
The Pincer Ligand Supported Ruthenium Catalysts for Acetylene Hydrochlorination: Molecular Mechanisms from Theoretical Insights

Abstract
:1. Introduction
2. Results and Discussions
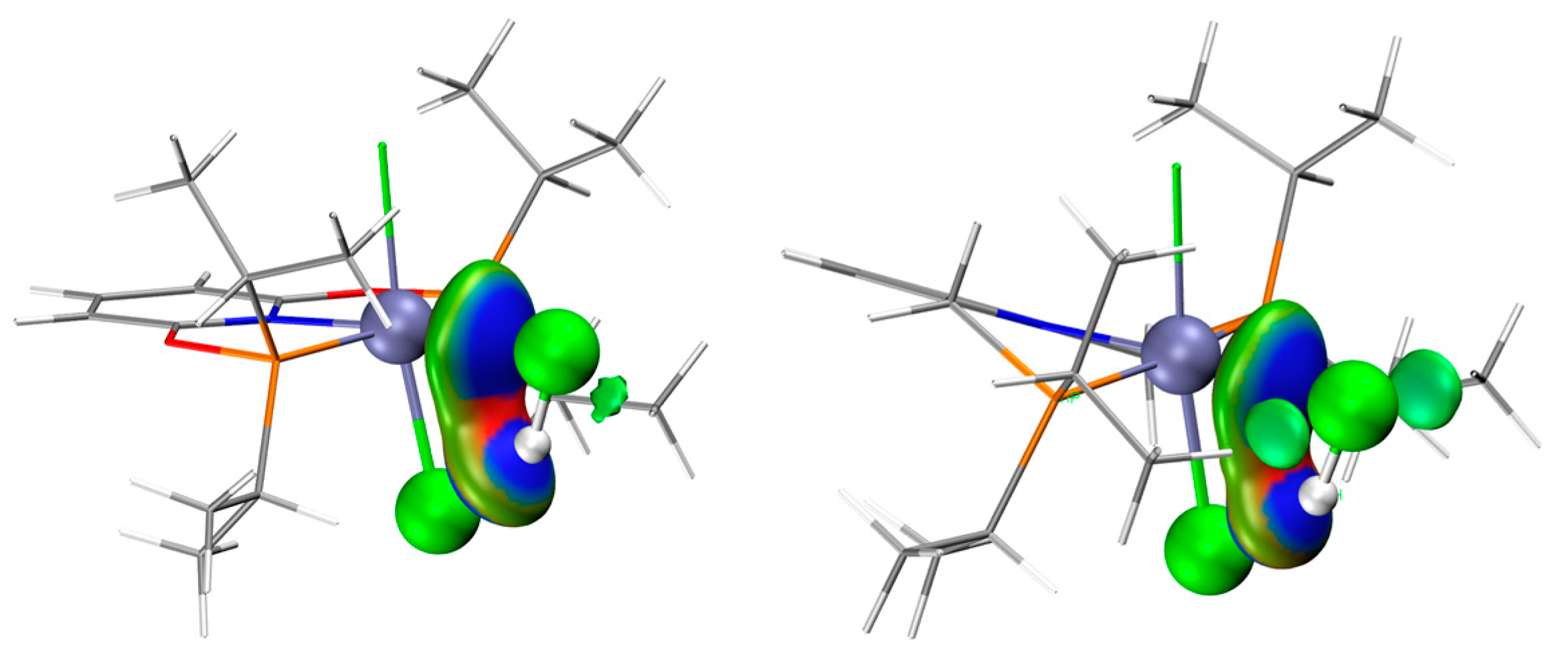
2.1. The Interaction between Catalysts and Reactants
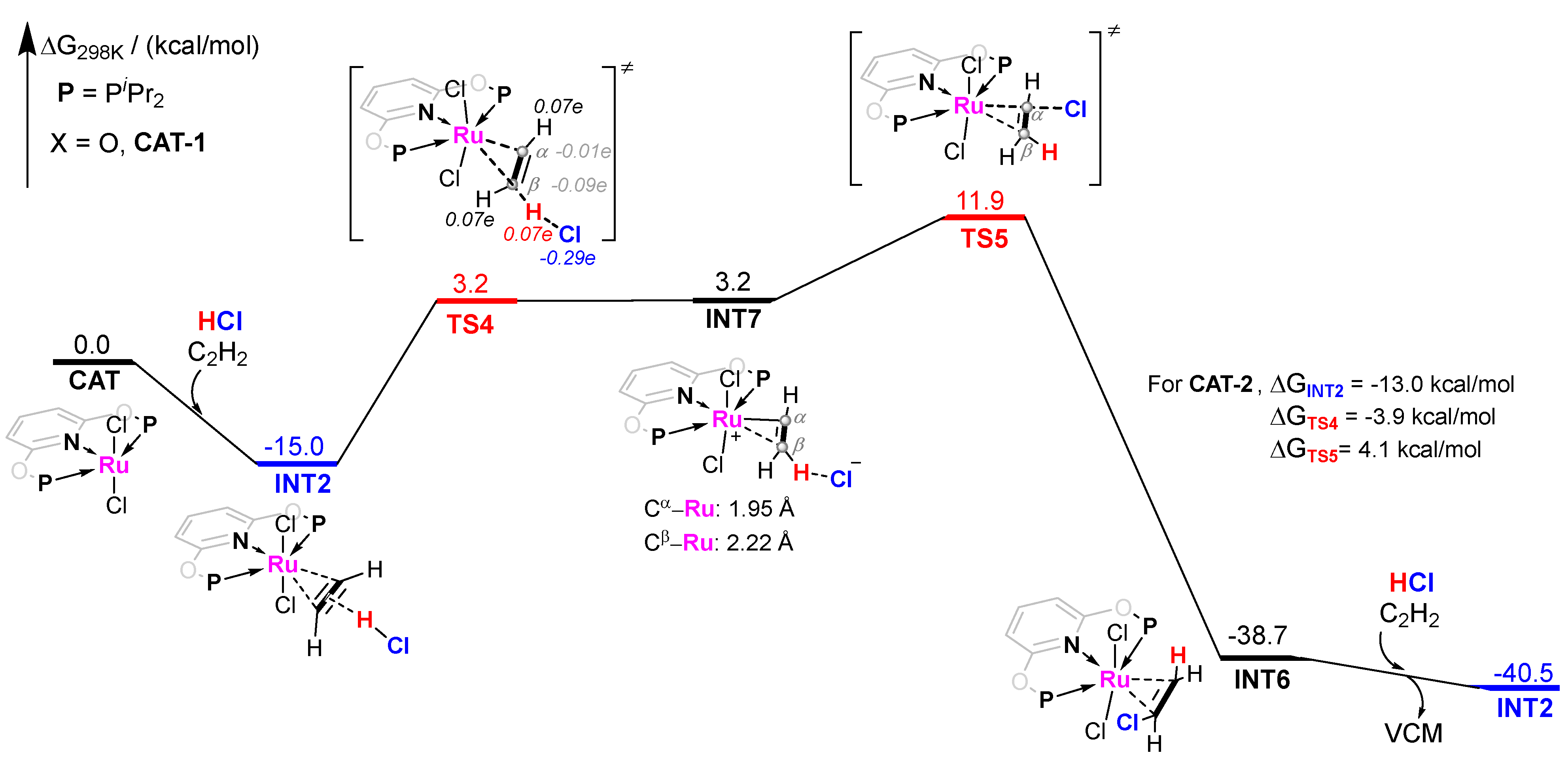
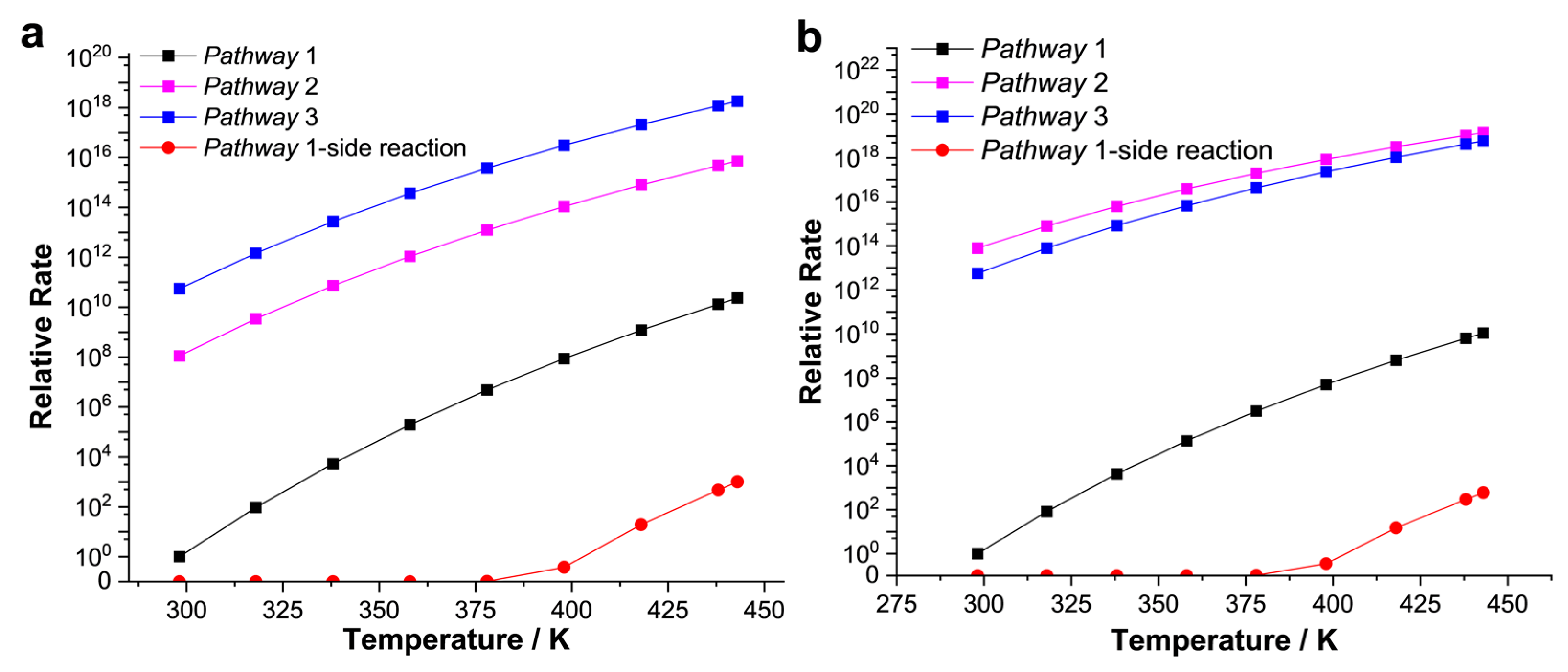
2.2. The Gibbs Free Energy of Intermediates and Transition States
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saeki, Y.; Emura, T. Technical progresses for PVC production. Prog. Polym. Sci. 2002, 27, 2055–2131. [Google Scholar] [CrossRef]
- Lin, R.; Amrute, A.P.; Pérez-Ramírez, J. Halogen-Mediated Conversion of Hydrocarbons to Commodities. Chem. Rev. 2017, 117, 4182. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.; Carthey, N.; Hutchings, G.J. Discovery, Development, and Commercialization of Gold Catalysts for Acetylene Hydrochlorination. J. Am. Chem. Soc. 2015, 137, 14548–14557. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.J.; Miedziak, P.J.; Brett, G.L.; Hutchings, G.J. Vinyl chloride monomer production catalyzed by gold: A review. Chin. J. Catal. 2016, 37, 1600–1607. [Google Scholar] [CrossRef]
- Minamata Convention on Mercury. Available online: https://www.mercuryconvention.org/ (accessed on 24 September 2017).
