
Carbon Dioxide Conversion on Supported Metal Nanoparticles: A Brief Review


Abstract
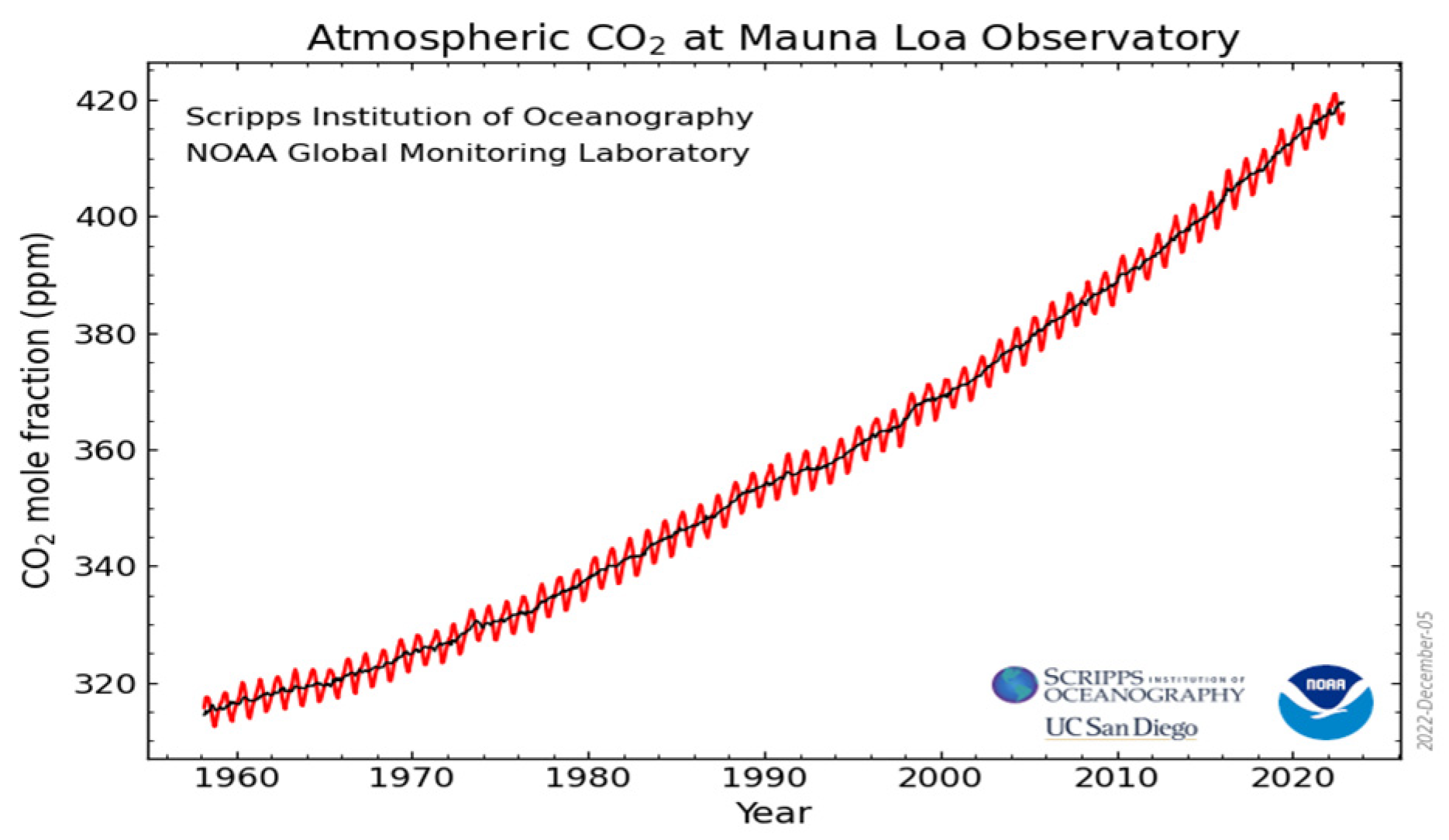
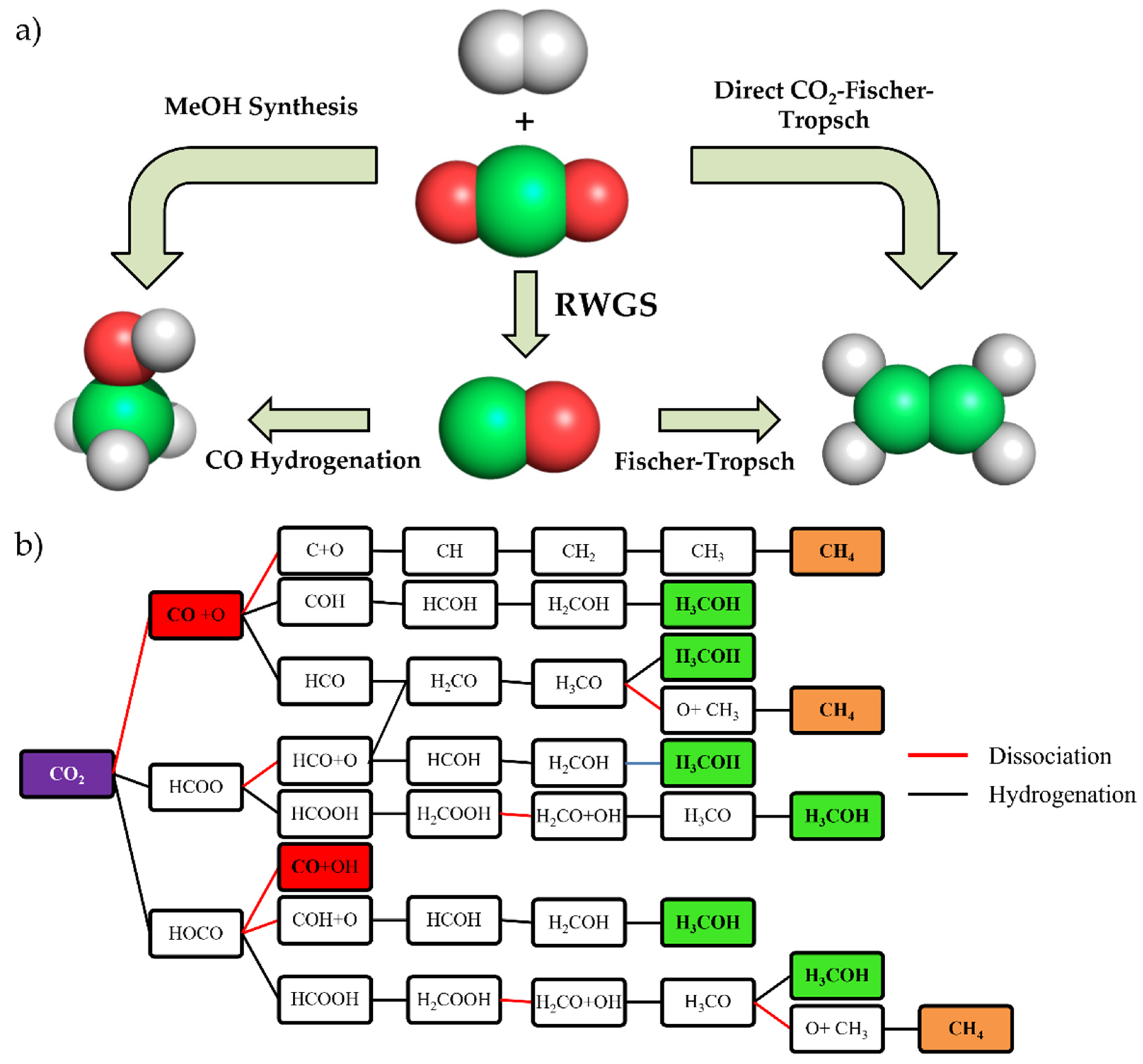
:1. Introduction
2. Metal Nanoparticles Supported on Metal Oxides
2.1. Al2O3
2.2. ZrO2
2.3. SiO2
2.4. TiO2
2.5. ZnO
2.6. CeO2
3. Transition Metal Carbides
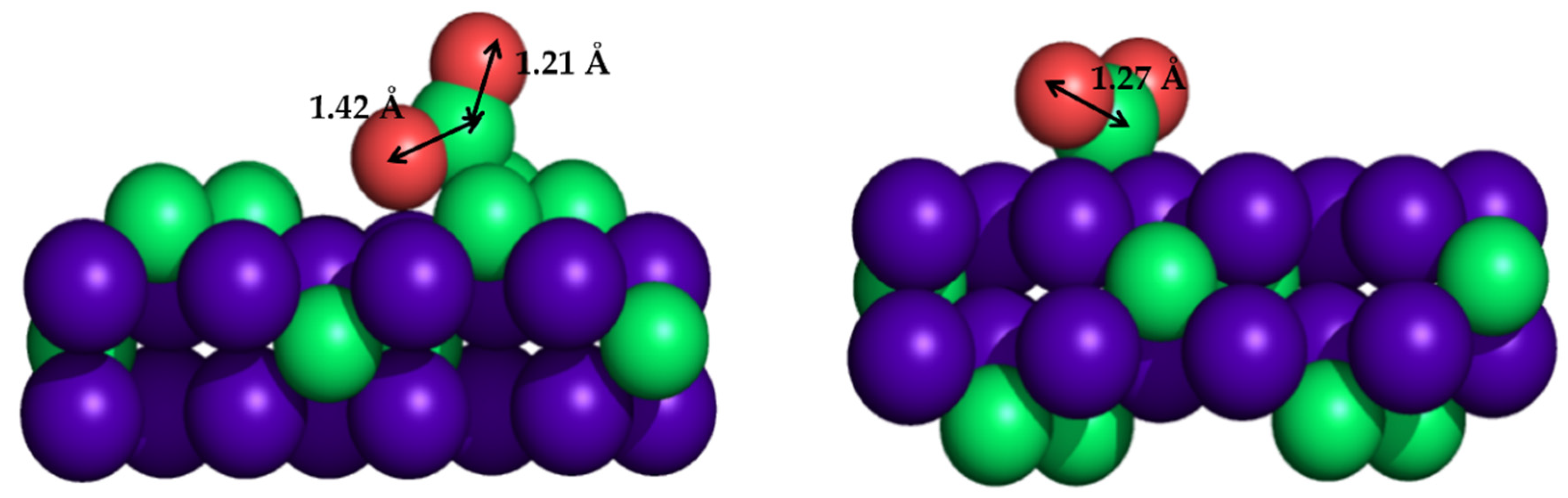
3.1. Metal Nanoparticles on TiC(001)
3.2. Metal Nanoparticles on Molybdenum Carbides
4. Perspective: Single-Metal-Atom Catalysts
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CCS | CO2 capture and storage |
| MOF | Metal organic framework |
| NP | Nanoparticle |
| RWGS | Reverse water gas shift reaction |
| SAC | Single-atom catalyst |
| TMC | Transition metal carbides |
| WGSR | Water gas shift reaction |
References
- Lim, L. How to make the most of carbon dioxide. Nature 2015, 526, 628–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intergovernmental Panel on Climate Change. Climate Change 2013—The Physical Science Basis, 1st ed.; Cambridge University Press: Cambridge, UK, 2014.
