
Guaiacol to Aromatics: Efficient Transformation over In Situ-Generated Molybdenum and Tungsten Oxides

A.V. Topchiev Institute of Petrochemical Synthesis, Russian Academy of Sciences (TIPS RAS), Moscow 119991, Russia
*
Authors to whom correspondence should be addressed.
Catalysts 2023, 13(2), 263; https://doi.org/10.3390/catal13020263
Submission received: 25 December 2022
/
Revised: 13 January 2023
/
Accepted: 17 January 2023
/
Published: 23 January 2023
(This article belongs to the Special Issue Transition-Metal-Containing Bifunctional Catalysts: Design and Catalytic Applications)
Abstract
:The development of catalysts for the hydrodeoxygenation of bio-based feedstocks is an important step towards the production of fuels and chemicals from biomass. This paper describes in situ-generated bulk molybdenum and tungsten oxides in the hydrodeoxygenation of the lignin-derived compound guaiacol. The catalysts obtained were studied using powder X-ray diffraction, X-ray photoelectron spectroscopy, scanning electron microscopy, high-resolution transition electron microscopy, diffuse reflectance infrared Fourier transform spectroscopy, and Raman spectroscopy. The use of metal carbonyls as precursors was shown to promote the formation of amorphous molybdenum oxide and crystalline tungsten phosphide under hydrodeoxygenation conditions. The catalysts’ activity was investigated under various reaction conditions (temperature, H2 pressure, solvent). MoOx was more active in the partial and full hydrodeoxygenation of guaiacol at temperatures of 200–380 °C (5 MPa H2, 6 h). However, cyclohexane, which is an undesirable product, was formed in significant amounts using MoOx (5 MPa H2, 6 h), while WOx was more selective to aromatics. When using dodecane as a solvent (380 °C, 5 MPa H2, 6 h), the benzene-toluene-xylenes fraction was obtained with a 96% yield over the WOx catalyst.
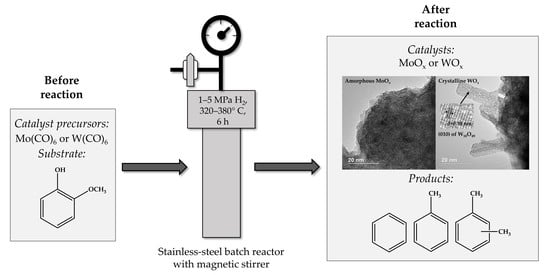
1. Introduction
Lignocellulosic biomass is a promising and widespread source of renewable energy [1,2]. One of the components of lignocellulosic biomass is lignin, which consists of oxygen-containing derivatives of phenylpropane. The structure of lignin allows it to be used as a source for the production of phenols and various aromatic compounds [3].
The main method for the processing of low-molecular-weight lignin derivatives is catalytic hydrodeoxygenation (HDO). Supported noble metals are actively involved in the HDO of different oxygenated compounds and show high catalytic activity. Nevertheless, the high cost of noble metals is a significant disadvantage in their application [4]. Transition metal phosphides, carbides, and nitrides are reported to be potential candidates to replace expensive catalysts (Table 1) [4].
However, the activity of such catalysts is partially related to the presence of oxygen on their surface, including in the metal oxide form. Therefore, transition metal oxides can also be considered as alternative HDO catalysts. Oxygen vacancies (coordinatively unsaturated metal sites) in oxides are reported to be active sites in HDO reactions [11,12]. Vacancies are formed as a result of H2 adsorption on the oxide surface and the subsequent removal of oxygen as a part of water [13]. The electron-lone-pair oxygen in a substrate is adsorbed on the active site which leads to HDO as a result of the cleavage of the C–O bond [11].
Mo- and W-containing compounds are well-known in the field of catalysis. It has been reported that Mo and W are resistant to oxygen, acids, and alkalis and, therefore, they are used for oxygen removal [14]. Efficient hydrodeoxygenation of various substrates over molybdenum oxides have been previously reported in a number of works [11,13], while pure tungsten oxides have not provided high selectivity for hydrodeoxygenation products [15]. Prasomsri et al. found that MoO3 can be used for the effective HDO of linear and cyclic ketones, phenolics, and cyclic ethers at 250–400 °C under low H2 pressure with the formation of olefins and aromatics [13]. Selectivity for hydrocarbons was reportedly more than 97% at 400 °C. Zhang et al. carried out the HDO of guaiacol, o-cresol, eugenol, trans-anethole, vanillin, and diphenyl ether over MoO3 under mixed H2 + N2 pressure at 300–360 °C [11]. Aromatics were obtained with the selectivity of 45.3–93.8% at 340 °C. Whiffen et al. tested MoO3 and MoO2 in the HDO of p-cresol at 325–375 °C, under 4.14–4.83 MPa H2 in a batch reactor [16]. Toluene was the main reaction product over both catalysts. MoO2 was noted to be more active in the HDO than MoO3. Several studies were devoted to m-cresol HDO to toluene over WOx promoted by other metals [17,18,19]. Wang et al. demonstrated that Pt-WOx/C catalyst can convert m-cresol into toluene with 94% selectivity (350 °C, 0.1 MPa H2, H2 flow rate: 12 mL/min, WHSV = 60 h−1) [17]. In another work, they used n-hexane which produced H2 by dehydration [18]. Direct bonding of Pt and WOx contributed to oxide stabilization and oxygen vacancies formation, which are responsible for direct hydrogenolysis of the C–O bond in m-cresol [17]. Chen et al. reported similar regularities using a Ni-WOx/C catalyst [19]. Toluene was obtained from m-cresol with 89% selectivity at 350 °C, under 0.1 MPa H2, WHSV of 1 h−1.
Previously, the in situ formation of transition metal catalysts during hydroprocessing of various model compounds and real feedstocks were studied [9,20,21,22,23,24]. In our recent works, the in situ formation of phosphide catalysts, including molybdenum and tungsten phosphides in the HDO of guaiacol, was investigated for the first time [9,20,21]. The absence of a catalyst synthesis stage, and therefore a simplification of the process, is one of the advantages of catalyst in situ formation. The use of metal carbonyls in combination with an oil-soluble phosphorus precursor made it possible to obtain the corresponding phosphides. It was found that, in addition to phosphides, the catalyst surface also contains metal oxides.
In the present work, molybdenum and tungsten oxides were obtained in situ from carbonyls during the HDO of guaiacol (a low-molecular-weight lignin derivative) and investigated in this process for the first time. We report that these bulk catalysts are highly active in the HDO of guaiacol to aromatics, in particular, to a benzene-toluene-xylenes (BTX) fraction. MoOx converted guaiacol into BTX with the highest selectivity of 89% (380 °C, 1 MPa H2, 6 h) at full conversion, while using WOx the highest selectivity for BTX was 96% (380 °C, 5 MPa H2, 6 h). In comparison with oxides obtained in the present work, phosphides and carbides promoted the obtaining of phenol as a main reaction product of guaiacol conversion (Table 1). In situ-generated MoP and WP from the same metal precursors in the HDO of guaiacol allows the obtaining of phenol as a main reaction product [9]. The highest phenol selectivity rates were 80% and 78% using MoP and WP, respectively. Ex situ-generated bulk MoP and WP also made a contribution to the phenol obtained from guaiacol (with 66% and 84% selectivity over MoP and WP, respectively) [10]. Kurisingal et al. made a report about selective guaiacol HDO to phenol using bimetallic Zn-Co zeolitic imidazolate framework (BMZIF)-decorated molybdenum carbide [7]. The distribution of active species, such as Mo2C, MoO3, ZnO, and CoO was found to have a high influence on guaiacol conversion and phenol selectivity. The most active Mo2C@BMZIF-700 °C (4 h) catalyst promoted a 97% guaiacol conversion and 70% phenol selectivity. Wang et al. demonstrated that an SBA-15-supported ultrastable Mo2N@CN catalyst showed similar catalytic activity in the guaiacol HDO to the catalysts reported in the present work [8]. A benzene and toluene mixture was obtained with 92% selectivity at 380 °C, while at 320 °C phenol was the main product. However, the synthesis of nitrides is a laborious procedure, while we report an easy method for catalyst synthesis.
2. Results and Discussion
2.1. Catalyst Characterization
In the present work, thein situ-formation of molybdenum and tungsten oxides from molybdenum and tungsten carbonyls during guaiacol hydrodeoxygenation was investigated. Metal carbonyl complexes are known to react with water, other sources of OH-groups, and oxygen with the formation of oxides [25,26,27,28,29,30,31]. A substrate used in this work has an OH-group, and the HDO process involves the formation of water. The catalysts obtained were called MoOx and WOx, and were studied using powder X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), high-resolution transmission electron microscopy (HRTEM) with energy-dispersive X-ray spectroscopy (EDX), scanning electron microscopy (SEM), diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), and Raman spectroscopy.
