
Samarium-Mediated Asymmetric Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Samarium-Mediated Ionic and Radical Asymmetric Reactions
2.1. Ketyl Radical-Olefin/Allene/Allyl Cyclization
2.1.1. Three-Membered Ring Forming Reactions
2.1.2. Five-Membered Ring Forming Cyclization and Cascade Reactions
2.1.3. Seven-Membered Ring Forming Cyclization Reactions
2.2. Ketyl Radical-α,β Unsaturated Carbonyl Cyclization
2.3. Ketyl Radical-α,β Unsaturated Sulfonyl Cyclization
2.4. Amino Ketyl Radical Olefin Cyclization
Sequential and Dearomatizing Cyclization Reactions
2.5. Ester Ketyl Radical-Allene/Olefin Cyclization and Cascades
2.6. Pinacol-Type Radical Cyclizations
2.6.1. Ketyl-Carbonyl Diradical Cyclizations
2.6.2. Ketyl Radical-Carbonyl Cyclizations
2.7. Barbier-Type π-Allyl Radical Cyclizations
2.8. Sm(III) Relay Catalytic Three-Component Tandem [4 + 3]-Cycloadditions
2.9. Stereoselective Reduction of Ketone
2.10. Chiral Ligand Promoted Hydroamination of Cyclopropene
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beemelmanns, C.; Reissig, H.U. Samarium Diiodide Induced Ketyl-(Het)Arene Cyclisations towards Novel N-Heterocycles. Chem. Soc. Rev. 2011, 40, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Sautier, B.; Procter, D.J. Recent Advances in the Chemistry of SmI2–H2O. Chimia 2012, 66, 399. [Google Scholar] [CrossRef] [PubMed]
- Szostak, M.; Procter, D.J. Beyond Samarium Diiodide: Vistas in Reductive Chemistry Mediated by Lanthanides(II). Angew. Chem. Int. Ed. 2012, 51, 9238–9256. [Google Scholar] [CrossRef] [PubMed]
- Harb, H.; Procter, D. SmI2-Mediated Carbonyl-Alkene Couplings for the Synthesis of Small Carbocyclic Rings. Synlett 2012, 2012, 6–20. [Google Scholar] [CrossRef]
- Streuff, J. The Electron-Way: Metal-Catalyzed Reductive Umpolung Reactions of Saturated and α,β-Unsaturated Carbonyl Derivatives. Synthesis 2013, 45, 281–307. [Google Scholar] [CrossRef]
- Concellón, J.M.; Rodríguez-Solla, H.; Concellón, C.; del Amo, V. Stereospecific and Highly Stereoselective Cyclopropanation Reactions Promoted by Samarium. Chem. Soc. Rev. 2010, 39, 4103. [Google Scholar] [CrossRef]