- Aresta, A. (Ed.) Carbon Dioxide as Chemical Feedstock; Wiley-VCH: New York, NY, USA, 2010. [Google Scholar]
- Gao, W.; Liang, S.; Wang, R.; Jiang, Q.; Zhang, Y.; Zheng, Q.; Xie, B.; Toe, C.Y.; Zhu, X.; Wang, J.; et al. Industrial carbon dioxide capture and utilization: State of the art and future challenges. Chem. Soc. Rev. 2020, 49, 8584–8686. [Google Scholar] [CrossRef] [PubMed]
- U.E.I. Administration. International Energy Outlook 2013. 2013. Available online: http://www.eia.gov/forecasts/ieo/pdf/0484%282013%29.pdf (accessed on 24 December 2022).
- Vummaleti, S.V.C.; Nolan, S.P.; Cavallo, L.; Talarico, G.; Poater, A. How easy is CO2 fixation by M–C bond containing complexes (M = Cu, Ni, Co, Rh, Ir)? Org. Chem. Front. 2016, 3, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Vummaleti, S.V.C.; Nolan, S.P.; Cavallo, L.; Talarico, G.; Poater, A. Mechanism of CO2 fixation by Ir–X Bonds (X = OH, OR, N, C). Eur. J. Inorg. Chem. 2015, 4653–4657. [Google Scholar] [CrossRef]
- Jacobson, M.Z. Review of solutions to global warming, air pollution, and energy security. Energy Environ. Sci. 2009, 2, 148–173. [Google Scholar] [CrossRef]
- D'Alessandro, D.M.; Smit, B.; Long, J.R. Carbon dioxide capture: Prospects for new materials. Angew. Chem. Int. Ed. 2010, 49, 6058–6082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IPCC. IPCC Special Report on Carbon Dioxide Capture and Storage; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Figueroa, J.D.; Fout, T.; Plasynski, S.; McIlvried, H.; Srivastava, R.D. Evaluation of Carbon Dioxide Absorption Characteristics Lithium Ortho-Silicate in Chemical Heat Storage. Int. J. Greenh. Gas Control 2008, 2, 9–20. [Google Scholar] [CrossRef]
- Poater, J.; Gimferrer, M.; Poater, A. Covalent and Ionic Capacity of MOFs To Sorb Small Gas Molecules. Inorg. Chem. 2018, 57, 6981–6990. [Google Scholar] [CrossRef]
- Shaikh, A.R.; Posada-Pérez, S.; Brotons-Rufes, A.; Pajski, J.J.; Vajiha; Kumar, G.; Mateen, A.; Poater, A.; Solà, M.; Chawla, M.; et al. Selective absorption of H2S and CO2 by azole based protic ionic liquids: A combined Density Functional Theory and Molecular Dynamics study. J. Mol. Liq. 2022, 367, 120558. [Google Scholar] [CrossRef]
- Shaikh, A.R.; Ashraf, M.; AlMayef, T.; Chawla, M.; Poater, A.; Cavallo, L. Amino acid ionic liquids as potential candidates for CO2 capture: Combined density functional theory and molecular dynamics simulations. Chem. Phys. Lett. 2020, 745, 137239. [Google Scholar] [CrossRef]
- Cavenati, S.; Grande, C.A.; Rodrigues, A.E. Adsorption Equilibrium of Methane, Carbon Dioxide, and Nitrogen on Zeolite 13X at High Pressures. J. Chem. Eng. Data 2004, 49, 1095–1101. [Google Scholar] [CrossRef]
- Millward, A.R.; Yaghi, O.M. Metal−Organic Frameworks with Exceptionally High Capacity for Storage of Carbon Dioxide at Room Temperature. J. Am. Chem. Soc. 2005, 127, 17998–17999. [Google Scholar] [CrossRef] [PubMed]
- Preti, D.; Resta, C.; Squarcialupi, S.; Fachinetti, G. Carbon dioxide hydrogenation to formic acid by using a heterogeneous gold catalyst. Angew. Chem. Int. Ed. 2011, 50, 12551–12554. [Google Scholar] [CrossRef]
- Pomelli, C.S.; Tomasi, J.; Solà, M. Theoretical Study on the Thermodynamics of the Elimination of Formic Acid in the Last Step of the Hydrogenation of CO2 Catalyzed by Rhodium Complexes in the Gas Phase and Supercritical CO2. Organometallics 1998, 17, 3164–3168. [Google Scholar] [CrossRef]
- Wang, Y.; Darensbourg, D.J. Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. [Google Scholar] [CrossRef]
- Aomchad, V.; Del Globo, S.; Poater, A.; D’Elia, V. Exploring the potential of Group III salen complexes for the conversion of CO2 under ambient conditions. Catal. Today 2021, 375, 324–334. [Google Scholar] [CrossRef]
- Natongchai, W.; Posada-Pérez, S.; Phungpanya, C.; Luque Urrutia, J.A.; Solà, M.; D'Elia, V.; Poater, A. Enhancing the catalytic performance of group I, II metal halides in the cycloaddition of CO2 to epoxides under atmospheric conditions by cooperation with homogeneous and heterogeneous highly nucleophilic aminopyridines: Experimental and theoretical study. J. Org. Chem. 2022, 87, 2873–2886. [Google Scholar] [CrossRef] [PubMed]
- Arayachukiat, S.; Yingcharoen, P.; Vummaleti, S.V.C.; Cavallo, L.; Poater, A.; D’Elia, V. Cycloaddition of CO2 to challenging N-tosyl aziridines using a halogen-free niobium complex: Catalytic activity and mechanistic insights. Mol. Catal. 2017, 443, 280–285. [Google Scholar] [CrossRef]
- Al Maksoud, W.; Saidi, A.; Samantaray, M.K.; Abou-Hamad, E.; Poater, A.; Ould-Chikh, S.; Guo, X.; Guan, E.; Ma, T.; Gates, B.C.; et al. Docking of tetra-methyl zirconium to the surface of silica: A well-defined pre-catalyst for conversion of CO2 to cyclic carbonate. Chem. Commun. 2020, 56, 3528–3531. [Google Scholar] [CrossRef] [Green Version]
- Natongchai, W.; Luque-Urrutia, J.A.; Phungpanya, C.; Solà, M.; D’Elia, V.; Poater, A.; Zipse, H. Cycloaddition of CO2 to epoxides by highly nucleophilic 4-aminopyridines: Establishing a relationship between carbon basicity and catalytic performance by experimental and DFT investigations. Org. Chem. Front. 2021, 8, 613–627. [Google Scholar] [CrossRef]
- Sodpiban, O.; Del Gobbo, S.; Barman, S.; Aomchad, V.; Kidkhunthod, P.; Ould-Chikh, S.; Poater, A.; D’Elia, V.; Basset, J.-M. Synthesis of Well-defined Yttrium-based Lewis Acids by Capture of a Reaction Intermediate and Catalytic Application for cycloaddition of CO2 to Epoxides Under Atmospheric Pressure. Catal. Sci. Technol. 2019, 9, 6152–6165. [Google Scholar] [CrossRef]
- Coufourier, S.; Gaignard-Gaillard, Q.; Lohier, J.-F.; Poater, A.; Gaillard, S.; Renaud, J.-L. Hydrogenation of CO2, Hydrogenocarbonate, and Carbonate to Formate in Water using Phosphine Free Bifunctional Iron Complexes. ACS Catal. 2020, 10, 2108–2116. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [Green Version]
- Perathoner, S.; Centi, G. CO2 recycling: A key strategy to introduce green energy in the chemical production chain. ChemSusChem 2014, 7, 1274–1282. [Google Scholar] [CrossRef]
- Song, C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yang, X.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G.Q.; Millar, G.J. Carbon Dioxide Reforming of Methane to Produce Synthesis Gas over Metal-Supported Catalysts: State of the Art. Energy Fuels 1996, 10, 896–904. [Google Scholar] [CrossRef]
- Caballero, A.; Pérez, P.J. Methane as raw material in synthetic chemistry: The final frontier. Chem. Soc. Rev. 2013, 42, 8809–8820. [Google Scholar] [CrossRef]
- Liu, X.M.; Lu, G.Q.; Yan, Z.F.; Beltramini, J. Recent Advances in Catalysts for Methanol Synthesis via Hydrogenation of CO and CO2. Ind. Eng. Chem. Res. 2003, 42, 6518–6530. [Google Scholar] [CrossRef]
- Xiaoding, X.; Moulijn, J.A. Mitigation of CO2 by Chemical Conversion: Plausible Chemical Reactions and Promising Products. Energy Fuels 1996, 10, 305–325. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Beyond Oil and Gas: The Methanol Economy; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. (Eds.) Green Carbon Dioxide: Advances in CO2 Utilization; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Baig, N.; Kammakakam, I.; Falath, W. Nanomaterials: A review of synthesis, methods, properties, recent progress, and challenges. Mater. Adv. 2021, 2, 1821–1871. [Google Scholar] [CrossRef]
- Lu, B.W.; Kawamoto, K. Preparation of mesoporous CeO2 and monodispersed NiO particles in CeO2, and enhanced selectivity of NiO/CeO2 for reverse water gas shift reaction. Mater. Res. Bull. 2014, 53, 70–78. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Mechanism of CO formation in reverse water–gas shift reaction over Cu/Al2O3 catalyst. Catal. Lett. 2000, 68, 45–48. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, D. Study of bimetallic Cu–Ni/γ-Al2O3 catalysts for carbon dioxide hydrogenation. Int. J. Hydrogen Energy 1999, 24, 351–354. [Google Scholar] [CrossRef]