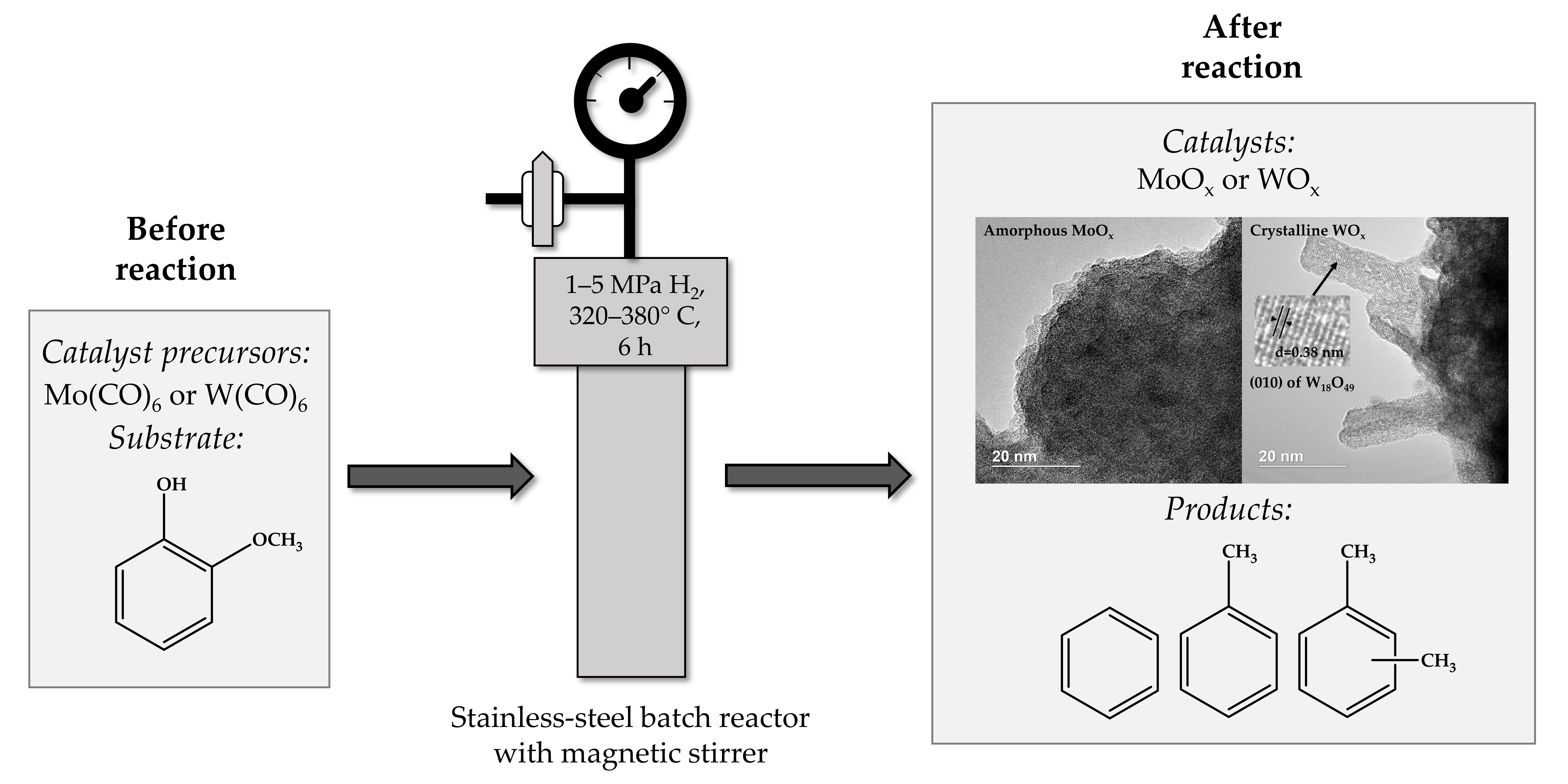
The XRD patterns of catalyst precursors are shown in Figure S1 and correspond to crystalline Mo(CO)6 (PDF №75-1336) and W(CO)6 (PDF № 84-2314). Figure 1a shows the XRD of MoOx obtained at temperatures of 120–360 °C. MoOx is shown to be amorphous over the whole temperature range. The evolution of the WOx catalyst with temperature can be followed using XRD (Figure 1b). At 120 °C the WOx diffractogram has characteristic peaks at 15.52° and 16.68°, which are assigned to W(CO)6. When temperature rises to 180 °C, carbonyl partially decomposes with the formation of a WO3 phase (PDF № 5-363), of which characteristic peaks are observed at 70.64° and 72.88°. With a further increase in temperature, the peaks related to carbonyl disappear, which characterizes its complete decomposition and oxidation. In addition, at 240 °C, WO3 is converted to W18O49 (PDF № 71-2450). This phase was identified in the sample obtained at 240–360 °C. The characteristic peaks at 23.52° and 48.12° correspond to the (010) and (020) planes in W18O49 [32]. The average crystallite sizes of W18O49 were 2.0 ± 0.6 nm, 13.3 ± 0.9 nm and 14.6 ± 0.1 nm at 240, 300, and 360 °C, respectively. Therefore, a temperature increase affects the growth of W18O49 crystallites. Thus, the XRD of WOx catalyst shows W(CO)6 decomposition with the formation of WO3 at 180 °C, and further WO3 reduction at 240 °C to W18O49.
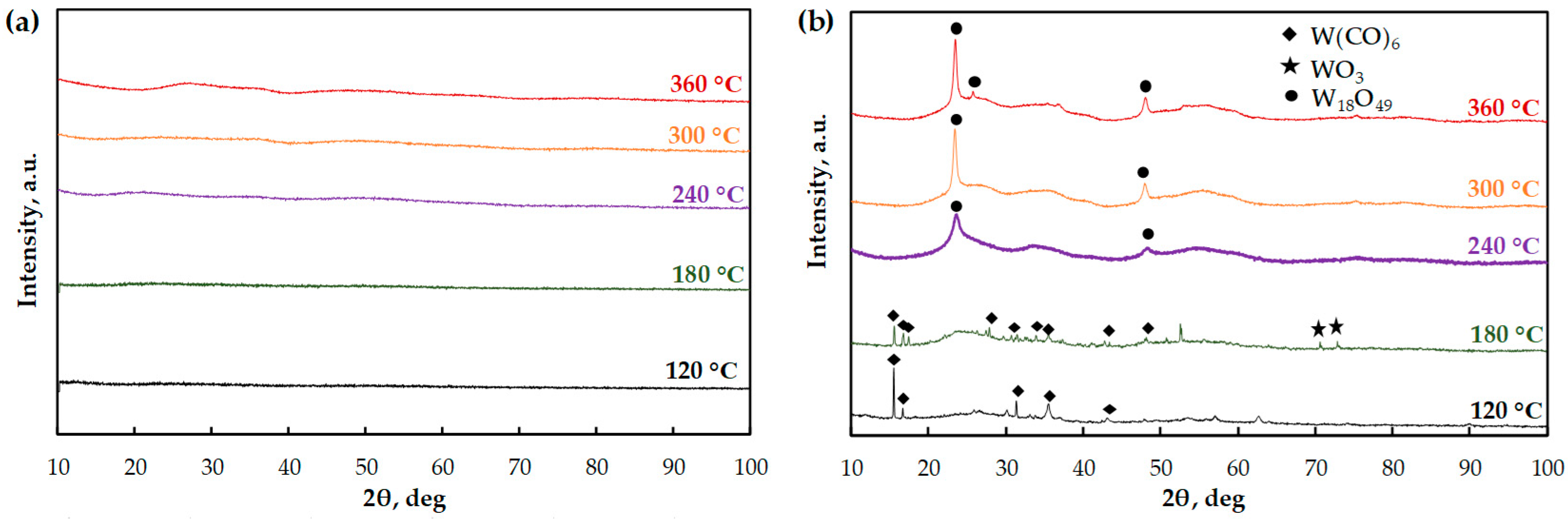
To identify electronic states on the surface of the catalysts, the XPS technique was applied. For MoOx obtained at 120 °C (Figure 2a), the doublet at 232.5 (3d5/2) and 235.7 (3d3/2) eV in the Mo 3d region can be assigned to Mo6+ in MoO3 [33]. The peaks at 231.8 (3d5/2) and 234.8 (3d3/2) eV are ascribed to Mo5+ [34]. The presence of Mo5+ species indicates the existence of the oxygen vacancies in MoO3 [35,36]. Choi and Thompson noted that the reduction of Mo6+ to Mo4+ can proceed through a +5 intermediate oxidation state [37]. Figure 2b shows the XP-spectrum of WOx in the W 4f region. The peaks with binding energies of 34.7 (4f7/2) and 35.6 (4f5/2) eV correspond to W4+ in WO2 [38]. The other two peaks observed at 35.9 (4f7/2) and 38.0 (4f5/2) eV are attributed to W6+ in WO3 [39].
The XP-spectrum of MoOx obtained at 240 °C is presented in Figure 2c. The peaks located at 230.6 (3d5/2) and 234.0 (3d3/2) eV correspond to Mo5+ species [34]. The doublet at 232.7 (3d5/2) and 235.5 (3d3/2) eV is attributed to Mo6+ [33]. The doublet at 230.5 (3d5/2) and 231.9 (3d3/2) eV is assigned to Mo4+ in MoO2 [33]. Therefore, with a temperature increase from 120 to 240 °C, Mo6+ and Mo5+ begin to reduce to Mo4+. The XP-spectrum of WOx obtained at 240 °C is presented in Figure 2d. The binding energies of 34.7 and 35.3 eV are defined as W4+ in the 4f7/2 and 4f5/2 regions, respectively. The doublet observed at 35.7 (4f7/2) and 37.8 (4f5/2) eV is attributed to W6+ in WO3 [39].
The XP-spectrum of MoOx (Figure 2e) obtained at 360 °C shows two peaks located at 230.5 (3d5/2) and 232.3 (3d3/2) eV and assigned to Mo4+ in MoO2 [33]. The predominant doublet with peaks at 232.8 (3d5/2) and 235.5 (3d3/2) eV corresponds to Mo6+ in MoO3 [33]. It is important to note that the presence of Mo5+ is not observed at 360 °C. Thus, at this temperature Mo5+ is completely reduced to Mo4+. WOx XP-spectrum (Figure 2f) shows the peaks with binding energies of 34.3 and 35.4 eV corresponding to W4+ in WO2 in 4f7/2 and 4f5/2 regions, respectively [38]. The peaks observed at 35.9 (4f7/2) and 37.8 (4f5/2) eV are attributed to W6+ in WO3 [39].
According to the XPS data, the total content of W4+ in the WOx catalyst increases with a temperature rise from 240 to 360 °C, which is associated with the reduction of W6+ to W4+ (Table S1). In the case of MoOx, Mo6+ and Mo5+ species are also reduced to Mo4+ with a temperature increase.