- Kiyonori, S. The vapor-phase hidrochlorination of acetylene over metal chlorides supported on activated carbon. Chem. Lett. 1975, 4, 219–220. [Google Scholar]
- Hutchings, G.J. Vapor phase hydrochlorination of acetylene: Correlation of catalytic activity of supported metal chloride catalysts. J. Catal. 1985, 96, 292–295. [Google Scholar] [CrossRef]
- Malta, G.; Freakley, S.J.; Kondrat, S.A.; Hutchings, G.J. Acetylene hydrochlorination using Au/carbon: A journey towards single site catalysis. Chem. Commun. 2017, 53, 11733. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Wang, Q.; Chen, K.; Wang, Y.; Huang, C.; Dai, H.; Yu, F.; Kang, L.; Dai, B. Development of a Heterogeneous Non-Mercury Catalyst for Acetylene Hydrochlorination. ACS Catal. 2015, 5, 5306–5316. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, N.; Li, W.; Dai, B. Progress on cleaner production of vinyl chloride monomers over non-mercury catalysts. Front. Chem. Sci. Eng. 2011, 5, 514–520. [Google Scholar] [CrossRef]
- Conte, M.; Carley, A.F.; Heirene, C.; Willock, D.J.; Johnston, P.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Hydrochlorination of acetylene using a supported gold catalyst: A study of the reaction mechanism. J. Catal. 2007, 250, 231–239. [Google Scholar] [CrossRef]
- Zhong, J.; Xu, Y.; Liu, Z. Heterogeneous non-mercury catalysts for acetylene hydrochlorination: Progress, challenges, and opportunities. Green Chem. 2018, 20, 2412–2427. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, L.; Zhang, Y.; Zhang, L.; Zan, X. Progress and Challenges of Mercury-Free Catalysis for Acetylene Hydrochlorination. Catalysts 2020, 10, 1218. [Google Scholar] [CrossRef]
- Mitchenko, S.A.; Krasnyakova, T.V.; Mitchenko, R.S.; Korduban, A.N. Acetylene catalytic hydrochlorination over powder catalyst prepared by pre-milling of K2PtCl4 salt. J. Mol. Cat. A Chem. 2007, 275, 101–108. [Google Scholar] [CrossRef]
- Mitchenko, S.A.; Khomutov, E.V.; Shubin, A.A.; Shul’ga, Y.M. Catalytic hydrochlorination of acetylene by gaseous HCl on the surface of mechanically pre-activated K2PtCl6 salt. J. Mol. Cat. A Chem. 2004, 212, 345–352. [Google Scholar] [CrossRef]
- Mitchenko, S.A.; Krasnyakova, T.V.; Zhikharev, I.V. Catalytic hydrochlorination of acetylene on mechanochemically-activated K2PdCl4. Theor. Exp. Chem. 2008, 44, 316–319. [Google Scholar] [CrossRef]
- Zhao, J.; Gu, S.; Xu, X.; Zhang, T.; Yu, Y.; Di, X.; Ni, J.; Pan, Z.; Li, X. Supported ionic-liquid-phase-stabilized Au(iii) catalyst for acetylene hydrochlorination. Catal. Sci. Technol. 2016, 6, 3263–3270. [Google Scholar] [CrossRef]
- Li, H.; Wu, B.; Wang, J.; Wang, F.; Zhang, X.; Wang, G.; Li, H. Efficient and stable Ru(III)-choline chloride catalyst system with low Ru content for non-mercury acetylene hydrochlorination. Chin. J. Catal. 2018, 39, 1770–1781. [Google Scholar] [CrossRef]
- Ren, Y.; Wu, B.; Wang, F.; Li, H.; Lv, G.; Sun, M.; Zhang, X. Chlorocuprate(i) ionic liquid as an efficient and stable Cu-based catalyst for hydrochlorination of acetylene. Catal. Sci. Technol. 2019, 9, 2868–2878. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhao, J.; Di, X.; Di, S.; Wang, B.; Yue, Y.; Sheng, G.; Lai, H.; Guo, L.; Wang, H.; et al. Carbon-supported perovskite-like CsCuCl3 nanoparticles: A highly active and cost-effective heterogeneous catalyst for the hydrochlorination of acetylene to vinyl chloride. Catal. Sci. Technol. 2018, 8, 2901–2908. [Google Scholar] [CrossRef]