- Kharaji, A.G.; Shariati, A.; Takassi, M.A. A novel γ-alumina supported Fe-Mo bimetallic catalyst for reverse water gas shift reaction. Chin. J. Chem. Eng. 2013, 21, 1007–1014. [Google Scholar] [CrossRef]
- Kharaji, A.G.; Shariati, A.; Ostadi, M. Development of Ni-Mo/Al2O3 Catalyst for Reverse Water Gas Shift (RWGS) Reaction. J. Nanosci. Nanotechnol. 2014, 14, 6841–6847. [Google Scholar] [CrossRef]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A study on the effect of support's reducibility on the reverse water-gas shift reaction over Pt catalysts. Appl. Catal. A 2012, 423–424, 100–107. [Google Scholar] [CrossRef]
- Xu, W.; Ramírez, P.J.; Stacchiola, D.; Brito, J.L.; Rodriguez, J.A. The Carburization of Transition Metal Molybdates (MxMoO4, M = Cu, Ni or Co) and the Generation of Highly Active Metal/Carbide Catalysts for CO2 Hydrogenation. Catal. Lett. 2015, 145, 1365–1373. [Google Scholar] [CrossRef]
- Laudenschleger, D.; Ruland, H.; Muhler, M. Identifying the nature of the active sites in methanol synthesis over Cu/ZnO/Al2O3 catalysts. Nat. Commun. 2020, 11, 3898. [Google Scholar] [CrossRef]
- Beck, A.; Zabilskiy, M.; Newton, M.A.; Safonova, O.; Willinger, M.G.; van Bokhoven, J.A. Following the structure of copper-zinc-alumina across the pressure gap in carbon dioxide hydrogenation. Nat. Catal. 2021, 4, 488–497. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kuhl, S.; Havecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Chen, C.S.; Lin, J.H.; You, J.H.; Chen, C.R. Properties of Cu(thd)2 as a precursor to prepare Cu/SiO2 catalyst using the atomic layer epitaxy technique. J. Am. Chem. Soc. 2006, 128, 15950–15951. [Google Scholar] [CrossRef]
- Niu, J.; Liu, H.; Jin, Y.; Fan, B.; Qi, W.; Ran, J. Comprehensive review of Cu-based CO2 hydrogenation to CH3OH: Insights from experimental work and theoretical analysis. Int. J. Hydrogen Energy 2022, 47, 9183–9200. [Google Scholar] [CrossRef]
- Liu, Y.M.; Liu, J.T.; Liu, S.Z.; Li, J.; Gao, Z.H.; Zuo, Z.J.; Huang, W. Reaction mechanisms of methanol synthesis from CO/CO2 hydrogenation on Cu2O(111): Comparison with Cu(111). J. CO2 Util. 2017, 20, 59–65. [Google Scholar] [CrossRef]
- Samantaray, M.K.; D'Elia, V.; Pump, E.; Falivene, L.; Harb, M.; Ould Chikh, S.; Cavallo, L.; Basset, J.-M. The Comparison between Single Atom Catalysis and Surface Organometallic Catalysis. Chem. Rev. 2020, 120, 734–813. [Google Scholar] [CrossRef]
- Yang, Y.; Mims, C.A.; Mei, D.H.; Peden, C.H.F.; Campbell, C.T. Mechanistic studies of methanol synthesis over Cu from CO/CO2/H2/H2O mixtures: The source of C in methanol and the role of water. J. Catal. 2013, 298, 10–17. [Google Scholar] [CrossRef]
- Lee, J.S.; Lee, K.H.; Lee, S.Y.; Kim, Y.G. A comparative study of methanol synthesis from CO2/H2 and CO/H2 over a Cu/ZnO/Al2O3 catalyst. J. Catal. 1993, 144, 414–424. [Google Scholar] [CrossRef]
- Grabow, L.C.; Mavrikakis, M. Mechanism of methanol synthesis on Cu through CO2 and CO hydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Kasatkin, I.; Kurr, P.; Kniep, B.; Trunschke, A.; Schlögl, R. Role of lattice strain and defects in copper particles on the activity of Cu/ZnO/Al2O3 catalysts for methanol synthesis. Angew. Chem. Int. Ed. 2007, 46, 7324–7327. [Google Scholar] [CrossRef]
- Jalama, K. Carbon dioxide hydrogenation over nickel-, ruthenium-, and copper-based catalysts: Review of kinetics and mechanism. Catal. Rev. 2017, 59, 95–164. [Google Scholar] [CrossRef]
- Schmider, D.; Maier, L.; Deutschmann, O. Reaction Kinetics of CO and CO2 Methanation over Nickel. Ind. Eng. Chem. Res. 2021, 60, 5792–5805. [Google Scholar] [CrossRef]
- Conner, W.C.; Falconer, J.L. Spillover in Heterogeneous Catalysis. Chem. Rev. 1995, 95, 759–788. [Google Scholar] [CrossRef]
- Shen, H.; Li, H.; Yang, Z.; Li, C. Magic of hydrogen spillover: Understanding and application. Green Energy Environ. 2022, 7, 1161–1198. [Google Scholar] [CrossRef]
- Massaro, A.; Pecoraro, A.; Hernandez, S.; Talarico, G.; Munoz-Garcia, A.B.; Pavone, M. Oxygen evolution reaction at the Mo/W-doped bismuth vanadate surface: Assessing the dopant role by DFT calculations. Mol. Catal. 2022, 517, 112036. [Google Scholar] [CrossRef]
- Zheng, Y.; Fu, K.; Yu, Z.; Su, Y.; Han, R.; Liu, Q. Oxygen vacancies in a catalyst for VOCs oxidation: Synthesis, characterization, and catalytic effects. J. Mater. Chem. A 2022, 10, 14171–14186. [Google Scholar] [CrossRef]
- Baiano, C.; Schiavo, E.; Gerbaldi, C.; Bella, F.; Meligrana, G.; Talarico, G.; Maddalena, P.; Pavone, M.; Muñoz-García, A.B. Role of surface defects in CO2 adsorption and activation on CuFeO2 delafossite oxide. Mol. Catal. 2020, 496, 111181. [Google Scholar] [CrossRef]
- Flórez, E.; Feria, L.; Viñes, F.; Rodriguez, J.A.; Illas, F. Effect of the Support on the Electronic Structure of Au Nanoparticles Supported on Transition Metal Carbides: Choice of the Best Substrate for Au Activation. Phys. Chem. Chem. Phys. 2012, 14, 427–438. [Google Scholar] [CrossRef]
- Anderson, J.A.; Fernández-García, M. (Eds.) Supported Metals in Catalysis; Catalytic Science Series; Imperial College Press: London, UK, 2005; Volume 5. [Google Scholar]
- Rodriguez, J.A.; Stacchiola, D. Catalysis and the nature of mixed-metal oxides at the nanometer level: Special properties of MOx/TiO2(110) {M= V, W, Ce} surfaces. Phys. Chem. Chem. Phys. 2010, 12, 9557–9565. [Google Scholar] [CrossRef]
- Samanta, B.; Morales-García, A.; Illas, F.; Goga, N.; Anta, J.A.; Calero, S.; Bieberle-Hütter, A.; Libisch, F.; Muñoz-García, A.B.; Pavone, M.; et al. Challenges of modeling nanostructured materials for photocatalytic water splitting. Chem. Soc. Rev. 2022, 51, 3794–3818. [Google Scholar] [CrossRef]
- Lam, E.; Corral-Pérez, J.J.; Larmier, K.; Noh, G.; Wolf, P.; Comas-Vives, A.; Urakawa, A.; Copéret, C. CO2 Hydrogenation on Cu/Al2O3: Role of the Metal/Support Interface in Driving Activity and Selectivity of a Bifunctional Catalyst. Angew. Chem. Int. Ed. 2019, 58, 13989–13996. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Yang, C.; Li, X.; Wang, Z.; Pei, C.; Zhao, Z.J.; Gong, J. On the Role of Hydroxyl Groups on Cu/Al2O3 in CO2 Hydrogenation. ACS Catal. 2022, 12, 14162–14172. [Google Scholar] [CrossRef]
- Bansode, A.; Tidona, B.; von Rohr, P.R.; Urakawa, A. Impact of K and Ba promoters on CO2 hydrogenation over Cu/Al2O3 catalysts at high pressure. Catal. Sci. Technol. 2013, 3, 767–778. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Baibars, F.; Le Sache, E.; Arellano-García, H.; Gu, S.; Reina, T.R. CO2 valorisation via Reverse Water-Gas Shift reaction using advanced Cs doped Fe-Cu/Al2O3 catalysts. J. CO2 Util. 2017, 21, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Garbarino, G.; Bellotti, D.; Riani, P.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3 and Ni/Al2O3 catalysts at atmospheric pressure: Catalysts activation, behaviour and stability. Int. J. Hydrogen Energy 2015, 40, 9171–9182. [Google Scholar] [CrossRef]
- Falbo, L.; Visconti, C.G.; Lietti, L.; Szanyi, J. The effect of CO on CO2 methanation over Ru/Al2O3 catalysts: A combined steady-state reactivity and transient DRIFT spectroscopy study. Appl. Catal. B Environ. 2019, 256, 117791. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szany, J. CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity. ACS Catal. 2013, 3, 2449–2455. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, C.; Chu, W.; Zhou, Y.; Köhler, K. CO2 Methanation over Supported Ru/Al2O3 Catalysts: Mechanistic Studies by In situ Infrared Spectroscopy. ChemistrySelect 2016, 1, 3197–3203. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Magistri, L.; Busca, G. A study of the methanation of carbon dioxide on Ni/Al2O3 catalysts at atmospheric pressure. Int. J. Hydrogen Energy 2014, 39, 11557–11565. [Google Scholar] [CrossRef]
- Garbarino, G.; Kowalik, P.; Riani, P.; Antoniak-Jurak, K.; Pieta, P.; Lewalska-Graczyk, A.; Lisowski, W.; Nowakowski, R.; Busca, G.; Pieta, I.S. Improvement of Ni/Al2O3 Catalysts for Low-Temperature CO2 Methanation by Vanadium and Calcium Oxide Addition. Ind. Eng. Chem. Res. 2021, 60, 6554–6564. [Google Scholar] [CrossRef]
