
Poly(ε-caprolactones) Initiated by Chiral Compounds: A New Protocol to Support Organocatalysts
 , ,
, ,
Abstract
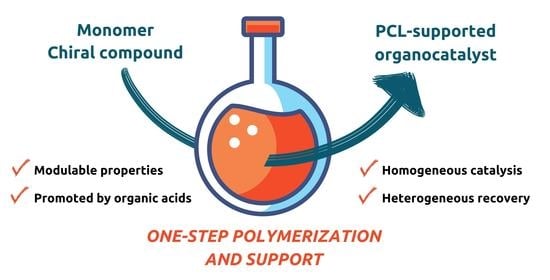
:
1. Introduction
2. Results and Discussion
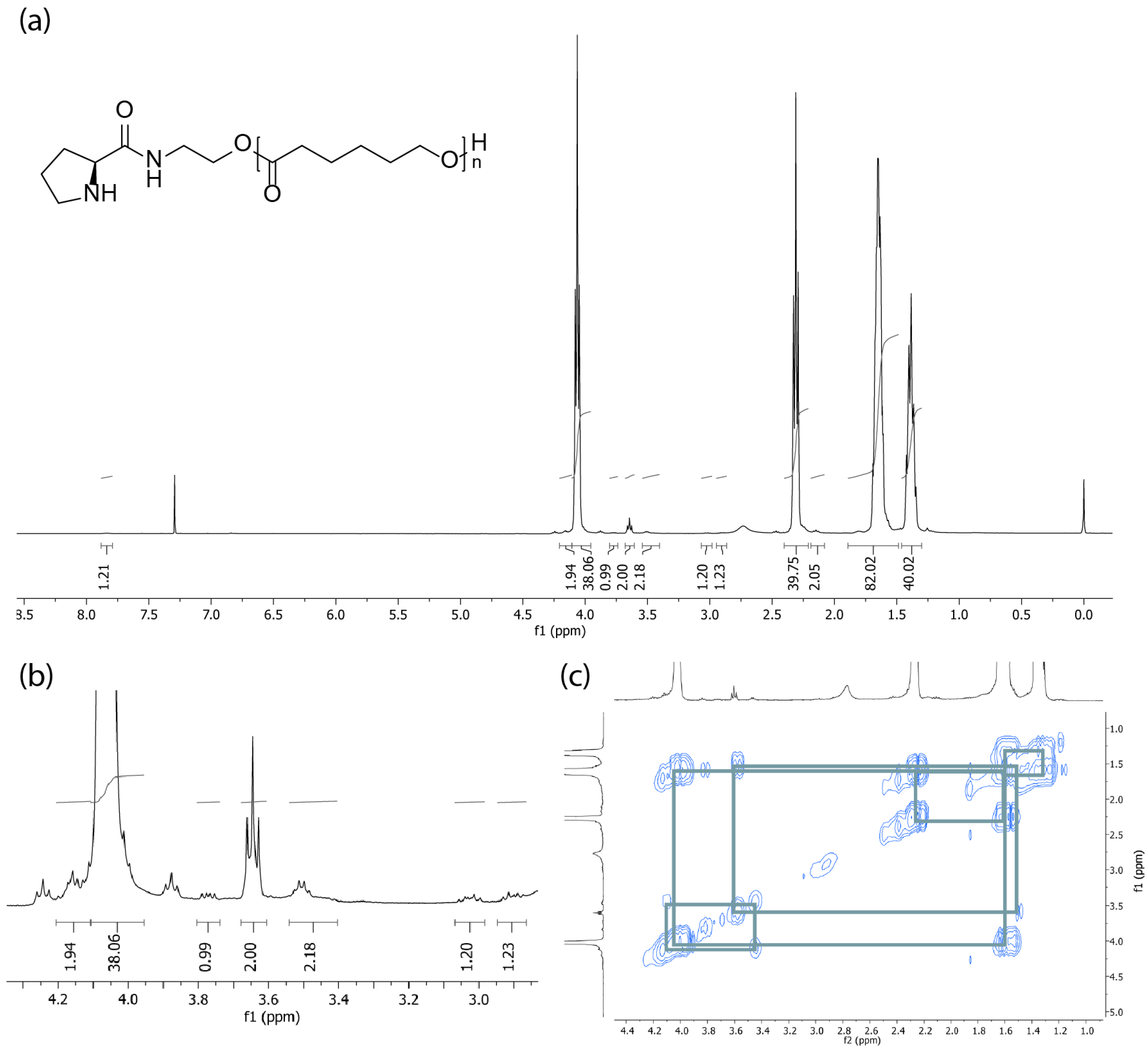
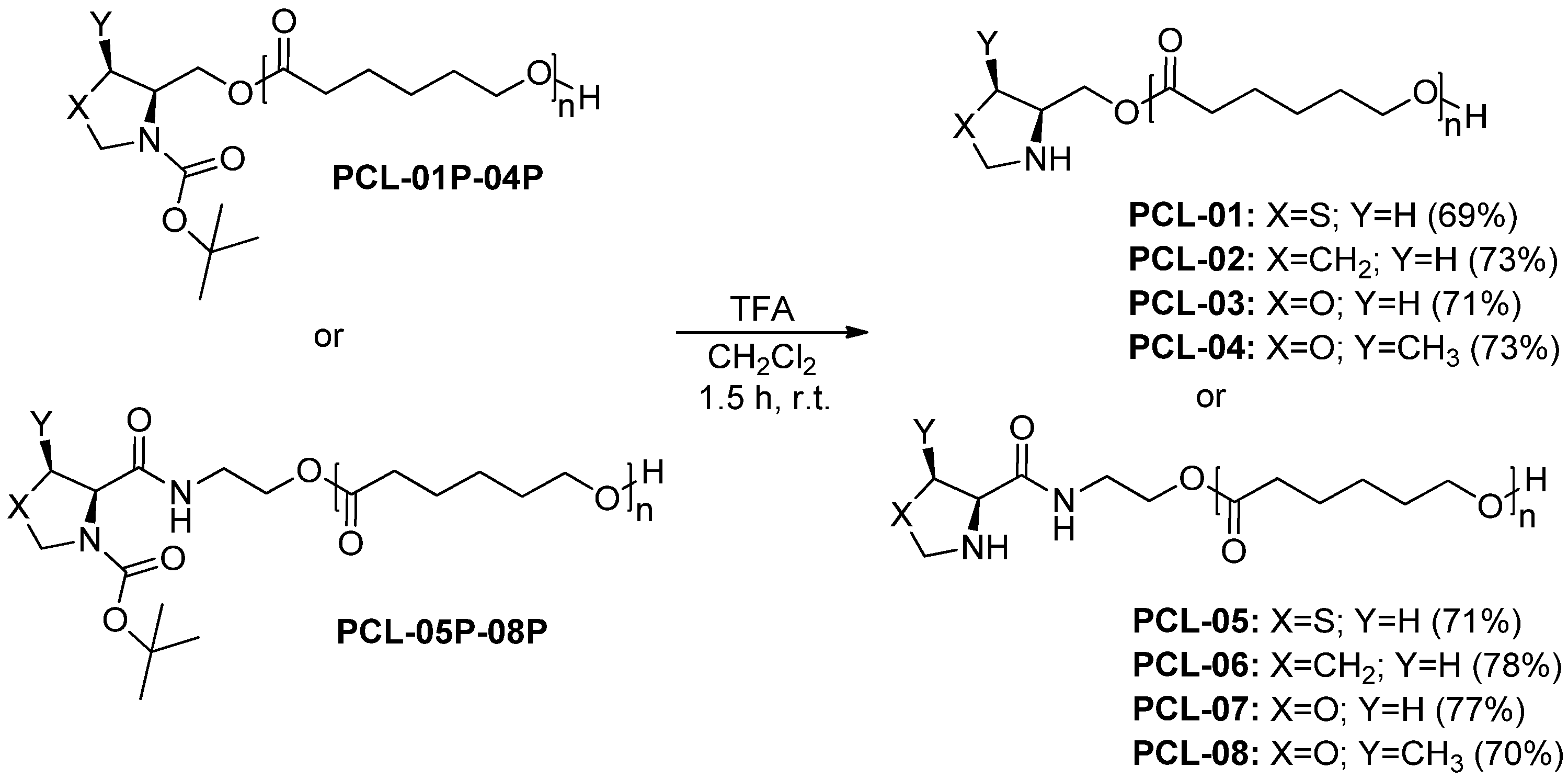
2.1. Synthesis and Characterization of Polymers
2.2. Application of Polymers in Organocatalysis
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Synthetic Procedures and Characterization Data
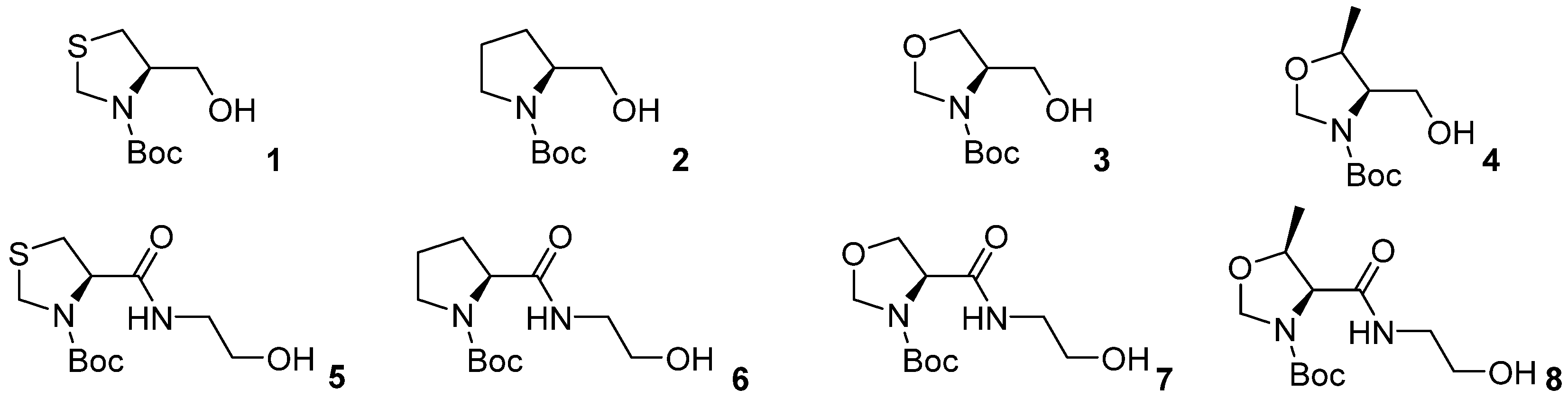
4.2.1. General Procedure and Characterization Data for the Preparation of Compounds 1 to 8, [16,42] and PCL-01P to PCL-08P
4.2.2. General Procedure for the Preparation of PCL-01 to PCL-08
4.2.3. Characterization Data of PCL-01 to PCL-08
4.2.4. General Procedure for Organocatalytic Asymmetric Direct Aldol Addition
4.2.5. Characterization Data of (S)-2-((R)-hydroxy(4-nitrophenyl)methyl)cyclohexanone (11) [43]
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- List, B. Introduction: Organocatalysis. Chem. Rev. 2007, 107, 5413. [Google Scholar] [CrossRef] [Green Version]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Recent Developments in Enantioselective Organocatalytic Michael Reactions in Aqueous Media. Curr. Org. Chem. 2018, 21, 323. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.C.; Bergbreiter, D.E. Soluble polymer-supported organocatalysts. Pure Appl. Chem. 2013, 85, 493. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395. [Google Scholar] [CrossRef]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471. [Google Scholar] [CrossRef] [PubMed]
- Agirre, M.; Arrieta, A.; Arrastia, I.; Cossío, F.P. Organocatalysts Derived from Unnatural α-Amino Acids: Scope and Applications. Chem. Asian J. 2019, 14, 44. [Google Scholar] [CrossRef]
- Albrecht, L.; Jiang, H.; Jørgensen, K.A. Hydrogen-Bonding in Aminocatalysis: From Proline and Beyond. Chem. Eur. J. 2014, 20, 358. [Google Scholar] [CrossRef]
