1. Introduction
Suzuki-Miyaura cross-coupling is an important and popular reaction with many applications across a wide field, including the synthesis of ligands for catalysis, natural products and pharmaceuticals [
1,
2]. This wide applicability is due to the high tolerance of the reaction to a wide variety of functional groups and the mild reaction conditions that are required [
1,
3,
4,
5,
6]. The reaction typically occurs through the reaction of an aryl boronic acid with a haloarene in the presence of a base with many different catalysts available [
7]. These catalysts are typically Pd and range from heterogeneous nanoparticles to homogeneous catalysts [
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14]. Early catalysts for Suzuki-Miyaura coupling reactions were based on phosphorus ligands, of which [Pd(PPh
3)
4] is a typical example, with the choice of Pd(II) pre-catalysts based on their air stability and their ready reduction to form the active Pd(0) species [
1,
3,
14]. The choice of ligand was also observed to have a major effect. This led to much work being carried out into monoligated species through the development of bulky, electron-rich phosphines or sterically demanding N–Heterocyclic carbene (NHC) ligands [
15,
16]. An important advance in Suzuki-Miyaura cross-coupling reactions is the activation of aryl chlorides due to their decreased cost compared to aryl bromides, which was first achieved by Shen et al. using Pd(OAc)
2 with 1,3 bis(diphenylphosphino)propane (dppp) [
17]. Through tuning the phosphorus ligands, it has been possible to cross-couple a wide variety of coupling partners with aryl chlorides [
18]. Whilst the yields for aryl chlorides are typically poor, high reaction temperatures, such as refluxing
ortho-xylene for 5–20 h, can be used to improve the yields [
19].
Another main class of ligands in this field are NHCs, first utilised for the Suzuki–Miyaura coupling of 4–chloroacetophenone with arylboronic acid by Herrmann and co-workers [
20]. Following this work, a wide range of NHCs have been utilized as ligands for catalytic cross-coupling reactions owing to the strong
σ-donating properties and steric shielding [
16]. A recent development in cross-coupling catalysis is the utilization of well-defined pre-catalysts that do not require the addition of ancillary ligands to metal salts. These catalysts can allow for transformations under milder conditions or at lower catalytic loadings [
21]. These advantages have been demonstrated by Beller and co-workers where the use of a pre-formed catalyst improved performance over the same catalytic system generated in situ [
22].
There is a large drive within the field of chemistry to work towards ‘greener’ reactions. These are summarized as the twelve principles of green chemistry introduced by Anastas and Warner in 1998 [
23]. One of these principles is the choice of safer solvents and auxiliaries [
24]. With many cross-coupling reactions requiring DMF, DMA or toluene heated under reflux, there is space here to improve these conditions and move to more sustainable alternatives since these solvents are classified by several companies as undesirable or in need of substitution where available following environmental, health and safety property analysis [
25,
26]. The use of better media for Suzuki-Miyaura reactions was reviewed recently across a range of solvents and catalyst types [
27,
28]. The switch away from solvents such as DMF is also beneficial when solvents such as methanol are used, as a simple extraction is required rather than a lengthy aqueous workup.