- Italiano, C.; Llorca, J.; Pino, L.; Ferraro, M.; Antonucci, V.; Vita, A. CO and CO2 methanation over Ni catalysts supported on CeO2, Al2O3 and Y2O3 oxides. Appl. Catal. B Environ. 2020, 264, 118494. [Google Scholar] [CrossRef]
- Quindimil, A.; De-La-Torre, U.; Pereda-Ayo, B.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; González-Marcos, J.A.; Bueno-López, A.; González-Velasco, J.R. Effect of metal loading on the CO2 methanation: A comparison between alumina supported Ni and Ru catalysts. Catal. Today 2020, 356, 419–432. [Google Scholar] [CrossRef]
- Mihet, M.; Lazar, M.D. Methanation of CO2 on Ni/γ-Al2O3: Influence of Pt, Pd or Rh promotion. Catal. Today 2018, 306, 294–299. [Google Scholar] [CrossRef]
- Schubert, M.; Pokhrel, S.; Thomé, A.; Zielasek, V.; Gesing, T.M.; Roessner, F.; Madler, L.; Baumer, M. Highly active Co–Al2O3-based catalysts for CO2 methanation with very low platinum promotion prepared by double flame spray pyrolysis. Catal. Sci. Technol. 2016, 6, 7449–7460. [Google Scholar] [CrossRef] [Green Version]
- Wambach, J.; Baiker, A.; Wokaun, A. CO2 hydrogenation over metal/zirconia catalysts. Phys. Chem. Chem. Phys. 1999, 1, 5071–5080. [Google Scholar] [CrossRef]
- Denise, B.; Cherifi, O.; Bettahar, M.M.; Sneeden, R.P.A. Supported Copper Catalysts Prepared from Copper(II) Formate: Hydrogenation of Carbon Dioxide Containing Feedstocks. Appl. Catal. 1989, 48, 365–372. [Google Scholar] [CrossRef]
- Nitta, Y.; Suwata, O.; Ikeda, Y.; Okamoto, Y.; Imanaka, T. Copper-zirconia catalysts for methanol synthesis from carbon dioxide: Effect of ZnO addition to Cu-ZrO2 catalysts. Catal. Lett. 1994, 26, 345–354. [Google Scholar] [CrossRef]
- Nitta, Y.; Fujimatsu, T.; Okamoto, Y.; Imanaka, T. Effect of starting salt on catalytic behaviour of Cu-ZrO2 catalysts in methanol synthesis from carbon dioxide. Catal. Lett. 1993, 17, 157–165. [Google Scholar] [CrossRef]
- Meunier, F.C.; Dansette, I.; Eng, K.; Schuurman, Y. Differentiating the Reactivity of ZrO2-Bound Formates Formed on Cu/ZrO2 during CO2 Hydrogenation. Catalysts 2022, 12, 793. [Google Scholar] [CrossRef]
- Bogdan, V.I.; Koklin, A.E.; Nikolaev, S.A.; Kustov, L.M. Carbon dioxide hydrogenation on Au nanoparticles supported on TiO2, ZrO2 and sulphated ZrO2 under supercritical conditions. Top. Catal. 2016, 59, 1104–1109. [Google Scholar] [CrossRef]
- Baiker, A.; Kilo, M.; Maciejewski, M.; Menzi, S.; Wokaun, A.; In Guczi, L.; Solymosi, F. Hydrogenation of CO2 over copper, silver and gold/zirconia catalysts: Comparative study of catalyst properties and reaction pathways. Stud. Surf. Sci. Catal. 1993, 75, 1257–1272. [Google Scholar]
- Wu, C.Y.; Zhang, P.; Zhang, Z.F.; Zhang, L.J.; Yang, G.Y.; Han, B.X. Efficient hydrogenation of CO2 to methanol over supported subnanometer gold catalysts at low temperature. ChemCatChem 2017, 9, 3691–3696. [Google Scholar] [CrossRef]
- Grabowski, R.; Sloczynski, J.; Sliwa, M.; Mucha, D.; Socha, R.P.; Lachowska, M.; Skrzypek, J. Influence of polymorphic ZrO2 phases and the silver electronic state on the activity of Ag/ZrO2 catalysts in the hydrogenation of CO2 to methanol. ACS Catal. 2011, 1, 266–278. [Google Scholar] [CrossRef]
- Fröhlich, C.; Köppel, R.A.; Baiker, A.; Kilo, M.; Wokaun, A. Hydrogenation of carbon dioxide over silver promoted copper/ zirconia catalysts. Appl. Catal. A 1993, 106, 275–293. [Google Scholar] [CrossRef]
- Tada, S.; Watanabe, F.; Kiyota, K.; Shimoda, N.; Hayashi, R.; Takahashi, M.; Nariyuki, A.; Igarashi, A.; Satokawa, S. Ag addition to CuO-ZrO2 catalysts promotes methanol synthesis via CO2 hydrogenation. J. Catal. 2017, 351, 107–118. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, I. Methanol synthesis from CO and CO2 hydrogenations over supported palladium catalysts. Bull. Chem. Soc. Jpn. 2002, 75, 1393–1398. [Google Scholar] [CrossRef]
- Kattel, S.; Yu, W.T.; Yang, X.F.; Yan, B.H.; Huang, Y.Q.; Wan, W.M.; Liu, P.; Chen, J.G.G. CO2 Hydrogenation over oxide supported PtCo catalysts: The role of the oxide support in determining the product selectivity. Angew. Chem. Int. Ed. 2016, 55, 7968–7973. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Xu, Z.-N.; Peng, S.-Y.; Zhang, M.-J.; Lu, G.; Cehn, Q.-S.; Chen, Y.; Guo, G.-C. High-Performance and Long-Lived Cu/SiO2 Nanocatalyst for CO2 Hydrogenation. ACS Catal. 2015, 5, 4255–4259. [Google Scholar] [CrossRef]
- Shawabkeh, R.A.; Faqir, N.M.; Rawajfeh, K.M.; Hussein, I.A.; Hamza, A. Adsorption of CO2 on Cu/SiO2 nano-catalyst: Experimental and theoretical study. Appl. Surf. Sci. 2022, 586, 152726. [Google Scholar] [CrossRef]
- Yu, J.; Yang, M.; Zhang, J.; Ge, Q.; Zimina, A.; Pruessmann, T.; Zheng, L.; Grunwaldt, J.D.; Sun, J. Stabilizing Cu+ in Cu/SiO2 Catalysts with a Shattuckite-Like Structure Boosts CO2 Hydrogenation into Methanol. ACS Catal. 2020, 10, 14694–14706. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Xu, Z.-N.; Zhang, M.-J.; Chen, Q.-S.; Chen, Y.; Guo, G.-C. Insight into composition evolution in the synthesis of high-performance Cu/SiO2 catalysts for CO2 hydrogenation. RSC Adv. 2016, 6, 25185–25190. [Google Scholar] [CrossRef]
- Lam, E.; Noh, G.; Larmier, K.; Safonova, O.V.; Copéret, C. CO2 Hydrogenation on Cu-Catalysts Generated from ZnII Single-Sites: Enhanced CH3OH Selectivity Compared to Cu/ZnO/Al2O3. J. Catal. 2021, 394, 266–272. [Google Scholar] [CrossRef]
- Fayisa, B.A.; Xi, Y.; Yang, Y.; Gao, Y.; Li, A.; Wang, M.-Y.; Lv, J.; Huang, S.; Wang, Y.; Ma, X. Pt-modulated Cu/SiO2 catalysts for efficient hydrogenation of CO2-derived ethylene carbonate to methanol and ethylene glycol. Chin. J. Chem. Eng. 2022, 41, 366–373. [Google Scholar] [CrossRef]
- Dias, Y.R.; Perez-Lopez, O.W. Carbon dioxide methanation over Ni-Cu/SiO2 catalysts. Energy Convers. Manag. 2020, 203, 112214. [Google Scholar] [CrossRef]
- Le, T.A.; Kang, J.K.; Park, E.D. CO and CO2 Methanation Over Ni/SiC and Ni/SiO2 Catalyst. Top. Catal. 2018, 61, 1537–1544. [Google Scholar] [CrossRef]
- Ye, R.-P.; Gong, W.; Sun, Z.; Sheng, Q.; Shi, X.; Wang, T.; Yao, Y.; Razink, J.J.; Lin, L.; Zhou, Z.; et al. Enhanced stability of Ni/SiO2 catalyst for CO2 methanation: Derived from nickel phyllosilicate with strong metal-support interactions. Energy 2019, 188, 116059. [Google Scholar] [CrossRef]
- Ye, R.-P.; Liao, L.; Ramirez Reina, T.; Liu, J.; Chevella, D.; Jin, Y.; Fan, M.; Liu, J. Engineering Ni/SiO2 catalysts for enhanced CO2 methanation. Fuel 2021, 285, 119151. [Google Scholar] [CrossRef]
- Wu, H.C.; Chang, Y.C.; Wu, J.H.; Lin, J.H.; Lin, I.K.; Chen, C.S. Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: The influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 2015, 5, 4154–4163. [Google Scholar] [CrossRef]
- Dias, Y.R.; Perez-Lopez, O.W. CO2 conversion to methane using Ni/SiO2 catalysts promoted by Fe, Co and Zn. J. Environ. Chem. Eng. 2021, 9, 104629. [Google Scholar] [CrossRef]
- Huang, Z.; Yuan, Y.; Song, M.; Hao, Z.; Xiao, J.; Cai, D.; Ibrahim, A.-R.; Zhan, G. CO2 hydrogenation over mesoporous Ni-Pt/SiO2 nanorod catalysts: Determining CH4/CO selectivity by surface ratio of Ni/Pt. Chem. Eng. Sci. 2022, 247, 117106–117119. [Google Scholar] [CrossRef]
- Pantaleo, G.; La Parola, V.; Testa, M.L.; Venezia, A.M. CO2 Reforming of CH4 over SiO2-Supported Ni Catalyst: Effect of Sn as Support and Metal Promoter. Ind. Eng. Chem. Res. 2021, 60, 18684–18694. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, S.; Julkapli, N.M.; Hamid, S.B.A. Titanium Dioxide as a Catalyst Support in Heterogeneous Catalysis. Sci. World J. 2014, 727496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- In, S.I.; Vaughn, D.D.; Schaak, R.E. Hybrid CuO-TiO2−xNx Hollow Nanocubes for Photocatalytic Conversion of CO2 into Methane under Solar Irradiation. Angew. Chem. Int. Ed. 2012, 51, 3915–3918. [Google Scholar] [CrossRef]