- Shajahan, R.; Sarang, R.; Saithalavi, A. Polymer Supported Proline-Based Organocatalysts in Asymmetric Aldol Reactions: A Review. Curr. Organocatalysis 2022, 9, 4. [Google Scholar] [CrossRef]
- Braga, A.L.; Silveira, C.C.; Bolster, M.W.G.; Schrekker, H.S.; Wessjohann, L.A.; Schneider, P.H. Microwave-accelerated asymmetric allylations using cysteine derived oxazolidine and thiazolidine palladium complexes. J. Mol. Catal. A Chem. 2005, 239, 235. [Google Scholar] [CrossRef]
- Braga, A.L.; Lüdtke, D.S.; Wessjohann, L.A.; Paixão, M.W.; Schneider, P.H. A chiral disulfide derived from (R)-cysteine in the enantioselective addition of diethylzinc to aldehydes: Loading effect and asymmetric amplification. J. Mol. Catal. A 2005, 229, 47. [Google Scholar] [CrossRef]
- Rambo, R.S.; Schneider, P.H. Thiazolidine-based organocatalysts for a highly enantioselective direct aldol reaction. Tetrahedron Asymmetry 2010, 21, 2254. [Google Scholar] [CrossRef]
- Rambo, R.S.; Jacoby, C.G.; Silva, T.L.; Schneider, P.H. A highly enantio- and diastereoselective direct aldol reaction in aqueous medium catalyzed by thiazolidine-based compounds. Tetrahedron Asymmetry 2015, 26, 632. [Google Scholar] [CrossRef]
- Bach, M.F.; Griebeler, C.H.; Jacoby, C.G.; Schneider, P.H. Design of a Chiral Ionic Liquid System for the Enantioselective Addition of Diethylzinc to Aldehydes. Eur. J. Org. Chem. 2017, 46, 6997. [Google Scholar] [CrossRef]
- Jacoby, C.G.; Vontobel, P.H.V.; Bach, M.F.; Schneider, P.H. Highly efficient organocatalysts for the asymmetric aldol reaction. New J. Chem. 2018, 42, 7416. [Google Scholar] [CrossRef]
- Silva, T.L.; Rambo, R.S.; Jacoby, C.G.; Schneider, P.H. Asymmetric Michael reaction promoted by chiral thiazolidine-thiourea catalyst. Tetrahedron 2020, 76, 130874. [Google Scholar] [CrossRef]
- Benaglia, M.; Celentano, G.; Cozzi, F. Enantioselective aldol condensations catalyzed by poly(ethylene glycol)-supported proline. Adv. Synth. Catal. 2001, 343, 171. [Google Scholar] [CrossRef]