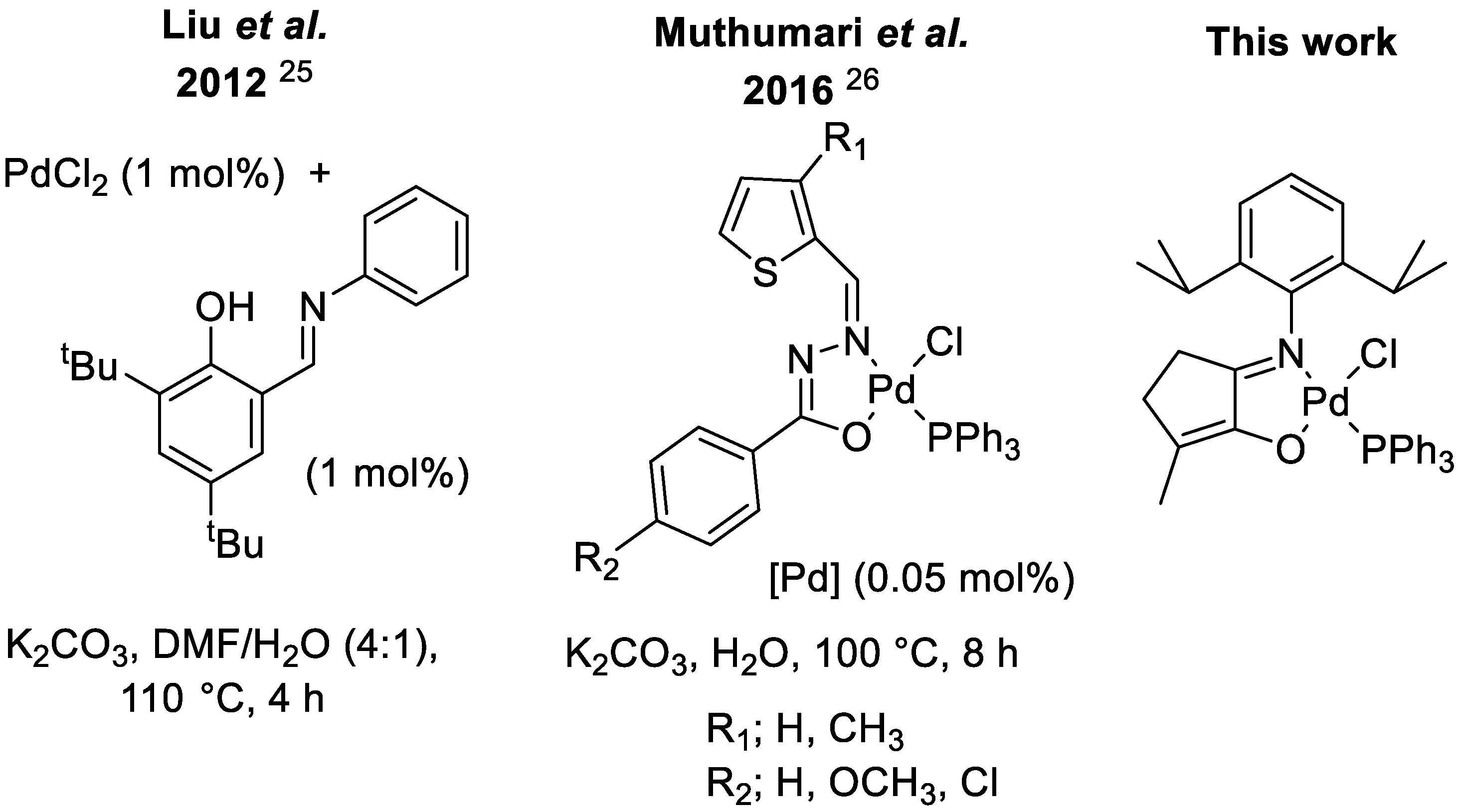
Schiff base-derived ligands have been shown to provide alternatives to classic phosphine-based catalysts with similar easy tuning of their steric and electronic properties [
8,
29,
30,
31,
32]. One catalyst design of note was developed by Liu et al. using salicylaldimine ligands with PdCl
2 to form catalytic species in situ, which were capable of coupling a wide range of aryl bromides between room temperature and 60 °C under 6 h (
Figure 1) [
33]. The use of pre-formed neutral Pd [N,O] catalysts in green solvents has also been observed in recent work by Muthumari et al. which was capable of coupling alkyl bromides with a range of boronic acids in yields of approximately 90% after 3 h at 100 °C in water. The catalyst was also capable of coupling 4–chloroacetophenone to phenyl boronic acid, but this required 8 h at 100 °C [
34].
We have previously developed [N,O]-chelating ligands based on maple lactone, 2-hydroxy–3–methylcyclopent–2–enone, also called cyclotene [
35,
36], which when condensed with a substituted aniline, gave ligands featuring a 5-membered chelate ring and a correspondingly smaller bite-angle than salicylaldimines [
37]. Complexation with [Ni(Ar)(PPh
3)] fragments gave catalysts for ethylene polymerization generating different polyethylene properties depending upon the initiator [
38]. These ligands are based on maple lactone, a cheap, bio-sourced material that is useful in the development of catalysts with inexpensive ligand systems. With limited work into the development of [N,O]-ligated catalysts in cross-coupling, we set out to investigate the use of a Pd catalyst with a 5-membered chelating ring in Suzuki-Miyaura cross-coupling reactions using benign conditions. The work presented herein includes the synthesis, complexation and characterization of an active catalyst, optimization studies for the Suzuki–Miyaura cross-coupling reaction and a substrate scope for both cross-coupling partners.
