
Development of L-Proline-Based Chiral Ionic Liquids for Asymmetric Michael Reaction
 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
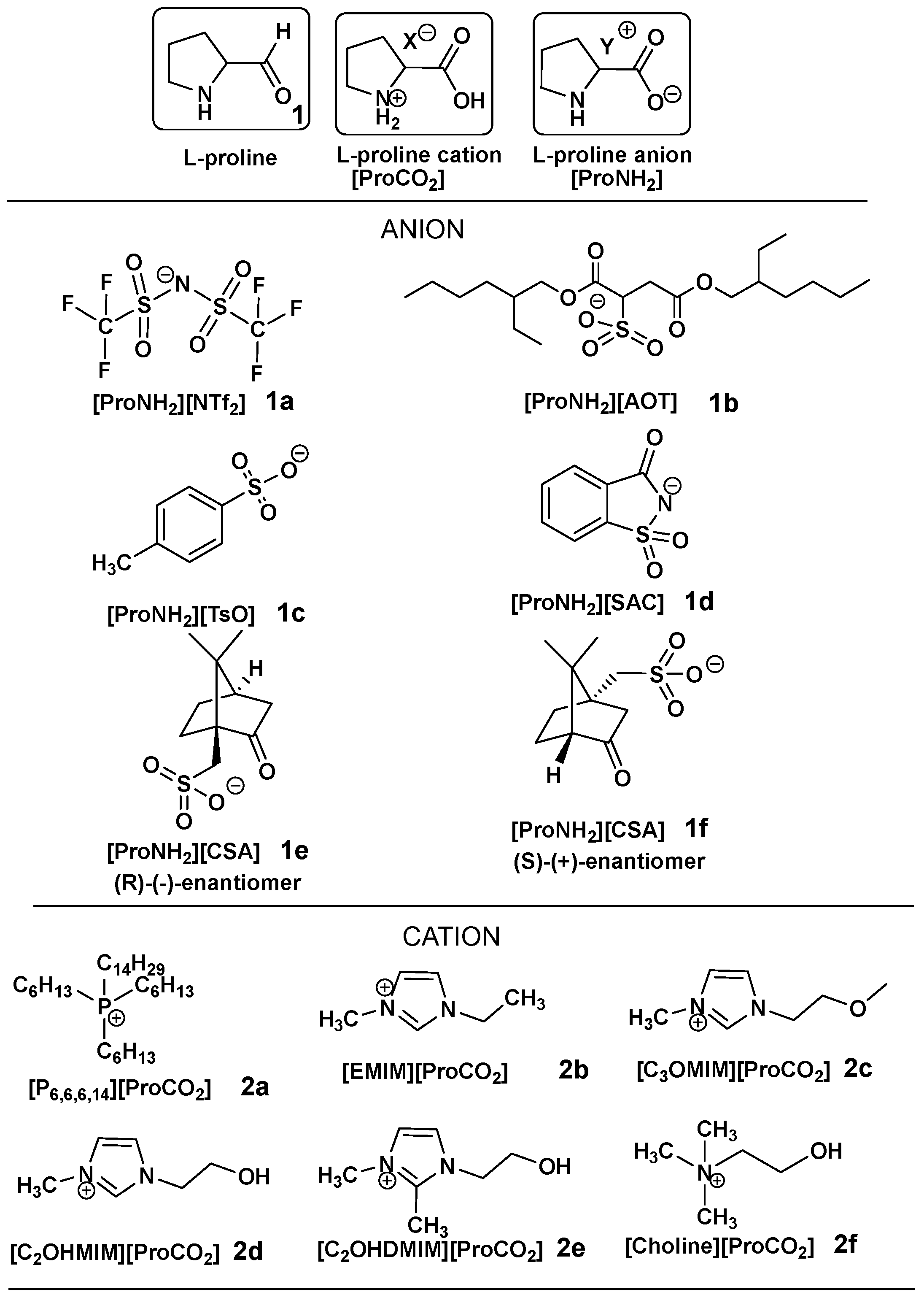
2.1. Design of CILs Based on L-Proline
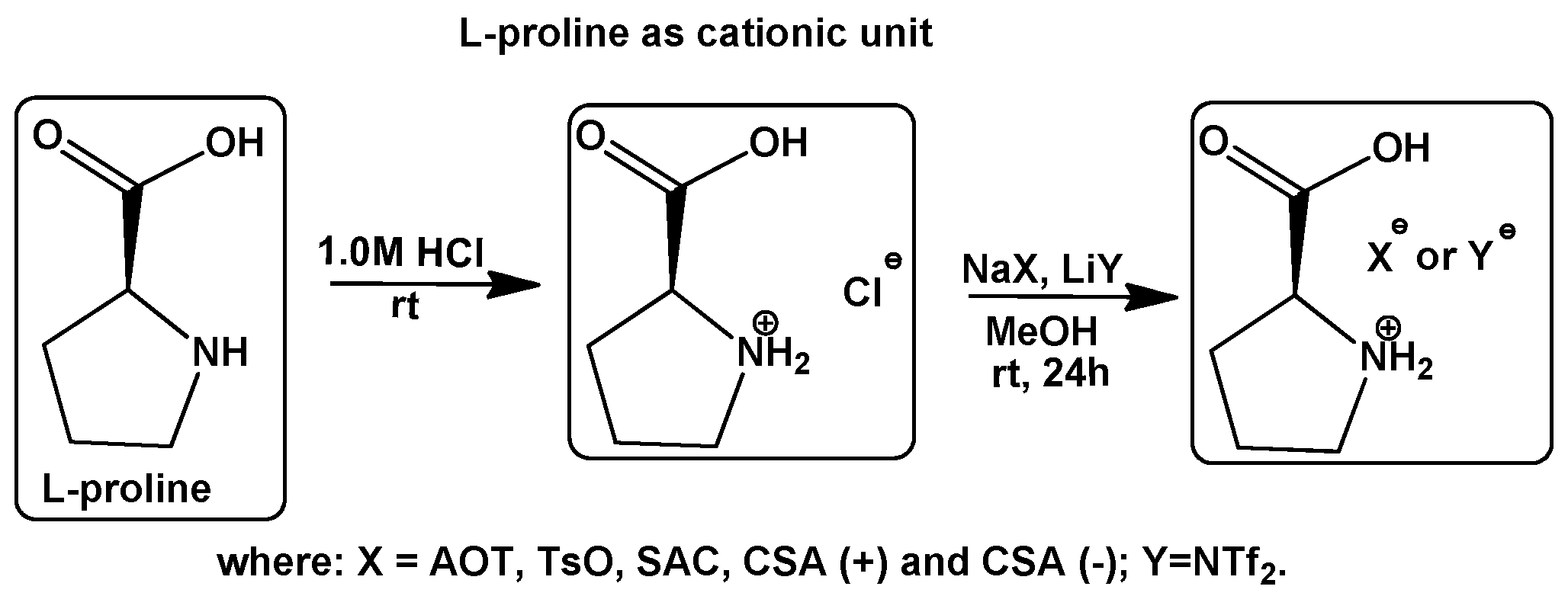
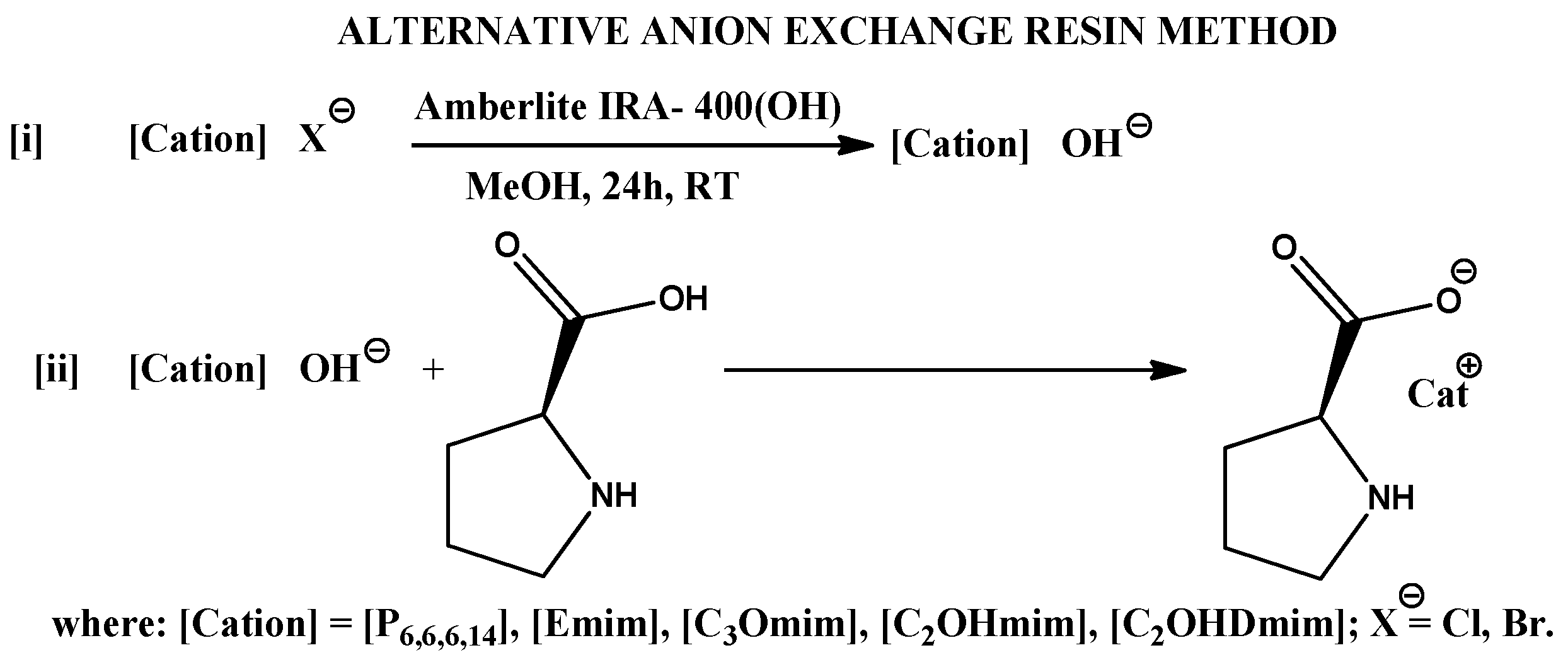
Synthesis and Characterization of CILs
2.2. Physical and Thermal Properties of CILs Based on L-Proline
2.3. Application of Novel CILs Based on L-Proline as a Catalyst for the Asymmetric Michael Reaction
2.4. Extraction of Michael Adduct with Supercritical Carbon Dioxide (scCO2)
3. Materials and Methods
3.1. Synthesis of (L)-Proline Hydrochloride—Cationic Approach
3.1.1. General Procedure for the Michael Reaction of Cyclohexanone and β–Nitrostyrene
3.1.2. General Procedure for Extraction Using scCO2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Freemantle, M. An Introduction to Ionic Liquids; RSC Publishing: London, UK, 2010. [Google Scholar]
- Wasserscheid, P.; Welton, T. (Eds.) Ionic Liquids in Synthesis; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Dupont, J.; Suarez, P.A.Z.; Souza, R.F.; Burrow, R.A.; Kintzinger, J.P. C-H-π Interactions in 1-n-Butyl-3-methylimidazolium Tetraphenylborate Molten Salt: Solid and Solution Structures. Chem. Eur. J. 2000, 6, 2377–2381. [Google Scholar] [CrossRef] [PubMed]
- Branco, L.C.; Serbanovic, A.; Ponte, M.N.; Afonso, C.A.M. Chiral Guanidinium Ionic Liquids for Asymmetric Dihydroxylation of Olefins with Recycling of the Catalytic System by Supercritical CO2. ACS Catal. 2011, 1, 1408–1413. [Google Scholar] [CrossRef]
- Qian, W.; Texter, J.; Yan, F. Frontiers in poly(ionic liquid)s: Syntheses and applications. Chem. Soc. Rev. 2017, 46, 1124–1159. [Google Scholar] [CrossRef] [PubMed]