The XP-spectra in C 1 s and O 1 s regions of the catalysts obtained at various temperatures are shown in Figures S2 and S3. The same peaks are shown to be observed in the spectra of both catalysts. For example, there are two peaks located at 283.7 and 284.3 eV in the spectrum of MoOx (C 1 s region) obtained at 120 °C (Figure S2). These peaks correspond to the C-C bond [40,41]. The other peaks observed at 285.2 and 288.8 eV can be assigned to the C-O and O = C-O bonds, respectively [42,43]. The presence of these peaks is associated with the adsorption of reaction products on the catalyst surface. In addition, the peaks corresponding to C-O can also be associated with the carbonyl groups in Mo(CO)6 and W(CO)6.
As shown in Figure S3, there are three peaks in the XP-spectra of MoOx and WOx (O 1 s region) obtained at 120 and 240 °C. In the case of MoOx obtained at 120 °C, the binding energies of 530.5 and 531.7 eV are assigned to the Mo-O and Mo-OH bonds [44]. The XP-spectrum of WOx obtained at 120 °C has two peaks at 530.7 and 532.0 eV, which can be attributed to W-O and W-OH, respectively [45]. Thus, at 120 and 240 °C, the presence of molybdenum and tungsten oxides and OH-groups on the surface of the catalysts is observed. The peaks located at 533.0 and 533.7 eV correspond to the C = O/C-OH bonds [41]. These peaks can be associated with the presence of water and oxygenated compounds on the surface of catalysts [46]. There are only two peaks corresponding to Me-O and Me-OH in XP-spectra of MoOx and WOx obtained at 360 °C. It can be concluded that at 360 °C carbonyls are completely converted into the corresponding molybdenum and tungsten oxides.
Figure 3 presents the SEM images of MoOx and WOx obtained at different reaction temperatures (120, 240, 360 °C). The MoOx catalyst is represented by close-to-spherical particles of different sizes. The morphology of MoOx (Figure 3a,c,e) is shown to not undergo significant changes with a temperature increase. The various morphology of the WOx catalyst (Figure 3b,d,f) is observed at different temperatures. At 120 °C, the catalyst particles are close to spherical; at 240 °C, the particles do not have a definite morphology; at 360 °C, the particles are urchin-like spheres.
HRTEM, DRIFTS and Raman spectroscopy was used to study catalysts formed at 360 °C. HRTEM was used to observe the morphology of the catalysts (Figure 4). As shown in Figure 4a, MoOx represents agglomerates of particles. Moreover, the sample of MoOx is amorphous (Figure 4b), which confirms the powder XRD results. Particle sizes are difficult to calculate due to agglomeration, since the edges of individual particles are not clearly visible. WOx consists of urchin-like spheres, which are made up of nanorods with an average length of ~70–80 nm and width of ~8–10 nm (Figure 4c,d). According to the HRTEM, WOx has a crystal lattice with the interplanar spacing of 0.38 nm, which corresponds to the (010) plane in W18O49 [47,48]. Figures S4 and S5 show the elemental maps of MoOx and WOx, respectively. Uniform distribution of metal and oxygen is observed for both samples. Energy-dispersive X-ray spectroscopy was used to study the elemental content of the catalysts. The ratio of O/Me calculated from the EDX spectra was ~1.5 for MoOx and ~2.3 for WOx (Figure S6).
The DRIFT spectra of MoOx are shown in Figure S7. The intense bands of Me-O at 1000–900 cm−1 (at room temperature) are attributed to molybdenum oxide (Figure S7) [49]. There is also a broad band at 3400–3200 cm−1 from the stretching vibration, and at 1630 cm−1 from the bending vibration of OH-groups in the spectra. This is caused by the presence of water on the catalyst surface. The band growth from 3300 to 1630 cm−1 occurs due to water removal from a surface layer of the sample with an increase in temperature from 100 to 300 °C [50]. The presence of the bands at 3080 and 2950–2850 cm−1 is assigned to the C-H bonds of aryl and alkyl groups, respectively. In addition, the bands at 1392 cm−1 and 1425 cm−1 are attributed to the bending vibration of C-H bonds of the alkyl group. A further increase and decrease in temperature also shows the presence of aromatic compounds (the bands at 3080 and 1510 cm−1) [51] and alkyl groups (the bands at 2950–2850 cm−1) [52]. The band at 3740 cm−1 is attributed to the vibrations of OH-groups in reaction products.
The DRIFT spectra of WOx are shown in Figure S8. The intense band at 990 cm−1 at a room temperature is attributed to the Me-O bond, which indicates the presence of tungsten oxide [52]. Water (the bands at 3300 and 1620 cm−1 from the stretching and bending vibrations of O-H, respectively) [50] and organic compounds are also present in small amounts (the bands at 3080 cm−1 from the bending vibrations of C-H in aromatic ring and at 1510 cm−1 from bending vibrations of aromatic ring) on the catalyst surface [51]. When the sample is calcined from room temperature to 300 °C, water is actively released from the sample layer to the surface, as evidenced by the growth of the bands at 3400 and 1620 cm−1. The bands from the vibrations of the C-H bonds in aromatic compounds (3080 and 1510 cm−1) and the C-H bonds in alkyl groups (CH2, CH3), and the bands at 2950–2850 cm−1 from stretching vibrations and 1460 cm−1 from bending vibrations also become more intense [53]. At 450 °C, the band at 3150 cm−1, characteristic of condensed aromatic rings, appears on the surface. The band at 3750 cm−1 exists over a whole temperature range and is attributed to OH-groups in different aromatic products of guaiacol HDO (cresols and phenol) [51]. Thus, according to the DRIFTS data, the reaction products are absorbed on the surface of the catalysts, which is also confirmed by the XPS in the C 1 s and O 1 s regions.
The Raman spectra of MoOx and WOx is presented in Figure S9. According to the results of Raman spectroscopy, MoOx represents crystalline MoO3. The Raman bands at 995, 820, 670, 289 cm−1 are attributed to the Mo = O bond and the band at 210 cm−1 corresponds to the Mo–O–Mo bond. According to the XRD, the sample of MoOx obtained at 360 °C is amorphous, which does not match with the results of Raman spectroscopy. Ajito et al. demonstrated that amorphous MoO3 can be crystallized by the laser used to obtain the Raman spectrum [54]. There are two intense peaks in the Raman spectra of WOx. The G peak at 1500–1600 cm−1 refers to the C–C bond, which is assigned to sp2 hybridized carbon atoms, and the D peak at 1330–1390 cm−1, which refers to the C–C bond (sp3 hybridized carbon atoms). The presence of these peaks is associated with the adsorption of reaction products on the WOx surface [55]. The peaks at 790, 700 and 250 cm−1 are broad, blurry and non-intense and refer to amorphous WO3. However, according to the XRD, this catalyst has the crystal structure of W18O49. These differences can be explained by the low stability of W18O49. Lu et al. noted that W18O49 can be transformed into WO3 by the laser used to carry out the Raman spectra [38].
2.2. Catalytic Activity
2.2.1. The Effect of Temperature in Toluene
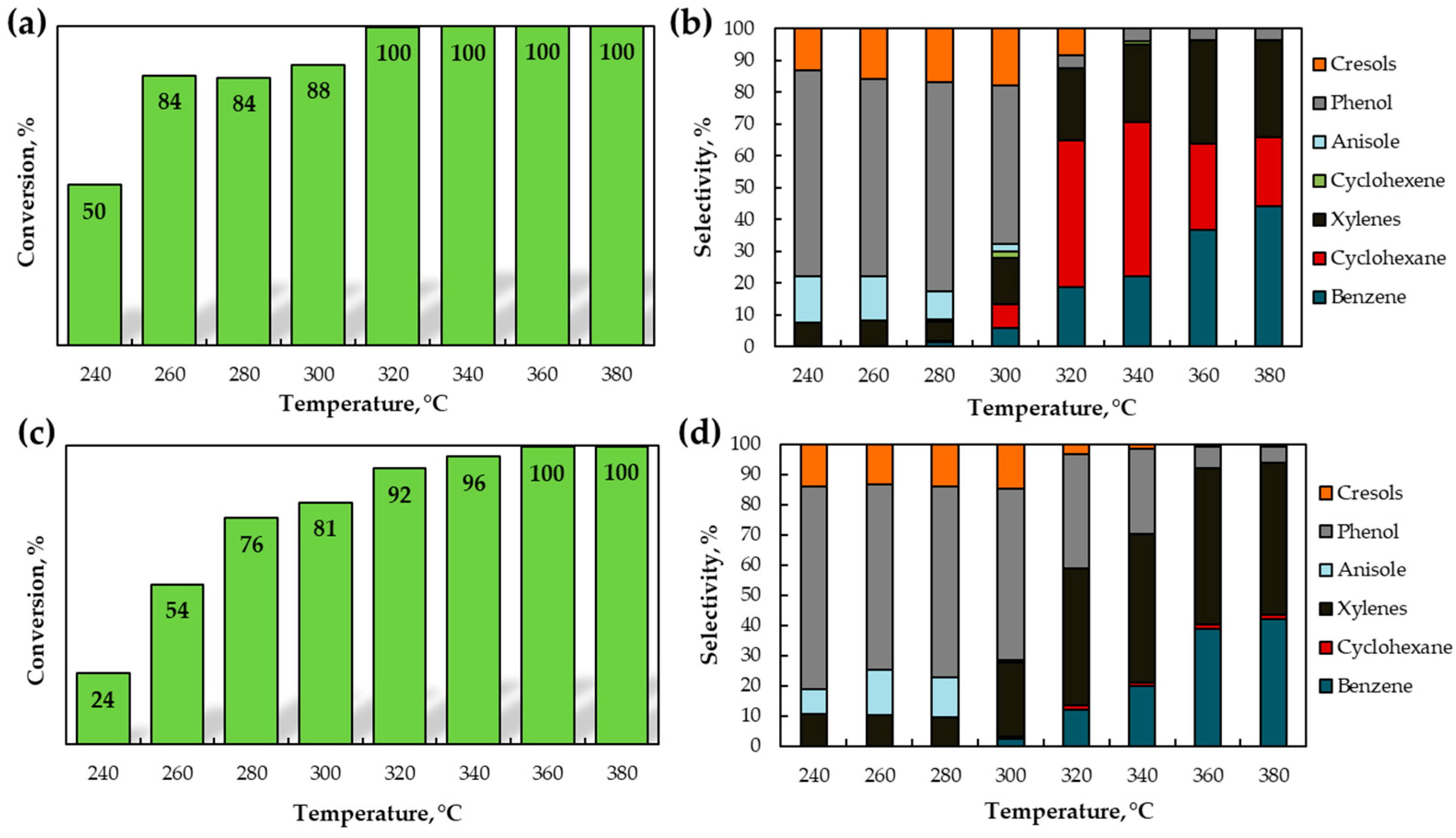
The activity of catalysts obtained in situ was investigated in the HDO of guaiacol. The effect of temperature on the catalytic activity was evaluated over a wide temperature range of 120–380 °C, under an initial H2 pressure of 5 MPa. The reaction time was 6 h, and toluene was used as a solvent. The conversion of guaiacol over MoOx was noted to be extremely low and reached 1–4% at 120–200 °C (Figure S10a). A rise in temperature from 200 to 240 °C promoted a sharp increase in guaiacol conversion from 4 to 18%—which may be related to the full precursor transformation to oxides—the partial reduction of the catalyst, and, as a result, an increase in the number of oxygen vacancies. As shown in Figure 5a, the full conversion of guaiacol was reached at 320 °C. At 240–300 °C, phenol was observed as a main reaction product (Figure 5b). Anisole, cresols, and cyclohexene were also obtained. Since the bond dissociation energy of CAR-OCH3 is 45 kJ/mol and lower than that of the CAr–OH bond, the conversion of guaiacol to phenol was more favored than conversion to anisole [56]. The highest selectivity for phenol was 66% at 280 °C. A following temperature increase led to direct deoxygenation (DDO) of phenol to benzene. Therefore, the cleavage of the CAr–OH bond was favored at higher temperatures [11]. In this way, the selectivity for benzene gradually increased with a rise in temperature from 280 to 380 °C. Cyclohexane and xylenes were also detected in large amounts at 320–380 °C.