3. Experimental
Reactions were either performed under an oxygen-free (H2O, O2 < 0.5 ppm) nitrogen atmosphere using standard Schlenk line techniques and an MBRAUN UNIlab Plus glovebox or in the open laboratory, as indicated. Anhydrous toluene, anhydrous DCM and anhydrous THF were obtained from an MBRAUN SPS–800,(MBRAUN, Munich, Germany) and petroleum ether 40–60 was distilled from sodium wire; benzene and benzene-d6 were dried over molten potassium and distilled. All anhydrous solvents were degassed before use and stored over activated molecular sieves.
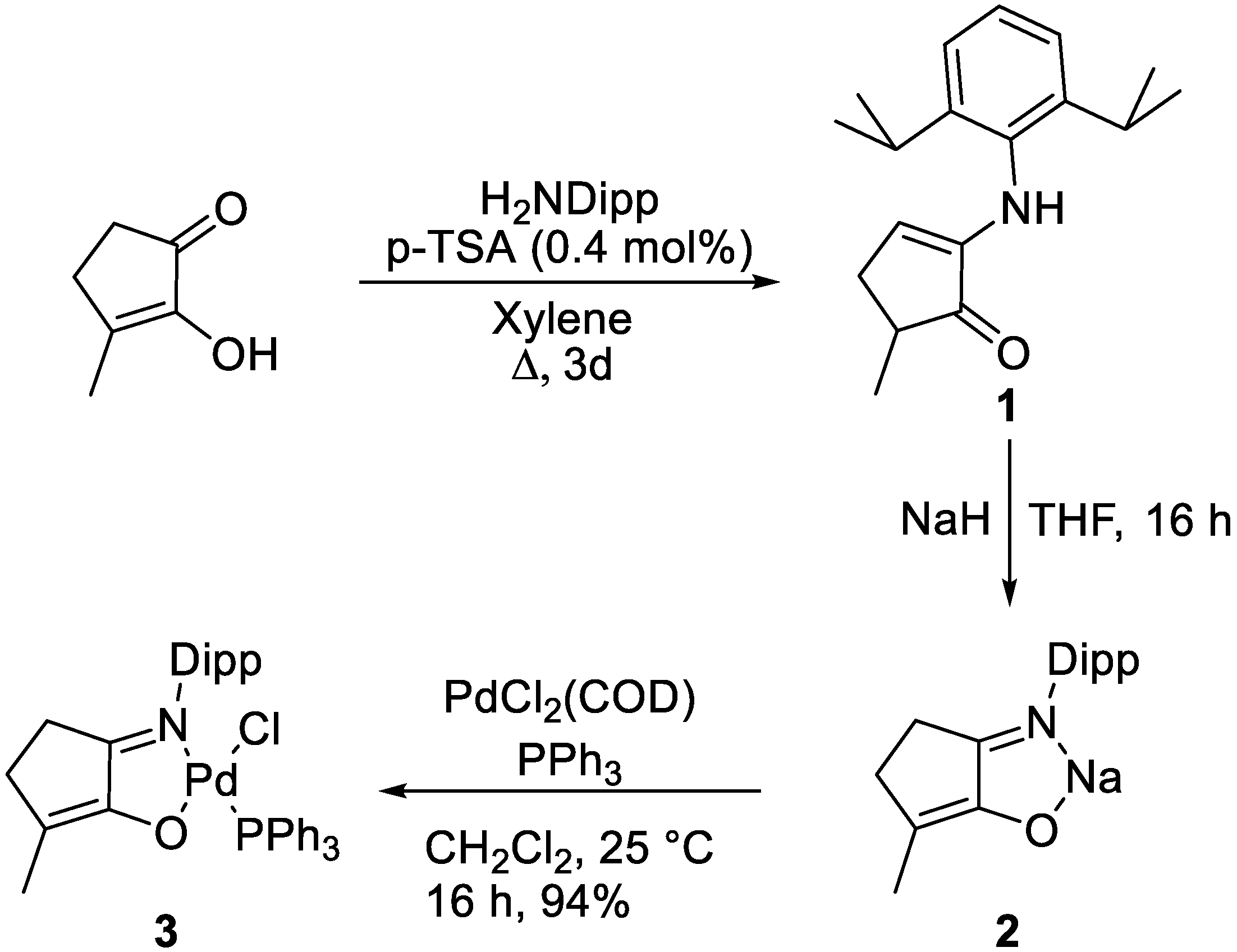
The following compounds were prepared according to the literature methods: 2–((2,6-diisopropylphenyl)amino)–5–methylcyclopent–2–en–1–one (
1),
2, [
38] and PdCl
2(COD) [
46]. The following were purchased from commercial suppliers and used without further purification: maple lactone,
p–toluenesulfonic acid, sodium hydride (95%), palladium chloride, 1,5–cyclooctadiene and triphenylphosphine. 2,6–Diisopropylaniline was distilled under reduced pressure before use. Air-sensitive samples for NMR spectroscopy were prepared in NMR tubes equipped with a J. Young tap. The NMR spectra were recorded on a Bruker AV300 (Bruker, MA, USA) (300 MHz), AVI400 (400 MHz), AVIII400 (400 MHz) or AVHDIII (400 MHz) spectrometer at 25 °C unless specified. Chemical shifts