- Earle, M.J.; McCormac, P.B.; Seddon, K.R. Diels–Alder reactions in ionic liquids. A safe recyclable alternative to lithium perchlorate–diethyl ether mixtures. Green Chem. 1999, 1, 23–25. [Google Scholar] [CrossRef]
- Park, J.K.; Sreekanth, P.; Kim, B.M. Recycling Chiral Imidazolidin-4-one Catalyst for Asymmetric Diels—Alder Reactions: Screening of Various Ionic Liquids. Adv. Synth. Catal. 2004, 346, 49–52. [Google Scholar] [CrossRef]
- Goodrich, P.; Nimal Gunaratne, H.Q.; Hall, L.; Wang, Y.; Jin, L.; Muldoon, M.J.; Ribeiro, A.P.C.; Pombeiro, A.J.L.; Pârvulescu, V.I.; Daveye, P.; et al. Using chiral ionic liquid additives to enhance asymmetric induction in a Diels-Alder reaction. Dalton Trans. 2017, 46, 1704–1713. [Google Scholar] [CrossRef]
- Lombardo, M.; Pasi, F.; Easwar, S.; Trombini, C. An improved protocol for the direct asymmetric aldol reaction in ionic liquids. Adv. Synth. Catal. 2007, 349, 2061–2065. [Google Scholar] [CrossRef]
- González, L.; Altava, B.; Bolte, M.; Burguete, M.I.; García-Verdugo, E.; Luis, S.V. Synthesis of Chiral Room Temperature Ionic Liquids from Amino Acids—Application in Chiral Molecular Recognition. Eur. J. Org. Chem. 2012, 2012, 4996–5009. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y. Asymmetric Michael Addition Mediated by Chiral Ionic Liquids. Mini Rev. Org. Chem. 2018, 15, 236–245. [Google Scholar] [CrossRef]
- Luo, S.Z.; Mi, X.L.; Xu, H.; Zhang, L.; Liu, S.; Cheng, J.P. Functionalized chiral ionic liquids as highly efficient asymmetric organocatalysts for Michael addition to nitroolefins. Angew. Chem. Int. Ed. 2006, 118, 3165–3169. [Google Scholar] [CrossRef]