At 120–220 °C, guaiacol conversion over WOx was very low and reached 1–5% (Figure S10b). The full conversion of guaiacol was reached at 360 °C (Figure 5c). Phenol was a major product at 240–300 °C. The decrease in phenol selectivity with a temperature increase from 240 °C to 380 °C is associated with its conversion to benzene (Figure 5d). Compared to MoOx, WOx demonstrated low catalytic activity in the hydrogenation of benzene to cyclohexene and cyclohexane. Oxygen vacancies in metal oxides are noted to act as active sites in hydrogenation reactions under typical HDO conditions [16,57]. With the temperature increase from 240 to 360 °C the selectivity for xylenes increased from 11 to 52%. As is the case with MoOx, anisole was not detected among the reaction products at 320–380 °C. Most likely, it is caused by the conversion of anisole to phenol or cresols.
Phenol was the major product of guaiacol HDO at 240–300 °C over both catalysts. With a temperature increase to 320–340 °C, phenol selectivity over MoOx decreased, and cyclohexane became the main product. In the case of WOx, phenol was also obtained in significant amounts at 320–340 °C. However, xylenes formed with higher selectivity. With a further increase in temperature to 360–380 °C, aromatics, such as benzene and xylenes, were obtained in large amounts over both catalysts. At this temperature, WOx is more selective to aromatics than MoOx.
The comparison of catalyst activities in the formation of HDO products (benzene, xylenes, cyclohexene, cyclohexane) is presented in Figure S11. The highest yield of HDO products (96%) was shown to be reached at 360 °C using MoOx. Compared to MoOx, the highest yield of HDO products over WOx was 94% and was reached at 380 °C.
2.2.2. The Effect of Pressure in Toluene
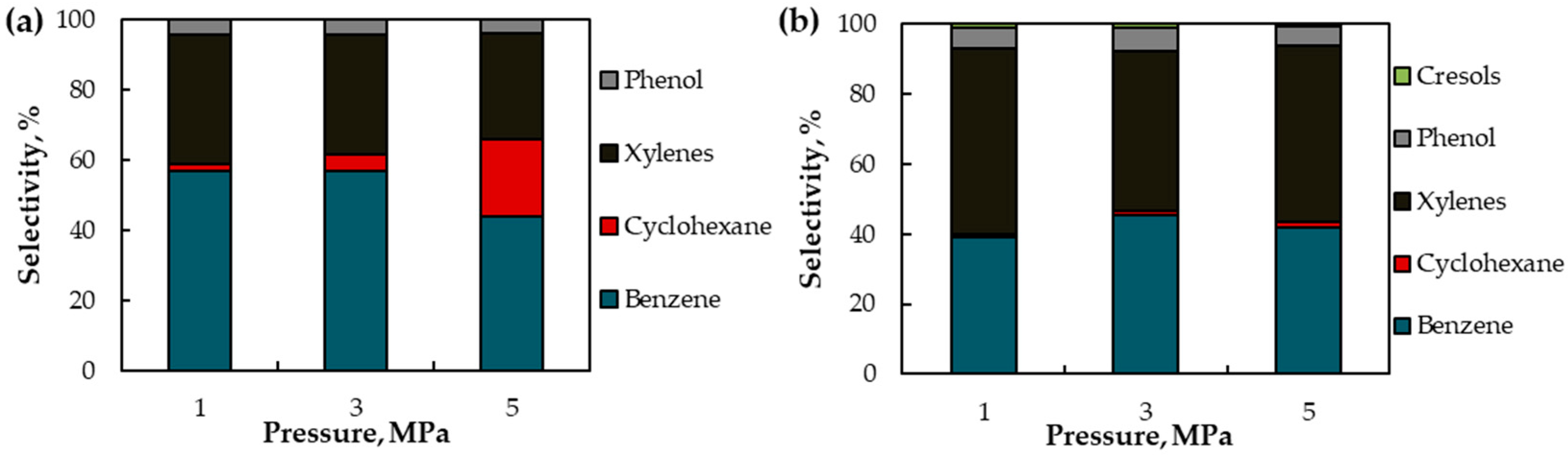
Because cyclohexane was formed in significant amounts when MoOx was used as a catalyst, the effect of H2 pressure (1–5 MPa) on product formation was studied (Figure 6). Guaiacol conversion was 100% in all cases. In the case of MoOx, benzene, xylenes, cyclohexane and phenol were formed as products (Figure 6a). An increase in pressure led to a decrease in selectivity for benzene and an increase in selectivity for cyclohexane. Thus, carrying out the HDO of guaiacol over MoOx under low H2 pressure contributes to higher benzene selectivity. At the same time, H2 pressure is shown to have almost no effect on benzene selectivity when WOx is used as a catalyst (Figure 6b).
2.2.3. Reaction Pathways
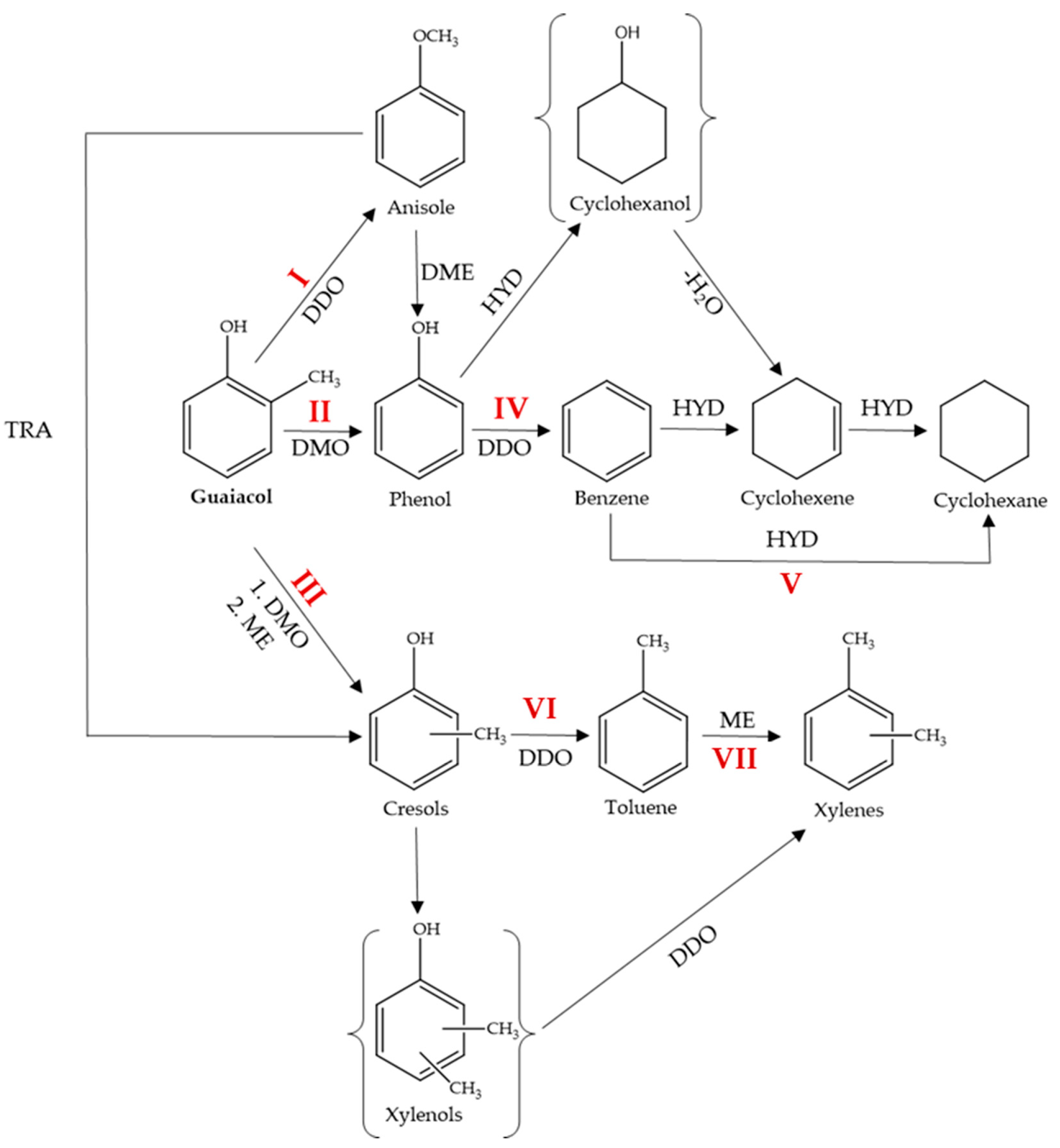
The HDO of guaiacol over MoOx and WOx can occur via three routes, i.e., direct deoxygenation to anisole (DDO), demethoxylation to phenol (DMO), and DMO followed by methylation to cresols (Figure 7). The formation of cresols also occurs through transalkylation (TRA) of anisole [58]. Cresols can then convert to xylenes via two pathways: methylation (ME) to xylenols followed by DDO, or DDO to toluene followed by ME. However, xylenols were not detected, and toluene was used as a solvent. Phenol is formed not only by DMO of guaiacol but also by demethylation (DME) of anisole. Phenol then converts to benzene through DDO. Benzene is transformed into cyclohexene and cyclohexane by hydrogenation (HYD). Cyclohexene may also be formed by dehydratation of cyclohexanol, however, cyclohexanol was not identified in the reaction medium. Cyclohexane is also obtained by the HYD of cyclohexene.