- Wang, Y.; Nian, Y.; Zhang, J.; Li, W.; Han, Y. MOMTPPC improved Cu-based heterogeneous catalyst with high efficiency for acetylene hydrochlorination. Mol. Catal. 2019, 479, 110612. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, Z.; Wang, T.; Tang, Q.; Yu, M.; Feng, T.; Tian, M.; Chang, R.; Yue, Y.; Pan, Z.; et al. Controllable Synthesis of Vacancy-Defect Cu Site and Its Catalysis for the Manufacture of Vinyl Chloride Monomer. ACS Catal. 2021, 11, 11016–11028. [Google Scholar] [CrossRef]
- Han, Y.; Wang, Y.; Wang, Y.; Hu, Y.; Nian, Y.; Li, W.; Zhang, J. Pyrrolidone ligand improved Cu-based catalysts with high performance for acetylene hydrochlorination. Appl. Organomet. Chem. 2021, 35, e6066. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, T.; Liu, Y.; Li, W.; Zhang, H.; Zhang, J. Phosphine-oxide organic ligand improved Cu-based catalyst for acetylene hydrochlorination. Appl. Catal. A Gen. 2022, 630, 118461. [Google Scholar] [CrossRef]
- Chen, X.; Liu, X.; Hu, R.; Xu, C. Complexation effect between Cu-based catalyst and DESMP in acetylene hydration. Mol. Catal. 2022, 531, 112659. [Google Scholar] [CrossRef]
- Wang, B.; Jin, C.; Shao, S.; Yue, Y.; Zhang, Y.; Wang, S.; Chang, R.; Zhang, H.; Zhao, J.; Li, X. Electron-deficient Cu site catalyzed acetylene hydrochlorination. Green Energy Environ. 2022; in press. [Google Scholar] [CrossRef]
- Li, J.; Zhang, H.; Cai, M.; Li, L.; Li, Y.; Zhao, R.; Zhang, J. Enhanced catalytic performance of activated carbon-supported ru-based catalysts for acetylene hydrochlorination by azole ligands. Appl. Catal. A Gen. 2020, 592, 117431. [Google Scholar] [CrossRef]
- Li, Y.; Wang, F.; Hu, J.; Sun, M.; Wang, J.; Zhang, X. A study on the rules of ligands in highly efficient Ru–amide/AC catalysts for acetylene hydrochlorination. Catal. Sci. Technol. 2021, 11, 7347–7358. [Google Scholar] [CrossRef]
- Li, Y.; Wang, F.; Wu, B.; Wang, X.; Sun, M.; Zhang, Z.; Zhang, X. Competing on the same stage: Ru-based catalysts modified by basic ligands and organic chlorine salts for acetylene hydrochlorination. Catal. Sci. Technol. 2022, 12, 5086–5096. [Google Scholar] [CrossRef]
- Cai, M.; Zhang, H.; Man, B.; Li, J.; Li, L.; Li, Y.; Xie, D.; Deng, R.; Zhang, J. Synthesis of a vinyl chloride monomer via acetylene hydrochlorination with a ruthenium-based N-heterocyclic carbene complex catalyst. Catal. Sci. Technol. 2020, 10, 3552–3560. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, M.; Dai, B. Effect of Phosphorus Ligand on Cu-Based Catalysts for Acetylene Hydrochlorination. ACS Sustain. Chem. Eng. 2019, 7, 6170–6177. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, C.; Qiao, X.; Guan, Q.; Li, W. Regulating the coordination environment of Ru single-atom catalysts and unravelling the reaction path of acetylene hydrochlorination. Green Energy Environ. 2022; in press. [Google Scholar] [CrossRef]