- Liu, Q.; Janssen, M.; Rantwijk, F.; Sheldon, R.A. Room-temperature ionic liquids that dissolve carbohydrates in high concentrations. Green Chem. 2005, 7, 39–42. [Google Scholar] [CrossRef]
- Prechtl, M.H.G.; Scholtena, J.D.; Neto, B.A.D.; Dupont, J. Application of Chiral Ionic Liquids for Asymmetric Induction in Catalysis. Curr. Org. Chem. 2009, 13, 1259–1277. [Google Scholar] [CrossRef] [Green Version]
- Vasiloiu, M.; Cervenk, I.; Gaertner, P.; Weil, M.; Schröder, C.; Bica, K. Amino alcohol-derived chiral ionic liquids: Structural investigations toward chiral recognition. Tetrahedron Asymmetry 2015, 26, 1069–1082. [Google Scholar] [CrossRef]
- Bouchardy, L.; Rodriguez-Ruiz, V.; Bournaud, C.; Boyer, F.D.; Toffano, M.; Judeinstein, P.; Vo-Thanh, G. Novel Class of Reversible Chiral Ionic Liquids Derived from Natural Amino Acids: Synthesis and Characterization. Chem. Select. 2018, 3, 958–962. [Google Scholar] [CrossRef]
- Zalewska, K.; Branco, L.C. Organocatalysis with chiral ionic liquids. Mini Rev. Org. Chem. 2014, 11, 141–153. [Google Scholar] [CrossRef]
- Wu, G.; Bazer, F.W.; Burghardt, R.C.; Johnson, G.A.; Kim, S.W.; Knabe, D.A.; Li, P.; Li, X.; McKnight, J.R.; Satterfield, M.C.; et al. Proline and hydroxyproline metabolism: Implications for animal and human nutrition. Amino Acids 2011, 40, 1053–1063. [Google Scholar] [CrossRef] [Green Version]
- Obregón-Zúñiga, A.; Milán, M.; Juaristi, E. Improving the Catalytic Performance of (S)-Proline as Organocatalyst in Asymmetric Aldol Reactions in the Presence of Solvate Ionic Liquids: Involvement of a Supramolecular Aggregate. Org. Lett. 2017, 19, 1108–1111. [Google Scholar] [CrossRef]