2.2.4. The Effect of Temperature and Pressure in Dodecane
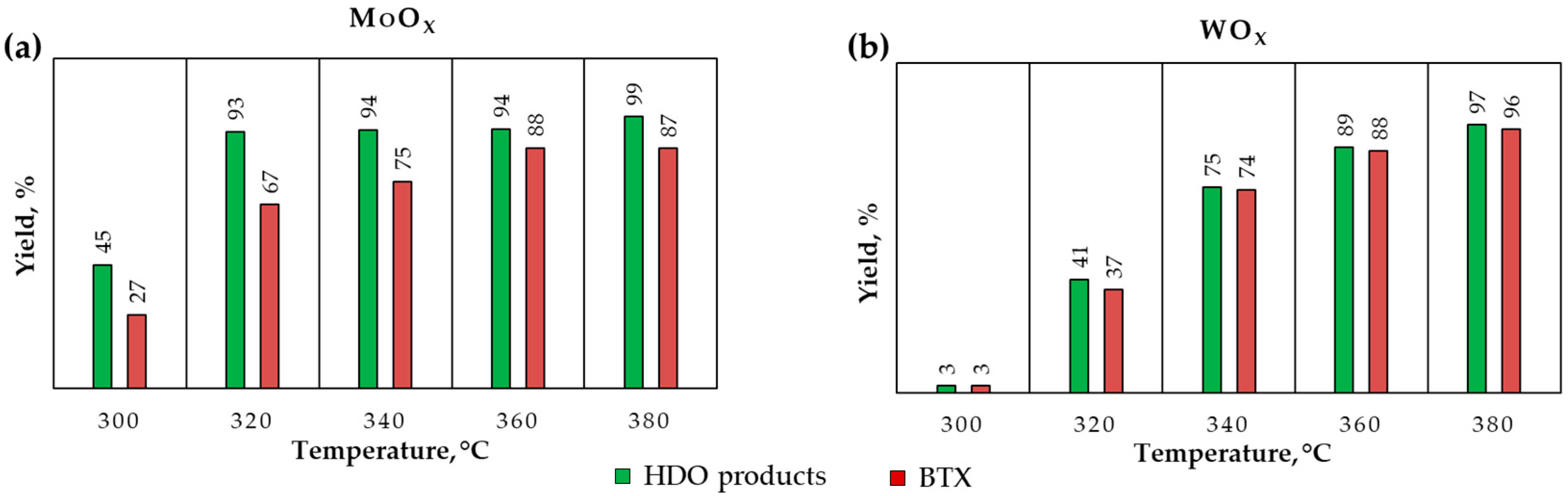
Since xylenes were one of the reaction products, toluene was also probably formed. Thereby, the solvent was changed from toluene to dodecane. The catalytic tests were carried out at 300–380 °C, 5 MPa H2, and 6 h of reaction. The yield of HDO products (benzene, toluene, xylenes, cyclohexene, cyclohexane) and the BTX fraction for both catalysts are shown in Figure 8. For both catalysts, the rise in the reaction temperature contributes to an increase in the yield of HDO products and the BTX fraction. At the same time, WOx is more selective in the formation of BTX fraction, while in the presence of MoOx, cyclohexane is also one of the HDO products. In addition, the catalytic tests were carried out at 380 °C, 1–5 MPa H2 and 6 h of reaction. As shown in Figure S12, the selectivity for BTX products changes insignificantly, however, higher pressure results in more cyclohexane selectivity, especially when MoOx is used.
2.2.5. The Recycling Tests
Five runs of recycling tests were carried out to estimate the stability of MoOx and WOx catalysts (Figure S13). The stability tests were carried out at average conversions (320 °C, 5 MPa H2, 1 h). The conversion of guaiacol over MoOx was 73% at the first run. It gradually decreased and was 56% after the fifth run. Such a decrease in conversion indicates insignificant deactivation of the MoOx catalyst during the recycling test runs. Phenol and HDO product selectivity remained almost unchanged. At the same conditions, the conversion of guaiacol over WOx changed significantly from 43% at the first run to 8% at the fifth run. The reason for this may be the adsorption on the catalyst surface of significant amounts of reaction products (confirmed by DRIFTS and Raman spectroscopy), and, as a consequence, the blocking of active sites. Phenol and HDO product selectivity changed insignificantly.
3. Materials and Methods
3.1. Materials
All reagents were purchased from commercial suppliers and used as received. Molybdenum hexacarbonyl (LLC “Redkino experimental plant,” Redkino, Russia, wt. % of Mo 36.5–37.5%), and tungsten hexacarbonyl (LLC “Redkino experimental plant,” Redkino, Russia, wt. % of W 51–52%) were used as precursors for catalyst preparation. Guaiacol (Acros organics, Geel, Belgium, >99%) was used as a substrate for hydrodeoxygenation. Toluene (Component-reaktiv, Moscow, Russia, >98.5%) or dodecane (Sigma-Aldrich, Burlington, VT, USA, >99%) were used as solvents. Acetone (Component-reaktiv, Moscow, Russia, >99.5%,) and petroleum ether 40/70 (Component-reaktiv, Moscow, Russia, tech.) were used for catalyst washing. H2 (≥98%, Air Liquide, Paris, France) and Ar (≥98%, Air Liquide, Paris, France) gases were also used.
3.2. Catalyst Preparation and Catalytic Tests
The catalysts were prepared in a 45 mL stainless-steel batch autoclave reactor during the HDO of guaiacol. A mixture of 84.4 mg Mo(CO)6 (or 113.7 mg W(CO)6) and guaiacol (2 g of 10 wt. % solution in toluene or dodecane) was added to the reactor. The HDO of guaiacol was investigated at 120–400 °C for 6 h while undergoing constant magnetic stirring (7000 rpm). The initial pressure of H2 was 5 MPa. After the catalytic test, the reactor was cooled to a room temperature. Liquid products were isolated from the reactor and separated from the catalysts by centrifugation (5000× g rpm). Catalysts obtained during the reaction were washed by petroleum ether and acetone until the solution over the catalysts became colorless. Then, the catalysts were dried in an argon atmosphere at room temperature.
3.3. Characterization
Powder X-ray diffraction was used to determine the phase composition of the catalysts. The X-ray diffractograms were obtained for a range of 5−100° 2θ by using a Rigaku Rotaflex RU-200 diffractometer (CuKα radiation) equipped with a Rigaku D/Max-RC goniometer (a rotation speed of 1°/min; a step 0.04°). Qualitative phase analysis of the samples was carried out using the PDF-2 ICDD database of powder diffraction patterns. The average crystallite sizes were estimated using the Scherrer equation.
X-ray photoelectron spectra were obtained using a PREVAC EA15 electronic spectrometer (AlKα radiation, hν = 1486.74 eV, 150 W). Spectra were recorded at a pressure not exceeding 5 × 10–9 mbar. Spectra deconvolution was carried out using a PeakFit software.
The morphology of the catalysts obtained was observed using scanning electron microscopy and high-resolution transmission electron microscopy. Scanning electron microscopy with energy-dispersive X-ray spectroscopy was performed using a Carl Zeiss NVision 40 microscope equipped with an Oxford Instruments X-Max EDX detector operated at 20 kV. High-resolution transmission electron microscopy with energy-dispersive X-ray spectroscopy were performed using a FEI Tecnai Osiris transmission electron microscope equipped with a field emission electron gun operated at 200 kV.
Diffuse reflectance infrared Fourier transform spectroscopy was carried out in a high-temperature PIKE Diffus IR cell coupled with a VERTEX-70 Bruker Fourier transform IR spectrometer. DRIFT spectra of catalysts (194 scans at a resolution of 2 cm−1 in a range of 600–4000 cm−1) were investigated in an argon flow in a temperature range of 25–450 °C. Spectra processing was carried out using OPUS-7 software. Raman spectra were obtained using a Senterra II spectrometer (laser power is 0.25 mW, a wavelength is 532 nm; a magnification is 50×).
