 h−1 CH4/CO2 = 1:1 T = 700 °C Max CH4 Conv. = 55% |
| [182] |
| Ni3Co9/Mg(Al)O (2.38 wt.%/7.13 wt.%) | Co-precipitation Support: Co-precipitation, Calcination: 800 °C, for 15 h, Reduction: 800 °C | GHSV = 120 L g−1 h−1 CH4/CO2 = 1:1 T = 750 °C Max CH4 Conv. = 88% |
| [184] |
| Ru-Ni/Mg(Al)Ox (0.1 wt.%/5.0 wt.%) | Reconstruction reaction using aqueous solutions of pre-chelated metals; Support: Co-precipitation, calcination: 500 °C, for 16 h | GHSV = 35 L g−1 h−1 CH4/CO2 = 1:1 T = 800 °C Max CH4 Conv. = 94% |
| [186] |
| Ru-Ni/Al2O3 (0.6 wt.%/8 wt.%) Ru-Ni/Mg(Al)O (0.6 wt.%/7 wt.%) | Impregnation; Support: Co-precipitation, Calcined at 400 °C, for 6 h | GHSV = NR CH4/CO2 = 1:1 T = 800 °C Max CH4 Conv. = 100% |
| [189] |
| Rh-NiAl (1 wt.%/11.5 wt.%) Rh-Ni/Mg(Al)O (1.5 wt.%/12 wt.%) | Wet impregnation Support: Co-precipitation, Calcined at 800 °C for 3 h | GHSV = 22 L g−1 h−1 CH4/CO2 = 1:1 T = 800 °C Max CH4 Conv. = 85% |
| [190] |
| Ni-Fe/Mg(Al)O (8 wt.%/5 wt.%) (8 wt.%/8 wt.%) (8 wt.%/11 wt.%) | Incipient wetness impregnation; Support: Co-precipitation, Calcined at 750 °C for 4 h | GHSV = 166 L g−1 h−1 CH4/CO2 = - T = 750 °C Max CH4 Conv. = NR |
| [191] |
| Ni-Fe/Mg(Al)O (8 wt.%/5 wt.%) | Incipient wetness impregnation; Support: Co-precipitation, Calcined: 750 °C for 4 h | GHSV = 30 L g−1 h−1 CH4/CO2 = 1:1 T = 750 °C Max CH4 Conv. = NR |
| [192] |
| Ni-Fe/Mg(Al)O (12 wt.%/3 wt.%) | Co-precipitation | GHSV = 60 L g−1 h−1 CH4/CO2 = 1:1 T = 800 °C CH4 Conv. = 95% |
| [193] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeedi, S.; Nguyen, X.T.; Bossola, F.; Evangelisti, C.; Dal Santo, V. Methane Reforming Processes: Advances on Mono- and Bimetallic Ni-Based Catalysts Supported on Mg-Al Mixed Oxides. Catalysts 2023, 13, 379. https://doi.org/10.3390/catal13020379
Saeedi S, Nguyen XT, Bossola F, Evangelisti C, Dal Santo V. Methane Reforming Processes: Advances on Mono- and Bimetallic Ni-Based Catalysts Supported on Mg-Al Mixed Oxides. Catalysts. 2023; 13(2):379. https://doi.org/10.3390/catal13020379
Chicago/Turabian StyleSaeedi, Soroosh, Xuan Trung Nguyen, Filippo Bossola, Claudio Evangelisti, and Vladimiro Dal Santo. 2023. "Methane Reforming Processes: Advances on Mono- and Bimetallic Ni-Based Catalysts Supported on Mg-Al Mixed Oxides" Catalysts 13, no. 2: 379. https://doi.org/10.3390/catal13020379