- Yang, C.C.; Yu, Y.H.; van der Linden, B.; Wu, J.C.; Mul, G. Artificial Photosynthesis over Crystalline TiO2-Based Catalysts: Fact or Fiction? J. Am. Chem. Soc. 2010, 132, 8398–8406. [Google Scholar] [CrossRef] [PubMed]
- Anzai, A.; Liu, M.-H.; Ura, K.; Noguchi, T.G.; Yoshizawa, A.; Kato, K.; Sugiyama, T.; Yamauchi, M. Cu Modified TiO2 Catalyst for Electrochemical Reduction of Carbon Dioxide to Methane. Catalysts 2022, 12, 478. [Google Scholar] [CrossRef]
- Tseng, I.-H.; Wu, J.C.S.; Chou, H.-Y. Effects of sol–gel procedures on the photocatalysis of Cu/TiO2 in CO2 photoreduction. J. Catal. 2004, 221, 432–440. [Google Scholar] [CrossRef]
- Slamet, N.; Nasution, H.; Purnama, E.; Kosela, S.; Gunlazuardi, J. Photocatalytic Reduction of CO2 on Copper-Doped Titania Catalysts Prepared by Improved-Impregnation Method. Catal. Commun. 2005, 6, 313–319. [Google Scholar] [CrossRef]
- Jiang, Z.; Sun, W.; Miao, W.; Yuan, Z.; Yang, G.; Kong, F.; Yan, T.; Chen, J.; Huang, B.; An, C.; et al. Living Atomically Dispersed Cu Ultrathin TiO2 Nanosheet CO2 Reduction Photocatalyst. Adv. Sci. 2019, 6, 1900289. [Google Scholar] [CrossRef] [Green Version]
- Gonell, F.; Puga, A.V.; Julián-López, B.; García, H.; Corma, A. Copper-doped titania photocatalysts for simultaneous reduction of CO2 and production of H2 from aqueous sulfide. Appl. Catal. B 2016, 180, 263–270. [Google Scholar] [CrossRef]
- López-Caballero, P.; Hauser, A.W.; de Lara-Castells, M.P. Exploring the Catalytic Properties of Unsupported and TiO2-Supported Cu5 Clusters: CO2 Decomposition to CO and CO2 Photoactivation. J. Phys. Chem. C 2019, 123, 23064–23074. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Xie, Y.; Chen, J.; Hu, Z.; Cui, D. Selective photocatalytic CO2 reduction on copper–titanium dioxide: A study of the relationship between CO production and H2 suppression. Chem. Commun. 2019, 55, 8068–8071. [Google Scholar] [CrossRef] [PubMed]
- Chao Liu, C.; Nauert, S.L.; Alsina, M.A.; Wang, D.; Grant, A.; He, K.; Weitz, E.; Nolan, M.; Gray, K.A.; Notestein, J.M. Role of surface reconstruction on Cu/TiO2 nanotubes for CO2 conversion. Appl. Catal. B Environ. 2019, 255, 117754. [Google Scholar]
- Xie, S.; Wang, Y.; Zhang, Q.; Deng, W.; Wang, Y. MgO- and Pt-Promoted TiO2 as an Efficient Photocatalyst for the Preferential Reduction of Carbon Dioxide in the Presence of Water. ACS Catal. 2014, 4, 3644–3653. [Google Scholar] [CrossRef]
- Jin, L.; Shaaban, E.; Bamonte, S.; Cintron, D.; Shuster, S.; Zhang, L.; Li, G.; He, J. Surface Basicity of Metal@TiO2 to Enhance Photocatalytic Efficiency for CO2 Reduction. ACS Appl. Mater. Interfaces 2021, 13, 38595–38603. [Google Scholar] [CrossRef]
- Li, N.; Liu, M.; Yang, B.; Shu, W.; Shen, Q.; Liu, M.; Zhou, J. Enhanced photocatalytic performance toward CO2 hydrogeneration over nanosized TiO2-loaded Pd under UV irradiation. J. Phys. Chem. C 2017, 121, 2923–2932. [Google Scholar] [CrossRef]
- Zhou, R.; Rui, N.; Fan, Z.; Liu, C.-J. Effect of the structure of Ni/TiO2 catalyst on CO2 methanation. Int. J. Hydrogen Energy 2016, 41, 22017–22025. [Google Scholar] [CrossRef]
- Li, Y.; Rao, Z.; Liu, Z.; Zeng, J.; Bao, W.; Wang, Z.; Li, J.; Yu, F.; Dai, B.; Zhou, Y. Photo-Assisted CO/CO2 Methanation over Ni/TiO2 Catalyst: Experiment and Density Functional Theory Calculation. ChemCatChem 2022, 14, e202200182. [Google Scholar] [CrossRef]
- Vrijburg, W.L.; Moioli, E.; Chen, W.; Zhang, M.; Terlingen, B.J.P.; Zijlstra, B.; Filot, I.A.W.; Zuttel, A.; Pidko, E.A.; Hensen, E.J.M. Efficient Base-Metal NiMn/TiO2 Catalyst for CO2 Methanation. ACS Catal. 2019, 9, 7823–7839. [Google Scholar] [CrossRef] [Green Version]
- Kohno, Y.; Hayashi, H.; Takenaka, S.; Tanaka, T.; Funabiki, T.; Yoshida, S. Photo-enhanced reduction of carbon dioxide with hydrogen over Rh/TiO2. J. Photochem. Photobiol. A 1999, 126, 117–123. [Google Scholar] [CrossRef]
- Zhou, J.; Gao, Z.; Xiang, G.; Zhai, T.; Liu, Z.; Zhao, W.; Liang, X.; Wang, L. Interfacial compatibility critically controls Ru/TiO2 metal-support interaction modes in CO2 hydrogenation. Nat. Commun. 2022, 13, 327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z.; Yang, X.; Wang, R.; Duan, H.; Shen, Z.; Li, L.; Su, Y.; Yang, R.; Zhang, Y.; et al. Tuning selectivity of CO2 hydrogenation by modulating the strong metal–support interaction over Ir/TiO2 catalysts. Green Chem. 2020, 22, 6855–6861. [Google Scholar] [CrossRef]
- Liu, Y.; Miao, C.; Yang, P.; He, Y.; Feng, J.; Li, D. Synergetic promotional effect of oxygen vacancy-rich ultrathin TiO2 and photochemical induced highly dispersed Pt for photoreduction of CO2 with H2O. Appl. Catal. B Environ. 2019, 244, 919–930. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, L.; Huang, C.; Ren, Z.; Wang, H.; Hu, J.; Zhang, H.; Jiang, Z.; Song, F. Exploring the CO2 reduction reaction mechanism on Pt/TiO2 with the ambient-pressure X-ray photoelectron spectroscopy. Appl. Surf. Sci. 2021, 568, 150933. [Google Scholar] [CrossRef]
- Qiu-ye, L.; Lan-Lan, Z.; Chen, L.; Yu-hui, C.; Xiao-Dong, W.; Jian-Jun, Y. Photocatalytic Reduction of CO2 to Methane on Pt/TiO2 Nanosheet Porous Film. Adv. Condens. Matter Phys. 2014, 316589. [Google Scholar]
- Permporn, D.; Khunphonoi, R.; Wilamat, J.; Khemthong, P.; Chirawatkul, P.; Butburee, T.; Sangkhun, W.; Wantala, K.; Grisdanurak, N.; Santatiwongchai, J.; et al. Insight into the Roles of Metal Loading on CO2 Photocatalytic Reduction Behaviors of TiO2. Nanomaterials 2022, 12, 474. [Google Scholar] [CrossRef]
- Neatu, S.; Maciá-Agulló, J.A.; Concepción, P.; Garcia, H. Gold–Copper Nanoalloys Supported on TiO2 as Photocatalysts for CO2 Reduction by Water. J. Am. Chem. Soc. 2014, 136, 15969–15976. [Google Scholar] [CrossRef]
- Reñones, P.; Collado, L.; Iglesias-Juez, A.; Oropeza, F.E.; Fresno, F.; de la Peña-O’Shea, V.A. Silver–Gold Bimetal-Loaded TiO2 Photocatalysts for CO2 Reduction. Ind. Eng. Chem. Res. 2020, 59, 9440–9450. [Google Scholar] [CrossRef]
- Singhal, N.; Kumar, U. Noble metal modified TiO2: Selective photoreduction of CO2 to hydrocarbons. Mol. Catal. 2017, 439, 91–99. [Google Scholar] [CrossRef]
- Ruland, H.; Song, H.; Laudenschleger, D.; Stürmer, S.; Schmidt, S.; He, J.; Kähler, K.; Muhler, M.; Schlögl, R. CO2 Hydrogenation with Cu/ZnO/Al2O3: A Benchmark Study. ChemCatChem 2020, 12, 3216–3222. [Google Scholar] [CrossRef]
- Kattel, S.; Ramirez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, H.; Nie, R.; Wu, G.; Hou, Z. Hydrogenation of CO2 to CH3OH over Cu/ZnO catalysts with different ZnO morphology. Fuel 2015, 154, 161–166. [Google Scholar] [CrossRef]
- Mahapatra, M.; Gutiérrez, R.A.; Kang, J.; Rui, N.; Hamlyn, R.; Liu, Z.; Orozco, I.; Ramírez, P.J.; Senanayake, S.D.; Rodriguez, J.A. The behavior of inverse oxide/metal catalysts: CO oxidation and water-gas shift reactions over ZnO/Cu(111) surfaces. Surf. Sci. 2019, 681, 116–121. [Google Scholar] [CrossRef]
- Le Valant, A.; Comminges, C.; Tisseraud, C.; Canaff, C.; Pinard, L.; Pouilloux, Y. The Cu–ZnO synergy in methanol synthesis from CO2, Part 1: Origin of active site explained by experimental studies and a sphere contact quantification model on Cu+ZnO mechanical mixtures. J. Catal. 2015, 324, 41–49. [Google Scholar] [CrossRef]
- Marcos, F.C.F.; Lin, L.; Betancourt, L.E.; Senanayake, S.D.; Rodriguez, J.A.; Assaf, J.M.; Giudici, R.; Assaf, E.M. Insights into the methanol synthesis mechanism via CO2 hydrogenation over Cu-ZnO-ZrO2 catalysts: Effects of surfactant/Cu-Zn-Zr molar ratio. J. CO2 Util. 2020, 41, 101215. [Google Scholar] [CrossRef]
- Liao, F.; Huang, Y.; Ge, J.; Zheng, W.; Tedsree, K.; Collier, P.; Hong, X.; Tsang, S.C.; Liao, F.; Huang, Y.; et al. Morphology-dependent interactions of ZnO with Cu nanoparticles at the materials' interface in selective hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2011, 50, 2162–2165. [Google Scholar] [CrossRef]
- Phongprueksathat, N.; Bansode, A.; Toyao, T.; Urakawa, A. Greener and facile synthesis of Cu/ZnO catalysts for CO2 hydrogenation to methanol by urea hydrolysis of acetates. RSC Adv. 2021, 11, 14323–14333. [Google Scholar] [CrossRef]