Liquid reaction products were identified by gas chromatography–mass spectrometry using a Thermo Scientific ISQ 7000 GC-MS system equipped with a Restek 5XI-17SIL MS CAP capillary column (30 m × 0.25 mm × 0.25 μm), with helium being used as a carrier gas. The quantitative analysis of liquid products was carried out using a Crystallux 4000 M gas chromatograph equipped with a flame ionization detector, an Optima-1 capillary column (25 m × 0.32 mm × 0.35 μm), with helium used as a carrier gas. Chromatograms were obtained and analyzed using a NetChromWin software. Guaiacol conversion (%), product selectivity (%), and yield of full HDO products (%) were calculated using the following equations:
4. Conclusions
Th possibility of obtaining in situ MoOx and WOx from the corresponding carbonyls during the HDO of guaiacol is shown in this study. According to XRD, MoOx is amorphous, while WOx displayed the crystalline patterns of WO3 at 180 °C and W18O49 at 240–360 °C. The HRTEM of the catalysts obtained at 360 °C confirms the results of the XRD. It was shown via XPS that an increase in reaction temperature promoted the reduction of the surface M6+ to M4+. In the case of MoOx, the reduction proceeded through the formation of an intermediate M5+. The presence of M5+ and M4+ may indicate vacancy sites on the surface of the catalysts.
The catalysts obtained were investigated in the HDO of guaiacol. Low guaiacol conversions at temperatures of 120–180 °C were associated with incomplete decomposition of the precursors. The active phase of the catalysts began to form at temperatures above 200 °C. An increase in the reaction temperature promoted the formation of vacancy sites on which hydrodeoxygenation could proceed. The full conversion of guaiacol was reached at 320 °C (5 MPa, 6 h) and 360 °C (5 MPa, 6 h) using MoOx and WOx, respectively. MoOx, compared to WOx, is shown to demonstrate higher activity in the full HDO of guaiacol. However, WOx is more selective in the production of aromatics, in particular the BTX fraction. The highest yield of the BTX was 96% (380 °C, 5 MPa, 6 h) using the WOx catalyst.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13020263/s1, Figure S1: X-ray diffractograms of (a) molybdenum and (b) tungsten carbonyls—the precursors of the catalysts; Table S1: Oxidation states (Mo 3d, W 4f regions) on the surface of the catalysts, identified by the XPS; Figure S2: The XPS spectra in C 1 s region of the catalysts obtained at different temperatures; Figure S3: The XPS spectra in O 1 s region of the catalysts obtained at different temperatures; Table S2: Oxidation states (C 1 s region) on the surface of the catalysts, identified by the XPS; Table S3: Oxidation states (O 1 s region) on the surface of the catalysts, identified by the XPS; Figure S4: The elemental mapping of MoOx. Maps are assigned to (a) molybdenum, (b) oxygen, (c) molybdenum and oxygen, and (d) the HAADF image; Figure S5: The elemental mapping of WOx. Maps are assigned to (a) tungsten, (b) oxygen, (c) tungsten and oxygen, and (d) the HAADF image; Figure S6: The EDX spectra of the catalysts: (a) MoOx (b) WOx; Figure S7: The DRIFT spectra of MoOx in a range of 4000–600 cm−1; Figure S8: The DRIFT spectra of WOx in a range of 4000–600 cm−1; Figure S9: The Raman spectra of MoOx and WOx; Figure S10: The results of the guaiacol HDO over in situ formed (a) MoOx (b) WOx. Reaction conditions: 120–220 °C, 5 MPa H2, 6 h; 10 wt. % guaiacol in toluene solution; Figure S11: The yield of HDO products over in situ-formed MoOx and WOx. Reaction conditions: 240–380 °C, 5 MPa H2, 6 h; 10 wt. % guaiacol in toluene solution.; Figure S12: The results of guaiacol HDO over in situ formed (a) MoOx (b) WOx. Reaction conditions: 380 °C, 1–5 MPa H2, 6 h; 10 wt. % guaiacol in dodecane solution.; Figure S13: The recycling test runs of (a) MoOx (b) WOx. Reaction conditions: 320 °C, 5 MPa H2, 1 h; 10 wt. % guaiacol in dodecane solution.
Author Contributions
Conceptualization, M.M., M.G. and A.M.; validation, M.M. and M.G.; formal analysis, M.M. and M.G.; investigation, M.M. and A.S.; resources, A.S.; writing—original draft preparation, M.M. and M.G.; writing—review and editing, M.G. and A.M.; visualization, M.M. and M.G.; supervision, M.G. and A.M.; project administration, M.G. and A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was carried out within the State Program of TIPS RAS.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Ivan Levin from TIPS RAS for the XRD analysis, Olga Arapova from TIPS RAS for the Raman spectroscopy and DRIFTs analysis, and Yury Grigoriev from A.V. Shubnikov Institute of Crystallography RAS for the HRTEM analysis. This work was performed using the equipment of the Shared Research Center Analytical center of deep oil processing and petrochemistry of the A.V. Topchiev Institute of Petrochemical Synthesis RAS and the JRC PMR IGIC RAS.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nanda, S.; Mohammad, J.; Reddy, S.N.; Kozinski, J.A.; Dalai, A.K. Pathways of lignocellulosic biomass conversion to renewable fuels. Biomass Convers. Biorefin. 2014, 4, 157–191. [Google Scholar] [CrossRef]
- Cherubini, F.; Strømman, A.H. Production of Biofuels and Biochemicals from Lignocellulosic Biomass: Estimation of Maximum Theoretical Yields and Efficiencies Using Matrix Algebra. Energy Fuels 2010, 24, 2657–2666. [Google Scholar] [CrossRef]
- Tribot, A.; Amer, G.; Abdou, A.M.; de Baynast, H.; Delattre, C.; Pons, A.; Mathias, J.; Callois, J.; Vial, C.; Michaud, P.; et al. Wood-lignin: Supply, extraction processes and use as bio-based material. Eur. Polym. J. 2019, 112, 228–240. [Google Scholar]
- Zhang, J.; Sun, J.; Wang, Y. Recent Advances in Selectively Catalytic Hydrodeoxygenation of Lignin-derived Oxygenates to Arenes. Green Chem. 2020, 22, 1072–1098. [Google Scholar] [CrossRef]
- Führer, M.; Van Haasterechta, T.; Bitter, J.H. Cinnamaldehyde hydrogenation over carbon supported molybdenum and tungsten carbide catalysts. Chem. Commun. 2022, 58, 13608–13611. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Xu, Y.; Gao, X.; Dai, Y.; Tang, Y. Palladium-Incorporated α-MoC Mesoporous Composites for Enhanced Direct Hydrodeoxygenation of Anisole. Catalysts 2021, 11, 370. [Google Scholar] [CrossRef]
- Kurisingal, J.F.; Lee, S.; Lee, J.G.; An, K. Zeolitic Imidazolate Framework Decorated Molybdenum Carbide Catalysts for Hydrodeoxygenation of Guaiacol to Phenol. Catalysts 2022, 12, 1605. [Google Scholar] [CrossRef]
- Wang, F.; Wen, C.; Lu, M.; Zhang, P.; Zhu, J.; Li, M.; Shan, Y.; Song, C. SBA-15-supported ultrastable Mo2N@CN catalysts for hydrodeoxygenation of guaiacol. Biomass Bioenergy 2023, 168, 106680. [Google Scholar] [CrossRef]
- Golubeva, M.A.; Mukhtarova, M.; Bugaev, A.L.; Naranov, E.R. In Situ Generated Dispersed Catalysts Based on Molybdenum and Tungsten Phosphides in Hydroprocessing of Guaiacol. Pet. Chem. 2023. [Google Scholar] [CrossRef]
- Golubeva, M.A.; Mukhtarova, M.; Sadovnikov, A.A.; Maximov, A.L. Bulk Molybdenum and Tungsten Phosphides for Selective Phenol Production from Guaiacol. ACS Omega 2022, 7, 40586–40595. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, J.; Zhang, Q.; Liu, Q.; Li, Y.; Chen, L.; Wang, C.; Ma, L. Hydrodeoxygenation of lignin-derived phenolic compounds into aromatic hydrocarbons under low hydrogen pressure using Molybdenum oxide as catalyst. Catal. Today 2019, 319, 41–47. [Google Scholar] [CrossRef]
- Jiang, S.; Ji, N.; Diao, X.; Li, H.; Rong, Y.; Lei, Y.; Yu, Z. Vacancy Engineering in Transition Metal Sulfide and Oxide Catalysts for Hydrodeoxygenation of Lignin-Derived Oxygenates. ChemSusChem 2021, 14, 4377–4396. [Google Scholar] [CrossRef] [PubMed]
- Prasomsri, T.; Nimmanwudipong, T.; Román-Leshkov, Y. Effective hydrodeoxygenation of biomass-derived oxygenates into unsaturated hydrocarbons by MoO3 using low H2 pressures. Energy Environ. Sci. 2013, 6, 1732. [Google Scholar] [CrossRef]
- Bagnato, G.; Sanna, A.; Paone, E.; Catizzone, E. Recent catalytic advances in hydrotreatment processes of pyrolysis bio-oil. Catalysts 2021, 11, 157. [Google Scholar] [CrossRef]
- Thibodeau, T.J.; Canney, A.S.; DeSisto, W.J.; Wheeler, M.C.; Amar, F.G.; Frederick, B.G. Composition of tungsten oxide bronzes active for Hydrodeoxygenation. Appl. Catal. A 2010, 388, 86–95. [Google Scholar] [CrossRef]
- Whiffen, V.M.L.; Smith, K.J. Hydrodeoxygenation of 4-Methylphenol over Unsupported MoP, MoS2 and MoOx Catalysts. Energy Fuels 2010, 24, 4728–4737. [Google Scholar] [CrossRef]
- Wang, C.; Mironenko, A.V.; Raizada, A.; Chen, T.; Mao, X.; Padmanabhan, A.; Vlachos, D.G.; Gorte, R.J.; Vohs, J.M. Mechanistic Study of the Direct Hydrodeoxygenation of m-Cresol over WOx-Decorated Pt/C Catalysts. ACS Catal. 2018, 8, 7749–7759. [Google Scholar] [CrossRef]
- Wang, C.; Wittreich, G.R.; Lin, C.; Huang, R.; Vlachos, D.G.; Gorte, R.J. Hydrodeoxygenation of m-Cresol Over Pt-WOx/C Using H2 Generated In Situ by n-Hexane Dehydrogenation. Catal. Lett. 2020, 150, 913–921. [Google Scholar] [CrossRef]
- Chen, T.; Kwon, O.; Huang, R.; Lin, C.; Vohs, J.M. WOx promoted nickel catalyst for hydrodeoxygenation of m-cresol. J. Catal. 2021, 400, 294–300. [Google Scholar] [CrossRef]