- Benaglia, M.; Cinquini, M.; Cozzi, F.; Puglisi, A.; Celentano, G. Poly(ethylene glycol)-supported proline: A versatile catalyst for the enantioselective aldol and iminoaldol reactions. Adv. Synth. Catal. 2002, 344, 533. [Google Scholar] [CrossRef]
- Benaglia, M.; Cinquini, M.; Cozzi, F.; Puglisi, A.; Celentano, G. Poly(ethylene-glycol)-supported proline: A recyclable aminocatalyst for the enantioselective synthesis of γ-nitroketones by conjugate addition. J. Mol. Catal. A 2003, 204, 157. [Google Scholar] [CrossRef]
- Fulgheri, T.; Della Penna, F.; Baschieri, A.; Carlone, A. Advancements in the recycling of organocatalysts: From classical to alternative approaches. Curr. Opin. Green Sustain. Chem. 2020, 25, 100387. [Google Scholar] [CrossRef]
- Susam, Z.D.; Tanyeli, C. Recyclable Organocatalysts in Asymmetric Synthesis. Asian J. Org. Chem. 2021, 10, 1251. [Google Scholar] [CrossRef]
- Bergbreiter, D.E. Using soluble polymers to recover catalysts and ligands. Chem. Rev. 2002, 102, 3345. [Google Scholar] [CrossRef]
- Bergbreiter, D.E.; Tian, J.; Hongfa, C. Using soluble supports to facilitate homogeneous catalysis. Chem. Rev. 2009, 109, 530. [Google Scholar] [CrossRef]
- Gruttadaria, M.; Giacalone, F.; Noto, R. Supported proline and proline-derivative as recyclable organocatalysts. Chem. Soc. Rev. 2008, 37, 1666. [Google Scholar] [CrossRef]
- Lu, A.; Cotanda, P.; Patterson, J.P.; Longbottom, D.A.; O’Reilly, R.K. Aldol reactions catalyzed by L-proline functionalized polymeric nanoreactors in water. Chem. Commun. 2012, 48, 9699. [Google Scholar] [CrossRef]
- Moore, B.L.; Lu, A.; Longbottom, D.A.; O’Reilly, R.K. Immobilization of MacMillan catalyst via controlled radical polymerization: Catalytic activity and reuse. Polym. Chem. 2013, 4, 2304. [Google Scholar] [CrossRef] [Green Version]
- Zayas, H.A.; Lu, A.; Valade, D.; Amir, F.; Jia, Z.; O’Reilly, R.K.; Monteiro, M.J. Thermoresponsive polymer-supported L-proline micelle catalysts for the direct asymmetric aldol reaction in water. ACS Macro Lett. 2013, 2, 327. [Google Scholar] [CrossRef]