δ are noted in parts per million (ppm).
1H and
13C spectra were calibrated to the residual proton resonances of the deuterated solvent.
31P NMR spectra were referenced to external samples of 85% H
3PO
4 at 0 ppm. The elemental analyses were performed by Brian Hutton at Heriot-Watt University using an Exeter CE440 elemental analyser. (Exeter Analytical, Coventry, UK)The mass spectrometry analysis was performed at the UK National Mass Spectrometry Facility at Swansea University using an Atmospheric Solids Analysis Probe interfaced to a Waters Xevo G2–S instrument (Waters, MA, USA).
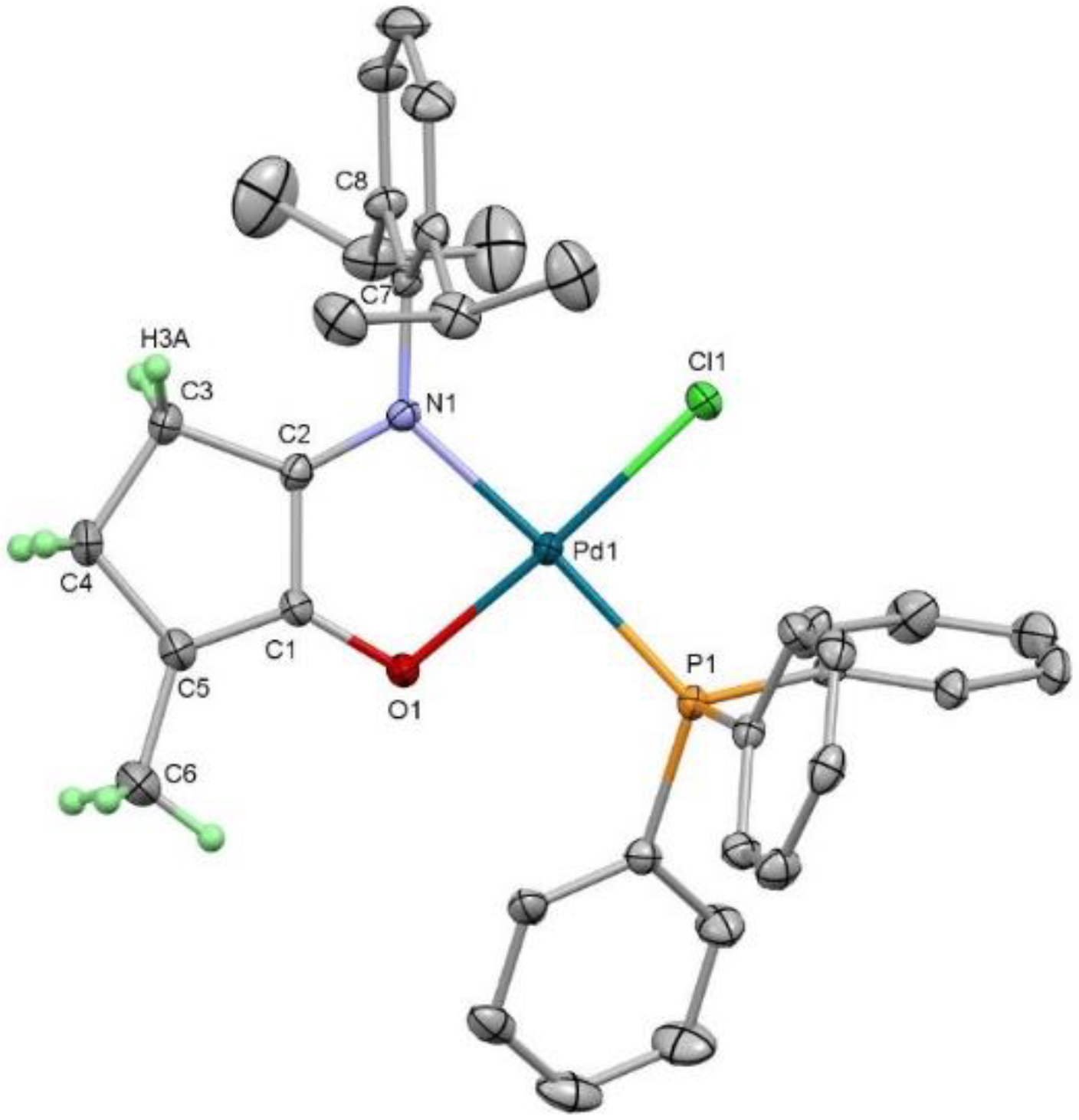
[Pd(Cl)(N,ODipp)(PPh3)] (3)
[PdCl2(cod)] (0.371 g, 1.30 mmol), 2 (0.400 g, 1.365 mmol, 1.05 eq) and PPh3 (0.341 g, 1.30 mmol) were weighed into a Schlenk tube in a glovebox before CH2Cl2 (0.8 mL) was added. The solution was then stirred at room temperature overnight, and the solvent was removed via rotatory evaporation yielding a dark-brown solid. The solid was then dissolved in toluene and