- Kuhlman, M.L.; Flowers, R.A. Aggregation State and Reducing Power of the Samarium Diiodide–DMPU Complex in Acetonitrile. Tetrahedron Lett. 2000, 41, 8049–8052. [Google Scholar] [CrossRef]
- Enemærke, R.J.; Hertz, T.; Skrydstrup, T.; Daasbjerg, K. Evidence for Ionic Samarium(II) Species in THF/HMPA Solution and Investigation of Their Electron-Donating Properties. Chem.-A Eur. J. 2000, 6, 3747–3754. [Google Scholar] [CrossRef]
- Enemærke, R.J.; Daasbjerg, K.; Skrydstrup, T. Is Samarium Diiodide an Inner- or Outer-Sphere Electron Donating Agent? Chem. Commun. 1999, 4, 343–344. [Google Scholar] [CrossRef]
- Szostak, M.; Fazakerley, N.J.; Parmar, D.; Procter, D.J. Cross-Coupling Reactions Using Samarium(II) Iodide. Chem. Rev. 2014, 114, 5959–6039. [Google Scholar] [CrossRef]
- Szostak, M.; Spain, M.; Procter, D.J. Recent Advances in the Chemoselective Reduction of Functional Groups Mediated by Samarium(Ii) Iodide: A Single Electron Transfer Approach. Chem. Soc. Rev. 2013, 42, 9155. [Google Scholar] [CrossRef]
- Szostak, M.; Spain, M.; Parmar, D.; Procter, D.J. Selective Reductive Transformations Using Samarium Diiodide-Water. Chem. Commun. 2012, 48, 330–346. [Google Scholar] [CrossRef]
- Dahlén, A.; Nilsson, Å.; Hilmersson, G. Estimating the Limiting Reducing Power of SmI 2/H2O/Amine and YbI2/H2O/Amine by Efficient Reduction of Unsaturated Hydrocarbons. J. Org. Chem. 2006, 71, 1576–1580. [Google Scholar] [CrossRef]
- Aspinall, H.C. Lanthanides: Chemistry and Use in Organic Synthesis Topics in Organometallic Chemistry; Kobayashi, S., Ed.; Springer: Berlin/Heidelberg, Germany, 1999; Volume 2, p. 307. ISBN 3-540-64526-8. [Google Scholar]
- Yeung, C.S.; Dong, V.M. Catalytic Dehydrogenative Cross-Coupling: Forming Carbon–Carbon Bonds by Oxidizing Two Carbon–Hydrogen Bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef]
- Lyons, T.W.; Sanford, M.S. Palladium-Catalyzed Ligand-Directed C−H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [Green Version]
- Alberico, D.; Scott, M.E.; Lautens, M. Aryl–Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation. Chem. Rev. 2007, 107, 174–238. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, H.; Shi, W.; Lei, A. Bond Formations between Two Nucleophiles: Transition Metal Catalyzed Oxidative Cross-Coupling Reactions. Chem. Rev. 2011, 111, 1780–1824. [Google Scholar] [CrossRef]