- Fan, Q.; Ren, C.; Yeung, C.; Hu, W.; Chan, A.S.C. Highly effective soluble polymer-supported catalysts for asymmetric hydrogenation. J. Am. Chem. Soc. 1999, 121, 7407. [Google Scholar] [CrossRef]
- Haraguchi, N.; Nguyen, T.L.; Itsuno, S. Polyesters containing chiral imidazolidinone salts in polymer main chain: Heterogeneous organocatalysts for asymmetric Diels-Alder reaction. ChemCatChem 2017, 9, 3786. [Google Scholar] [CrossRef]
- Labet, M.; Thielemans, W. Synthesis of polycaprolactone: A review. Chem. Soc. Rev. 2009, 38, 3484. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, P.; Pretula, J.; Kaluzynski, K.; Kazmierski, D.; Penczek, S. ε-Caprolactone: Activated monomer polymerization; controversy over the mechanism of polymerization catalyzed by phosphorus acids (diarylhydrogen phosphates). Do acids also act as initiators? J. Catal. 2019, 371, 305. [Google Scholar] [CrossRef]
- Lou, X.; Detrembleur, C.; Jérome, R. Living cationic polymerization of γ-valerolactone and synthesis of high molecular weight homopolymer and asymmetric telechelic and block copolymer. Macromolecules 2002, 35, 1190. [Google Scholar] [CrossRef]
- Gazeau-Bureau, S.; Delcroix, D.; Martín-Vaca, B.; Bourissou, D.; Navarro, C.; Magnet, S. Organo-catalyzed ROP of ε-caprolactone: Methanesulfonic acid competes with trifluoromethanesulfonic acid. Macromolecules 2008, 41, 3782. [Google Scholar] [CrossRef]
- Braibante, M.E.F.; Braibante, H.S.; Costenaro, E.R. The Use of Curtius Rearrangement in the Synthesis of 4-Aminothiazolidines. Synthesis 1999, 6, 943. [Google Scholar] [CrossRef]
- Falorni, M.; Conti, S.; Giacomelli, G.; Cossu, S.; Soccolini, F. Optically active 4-oxaproline derivatives: New useful chiral synthons derived from serine and threonine. Tetrahedron Asymmetry 1995, 6, 287. [Google Scholar] [CrossRef]
- Sanda, F.; Sanada, H.; Shibasaki, Y.; Endo, T. Star polymer synthesis from ε-caprolactone utilizing polyol/protonic acid initiator. Macromolecules 2002, 35, 680. [Google Scholar] [CrossRef]