filtered before evaporation of the solvent under reduced pressure. Recrystallisation was then carried out using THF/petroleum ether 40–60 to yield the desired product as dark red crystals (0.826 g, 1.22 mmol, 94%).
![Catalysts 13 00303 i008]()
1H NMR (400 Hz, 298 K, C6D6): δ 7.73 (6H, m, H20), 7.46 (m, 3H, H22), 7.38 (m, 6H, H21), 7.16 (3H, m, H9-11), 6.24 (1H, td, J = 7.3, 1.2 Hz, HNi-o-tolyl), 3.46 (2H, sept, J = 7.3 Hz, H13,16), 2.57 (bs, 2H, H4/3), 2.02 (bs, 2H, H3/4), 1.67 (s, 3H, H6), 1.44 (6H, d, J = 6.8 Hz, HiPr), 1.20 (6H, d, J = 6.8 Hz, HiPr). 13C{1H} NMR (100.6 Hz, 298 K, C6D6): δ 194.06 (C), 166.65 (C), 141.25 (C), 140.84 (C), 137.84 (C), 135.10 (CH), 135.00 (CH), 132.32 (CH), 130.80 (CH), 129.16 (C), 129.03 (CH), 128.62 (C), 128.21 (CH), 128.03 (CH), 127.92 (CH), 126.56 (CH), 125.29 (CH), 123.15 (CH), 34.44 (CH2), 28.31 (CH), 24.82 (CH2), 23.99 (CH3), 23.86 (CH2), 21.41 (CH3), 13.15 ppm (CH3). 31P{1H} NMR (162 Hz, 298 K, C6D6): δ 26.6 ppm. HRMS (ASAP): Calculated for [M+H]: 674.1581, Found: 674.1575 m/z; calculated for [M-Cl]: 638.1818, Found: 638.1816 m/z. Elemental analysis calcd (%) for C15H19NO: C; 64.10, H; 5.83, N; 2.08. Found: C; 63.82, H; 5.86, N; 2.21.