- Golubeva, M.A.; Maximov, A.L. Hydroprocessing of furfural over in situ generated nickel phosphide based catalysts in different solvents. Appl. Catal. A 2020, 608, 117890. [Google Scholar] [CrossRef]
- Golubeva, M.A.; Maximov, A.L. Investigations on the Formation of Transition Metal Phosphides during the Hydrotreating of Light Cycle Oil Russ. J. Appl. Chem. 2021, 94, 1536–1545. [Google Scholar]
- Vagvala, T.C.; Pandey, S.S.; Ogomi, Y.; Ma, T.; Hayase, S. Investigation of metal xanthates as latent curing catalysts for epoxy resin via formation of in-situ metal sulfides. Inorg. Chim. Acta 2015, 435, 292–298. [Google Scholar] [CrossRef]
- Vutolkina, A.V.; Baygildin, I.G.; Glotov, A.P.; Cherednichenko, K.A.; Maksimov, A.L.; Karakhanov, E.A. Dispersed Ni-Mo sulfide catalysts from water-soluble precursors for HDS of BT and DBT via in situ produced H2 under Water gas shift conditions. Appl. Catal. B 2021, 282, 119616. [Google Scholar] [CrossRef]
- Kuchinskaya, T.; Kniazeva, M.; Samoilov, V.; Maximov, A. In Situ Generated Nanosized Sulfide Ni-W Catalysts Based on Zeolite for the Hydrocracking of the Pyrolysis Fuel Oil into the BTX Fraction. Catalysts 2020, 10, 1152. [Google Scholar] [CrossRef]
- Zhang, K.; McCleese, C.; Lin, P.; Chen, X.; Morales, M.; Cao, W.; Seo, F.J.; Burda, C.; Baumgart, H. Synthesis of ALD Tungsten Trioxide Thin Films from W(CO)6 and H2O Precursors. ECS Trans. 2015, 69, 199–209. [Google Scholar] [CrossRef]
- Davazoglou, D.; Moutsakis, A.; Valamontes, V.; Psychorish, V. Tungsten Oxide Thin Films Chemically Vapor Deposited at Low Pressure by W(CO)6 Pyrolysis. J. Electrochem. Soc. 1997, 144, 595–599. [Google Scholar] [CrossRef]
- Suvanto, M.; Pakkanen, T. Deposition of tungsten hexacarbonyl on alumina: A diffuse reflectance infrared Fourier transform spectroscopy study. J. Mol. Catal. A Chem. 1999, 138, 211–220. [Google Scholar] [CrossRef]
- Ou, N.C.; Su, X.; Bock, D.C.; McElwee-White, L. Precursors for chemical vapor deposition of tungsten oxide and molybdenum oxide. Coord. Chem. Rev. 2020, 421, 213459. [Google Scholar] [CrossRef]
- Dhas, N.A.; Gedanken, A. Characterization of Sonochemically Prepared Unsupported and Silica-Supported Nanostructured Pentavalent Molybdenum Oxide. J. Phys. Chem. B 1997, 101, 9495–9503. [Google Scholar] [CrossRef]
- Dimitrova, Z.; Gogova, D. On the structure, stress and optical properties of CVD tungsten oxide films. Mater. Res. Bull. 2005, 40, 333–340. [Google Scholar] [CrossRef]
- Fu, J.; Liu, S.; Zheng, W.; Huang, R.; Wang, C.; Lawal, A.; Alexopoulos, K.; Liu, S.; Wang, Y.; Yu, K.; et al. Modulating the dynamics of Brønsted acid sites on PtWOx inverse catalyst. Nat. Catal. 2022, 5, 144–153. [Google Scholar] [CrossRef]
- Fang, Z.; Jiao, S.; Kang, Y.; Pang, G.; Feng, S. Photothermal Conversion of W18O49 with a Tunable Oxidation State. ChemistryOpen 2017, 6, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Baltrusaitis, J.; Mendoza-Sanchez, B.; Fernandez, V.; Veenstra, R.; Dukstiene, N.; Roberts, A.; Fairley, N. Generalized molybdenum oxide surface chemical state XPS determination via informed amorphous sample model. Appl. Surf. Sci. 2015, 326, 151–161. [Google Scholar] [CrossRef]
- Xie, F.; Choy, W.C.H.; Wang, C.; Li, X.; Zhang, S.; Hou, J. Low-Temperature Solution-Processed Hydrogen Molybdenum and Vanadium Bronzes for an Efficient Hole-Transport Layer in Organic Electronics. Adv. Mater. 2013, 25, 2051–2055. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Cook, J.B.; Lin, H.; Ko, J.S.; Tolbert, S.H.; Ozolins, V.; Dunn, B. Oxygen vacancies enhance pseudocapacitive charge storage properties of MoO3−x. Nat. Mater. 2017, 16, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Apergi, S.; Koch, C.; Brocks, G.; Olthof, S.; Tao, S. Decomposition of Organic Perovskite Precursors on MoO3: Role of Halogen and Surface Defects. Appl. Mater. Interfaces 2022, 14, 34208–34219. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Thompson, L.T. XPS study of as-prepared and reduced molybdenum oxides. Appl. Surf. Sci. 1996, 93, 143–149. [Google Scholar] [CrossRef]
- Lu, D.Y.; Chen, J.; Zhou, J.; Deng, S.Z.; Xu, N.S.; Xu, J.B. Raman spectroscopic study of oxidation and phase transition in W18O49 nanowires. J. Raman Spectrosc. 2007, 38, 176–180. [Google Scholar] [CrossRef]
- Katoh, M.; Takeda, Y. Chemical State Analysis of Tungsten and Tungsten Oxides Using an Electron Probe Microanalyzer. Jpn. J. Appl. Phys. 2004, 43, 7292–7295. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Lai, J.-Y.; Tsai, T.-H.; Chuang, P.-Y.; Chen, Y.-C. Effects of oxygen addition on electrochromic properties in low temperature plasma-enhanced chemical vapor deposition-synthesized MoOxCy thin films for flexible electrochromic devices. Thin Solid Films 2011, 519, 3875–3882. [Google Scholar] [CrossRef]
- Światowska-Mrowiecka, J.; de Diesbach, S.; Maurice, V.; Zanna, S.; Klein, L.; Briand, E.; Vickridge, I.; Marcus, P. Li-Ion Intercalation in Thermal Oxide Thin Films of MoO3 as Studied by XPS, RBS, and NRA. J. Phys. Chem. C 2008, 112, 11050–11058. [Google Scholar] [CrossRef]
- Ghasempour, R.; Iraji, A. Hybrid multiwalled carbon nanotubes and trioxide tungsten nanoparticles for hydrogen gas sensing. J. Phys. D Appl. Phys. 2009, 42, 165105. [Google Scholar] [CrossRef]
- Li, J.; Zhou, C.; Mu, J.; Yang, E.; Zhao, X. In situ synthesis of molybdenum carbide/N-doped carbon hybrids as an efficient hydrogen-evolution electrocatalyst. RSC Adv. 2018, 8, 17202–17208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajadi, M.; Ranjbar, M.; Rasuli, R. Two-step synthesis of Ag-decorated MoO3 nanotubes, and the effect of hydrogen doping. Appl. Surf. Sci. 2020, 527, 146675. [Google Scholar] [CrossRef]
- Vasilopoulou, M.; Soultati, A.; Georgiadou, D.G.; Stergiopoulos, T.; Palilis, L.C.; Kennou, S.; Stathopoulos, N.A.; Davazoglou, D.; Argitis, P. Hydrogenated under-stoichiometric tungsten oxide anode interlayers for efficient and stable organic photovoltaics. J. Mater. Chem. A 2014, 2, 1738. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; You, Z.; Fan, J.; Zhang, J.; Wang, S.; Xu, J. Laser Induced Nano and Micro Structures of Molybdenum Surface Applied in Multistage Depressed Collector for Secondary Electron Suppression. Appl. Sci. 2019, 9, 4374. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Tang, L.; Deng, Y.; Wang, J.; Tang, W.; Liu, Y.; Chen, Z.; Yu, J.; Wang, J.; Liang, Q. Synthesis of branched WO3@W18O49 homojunction with enhanced interfacial charge separation and full-spectrum photocatalytic performance. Chem. Eng. J. 2020, 389, 124474. [Google Scholar] [CrossRef]
- Chen, C.L.; Mori, H. In situ TEM observation of the growth and decomposition of monoclinic W18O49 nanowires. Nanotechnology 2009, 20, 285604. [Google Scholar] [CrossRef]
- Hahn, T.; Bentrup, U.; Armbrüster, M.; Kondratenko, E.V.; Linke, D. The Enhancing Effect of Brønsted Acidity of Supported MoOx Species on their Activity and Selectivity in Ethylene/trans-2-Butene Metathesis. ChemCatChem 2014, 6, 1664–1672. [Google Scholar] [CrossRef]
- Zafeiratos, S.; Papakonstantinou, G.; Jacksic, M.M.; Neophytides, S.G. The effect of Mo oxides and TiO2 support on the chemisorption features of linearly adsorbed CO on Pt crystallites: An infrared and photoelectron spectroscopy study. J. Catal. 2005, 232, 127–136. [Google Scholar] [CrossRef]
- Baes, A.U.; Bloom, P.R. Diffuse Reflectance and Transmission Fourier Transform Infrared (DRIFT) Spectroscopy of Humic and Fulvic Acids Soil. Sci. Soc. Am. J. 1989, 53, 695–700. [Google Scholar] [CrossRef]
- Barreto, M.S.C.; Reis, J.V.; Muraoka, T.; Jemo, M.; Vergutz, L.; Alleoni, L.R.F. Diffuse reflectance infrared Fourier transform spectroscopy for a qualitative evaluation of plant leaf pigment extraction. Analyst 2021, 146, 3440. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, C.; Zhang, L.; Gholizadeh, M.; Hu, X. Biochar catalyzing polymerization of the volatiles from pyrolysis of poplar wood. Int. J. Energy. Res. 2021, 45, 13936–13951. [Google Scholar] [CrossRef]
- Ajito, K.; Nagahara, L.A.; Tryk, D.A.; Hashimoto, K.; Fujishima, A. Study of the Photochromic Properties of Amorphous MoO3 Films Using Raman Microscopy. J. Phys. Chem. 1995, 99, 16383–16388. [Google Scholar] [CrossRef]
- Casiraghi, C.; Piazza, F.; Ferrari, A.C.; Grambole, D.; Robertson, J. Bonding in hydrogenated diamond-like carbon by Raman spectroscopy. Diamond Relat. Mater. 2005, 14, 1098–1102. [Google Scholar] [CrossRef]
- Tran, N.T.T.; Uemura, Y.; Chowdhury, S.; Ramli, A. Vapor-phase Hydrodeoxygenation of Guaiacol on Al−MCM−41 Supported Ni and Co Catalysts. Appl. Catal. A 2016, 512, 93–100. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Afanasiev, P.; Geantet, C. Hydrodeoxygenation of guaiacol with CoMo catalysts. Part I: Promoting effect of cobalt on HDO selectivity and activity. Appl. Catal. B 2011, 101, 239–245. [Google Scholar] [CrossRef]
- Venkatesan, K.; Krishna, J.V.J.; Anjana, S.; Selvam, P.; Vinu, R. Hydrodeoxygenation kinetics of syringol, guaiacol and phenol over H-ZSM-5. Catal. Commun. 2021, 148, 106164. [Google Scholar] [CrossRef]
Figure 1.