- Evans, W.J.; Gummersheimer, T.S.; Ziller, J.W. Coordination Chemistry of Samarium Diiodide with Ethers Including the Crystal Structure of Tetrahydrofuran-Solvated Samarium Diiodide, SmI2(THF)5. J. Am. Chem. Soc. 1995, 117, 8999–9002. [Google Scholar] [CrossRef]
- Hou, Z.; Wakatsuki, Y. Isolation and X-Ray Structures of the Hexamethylphosphoramide (Hmpa)-Coordinated Lanthanide(II) Diiodide Complexes [Sml2(Hmpa)4] and [Yb(Hmpa)4(Thf)2]I2. J. Chem. Soc. Chem. Commun. 1994, 10, 1205–1206. [Google Scholar] [CrossRef]
- Upadhyay, S.K.; Hoz, S. The Effect of Proton Donors on the Facial Stereoselectivity in SmI2 Reduction of Norcamphor. J. Org. Chem. 2011, 76, 1355–1360. [Google Scholar] [CrossRef]
- Chopade, P.R.; Prasad, E.; Flowers, R.A. The Role of Proton Donors in SmI2-Mediated Ketone Reduction: New Mechanistic Insights. J. Am. Chem. Soc. 2004, 126, 44–45. [Google Scholar] [CrossRef] [PubMed]
- Coote, S.C.; Li, R.A.F.; Skrydstrup, T.; Procter, D.J. Diiodide; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 9780470971253. [Google Scholar]
- Procter, D.J.; Flowers, R.A., II; Skrydstrup, T. Organic Synthesis Using Samarium Diiodide. A Practical Guide. Sciarium. Available online: https://sciarium.com/file/365254/ (accessed on 5 November 2022).
- Molander, G.A.; Cormier, E.P. Ketyl–Allene Cyclizations Promoted by Samarium(II) Iodide. J. Org. Chem. 2005, 70, 2622–2626. [Google Scholar] [CrossRef] [PubMed]
- Shabangi, M.; Flowers, R.A. Electrochemical Investigation of the Reducing Power of SmI2 in THF and the Effect of HMPA Cosolvent. Tetrahedron Lett. 1997, 38, 1137–1140. [Google Scholar] [CrossRef]
- Shabangi, M.; Kuhlman, M.L.; Flowers, R.A. Mechanism of Reduction of Primary Alkyl Radicals by SmI2–HMPA. Org. Lett. 1999, 1, 2133–2135. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Ellery, S.P.; Chen, J.S. Samarium Diiodide Mediated Reactions in Total Synthesis. Angew. Chem. Int. Ed. 2009, 48, 7140–7165. [Google Scholar] [CrossRef]
- Nakata, T. SmI2-Induced Cyclizations and Their Applications in Natural Product Synthesis. Chem. Rec. 2010, 10, 159–172. [Google Scholar] [CrossRef]