- Huang, L.; Li, Y.; Lin, Q.; Lou, B.; Chen, Y. Enantioselective permeations of amino acids through l-proline-modified gold nanochannel membrane: An experimental and theoretical study. Amino Acids 2018, 50, 1549–1556. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Fukumoto, K.; Yoshizawa, M.; Ohno, H. Room Temperature Ionic Liquids from 20 Natural Amino Acids. J. Am. Chem. Soc. 2006, 127, 2398–2399. [Google Scholar] [CrossRef]
- Fukaya, Y.; Iizuka, Y.; Sekikawa, K.; Ohno, H. Bio ionic liquids: Room temperature ionic liquids composed wholly of biomaterials. Green Chem. 2007, 9, 1155–1157. [Google Scholar] [CrossRef]
- Wagner, M.; Contie, Y.; Ferroud, C.; Revial, G. Enantioselective Aldol Reactions and Michael Additions Using Proline Derivatives as Organocatalysts. Int. J. Org. Chem. 2014, 4, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhang, Y.; Zhao, J.; Yang, Q.; Ma, Y.; Cao, X. A Recyclable Organocatalyst for Asymmetric Michael Addition. Catal. Lett. 2016, 146, 587–595. [Google Scholar] [CrossRef]
- Hayashi, Y.; Gotoh, H.; Tamura, T.; Yamaguchi, H.; Masui, R.; Shoji, M. Cysteine-Derived Organocatalyst in a Highly Enantioselective Intramolecular Michael Reaction. J. Am. Chem. Soc. 2005, 127, 16028–16029. [Google Scholar] [CrossRef]
- Naganaboina, R.T.; Nayak, A.; Peddinti, R.K. Trifluoroacetic acid-promoted Michael addition–cyclization reactions of vinylogous carbamates. Org. Biomol. Chem. 2014, 12, 3366–3370. [Google Scholar] [CrossRef]
- Qian, Y.; Xiao, S.; Liu, L.; Wang, Y. A mild and efficient procedure for asymmetric Michael additions of cyclohexanone to chalcones catalyzed by an amino acid ionic liquid. Tetrahedron Asymmetry 2008, 19, 1515–1518. [Google Scholar] [CrossRef]