Powder X-ray diffraction of (a) MoOx, (b) WOx formed in situ during guaiacol HDO (10 wt. % solution in toluene). Reaction conditions: 120–360 °C, 5 MPa H2, 6 h.
Figure 1.
Powder X-ray diffraction of (a) MoOx, (b) WOx formed in situ during guaiacol HDO (10 wt. % solution in toluene). Reaction conditions: 120–360 °C, 5 MPa H2, 6 h.

Figure 2.
X-ray photoelectron spectroscopy of MoOx and WOx catalysts formed in situ during guaiacol HDO (10 wt. % solution in toluene) at (a,b) 120 °C, (c,d) 240 °C and (e,f) 360 °C.
Figure 2.
X-ray photoelectron spectroscopy of MoOx and WOx catalysts formed in situ during guaiacol HDO (10 wt. % solution in toluene) at (a,b) 120 °C, (c,d) 240 °C and (e,f) 360 °C.

Figure 3.
The SEM images of MoOx and WOx catalysts formed in situ during guaiacol HDO (10 wt. % solution in toluene) at (a,b) 120 °C, (c,d) 240 °C and (e,f) 360 °C.
Figure 3.
The SEM images of MoOx and WOx catalysts formed in situ during guaiacol HDO (10 wt. % solution in toluene) at (a,b) 120 °C, (c,d) 240 °C and (e,f) 360 °C.

Figure 4.
The microphotographs of the catalysts obtained in situ during guaiacol HDO (10 wt. % solution in toluene) (a,b) MoOx, (c,d) WOx.
Figure 4.
The microphotographs of the catalysts obtained in situ during guaiacol HDO (10 wt. % solution in toluene) (a,b) MoOx, (c,d) WOx.

Figure 5.
The results of guaiacol HDO over in situ-formed (a,b) MoOx (c,d) WOx. Reaction conditions: 120–380 °C, 5 MPa H2, 6 h; 10 wt. % guaiacol in toluene solution.
Figure 5.
The results of guaiacol HDO over in situ-formed (a,b) MoOx (c,d) WOx. Reaction conditions: 120–380 °C, 5 MPa H2, 6 h; 10 wt. % guaiacol in toluene solution.

Figure 6.
The results of guaiacol HDO over in situ formed (a) MoOx (b) WOx. Reaction conditions: 380 °C, 1–5 MPa H2, 6 h; 10 wt. % guaiacol in toluene solution.
Figure 6.
The results of guaiacol HDO over in situ formed (a) MoOx (b) WOx. Reaction conditions: 380 °C, 1–5 MPa H2, 6 h; 10 wt. % guaiacol in toluene solution.

Figure 7.
Proposed reaction pathways of guaiacol HDO over in situ formed MoOx and WOx. DDO—direct deoxygenation, HYD—hydrogenation, DMO—demethoxylation, DME—demethylation, ME—methylation, TRA—transalkylation. The following conditions favored each path: I—MoOx or WOx, 240–280 °C, 5 MPa (anisole is a minor product); II—MoOx or WOx, 240–300 °C, 5 MPa; III—MoOx or WOx, 240–300 °C, 5 MPa (cresols are minor products); IV—MoOx, 320–360 °C, 5 MPa; 380 °C, 1–5 MPa; WOx, 360 °C, 5 MPa; 380 °C, 1–5 MPa; V—MoOx, 320–380 °C, 5 MPa; MoOx, 340 °C, 5 MPa, 6 h; VI, VII—MoOx or WOx, 320–360 °C, 5 MPa; 380 °C, 1–5 MPa.
Figure 7.
Proposed reaction pathways of guaiacol HDO over in situ formed MoOx and WOx. DDO—direct deoxygenation, HYD—hydrogenation, DMO—demethoxylation, DME—demethylation, ME—methylation, TRA—transalkylation. The following conditions favored each path: I—MoOx or WOx, 240–280 °C, 5 MPa (anisole is a minor product); II—MoOx or WOx, 240–300 °C, 5 MPa; III—MoOx or WOx, 240–300 °C, 5 MPa (cresols are minor products); IV—MoOx, 320–360 °C, 5 MPa; 380 °C, 1–5 MPa; WOx, 360 °C, 5 MPa; 380 °C, 1–5 MPa; V—MoOx, 320–380 °C, 5 MPa; MoOx, 340 °C, 5 MPa, 6 h; VI, VII—MoOx or WOx, 320–360 °C, 5 MPa; 380 °C, 1–5 MPa.

Figure 8.
The yield of HDO products and BTX over in situ-formed (a) MoOx (b) WOx. Reaction conditions: 300–380 °C, 5 MPa H2, 6 h; 10 wt. % guaiacol in dodecane solution.
Figure 8.
The yield of HDO products and BTX over in situ-formed (a) MoOx (b) WOx. Reaction conditions: 300–380 °C, 5 MPa H2, 6 h; 10 wt. % guaiacol in dodecane solution.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Some results from the literature of the lignin-derived compound HDO over Mo- and W-containing catalysts.
Table 1.
Some results from the literature of the lignin-derived compound HDO over Mo- and W-containing catalysts.
| Catalyst | Feedstock | Reaction Conditions a | Conversion, % | Selectivity, % | Ref. |
|---|---|---|---|---|---|
| MoxC/CNF | 0.68 g cinnamaldehyde in 50 mL toluene | 200 °C, 2 MPa H2, ~2.75 h a | 90 | 40.9 for hydrocinnamaldehyde 25.8 for β-methylstyrene | [5] |
| WxC/CNF | 200 °C, 2 MPa H2, ~5.75 h a | 90 | 43 for hydrocinnamaldehyde 34 for β-methylstyrene | ||
| Pd/α-MoC | anisole | 200 °C b | 21.5 | 94 for benzene | [6] |
| Mo2C@ BMZIF-700 °C (4 h) | 0.12 g guaiacol in 20 mL n-decane | 330 °C, 4 MPa H2, 4 h a | 97 | 70 for phenol | [7] |
| Mo2N@ NC500/SBA-15 | 3 wt. % guaiacol in n-decane | 380 °C, 2 MPa H2 c | 100 | 92 to benzene + toluene | [8] |
| in situ formed MoP | 10 wt. % guaiacol in n-dodecane | 360 °C, 5 MPa H2, 6 h a | 90 | 80 for phenol | [9] |
| in situ formed WP | 340 °C, 5 MPa H2, 1 h a | 53 | 78 for phenol | ||
| MoP | 340 °C, 5 MPa H2, 6 h a | 90 | 66 for phenol | [10] | |
| WP | 380 °C, 5 MPa H2, 6 h a | 89 | 84 for phenol | ||
| in situ formed MoOx | 380 °C, 1 MPa H2, 6 h a | 100 | 89 for BTX | This work | |
| in situ formed WOx | 380 °C, 5 MPa H2, 6 h a | 100 | 96 for BTX |
a a batch reactor was used; b a fixed bed quartz tubular reactor was used; c a micro-hydrogenation reactor was used.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mukhtarova, M.; Golubeva, M.; Sadovnikov, A.; Maximov, A. Guaiacol to Aromatics: Efficient Transformation over In Situ-Generated Molybdenum and Tungsten Oxides. Catalysts 2023, 13, 263. https://doi.org/10.3390/catal13020263
AMA Style
Mukhtarova M, Golubeva M, Sadovnikov A, Maximov A. Guaiacol to Aromatics: Efficient Transformation over In Situ-Generated Molybdenum and Tungsten Oxides. Catalysts. 2023; 13(2):263. https://doi.org/10.3390/catal13020263
Chicago/Turabian StyleMukhtarova, Mariyam, Maria Golubeva, Alexey Sadovnikov, and Anton Maximov. 2023. "Guaiacol to Aromatics: Efficient Transformation over In Situ-Generated Molybdenum and Tungsten Oxides" Catalysts 13, no. 2: 263. https://doi.org/10.3390/catal13020263
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.