3.1. Crystallographic Details
The X-ray diffraction experiments were performed on single crystals of
3 with the data collected on a Bruker X8 APEX II (Bruker, MA, USA) four-circle diffractometer. The crystal was kept at 100 K during the data collection. Indexing, data collection and absorption corrections were performed, and the structures were solved using direct methods (SHELXT [
47] and refined with full-matrix least-squares (SHELXL) interfaced with the programme OLEX2 [
48,
49].
Crystal data for C36H39ClNOPPd (M = 674.50 g/mol): orthorhombic, space group P212121 (no. 19), a = 8.9925(4) Å, b = 16.4896(8) Å, c = 22.0909(10) Å, V = 3275.7(3) Å3, Z = 4, T = 100.15 K, μ(synchrotron) = 0.765 mm−1, Dcalc = 1.368 g/cm3, 103,238 reflections measured (4.552° ≤ 2Θ ≤ 62.764°), 9994 unique (Rint = 0.0781, Rsigma = 0.0398), which were used in all calculations. The final R1 was 0.0298 (I > 2σ(I)), and wR2 was 0.0726 (all data). CCDC deposition number: 2102831.
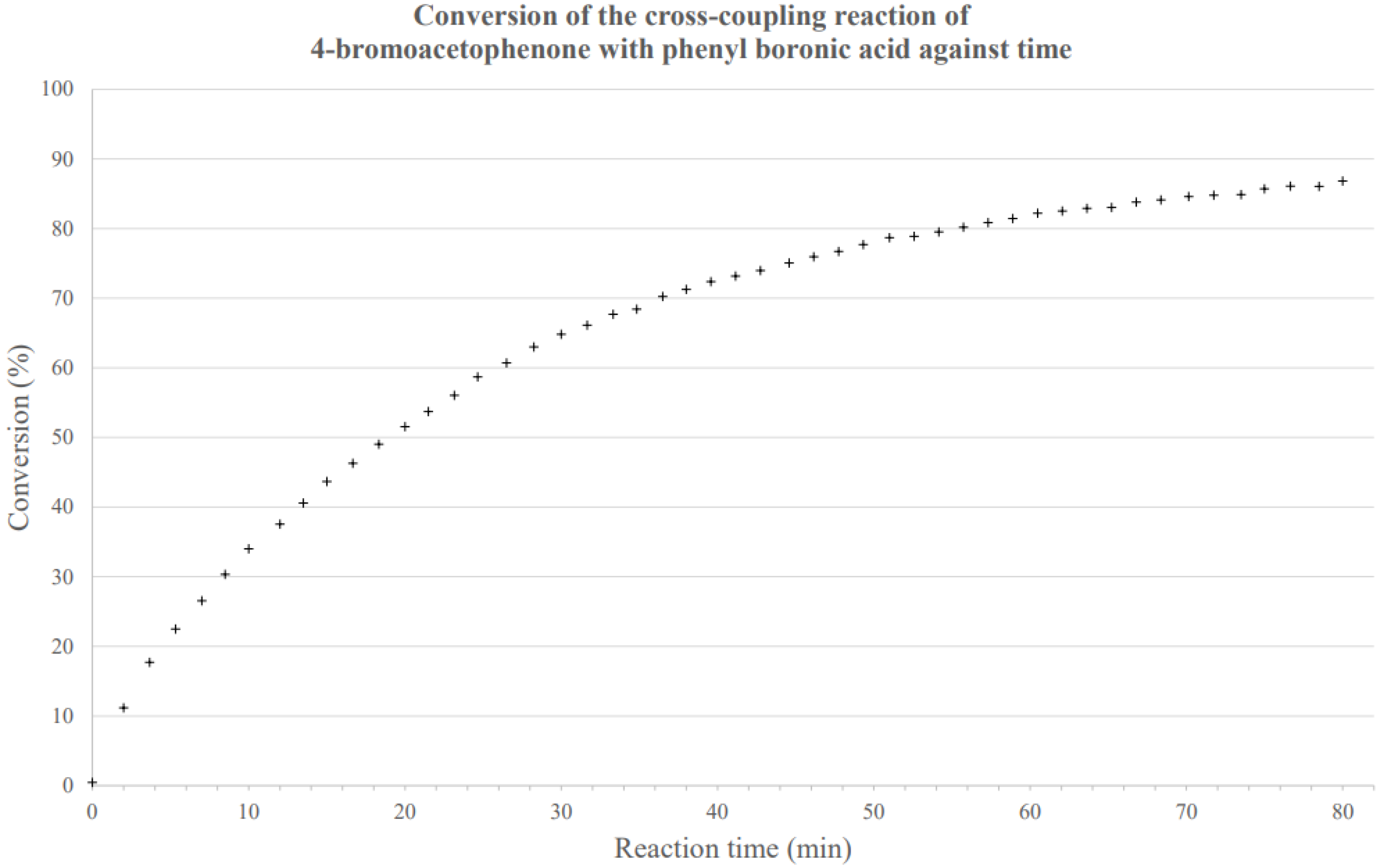
3.2. Suzuki-Miyaura Cross-Coupling Reactions
A typical cross-coupling reaction was carried out as follows:
4–bromoacetophenone (0.100 g, 0.5 mmol, 1 eq.), phenyl boronic acid (0.092 g, 0.754 mmol, 1.5 eq.), Na
2CO
3 (0.1065 g, 1.005 mmol, 2 eq.) and [PdCl(N,O
Dipp)(PPh
3)] (3.4 mg, 0.005 mmol, 1 mol%) were added to a 100 mL round-bottomed flask equipped with a stirrer bar. A solvent mixture of methanol:water 3:1 (10 mL) was added, and the mixture was stirred for 1 h at room temperature. Water (10 mL) was then added to the flask followed by diethyl ether (10 mL), and the organic phase was collected by separation. This was then dried over MgSO
4, filtered and the solvent removed by rotatory evaporation.