- Duan, S.H.; Kai, T.; Chowdhury, F.A.; Taniguchi, I.; Kazama, S. Effect of addition of Proline, ionic liquid [Choline][Pro] on CO2 separation properties of poly(amidoamine) dendrimer/poly(ethylene glycol) hybrid membranes. Mater. Sci. Eng. 2018, 292, 012040–012048. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CIL | Physical State | Density [a] [g·cm−3] | αD [b] [cm2 g−1] | Tg [°C] [c] (Tm) | Tdec [°C] [d] |
|---|---|---|---|---|---|
| L-Pro 1 | white powder | --- | −41.3 | (228 dec.) | 238.60 |
| [ProNH2] [NTf2] 1a | yellow liquid | 1.63 | −12.7 | −58.92 | 287.10 |
| [ProNH2] [AOT] 1b | yellow liquid | 1.32 | −12.0 | −61.88 | 258.40 |
| [ProNH2] [TsO] 1c | yellow liquid | 0.93 | −4.0 | −25.07 | 273.90 |
| [ProNH2] [SAC] 1d | white liquid | 0.98 | −4.5 | -- [e] | 299.50 |
| [ProNH2] [(-)CSA] 1e | white solid | --- | −44.0 | -- [e] (154) [f] | 292.90 |
| [ProNH2] [(+)CSA] 1f | white solid | --- | +66.0 | -- [e] (160) [f] | 292.30 |
| [P6,6,6,14] [ProCO2] 2a | orange liquid | 0.93 | −17.3 | −70.59 | 290.10 |
| [Emim] [ProCO2] 2b | orange liquid | 1.19 | −8.8 | −63.44 | 247.80 |
| [C3Omim] [ProCO2] 2c | orange liquid | 1.24 | −5.3 | −53.42 | 256.30 |
| [C2OHmim] [ProCO2] 2d | yellow liquid | 1.25 | −6.7 | −52.60 | 270.40 |
| [C2OHDmim] [ProCO2] 2e | yellow liquid | 1.28 | −19.0 | −40.30 | 277.50 |
| [Choline] [ProCO2] 2f | brown liquid | 0.97 | −12.7 | −70.24 | [g] |
 | ||||
|---|---|---|---|---|
| Catalyst | Loading Catalyst [mol%] | Conversion [%] | dr(syn: anti)[d] | ee [%] [e] |
| L-PRO [a] | 30 | ≥99 | 93:7 | 94 |
| L-PRO [b] | 60 | ≥99 | 91:9 | 96 |