- Guzmán, H.; Salomone, F.; Bensaid, S.; Castellino, M.; Russo, N.; Hernández, S. CO2 Conversion to Alcohols over Cu/ZnO Catalysts: Prospective Synergies between Electrocatalytic and Thermocatalytic Routes. ACS Appl. Mater. Interfaces 2022, 14, 517–530. [Google Scholar] [CrossRef]
- Sakurai, H.; Tsubota, S.; Haruta, M. Hydrogenation of CO2 over gold supported on metal oxides. Appl. Catal. A 1993, 102, 125–136. [Google Scholar] [CrossRef]
- Chen, S.; Abdel-Mageed, A.M.; Hauble, A.; Ishida, T.; Murayama, T.; Parlinska-Wojtan, M.; Behm, R.J. Performance of Au/ZnO catalysts in CO2 reduction to methanol: Varying the Au loading / Au particle size. Appl. Catal. A Gen. 2021, 624, 118318. [Google Scholar] [CrossRef]
- Hartadi, Y.; Widmann, D.; Behm, R.J. Methanol synthesis via CO2 hydrogenation over a Au/ZnO catalyst: An isotope labelling study on the role of CO in the reaction process. Phys. Chem. Chem. Phys. 2016, 18, 10781–10791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Mageed, A.M.; Klyushin, A.; Rezvani, A.; Knop-Gericke, A.; Schlögl, R.; Behm, R.J. Negative Charging of Au Nanoparticles during Methanol Synthesis from CO2 /H2 on a Au/ZnO Catalyst: Insights from Operando IR and Near-Ambient-Pressure XPS and XAS Measurements. Angew. Chem. Int. Ed. 2019, 58, 10325–10329. [Google Scholar] [CrossRef]
- Chen, S.; Abdel-Mageed, A.M.; Mochizuki, C.; Ishida, T.; Murayama, T.; Rabeah, J.; Parlinska-Wojtan, M.; Brückner, A.; Behm, R.J. Formation and Performance of Au/ZnO Catalysts in CO2 Reduction to Methanol by the ZnO Particle Size. ACS Catal. 2021, 11, 9022–9033. [Google Scholar] [CrossRef]
- Liao, W.; Tang, C.; Zheng, H.; Ding, J.; Zhang, K.; Wang, H.; Lu, J.; Huang, W.; Zhang, Z. Tuning activity and selectivity of CO2 hydrogenation via metal-oxide interfaces over ZnO-supported metal catalysts. J. Catal. 2022, 407, 126–140. [Google Scholar] [CrossRef]
- Dreyer, J.A.H.; Li, P.; Zhang, L.; Khai Beh, G.; Zhang, R.; Sit, P.H.-L.; Yang Teoh, W. Influence of the oxide support reducibility on the CO2 methanation over Ru-based catalysts. Appl. Catal. B Environ. 2017, 219, 715–726. [Google Scholar] [CrossRef]
- Wu, D.; Deng, K.; Hu, B.; Lu, Q.; Liu, G.; Hong, X. Plasmon-Assisted Photothermal Catalysis of Low-Pressure CO2 Hydrogenation to Methanol over Pd/ZnO Catalyst. ChemCatChem 2019, 11, 1598–1601. [Google Scholar] [CrossRef]
- Wang, F.; Wei, M.; Evans, D.G.; Duan, X. CeO2-based heterogeneous catalysts toward catalytic conversion of CO2. J. Mater. Chem. A 2016, 4, 5773–5783. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction. Catalysts 2022, 12, 1101. [Google Scholar] [CrossRef]
- Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A.E.; Evans, J.; Senanayake, S.D.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Fernández Sanz, J.; et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 2014, 345, 546–550. [Google Scholar] [CrossRef]
- Wang, M.; Shen, M.; Jin, X.; Tian, J.; Li, M.; Zhou, Y.; Zhang, L.; Li, Y.; Shi, J. Oxygen Vacancy Generation and Stabilization in CeO2–x by Cu Introduction with Improved CO2 Photocatalytic Reduction Activity. ACS Catal. 2019, 9, 4573–4581. [Google Scholar] [CrossRef]
- Lin, L.; Yao, S.; Liu, Z.; Zhang, F.; Li, N.; Vovchok, D.; Martínez-Arias, A.; Castañeda, R.; Lin, J.; Senanayake, S.D.; et al. In Situ Characterization of Cu/CeO2 Nanocatalysts for CO2 Hydrogenation: Morphological Effects of Nanostructured Ceria on the Catalytic Activity. J. Phys. Chem. C 2018, 122, 12934–12943. [Google Scholar] [CrossRef]
- Figueiredo, W.T.; Escudero, C.; Pérez-Dieste, V.; Ospina, C.A.; Bernardi, F. Determining the Surface Atomic Population of CuxNi1–x/CeO2 (0 < x ≤ 1) Nanoparticles during the Reverse Water–Gas Shift (RWGS) Reaction. J. Phys. Chem. C 2020, 124, 16868–16878. [Google Scholar]
- Yan, Y.; Wong, R.J.; Ma, Z.; Donat, F.; Xi, S.; Saqline, S.; Fan, Q.; Du, Y.; Borgna, A.; He, Q.; et al. CO2 hydrogenation to methanol on tungsten-doped Cu/CeO2 catalysts. Appl. Catal. B Environ. 2022, 306, 121098. [Google Scholar] [CrossRef]
- Li, M.; Pham, T.H.M.; Ko, Y.; Zhao, K.; Zhong, L.; Luo, W.; Züttel, A. Support-Dependent Cu–In Bimetallic Catalysts for Tailoring the Activity of Reverse Water Gas Shift Reaction. ACS Sustain. Chem. Eng. 2022, 10, 1524–1535. [Google Scholar] [CrossRef]
- Yang, L.; Pastor-Pérez, L.; Villora-Pico, J.J.; Sepúlveda-Escribano, A.; Tian, F.; Zhu, M.; Han, Y.-F.; Ramirez Reina, T. Highly Active and Selective Multicomponent Fe–Cu/CeO2–Al2O3 Catalysts for CO2 Upgrading via RWGS: Impact of Fe/Cu Ratio. ACS Sustain. Chem. Eng. 2021, 9, 12155–12166. [Google Scholar] [CrossRef]
- Wang, L.-C.; Khazaneh, M.T.; Widmann, D.; Behm, R.J. TAP reactor studies of the oxidizing capability of CO2 on a Au/CeO2 catalyst—A first step toward identifying a redox mechanism in the Reverse Water–Gas Shift reaction. J. Catal. 2013, 302, 20–30. [Google Scholar] [CrossRef]
- Wang, L.; Widmann, D.; Behm, R.J. Reactive removal of surface oxygen by H2, CO and CO/H2 on a Au/CeO2 catalyst and its relevance to the preferential CO oxidation (PROX) and reverse water gas shift (RWGS) reaction. Catal. Sci. Technol. 2015, 5, 925–941. [Google Scholar] [CrossRef]
- Lu, B.; Quan, F.; Sun, Z.; Jia, F.; Zhang, L. Photothermal reverse-water-gas-shift over Au/CeO2 with high yield and selectivity in CO2 conversion. Catal. Commun. 2019, 129, 105724. [Google Scholar] [CrossRef]
- Rezvani, A.; Abdel-Mageed, A.M.; Ishida, T.; Murayama, T.; Parlinska-Wojtan, M.; Behm, R.J. CO2 Reduction to Methanol on Au/CeO2 Catalysts: Mechanistic Insights from Activation/Deactivation and SSITKA Measurements. ACS Catal. 2020, 10, 3580–3594. [Google Scholar] [CrossRef]
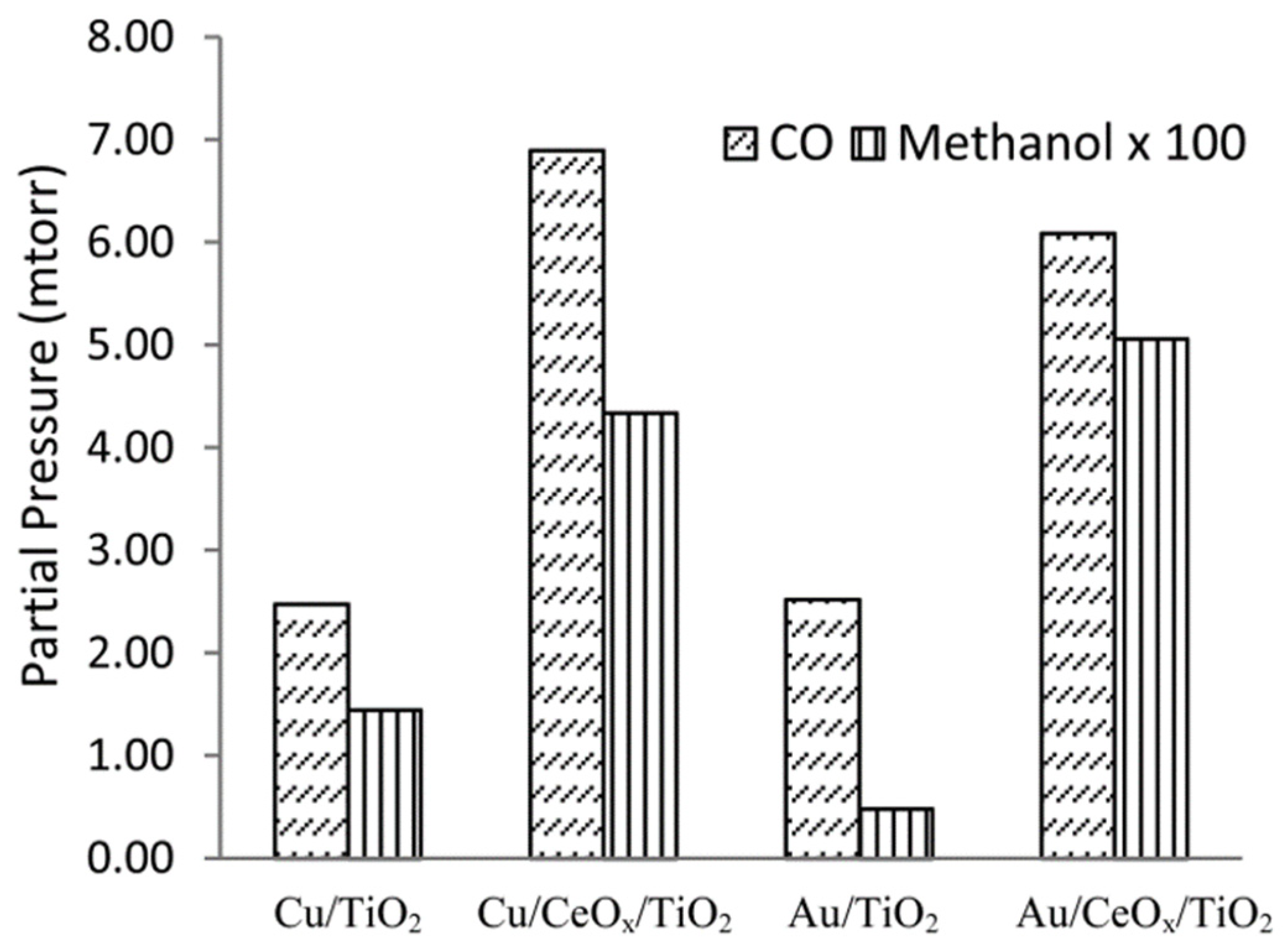
- Yang, X.; Kattel, S.; Senanayake, S.D.; Boscoboinik, J.A.; Nie, X.; Graciani, J.; Rodríguez, J.A.; Liu, P.; Stacchiola, D.J.; Chen, J.G. Low Pressure CO2 Hydrogenation to Methanol over Gold Nanoparticles Activated on a CeOx/TiO2 Interface. J. Am. Chem. Soc. 2015, 137, 10104–10107. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, J.; Chu, M.; Yue, J.; Cui, Y.; Xu, G. Cooperation between active metal and basic support in Ni-based catalyst for low-temperature CO2 methanation. Catal. Lett. 2020, 150, 1418–1426. [Google Scholar] [CrossRef]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrogen Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, M.S.; Lee, S.H.; Kim, T.W.; Park, E.D. CO and CO2 methanation over supported Ni catalysts. Catal. Today 2017, 293-294, 89–96. [Google Scholar] [CrossRef]
- Rui, N.; Zhang, X.; Zhang, F.; Liu, Z.; Cao, X.; Xie, Z.; Zou, R.; Senanayake, S.D.; Yang, Y.; Rodriguez, J.A.; et al. Highly active Ni/CeO2 catalyst for CO2 methanation: Preparation and characterization. Appl. Catal. B Environ. 2021, 282, 119581. [Google Scholar] [CrossRef]
- Bian, Z.; Chan, Y.M.; Yu, Y.; Kawi, S. Morphology dependence of catalytic properties of Ni/CeO2 for CO2 methanation: A kinetic and mechanism study. Catal. Today 2020, 347, 31–38. [Google Scholar] [CrossRef]
- Jomjaree, T.; Sintuya, P.; Srifa, A.; Koo-amornpattana, W.; Kiatphuengporn, S.; Assabumrungrat, S.; Sudoh, M.; Watanabe, R.; Fukuhara, C.; Ratchahat, S. Catalytic performance of Ni catalysts supported on CeO2 with different morphologies for low-temperature CO2 methanation. Catal. Today 2021, 375, 234–244. [Google Scholar] [CrossRef]
- Varvoutis, G.; Lykaki, M.; Stefa, S.; Binas, V.; Marnellos, G.E.; Konsolakis, M. Deciphering the role of Ni particle size and nickel-ceria interfacial perimeter in the low-temperature CO2 methanation reaction over remarkably active Ni/CeO2 nanorods. Appl. Catal. B Environ. 2021, 297, 120401. [Google Scholar] [CrossRef]
- Lin, L.; Gerlak, C.A.; Liu, C.; Llorca, J.; Yao, S.; Rui, N.; Zhang, F.; Liu, Z.; Zhang, S.; Deng, K.; et al. Effect of Ni particle size on the production of renewable methane from CO2 over Ni/CeO2 catalyst. J. Energy Chem. 2021, 61, 602–611. [Google Scholar] [CrossRef]