- Nakata, T. SmI2-Induced Reductive Cyclizations for the Synthesis of Cyclic Ethers and Applications in Natural Product Synthesis. Chem. Soc. Rev. 2010, 39, 1955. [Google Scholar] [CrossRef]
- Dibrell, S.E.; Tao, Y.; Reisman, S.E. Synthesis of Complex Diterpenes: Strategies Guided by Oxidation Pattern Analysis. Acc. Chem. Res. 2021, 54, 1360–1373. [Google Scholar] [CrossRef]
- Maity, S.; Hoz, S. Mechanistic Vistas of Trivalent Nitrogen Compound Reduction by Samarium Diiodide. Eur. J. Org. Chem. 2021, 2021, 1103–1112. [Google Scholar] [CrossRef]
- Namy, J.-L.; Colomb, M.; Kagan, H.B. Samarium Diiodide in Tetrahydropyran: Preparation and Some Reactions in Organic Chemistry. Tetrahedron Lett. 1994, 35, 1723–1726. [Google Scholar] [CrossRef]
- Machrouhi, F.; Hamann, B.; Namy, J.-L.; Kagan, H.B. Improved Reactivity of Diiodosamarium by Catalysis with Transition Metal Salts. Synlett 1996, 1996, 633–634. [Google Scholar] [CrossRef]
- Machrouhi, F.; Namy, J.-L.; Kagan, H.B. Nucleophilic Acylation of Esters by Acid Chlorides Mediated by Samarium Diiodide: Formation and Use of Samarium Enediolates. Tetrahedron Lett. 1997, 38, 7183–7186. [Google Scholar] [CrossRef]
- Girard, P.; Namy, J.L.; Kagan, H.B. Divalent Lanthanide Derivatives in Organic Synthesis. 1. Mild Preparation of Samarium Iodide and Ytterbium Iodide and Their Use as Reducing or Coupling Agents. J. Am. Chem. Soc. 1980, 102, 2693–2698. [Google Scholar] [CrossRef]
- Molander, G.A.; Etter, J.B. Lanthanides in Organic Synthesis. Synthesis of Bicyclicalcohols. Tetrahedron Lett. 1984, 25, 3281–3284. [Google Scholar] [CrossRef]
- Wu, J.; Panek, J.S. Total Synthesis of (−)-Virginiamycin M2: Application of Crotylsilanes Accessed by Enantioselective Rh(II) or Cu(I) Promoted Carbenoid Si–H Insertion. J. Org. Chem. 2011, 76, 9900–9918. [Google Scholar] [CrossRef]
- Fukuzawa, S.; Matsuzawa, H.; Yoshimitsu, S. Asymmetric Samarium-Reformatsky Reaction of Chiral α-Bromoacetyl-2-Oxazolidinones with Aldehydes. J. Org. Chem. 2000, 65, 1702–1706. [Google Scholar] [CrossRef]
- Namy, J.L.; Souppe, J.; Kagan, H.B. Efficient Formation of Pinacols from Aldehydes or Ketones Mediated by Samarium Diiodide. Tetrahedron Lett. 1983, 24, 765–766. [Google Scholar] [CrossRef]