- Liu, J.; Liu, L. Ring-opening polymerization of ε-caprolactone initiated by natural amino acids. Macromolecules 2004, 37, 2674. [Google Scholar] [CrossRef]
- Notz, W.; Tanaka, F.; Barbas III, C.F. Enamine-Based Organocatalysis with Proline and Diamines: The Development of Direct Catalytic Asymmetric Aldol, Mannich, Michael, and Diels−Alder Reactions. Accounts Chem. Res. 2004, 37, 580. [Google Scholar] [CrossRef]
- Jiang, H.; Albrecht, L.; Jorgensen, K.A. Aminocatalytic remote functionalization strategies. Chem. Sci. 2013, 4, 2287. [Google Scholar] [CrossRef]
- Mandelkern, L. Crystallization of Polymers; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Perrin, D.D.; Armarego, W.L. Purification of Laboratory Chemicals, 4th ed.; Pergamon: New York, NY, USA, 1997. [Google Scholar]
- Molander, G.A.; Romero, J.A.C. Investigations concerning the organolanthanide and group 3 metallocene-catalyzed cyclization–functionalization of nitrogen-containing dienes. Tetrahedron 2005, 61, 2631. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andres, J.M.; Gamarra, A.; Manzano, R.; Perez-Lopez, C. Novel sulfonylpolystyrene-supported prolinamides as catalysts for enantioselective aldol reaction in water. Tetrahedron 2013, 69, 10811. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry a | [ε-CL]0/[I]0 | [Acid]0/[I]0 | Temp. (°C) | Time (h) | Yield (%) b | Mn,t c | Mn,RMN d | Ɖ e |
|---|---|---|---|---|---|---|---|---|
| 1 | 20 | 2 | 90 | 24 | 94 | 2400 | 1900 | 1.45 |
| 2 | 100 | 2 | 90 | 24 | 84 | 9850 | 2400 | 1.74 |
| 3 | 20 | 2 | 90 | 48 | 71 | 1900 | 1500 | 1.89 |
| 4 | 50 | 2.5 | 90 | 24 | 95 | 5700 | 2200 | 1.32 |
| 5 | 50 | 5 | 90 | 24 | 98 | 5850 | 2400 | 1.22 |
| 6 | 50 | 10 | 90 | 24 | 98 | 5850 | 2000 | 1.17 |
| 7 | 50 | 10 | 110 | 24 | 98 | 5850 | 1750 | 2.00 |
 | ||||
|---|---|---|---|---|
| Entry | Polymer a | Initiator | Yield (%) b | Mn,RMN c |
| 1 | PCL-01P |  | 85 | 2500 |
| 2 | PCL-02P |  | 85 | 1750 |