- Winter, L.R.; Gomez, E.; Yan, B.; Yao, S.; Chen, J.G. Tuning Ni-catalyzed CO2 hydrogenation selectivity via Ni-ceria support interactions and Ni-Fe bimetallic formation. Appl. Catal. B Environ. 2018, 224, 442–450. [Google Scholar] [CrossRef]
- Sun, C.; Beaunier, P.; La Parola, V.; Liotta, L.F.; Da Costa, P. Ni/CeO2 Nanoparticles Promoted by Yttrium Doping as Catalysts for CO2 Methanation. ACS Appl. Nano Mater. 2020, 3, 12355–12368. [Google Scholar] [CrossRef]
- Alvarez-Galvan, C.; Lustemberg, P.G.; Oropeza, F.E.; Bachiller-Baeza, B.; Dapena Ospina, M.; Herranz, M.; Cebollada, J.; Collado, L.; Campos-Martin, J.M.; de la Peña-O’Shea, V.A.; et al. Highly Active and Stable Ni/La-Doped Ceria Material for Catalytic CO2 Reduction by Reverse Water-Gas Shift Reaction. ACS Appl. Mater. Interfaces 2022, 14, 50739–50750. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pastor-Pérez, L.; Gu, S.; Sepúlveda-Escribano, A.; Reina, T.R. Highly efficient Ni/CeO2-Al2O3 catalysts for CO2 upgrading via reverse water-gas shift: Effect of selected transition metal promoters. Appl. Catal. B Environ. 2018, 232, 464471. [Google Scholar] [CrossRef]
- Xie, F.; Xu, S.; Deng, L.; Xie, H.; Zhou, G. CO2 hydrogenation on Co/CeO2-δ catalyst: Morphology effect from CeO2 support. Int. J. Hydrogen Energy 2020, 45, 26938–26952. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Kim, H.B.; Park, E.D. CO and CO2 Methanation over CeO2-Supported Cobalt Catalysts. Catalysts 2022, 12, 212. [Google Scholar] [CrossRef]
- López-Rodríguez, S.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; Bueno-López, A. Effect of Ru loading on Ru/CeO2 catalysts for CO2 methanation. Mol. Catal. 2021, 515, 111911. [Google Scholar] [CrossRef]
- López-Rodríguez, S.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; Herrera, F.C.; Pellegrin, E.; Escudero, C.; García-Melchor, M.; Bueno-López, A. Elucidating the Role of the Metal Catalyst and Oxide Support in the Ru/CeO2-Catalyzed CO2 Methanation Mechanism. J. Phys. Chem. C 2021, 125, 25533–25544. [Google Scholar] [CrossRef]
- Guo, Y.; Mei, S.; Yuan, K.; Wang, D.-J.; Liu, H.-C.; Yan, C.-H.; Zhang, Y.-W. Low-Temperature CO2 Methanation over CeO2-Supported Ru Single Atoms, Nanoclusters, and Nanoparticles Competitively Tuned by Strong Metal–Support Interactions and H-Spillover Effect. ACS Catal. 2018, 8, 6203–6215. [Google Scholar] [CrossRef]
- Panaritis, C.; Edake, M.; Couillard, M.; Einakchi, R.; Baranova, E.A. Insight towards the role of ceria-based supports for reverse water gas shift reaction over RuFe nanoparticles. J. CO2 Util. 2018, 26, 350–358. [Google Scholar] [CrossRef]
- Yang, L.; Pastor-Pérez, L.; Villora-Pico, J.J.; Gu, S.; Sepúlveda-Escribano, A.; Reina, T.R. CO2 valorisation via reverse water-gas shift reaction using promoted Fe/CeO2-Al2O3 catalysts: Showcasing the potential of advanced catalysts to explore new processes design. Appl. Catal. A Gen. 2020, 593, 117442. [Google Scholar] [CrossRef]
- Lou, Y.; Jiang, F.; Zhu, W.; Wang, L.; Yao, T.; Wang, S.; Yang, B.; Yang, B.; Zhu, Y.; Liu, X. CeO2 supported Pd dimers boosting CO2 hydrogenation to ethanol. Appl. Catal. B Environ. 2021, 291, 120122. [Google Scholar] [CrossRef]
- Jiang, F.; Wang, S.; Liu, B.; Liu, J.; Wang, L.; Xiao, Y.; Xu, Y.; Liu, X. Insights into the Influence of CeO2 Crystal Facet on CO2 Hydrogenation to Methanol over Pd/CeO2 Catalysts. ACS Catal. 2020, 10, 11493–11509. [Google Scholar] [CrossRef]
- Zhang, F.; Gutiérrez, R.A.; Lustemberg, P.G.; Liu, Z.; Rui, N.; Wu, T.; Ramírez, P.J.; Xu, W.; Idriss, H.; Ganduglia-Pirovano, M.V.; et al. Metal–Support Interactions and C1 Chemistry: Transforming Pt-CeO2 into a Highly Active and Stable Catalyst for the Conversion of Carbon Dioxide and Methane. ACS Catal. 2021, 11, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Goguet, A.; Meunier, F.; Breen, J.P.; Burch, R.; Petch, M.I.; Ghenciu, A.F. Study of the origin of the deactivation of a Pt/CeO2 catalyst during reverse water gas shift (RWGS) reaction. J. Catal. 2004, 226, 382–392. [Google Scholar] [CrossRef]
- Zheng, K.; Li, Y.; Liu, B.; Jiang, F.; Xu, Y.; Liu, X. Ti-doped CeO2 Stabilized Single-Atom Rhodium Catalyst for Selective and Stable CO2 Hydrogenation to Ethanol. Angew. Chem. Int. Ed. 2022, 61, e202210991. [Google Scholar] [CrossRef] [PubMed]
- Toth, L.E. Transition Metal Carbides and Nitrides; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Oyama, S.T. The Chemistry of Transition Metal Carbides and Nitrides; Blackie Academic and Professional: Scotland, UK, 1996. [Google Scholar]
- Levy, R.B.; Boudart, M. Platinum-Like Behavior of Tungsten Carbide in Surface Catalysis. Science 1973, 181, 547–549. [Google Scholar] [CrossRef]
- Woo, H.C.; Park, K.Y.; Kim, Y.G.; Nam, I.S.; Chung, J.S.; Lee, J.S. Mixed alcohol synthesis from carbon monoxide and dihydrogen over potassium-promoted molybdenum carbide catalysts. Appl. Catal. 1991, 75, 267–280. [Google Scholar] [CrossRef]
- Kitchin, J.R.; Nørskov, J.K.; Barteau, M.A.; Chen, J.G. Trends in the chemical properties in early transition metal carbide surfaces: A density functional study. Catal. Today 2005, 105, 66–73. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Viñes, F.; Illas, F.; Liu, P.; Takahashi, Y.; Nakamura, K. Adsorption of gold on TiC(001): Au-C interactions and charge polarization. J. Chem. Phys. 2007, 127, 211102. [Google Scholar] [CrossRef]
- Ono, L.K.; Sudfeld, D.; Roldan-Cuenya, B. In situ gas-phase catalytic properties of TiC-supported size-selected gold nanoparticles synthesized by diblock copolymer encapsulation. Surf. Sci. 2006, 600, 5041. [Google Scholar] [CrossRef]
- Ono, L.K.; Roldan-Cuenya, B. Effect of interparticle interaction on the low temperature oxidation of CO over size-selected Au nanocatalysts supported on ultrathin TiC films. Catal. Lett. 2007, 113, 86–94. [Google Scholar] [CrossRef]
- Gomez, T.; Florez, E.; Rodriguez, J.A.; Illas, F. Reactivity of Transition Metals (Pd, Pt, Cu, Ag, Au) toward Molecular Hydrogen Dissociation: Extended Surfaces versus Particles Supported on TiC(001) or Small Is Not Always Better and Large Is Not Always Bad. J. Phys. Chem. C 2011, 115, 11666–11672. [Google Scholar] [CrossRef]
- Asara, G.G.; Feria, L.; Florez, E.; Ricart, J.M.; Liu, P.; Rodriguez, J.A.; Illas, F. Theoretical Study of the Interaction of CO on TiC(001) and Au Nanoparticles Supported on TiC(001): Probing the Nature of the Au/TiC Interface. J. Phys. Chem. C 2011, 115, 22495–22504. [Google Scholar] [CrossRef]
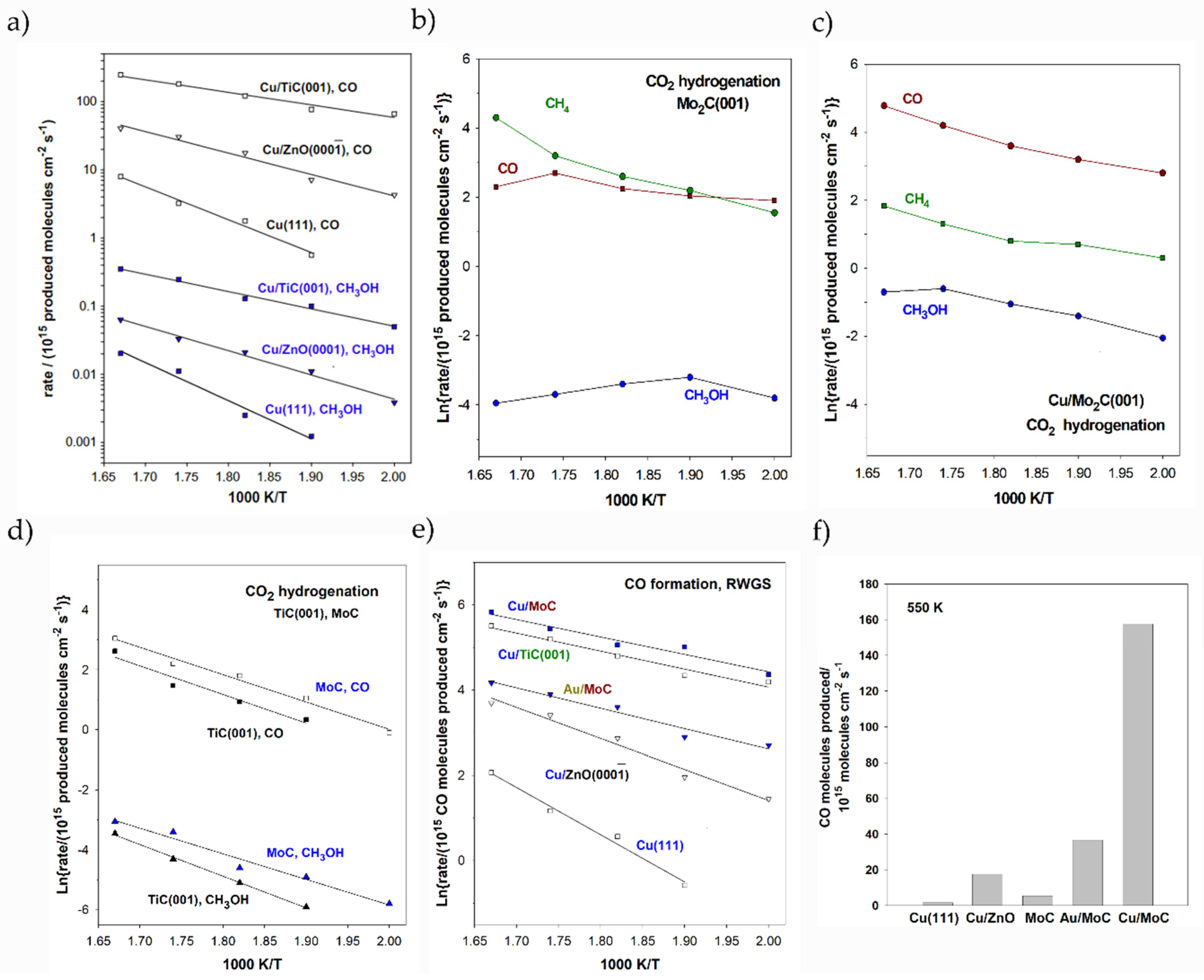
- Vidal, A.B.; Feria, L.; Evans, J.; Takahashi, Y.; Liu, P.; Nakamura, K.; Illas, F.; Rodriguez, J.A. CO2 Activation and Methanol Synthesis on Novel Au/TiC and Cu/TiC Catalysts. J. Phys. Chem. Lett. 2012, 3, 2275–2280. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Evans, J.; Feria, L.; Vidal, A.B.; Liu, P.; Nakamura, K.; Illas, F. CO2 hydrogenation on Au/TiC, Cu/TiC, and Ni/TiC catalysts: Production of CO, methanol, and methane. J. Catal. 2013, 307, 162–169. [Google Scholar] [CrossRef]
- Lozano-Reis, P.; Sayós, R.; Rodriguez, J.A.; Illas, F. Structural, electronic, and magnetic properties of Ni nanoparticles supported on the TiC(001) surface. Phys. Chem. Chem. Phys. 2020, 22, 26145–26154. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Reis, P.; Prats, H.; Sayós, R.; Rodriguez, J.A.; Illas, F. Assessing the Activity of Ni Clusters Supported on TiC(001) toward CO2 and H2 Dissociation. J. Phys. Chem. C 2021, 125, 12019–12027. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Ramirez, P.J.; Gutierrez, R.A.; Stacchiola, D.J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. The conversion of CO2 to methanol on orthorhombic β-Mo2C and Cu/β-Mo2C catalysts: Mechanism for admetal induced change in the selectivity and activity. Catal. Sci. Technol. 2016, 6, 6766–6777. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Ramírez, P.J.; Evans, J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. Highly active Au/δ-MoC and Cu/δ-MoC catalysts for the conversion of CO2: The metal/C ratio as a key factor defining activity, selectivity, and stability. J. Am. Chem. Soc. 2016, 138, 8269–8278. [Google Scholar] [CrossRef] [Green Version]