- Marnera, G.; D’Alarcao, M. Synthesis of Galactosaminyl D-Chiro-Inositols. Carbohydr. Res. 2006, 341, 1105–1116. [Google Scholar] [CrossRef]
- Sakakura, A.; Hori, M.; Fushimi, M.; Ishihara, K. Catalytic Enantioselective 1,3-Dipolar Cycloadditions of Nitrones with Propioloylpyrazoles and Acryloylpyrazoles Induced by Chiral π-Cation Catalysts. J. Am. Chem. Soc. 2010, 132, 15550–15552. [Google Scholar] [CrossRef]
- Banik, B.K.; Zegrocka, O.; Becker, F.F. Samarium-Mediated Iodine-Catalysed Reductive Amination of the Adamantyl Methyl Ketone. J. Chem. Res. 2000, 2000, 321–323. [Google Scholar] [CrossRef]
- Basu, M.K.; Banik, B.K. Samarium-Mediated Barbier Reaction of Carbonyl Compounds. Tetrahedron Lett. 2001, 42, 187–189. [Google Scholar] [CrossRef]
- Banik, B.K.; Venkatraman, M.S.; Banik, I.; Basu, M.K. Samarium-Induced Reductive Dimerization of Methyl Cinnamate: Synthesis of 2,8-Diamino Chrysene. Tetrahedron Lett. 2004, 45, 4737–4739. [Google Scholar] [CrossRef]
- Banik, B.K.; Mukhopadhyay, C.; Venkatraman, M.S.; Becker, F.F. A Facile Reduction of Aromatic Nitro Compounds to Aromatic Amines by Samarium and Iodine. Tetrahedron Lett. 1998, 39, 7243–7246. [Google Scholar] [CrossRef]
- Banik, B.K. Samarium Metal in Organic Synthesis. Eur. J. Org. Chem. 2002, 2002, 2431. [Google Scholar] [CrossRef]
- Collin, J.; Namy, J.L.; Jones, G.; Kagan, H.B. Reactions of Protected Amino Acid Chlorides Mediated by SmI2. Tetrahedron Lett. 1992, 33, 2973–2976. [Google Scholar] [CrossRef]
- Monovich, L.G.; Le Huérou, Y.; Rönn, M.; Molander, G.A. Total Synthesis of (−)-Steganone Utilizing a Samarium(II) Iodide Promoted 8-Endo Ketyl–Olefin Cyclization. J. Am. Chem. Soc. 2000, 122, 52–57. [Google Scholar] [CrossRef]
- Molander, G.A.; Machrouhi, F. Sequenced Reactions with Samarium(II) Iodide. A Complementary Annulation Process Providing Access to Seven-, Eight-, and Nine-Membered Carbocycles. J. Org. Chem. 1999, 64, 4119–4123. [Google Scholar] [CrossRef]
- Specht, K.M.; Harris, C.R.; Molander, G.A.; Kahne, D. SmI2 Cleavage of Chromomycin A3 Sugars. Tetrahedron Lett. 1999, 40, 1855–1856. [Google Scholar] [CrossRef]