1H NMR spectroscopic analysis was then carried out in CDCl
3, and the conversion was calculated through integration of the resonances at 2.60 ppm (starting material) and 2.65 ppm (product). The product was then isolated using column chromatography (199:1 petroleum ether 40–60:ethyl acetate) as an off-white solid (0.069 g, 0.35 mmol, 70%).
1H NMR (400 Hz, 298 K, C6D6): δ 7.92 (2H, d,
J = 8.8 Hz), 7.57 (2H, d,
J = 8.5 Hz), 7.51 (2H, m), 7.36 (2H, m), 7.29 (1H, m), 2.52 (3H, s). The data matches the literature values [
10].
3.3. Cross-Coupling Reaction of 4–Bromoaniline at Elevated Temperatures
4–bromoaniline (0.0864 g, 0.5 mmol, 1 eq.), phenyl boronic acid (0.092 g, 0.754 mmol, 1.5 eq.), Na
2CO
3 (0.1065 g, 1.005 mmol, 2 eq.) and [PdCl(N,O
Dipp)(PPh
3)] (3.4 mg, 0.005 mmol, 1 mol%) were added to a 100 mL round-bottomed flask equipped with a stirrer bar. A solvent mixture of methanol:water 3:1 (10 mL) was added, and the mixture was stirred at 60 °C for 24 h. The mixture was then cooled to room temperature before water (10 mL) was then added to the flask followed by diethyl ether (10 mL), and the organic phase was collected by separation. This was then dried over MgSO
4, filtered and the solvent removed by rotatory evaporation. The product was then isolated using column chromatography (1:1 petroleum ether 40–60:ethyl acetate) with a small amount of DCM added to improve solubility to yield a brown solid (0.0736 g, 0.43 mmol, 87%).
1H NMR (400 Hz, 298 K, C6D6): δ 7.47 (2H, d,
J = 8.3 Hz), 7.34 (4H, m), 7.20 (1H, m), 6.69 (2H, d,
J = 8.4 Hz), 3.66 (2H, br. s, NH
2). The data matches the literature values [
10].









{kind=link}
{kind=link}
{kind=link}
{kind=link}