| [ProNH2][Cl] [b] | 30 | 50 | 85:15 | 53 |
| [ProNH2][PTSA] [b] | 30 | 50 | 75:25 | n.d. [e] |
| [ProNH2][AOT] [c] | 30 | 19 | 73:27 | 55 |
| [ProNH2][AOT] [b] | 60 | 50 | 82:18 | 59 |
| [ProNH2][NTf2] [b] | 30 | 34 | 80:20 | 58 |
| [Na][ProCO2] [b] | 30 | ≥99 | 78:22 | 40 |
| [P6,6,6,14][ProCO2] [b] | 30 | ≥99 | 83:17 | 69 |
| [Emim][ProCO2] [b] | 30 | ≥99 | 86:14 | 78 |
| [Emim][ProCO2] [b] | 60 | ≥99 | 69:31 | 81 |
| [C3Omim][ProCO2] [a] | 30 | ≥99 | 72:28 | 74 |
| [C3Omim][ProCO2] [b] | 60 | ≥99 | 75:25 | 93 |
| [C2OHmim][ProCO2] [b] | 30 | ≥99 | 70:30 | 62 |
| [C2OHDmim][ProCO2] [b] | 30 | ≥99 | 65:35 | 55 |
| Catalyst | Solvent | Conv. [%] [c] | dr (syn: anti)[d] | ee [%] [e] |
|---|---|---|---|---|
| L-PRO [a] | [Emim][EtSO4] | ≥99 | 90:10 | 87 |
| L-PRO [a] | [Bmim][DCA] | ≥99 | 93:7 | 96 |
| [ProNH2][Cl] [a] | [Bmim][DCA] | 83 | 89:11 | 27 |
| [ProNH2][AOT] [b] | [Emim][EtSO4] | 92 | 78:22 | n.d. [f] |
| [ProNH2][AOT] [b] | [Bmim][DCA] | ≥99 | 90:10 | 65 |
| [ProNH2][NTf2] [b] | [Emim][EtSO4] | 92 | 64:36 | n.d. [f] |
| [ProNH2][NTf2] [b] | [Bmim][DCA] | ≥99 | 91:9 | 96 |
| [Emim][ProCO2] [b] | [Emim][EtSO4] | ≥99 | 94:6 | 94 |
| [Emim][ProCO2] [b] | [Bmim][DCA] | ≥99 | 92:8 | 94 |
| [C3Omim][ProCO2] [b] | [Emim][EtSO4] | ≥99 | 74:26 | 76 |
| [C3Omim][ProCO2] [b] | [Emim][EtSO4] | ≥99 | 75:25 | 83 |
| [Choline][ProCO2] [b] | [Emim][EtSO4] | ≥99 | 91:9 | 95 |
| [Choline][ProCO2] [b] | [Bmim][DCA] | ≥99 | 91:9 | 97 |
 | |||||
|---|---|---|---|---|---|
| Prod. | Catalyst | Solvent [a] | Conv. [%] [b] | dr (syn: anti)[c] | ee [%] [d] |
| A | L-PRO | EtOH | ≥99 | 93:7 | 94 |
| A | L-PRO | IL | ≥99 | 91:9 | 96 |
| A | [ProNH2][NTf2] | IL | ≥99 | 94:6 | 94 |