- Posada-Pérez, S.; Viñes, F.; Ramirez, P.J.; Vidal, A.B.; Rodriguez, J.A.; Illas, F. The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces. Phys. Chem. Chem. Phys. 2014, 16, 14912–14921. [Google Scholar] [CrossRef] [Green Version]
- Posada-Pérez, S.; Viñes, F.; Rodriguez, J.A.; Illas, F. Fundamentals of methanol synthesis on metal carbide based catalysts: Activation of CO2 and H2. Top. Catal. 2015, 58, 159–173. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Viñes, F.; Rodriguez, J.A.; Illas, F. Structure and electronic properties of Cu nanoclusters supported on Mo2C(001) and MoC(001) surfaces. J. Chem. Phys. 2015, 143, 114704. [Google Scholar] [CrossRef] [PubMed]
- Heracleous, E.; Koidi, V.; Lappas, A.A. CO2 conversion over Cu–Mo2C catalysts: Effect of the Cu promoter and preparation method. Catal. Sci. Technol. 2021, 11, 1467–1480. [Google Scholar] [CrossRef]
- Zhang, R.; Wei, C.; Guo, W.; Li, Z.; Wang, B.; Ling, L.; Li, D. Syngas Conversion to C2 Oxygenates over the Cu/β-Mo2C Catalyst: Probing into the Effect of the Interface between Cu and β-Mo2C on Catalytic Performance. J. Phys. Chem. C 2019, 123, 21022–21030. [Google Scholar] [CrossRef]
- Jing, H.; Li, Q.; Wang, J.; Liu, D.; Wu, K. Enhanced N2-Fixation by Engineering the Edges of Two-Dimensional Transition-Metal Disulfides. J. Phys. Chem. C 2019, 123, 1235–1251. [Google Scholar] [CrossRef]
- Zhang, Q.; Pastor-Pérez, L.; Jin, W.; Gu, S.; Reina, T.R. Understanding the promoter effect of Cu and Cs over highly effective β-Mo2C catalysts for the reverse water-gas shift reaction. Appl. Catal. B Environ. 2019, 244, 889–898. [Google Scholar] [CrossRef]
- Delporte, P.; Meunier, F.; Pham-Huu, C.; Vennegues, P.; Ledoux, M.J.; Guille, J. Physical characterization of molybdenum oxycarbide catalyst; TEM, XRD and XPS. Catal. Today 1995, 23, 251–267. [Google Scholar] [CrossRef]
- Dixit, M.; Peng, X.; Porosoff, M.D.; Willauer, H.D.; Mpourmpakis, G. Elucidating the role of oxygen coverage in CO2 reduction on Mo2C. Catal. Sci. Technol. 2017, 7, 5521–5529. [Google Scholar] [CrossRef]
- Yao, L.; Wang, Y.; Galvez, M.E.; Hu, C.; da Costa, P. g-Alumina-Supported Ni-Mo Carbides as Promising Catalysts for CO2 Methanation. Modern Res. Catal. 2017, 6, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Rodriguez, J.A.; Asakura, T.; Gomes, J.; Nakamura, K. Optimization and Application of Lithium Parameters for the Reactive Force Field, ReaxFF. J. Phys. Chem. B 2005, 109, 4575–4582. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A. Water-Gas-Shift Reaction on Molybdenum Carbide Surfaces: Essential Role of the Oxycarbide. J. Phys. Chem. B 2006, 110, 19418–19425. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, C.; Viñes, F.; Illas, F. Transition metal carbides as novel materials for CO2 capture storage, and activation. Energy Environ. Sci. 2016, 9, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Len, T.; Luque, R. Addressing the CO2 challenge through thermocatalytic hydrogenation to carbon monoxide, methanol and methane. Green Chem. 2023, 25, 490–521. [Google Scholar] [CrossRef]
- Nagai, M.; Matsuda, K. Low-temperature water–gas shift reaction over cobalt–molybdenum carbide catalyst. J. Catal. 2006, 238, 489–496. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ramírez, P.J.; Gutierrez, R.A. Highly Active Pt/MoC and Pt/TiC Catalysts for the Low-Temperature Water-Gas Shift Reaction: Effects of the Carbide Metal/Carbon Ratio on the Catalyst Performance. Catal. Today 2017, 289, 47–52. [Google Scholar] [CrossRef]
- Wan, W.; Tackett, B.M.; Chen, J.G. Reactions of water and C1 molecules on carbide and metal-modified carbide surfaces. Chem. Soc. Rev. 2017, 46, 1807–1823. [Google Scholar] [CrossRef]
- Kelly, T.G.; Stottlemyer, A.L.; Ren, H.; Chen, J.G. Comparison of O-H, C-H, and C-O bond scission sequence of methanol on tungsten carbide surfaces modified by Ni, Rh, and Au. J. Phys. Chem. C 2011, 115, 6644–6650. [Google Scholar] [CrossRef]
- Mehdizadeh, S.; Sadjadi, A.; Poater, A.; Mansouri, A.; Bahri-Laleh, N. Molecular modelling aided catalyst design for PAO oils hydrofinishing. J. Mol. Liq. 2022, 352, 118675. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Lang, R.; Du, X.; Huang, Y.; Jiang, X.; Zhang, Q.; Guo, Y.; Liu, K.; Qiao, B.; Wang, A.; Zhang, T. Single-Atom Catalysts Based on the Metal–Oxide Interaction. Chem. Rev. 2020, 120, 11986–12043. [Google Scholar] [CrossRef]
- He, H.; Wang, H.H.; Liu, J.; Liu, X.; Li, W.; Wang, Y. Research Progress and Application of Single-Atom Catalysts: A Review. Molecules 2021, 26, 6501. [Google Scholar] [CrossRef] [PubMed]
- Hannagan, R.T.; Giannakakis, G.; Flytzani-Stephanopoulos, M.; Sykes, E.C.H. Single-Atom Alloy Catalysis. Chem. Rev. 2020, 120, 12044–12088. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Zhang, L.; Doyle-Davis, K.; Sun, X. Single-Atom Catalysts: From Design to Application. Electrochem. Energy Rev. 2019, 2, 539–573. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, G.; Shi, L.; Ye, J. Single-Atom Catalysts: Emerging Multifunctional Materials in Heterogeneous Catalysis. Adv. Energy Mater. 2018, 8, 1701343. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, Z.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Guo, L.W.; Du, P.P.; Fu, X.P.; Ma, C.; Zeng, J.; Si, R.; Huang, Y.Y.; Jia, C.J.; Zhang, Y.W.; Yan, C.H. Contributions of Distinct Gold Species to Catalytic Reactivity for Carbon Monoxide Oxidation. Nat. Commun. 2016, 7, 13481. [Google Scholar] [CrossRef]
- Chen, J.; Iyemperumal, S.K.; Fenton, T.; Carl, A.; Grimm, R.; Li, G.; Deskins, N.A. Synergy between Defects, Photoexcited Electrons, and Supported Single Atom Catalysts for CO2 Reduction. ACS Catal. 2018, 8, 10464–10478. [Google Scholar] [CrossRef]
- Morales-García, A.; Calle-Vallejo, F.; Illas, F. MXenes: New Horizons in Catalysis. ACS Catal. 2020, 10, 13487–13503. [Google Scholar] [CrossRef]
- Zhao, D.; Chen, Z.; Yang, W.; Liu, S.; Zhang, X.; Yu, Y.; Cheong, W.-C.; Zheng, L.; Ren, F.; Ying, G.; et al. MXene (Ti3C2) Vacancy-Confined Single-Atom Catalyst for Efficient Functionalization of CO2. J. Am. Chem. Soc. 2019, 141, 4086–4093. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Metal catalysts for heterogeneous catalysis: From single atoms to nanoclusters and nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [Green Version]
- Jurado, L.; Esvan, J.; Luque-Álvarez, L.A.; Bobadilla, L.F.; Odriozola, J.A.; Posada-Pérez, S.; Poater, A.; Comas-Vives, A.; Axet, M.R. Highly dispersed Rh single atoms over graphitic carbon nitride as a robust catalyst for the hydroformylation reaction. Catal. Sci. Technol. 2023. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | H2:CO2 Ratio | Temperature (°C) | Pressure (MPa) | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|---|
| NiO/CeO2 [37] | 1:1 | 700 | 0.1 | ~40 | ~100 |
| Cu/Al2O3 [40] | 1:9 | 500 | N/A | ~60 | N/A |
| Cu-Ni/γ-Al2O3 [41] | 1:1 | 600 | 0.1 | 28.7 | 79.7 |
| Fe-Mo/γ-Al2O3 [42] | 1:1 | 600 | 1 | ~45 | ~100 |
| Mo/γ-Al2O3 [43] | 1:1 | 600 | 1 | 34.2 | 97 |
| Pt/TiO2 [44] | 1.4:1 | 400 | N/A | ~30 | N/A |
| Pt/Al2O3 [33] | 1.4:1 | 400 | N/A | ~20 | N/A |
| Ni-Mo2C [45] | 5:1 | 250 | 2 | 21 | 29 |
| Co-Mo2C [34] | 5:1 | 250 | 2 | 23 | 24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Posada-Pérez, S.; Solà, M.; Poater, A. Carbon Dioxide Conversion on Supported Metal Nanoparticles: A Brief Review. Catalysts 2023, 13, 305. https://doi.org/10.3390/catal13020305
Posada-Pérez S, Solà M, Poater A. Carbon Dioxide Conversion on Supported Metal Nanoparticles: A Brief Review. Catalysts. 2023; 13(2):305. https://doi.org/10.3390/catal13020305
Chicago/Turabian StylePosada-Pérez, Sergio, Miquel Solà, and Albert Poater. 2023. "Carbon Dioxide Conversion on Supported Metal Nanoparticles: A Brief Review" Catalysts 13, no. 2: 305. https://doi.org/10.3390/catal13020305