- Samantaray, M.K.; D’Elia, V.; Pump, E.; Falivene, L.; Harb, M.; Ould Chikh, S.; Cavallo, L.; Basset, J.-M. The Comparison between Single Atom Catalysis and Surface Organometallic Catalysis. Chem. Rev. 2020, 120, 734–813. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Kang, L.; Su, Y.; Zhang, S.; Dai, B. MClx (M = Hg, Au, Ru; x = 2, 3) catalyzed hydrochlorination of acetylene—A density functional theory study. Can. J. Chem. 2013, 91, 120–125. [Google Scholar] [CrossRef]
- Dérien, S.; Klein, H.; Bruneau, C. Selective Ruthenium-Catalyzed Hydrochlorination of Alkynes: One-Step Synthesis of Vinylchlorides. Angew. Chem. Int. Ed. 2015, 54, 12112–12115. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jiang, J.-l.; Yu, H.-Z.; Fu, Y. Mechanistic Study on the Ruthenium-Catalyzed Terminal Alkyne Hydrochlorination. Organometallics 2017, 36, 523–529. [Google Scholar] [CrossRef]
- Eisenstein, O.; Hoffmann, R. Activation of a coordinated olefin toward nucleophilic attack. J. Am. Chem. Soc. 1980, 102, 6148–6149. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, X.; He, Z.; Guan, Q.; Li, W. Demystifying the mechanism of NMP ligands in promoting Cu-catalyzed acetylene hydrochlorination: Insights from a density functional theory study. Inorg. Chem. Front. 2020, 7, 3204–3216. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, T.; Jia, Y.; Zhang, J.; Han, Y. Single-Atom Ruthenium Catalytic Sites for Acetylene Hydrochlorination. J. Phys. Chem. Lett. 2021, 12, 7350–7356. [Google Scholar] [CrossRef]
- Becke, A.D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted abinitio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Legault, C.Y. CYLview, 1.0b. Available online: http://www.cylview.org (accessed on 1 January 2009).
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Michalak, A.; Mitoraj, M.; Ziegler, T. Bond Orbitals from Chemical Valence Theory. J. Phys. Chem. A 2008, 112, 1933–1939. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Zou, J. MOKIT Program. Available online: https://gitlab.com/jxzou/mokit (accessed on 25 October 2022).
- Upadhyay, S.K. Chemical Kinetics and Reaction Dynamics; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar] [CrossRef]
- El-Nahas, A.M.; Mangood, A.H.; Takeuchi, H.; Taketsugu, T. Thermal Decomposition of 2-Butanol as a Potential Nonfossil Fuel: A Computational Study. J. Phys. Chem. A 2011, 115, 2837–2846. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorption Free Energy ΔGads 1/(kcal/mol) | C2H2 | HCl |
|---|---|---|
| CAT−1 | −11.53 | 0.90 |
| CAT−2 | −9.89 | −3.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhao, J.; Li, Y.; Zhang, X.; Wang, F.; Wu, B.; Wang, T. The Pincer Ligand Supported Ruthenium Catalysts for Acetylene Hydrochlorination: Molecular Mechanisms from Theoretical Insights. Catalysts 2023, 13, 31. https://doi.org/10.3390/catal13010031
Wang X, Zhao J, Li Y, Zhang X, Wang F, Wu B, Wang T. The Pincer Ligand Supported Ruthenium Catalysts for Acetylene Hydrochlorination: Molecular Mechanisms from Theoretical Insights. Catalysts. 2023; 13(1):31. https://doi.org/10.3390/catal13010031
Chicago/Turabian StyleWang, Xingtao, Jiangshan Zhao, Yongwang Li, Xubin Zhang, Fumin Wang, Botao Wu, and Tian Wang. 2023. "The Pincer Ligand Supported Ruthenium Catalysts for Acetylene Hydrochlorination: Molecular Mechanisms from Theoretical Insights" Catalysts 13, no. 1: 31. https://doi.org/10.3390/catal13010031