- Kerrigan, N.J.; Upadhyay, T.; Procter, D.J. The Samarium(II)-Mediated Intermolecular Couplings of Ketones and β-Alkoxyacrylates: A Short Asymmetric Synthesis of an Antifungal γ-Butyrolactone. Tetrahedron Lett. 2004, 45, 9087–9090. [Google Scholar] [CrossRef]
- Mikami, K.; Yamaoka, M. Chiral Ligand Control in Enantioselective Reduction of Ketones by SmI2 for Ketyl Radical Addition to Olefins. Tetrahedron Lett. 1998, 39, 4501–4504. [Google Scholar] [CrossRef]
- Gopalaiah, K.; Kagan, H.B. Use of Samarium Diiodide in the Field of Asymmetric Synthesis. New J. Chem. 2008, 32, 607. [Google Scholar] [CrossRef]
- Davies, S.G.; Rodríguez-Solla, H.; Tamayo, J.A.; Cowley, A.R.; Concellón, C.; Garner, A.C.; Parkes, A.L.; Smith, A.D. Asymmetric Conjugate Reductions with Samarium Diiodide. Org. Biomol. Chem. 2005, 3, 1435–1447. [Google Scholar] [CrossRef]
- Davies, S.G.; Rodríguez-Solla, H.; Tamayo, J.A.; Garner, A.C.; Smith, A.D. Diastereoselective Conjugate Reduction with Samarium Diiodide: Asymmetric Synthesis of Methyl (2S,3R)-N-Acetyl-2-Amino-2,3-Dideuterio-3-Phenylpropionate. Chem. Commun. 2004, 36, 2502–2503. [Google Scholar] [CrossRef]
- Molander, G.A. Reductions with Samarium(II) Iodide. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1994; pp. 211–367. [Google Scholar]
- Heravi, M.M.; Nazari, A. Samarium(II) Iodide-Mediated Reactions Applied to Natural Product Total Synthesis. RSC Adv. 2022, 12, 9944–9994. [Google Scholar] [CrossRef]
- Dahlén, A.; Hilmersson, G. Samarium(II) Iodide Mediated Reductions—Influence of Various Additives. Eur. J. Inorg. Chem. 2004, 2004, 3393–3403. [Google Scholar] [CrossRef]
- Martin-Fontecha, M.; Agarrabeitia, A.R.; Ortiz, M.J.; Armesto, D. SmI2-Mediated 3-Exo-Trig Cyclization of β,γ-Unsaturated Carbonyl Compounds: Diastereoselective Synthesis of Cyclopropanols. Org. Lett. 2010, 12, 4082–4085. [Google Scholar] [CrossRef]
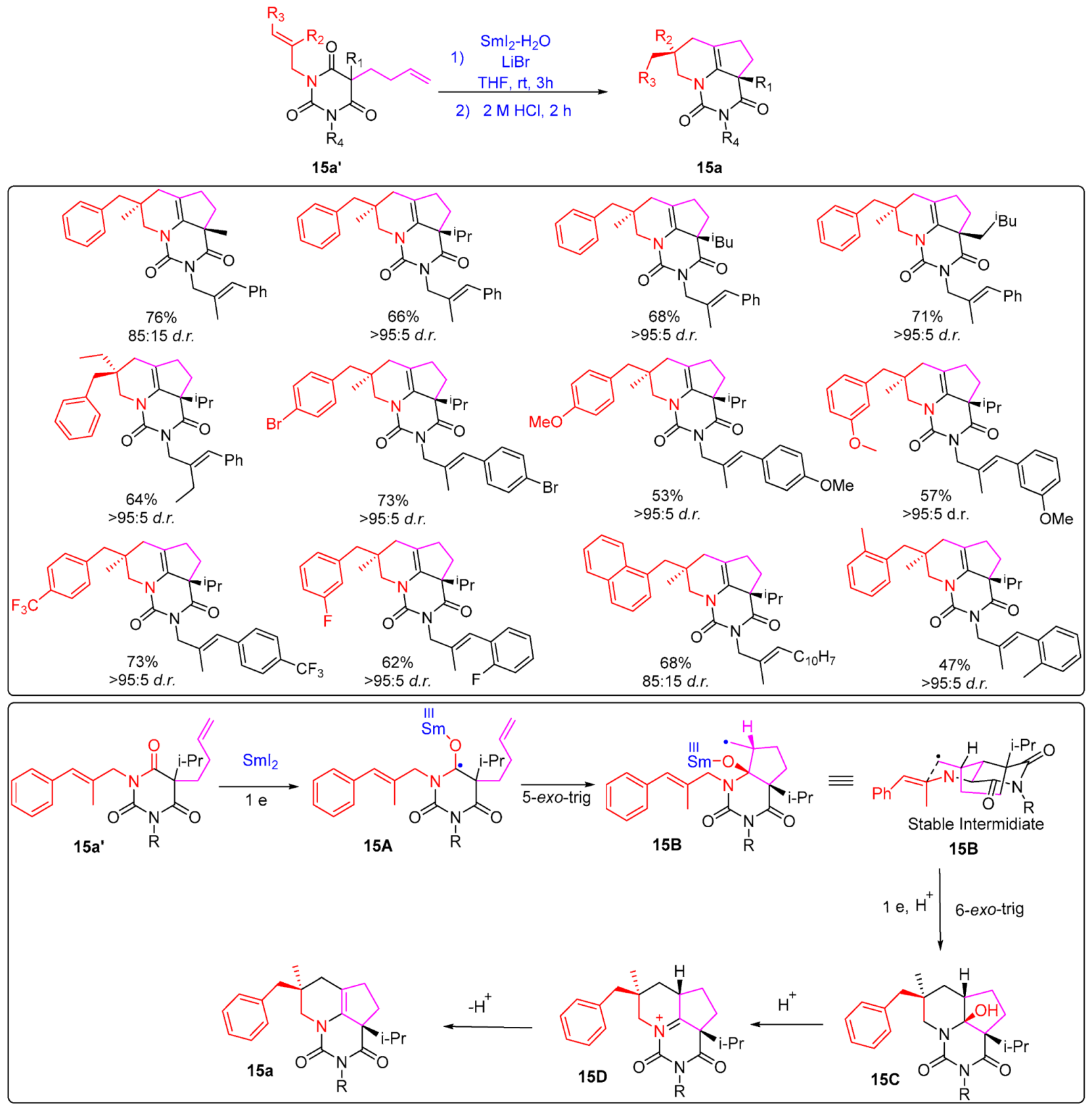
- Kern, N.; Plesniak, M.P.; McDouall, J.J.W.; Procter, D.J. Enantioselective Cyclizations and Cyclization Cascades of Samarium Ketyl Radicals. Nat. Chem. 2017, 9, 1198–1204. [Google Scholar] [CrossRef]
- Baker, T.M.; Sloan, L.A.; Choudhury, L.H.; Murai, M.; Procter, D.J. A Stereoselective Cyclisation Cascade Mediated by SmI2–H2O: Synthetic Studies towards Stolonidiol. Tetrahedron Asymmetry 2010, 21, 1246–1261. [Google Scholar] [CrossRef]