| 3 | PCL-03P |  | 79 | 1800 |
| 4 | PCL-04P |  | 77 | 1600 |
| 5 | PCL-05P |  | 79 | 2000 |
| 6 | PCL-06P |  | 98 | 2400 |
| 7 | PCL-07P |  | 76 | 1900 |
| 8 | PCL-08P |  | 71 | 2400 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (mol %) | Benzoic Acid (mol %) | Yield b (%) | e.e.c (%) | d.r.d (anti:syn) |
| 1 | Cyclohexanone | 5 | 5 | 76 | 87 | 6:1 |
| 2 | Cyclohexanone | 10 | 10 | 98 | 93 | 8:1 |
| 3 | Cyclohexanone | 15 | 15 | 90 | 86 | 6:1 |
| 4 | H2O | 10 | 10 | 88 | 89 | 7:1 |
| 5 | THF | 10 | 10 | 87 | 92 | 7:1 |
| 6 | CH2Cl2 | 10 | 10 | 98 | 96 | 7.5:1 |
| 7 | CH2Cl2 | 10 | 20 | 68 | 91 | 7:1 |
| 8 | CH2Cl2 | 10 | - | 76 | 93 | 7:1 |
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Yield b (%) | e.e.c (%) | d.r.d (anti:syn) |
| 1 |  | 13 | 21 | 1:1 |
| 2 |  | 74 | 35 | 3:1 |
| 3 |  | Traces | - | - |
| 4 |  | Traces | - | - |
| 5 |  | 19 | 82 | 3:1 |
| 6 |  | 98 | 96 | 7.5:1 |
| 7 |  | 26 | 28 | 1:1 |
| 8 |  | 28 | 83 | 4:1 |
 | |||
|---|---|---|---|
| Cycle a | Yield b (%) | e.e.c (%) | d.r. d (anti:syn) |
| 1 | 98 | 96 | 7.5:1 |
| 2 | 84 | 75 | 4:1 |
| 3 | 79 | 70 | 4:1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacoby, C.G.; Sbardelotto, J.H.; Daitx, T.d.S.; Dalberto, B.T.; Mauler, R.S.; Schneider, P.H. Poly(ε-caprolactones) Initiated by Chiral Compounds: A New Protocol to Support Organocatalysts. Catalysts 2023, 13, 164. https://doi.org/10.3390/catal13010164
Jacoby CG, Sbardelotto JH, Daitx TdS, Dalberto BT, Mauler RS, Schneider PH. Poly(ε-caprolactones) Initiated by Chiral Compounds: A New Protocol to Support Organocatalysts. Catalysts. 2023; 13(1):164. https://doi.org/10.3390/catal13010164
Chicago/Turabian StyleJacoby, Caroline Gross, Jorge Hugo Sbardelotto, Tales da Silva Daitx, Bianca Thaís Dalberto, Raquel Santos Mauler, and Paulo Henrique Schneider. 2023. "Poly(ε-caprolactones) Initiated by Chiral Compounds: A New Protocol to Support Organocatalysts" Catalysts 13, no. 1: 164. https://doi.org/10.3390/catal13010164