| A | [Emim][ProCO2] | IL | ≥99 | 91:9 | 97 |
| B | L-PRO | EtOH | 99 | 80:20 | 47 |
| B | L-PRO | IL | ≥99 | 75:25 | 31 |
| B | [Emim][ProCO2] | IL | 72 | 69:31 | 29 |
| B | [Choline][ProCO2] | IL | 81 | 66:34 | 28 |
| C | L-PRO | EtOH | 75 | -- | (nd) [e] |
| C | [Emim][ProCO2] | IL | 75 | -- | 20 |
| C | [Emim][ProCO2] | none | 71 | -- | 33 |
| C | [Choline][ProCO2] | EtOH | 79 | -- | 86 |
| D | L-PRO | EtOH | 98 | -- | 10 |
| D | L-PRO | IL | 94 | -- | rac. |
| D | [Emim][ProCO2] | IL | 93 | -- | 10 |
| D | [Choline][ProCO2] | IL | 94 | -- | 10 |
| E | L-PRO | EtOH | 86 | 84:16 | 40 |
| E | L-PRO | IL | ≥99 | 84:16 | 4 |
| E | [Emim][ProCO2] | IL | ≥99 | 78:22 | 4 |
| E | [Choline][ProCO2] | IL | ≥99 | 94:6 | 4 |
| F | L-PRO | EtOH | 94 | -- | 100 |
| F | L-PRO | IL | ≥99 | -- | 31 |
| F | [Emim][ProCO2] | EtOH | ≥99 | -- | 19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zalewska, K.; Pinto, I.; Cabrita, L.; Zakrzewska, M.E.; Noronha, J.P.; da Ponte, M.N.; Branco, L.C. Development of L-Proline-Based Chiral Ionic Liquids for Asymmetric Michael Reaction. Catalysts 2023, 13, 270. https://doi.org/10.3390/catal13020270
Zalewska K, Pinto I, Cabrita L, Zakrzewska ME, Noronha JP, da Ponte MN, Branco LC. Development of L-Proline-Based Chiral Ionic Liquids for Asymmetric Michael Reaction. Catalysts. 2023; 13(2):270. https://doi.org/10.3390/catal13020270
Chicago/Turabian StyleZalewska, Karolina, Isabel Pinto, Luis Cabrita, Małgorzata E. Zakrzewska, João P. Noronha, M. Nunes da Ponte, and Luis C. Branco. 2023. "Development of L-Proline-Based Chiral Ionic Liquids for Asymmetric Michael Reaction" Catalysts 13, no. 2: 270. https://doi.org/10.3390/catal13020270