- Saadi, J.; Brüdgam, I.; Reissig, H.-U. Highly Substituted Benzannulated Cyclooctanol Derivatives by Samarium Diiodide-Induced Cyclizations. Beilstein J. Org. Chem. 2010, 6, 1229–1245. [Google Scholar] [CrossRef] [Green Version]
- Hsu, D.S.; Hsu, C.W. Spiranes Synthesis Based on Samarium Diiodide-Mediated Reductive Cyclization. Tetrahedron Lett. 2012, 53, 2185–2188. [Google Scholar] [CrossRef]
- Li, Z.; Nakashige, M.; Chain, W.J. A Brief Synthesis of (−)-Englerin A. J. Am. Chem. Soc. 2011, 133, 6553–6556. [Google Scholar] [CrossRef]
- Komada, T.; Adachi, M.; Nishikawa, T. A Concise Synthesis of a Highly Strained Cyclobutane in Solanoeclepin A by Radical Cyclization. Chem. Lett. 2012, 41, 287–289. [Google Scholar] [CrossRef]
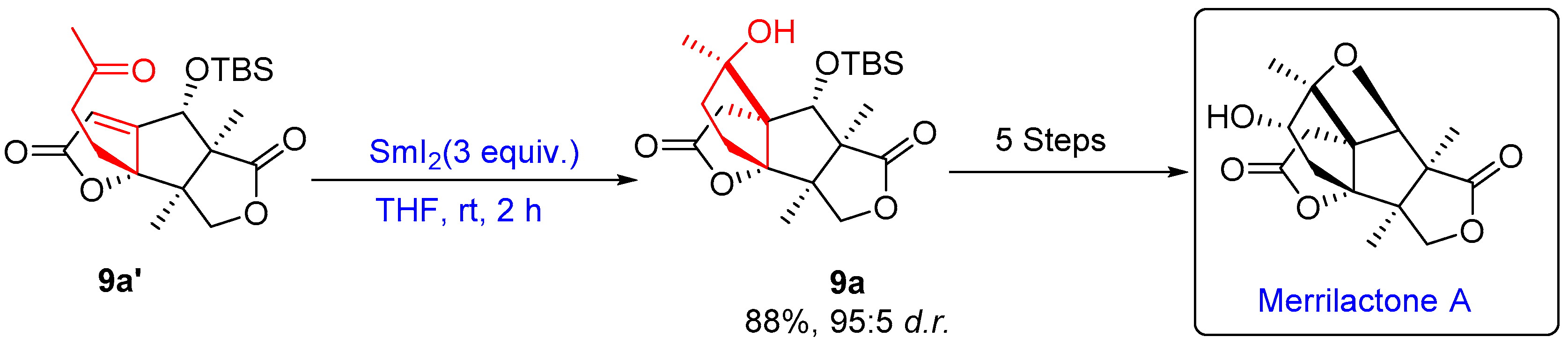
- Chen, J.; Gao, P.; Yu, F.; Yang, Y.; Zhu, S.; Zhai, H. Total Synthesis of (±)-Merrilactone A. Angew. Chem.-Int. Ed. 2012, 51, 5897–5899. [Google Scholar] [CrossRef]
- Zi, W.; Yu, S.; Ma, D. A Convergent Route to the Galbulimima Alkaloids (−)-GB 13 and (+)-GB 16. Angew. Chem. 2010, 122, 6023–6026. [Google Scholar] [CrossRef]
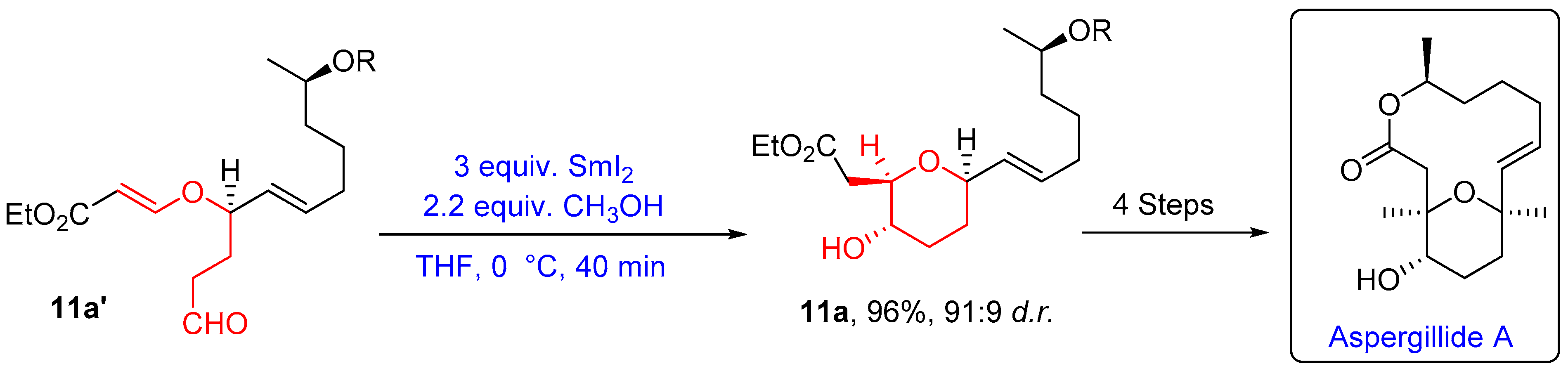
- Sabitha, G.; Vasudeva Reddy, D.; Senkara Rao, A.; Yadav, J.S. Stereoselective Formal Synthesis of Aspergillide A. Tetrahedron Lett. 2010, 51, 4195–4198. [Google Scholar] [CrossRef]
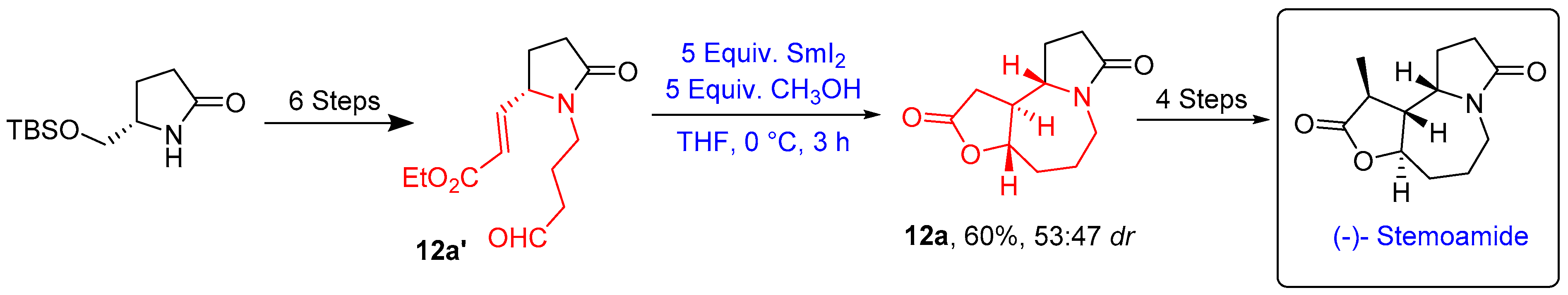
- Honda, T.; Matsukawa, T.; Takahashi, K. Efficient Total Synthesis of (−)-Stemoamide. Org. Biomol. Chem. 2011, 9, 673–675. [Google Scholar] [CrossRef]
- Shi, S.; Szostak, M. Aminoketyl Radicals in Organic Synthesis: Stereoselective Cyclization of Five- and Six-Membered Cyclic Imides to 2-Azabicycles Using SmI2–H2O. Org. Lett. 2015, 17, 5144–5147. [Google Scholar] [CrossRef]
- Huang, H.-M.; Bonilla, P.; Procter, D.J. Selective Construction of Quaternary Stereocentres in Radical Cyclisation Cascades Triggered by Electron-Transfer Reduction of Amide-Type Carbonyls. Org. Biomol. Chem. 2017, 15, 4159–4164. [Google Scholar] [CrossRef]
- Parmar, D.; Matsubara, H.; Price, K.; Spain, M.; Procter, D.J. Lactone Radical Cyclizations and Cyclization Cascades Mediated by SmI2–H2O. J. Am. Chem. Soc. 2012, 134, 12751–12757. [Google Scholar] [CrossRef]
- Sautier, B.; Lyons, S.E.; Webb, M.R.; Procter, D.J. Radical Cyclization Cascades of Unsaturated Meldrum’s Acid Derivatives. Org. Lett. 2012, 14, 146–149. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Ding, H.; Richard, J.-A.; Chen, D.Y.-K. Total Synthesis of Echinopines A and B. J. Am. Chem. Soc. 2010, 132, 3815–3818. [Google Scholar] [CrossRef]
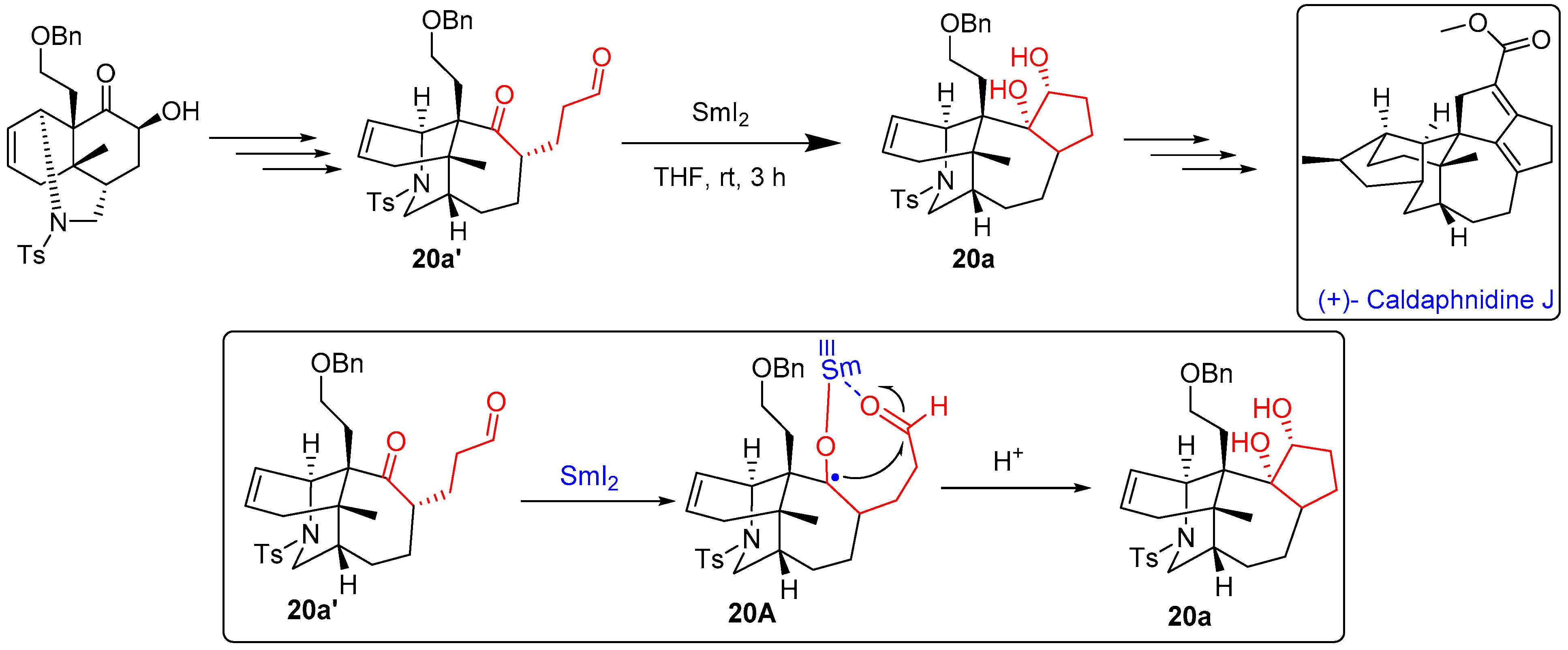
- Guo, L.D.; Zhang, Y.; Hu, J.; Ning, C.; Fu, H.; Chen, Y.; Xu, J. Asymmetric Total Synthesis of Yuzurimine-Type Daphniphyllum Alkaloid (+)-Caldaphnidine J. Nat. Commun. 2020, 11, 3538. [Google Scholar] [CrossRef]
- Nishikawa, K.; Kikuta, K.; Tsuruta, T.; Nakatsukasa, H.; Sugahara, S.; Kume, S.; Morimoto, Y. Asymmetric Total Synthesis of Toxicodenane A by Samarium-Iodide-Induced Barbier-Type Cyclization and Its Cell-Protective Effect against Lipotoxicity. Org. Lett. 2022, 24, 531–535. [Google Scholar] [CrossRef]
- Xu, C.; Qiao, J.; Dong, S.; Zhou, Y.; Liu, X.; Feng, X. Asymmetric Synthesis of Dihydro-1,3-Dioxepines by Rh(II)/Sm(III) Relay Catalytic Three-Component Tandem [4 + 3]-Cycloaddition. Chem. Sci. 2021, 12, 5458–5463. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Z.; Fu, S.; Liu, B. Asymmetric Total Synthesis of Rumphellclovane E. Org. Lett. 2021, 23, 290–295. [Google Scholar] [CrossRef]
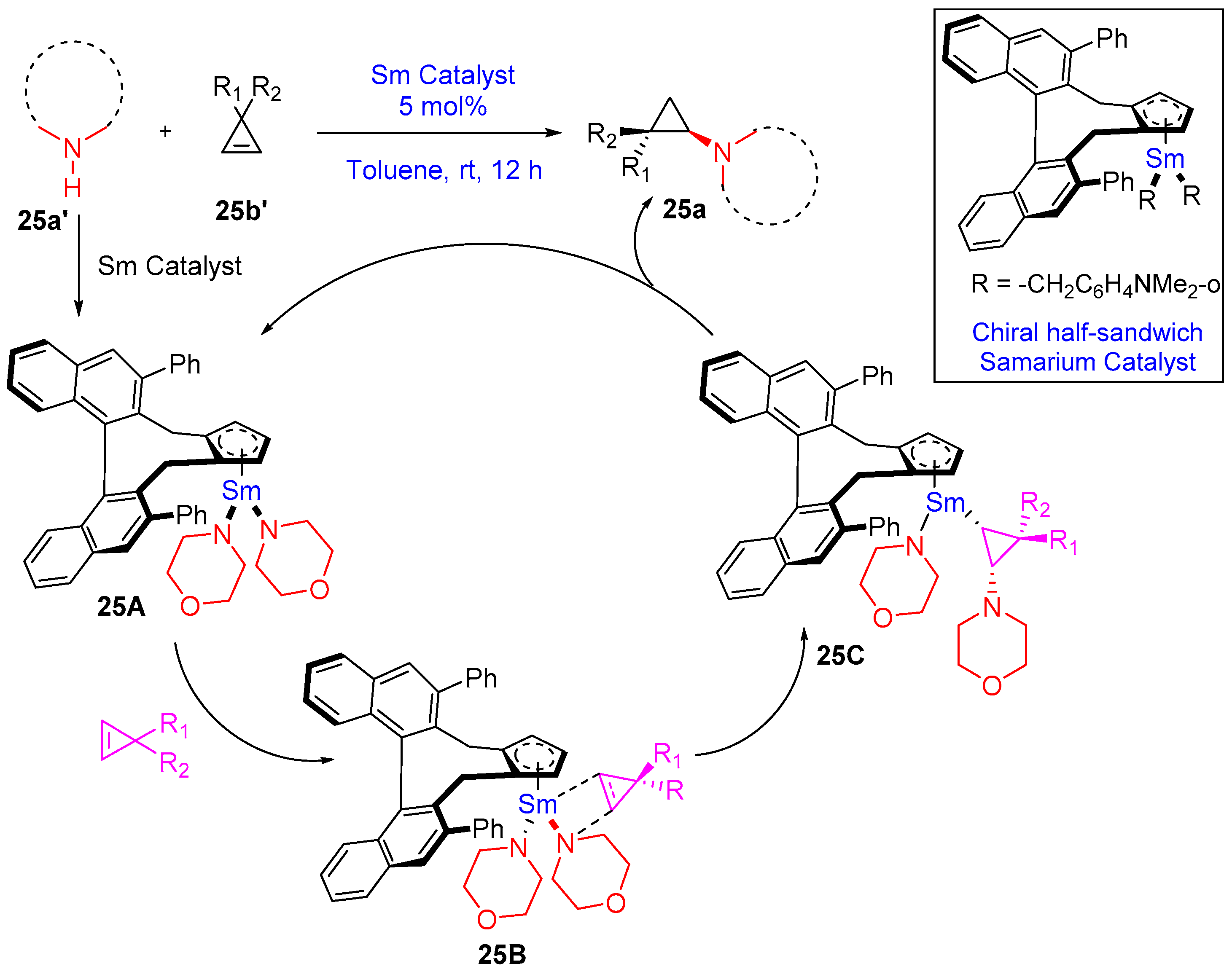
- Teng, H.-L.; Luo, Y.; Wang, B.; Zhang, L.; Nishiura, M.; Hou, Z. Synthesis of Chiral Aminocyclopropanes by Rare-Earth-Metal-Catalyzed Cyclopropene Hydroamination. Angew. Chem. Int. Ed. 2016, 55, 15406–15410. [Google Scholar] [CrossRef]





























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majee, S.; Ray, D.; Banik, B.K. Samarium-Mediated Asymmetric Synthesis. Catalysts 2023, 13, 24. https://doi.org/10.3390/catal13010024
Majee S, Ray D, Banik BK. Samarium-Mediated Asymmetric Synthesis. Catalysts. 2023; 13(1):24. https://doi.org/10.3390/catal13010024
Chicago/Turabian StyleMajee, Suman, Devalina Ray, and Bimal Krishna Banik. 2023. "Samarium-Mediated Asymmetric Synthesis" Catalysts 13, no. 1: 24. https://doi.org/10.3390/catal13010024