

Novel Highly Efficient Green and Reusable Cu(II)/Chitosan-Based Catalysts for the Sonogashira, Buchwald, Aldol, and Dipolar Cycloaddition Reactions
 , , , , and
, , , , and
Abstract
:1. Introduction
- (i)
- (ii)
- (iii)
- (iv)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
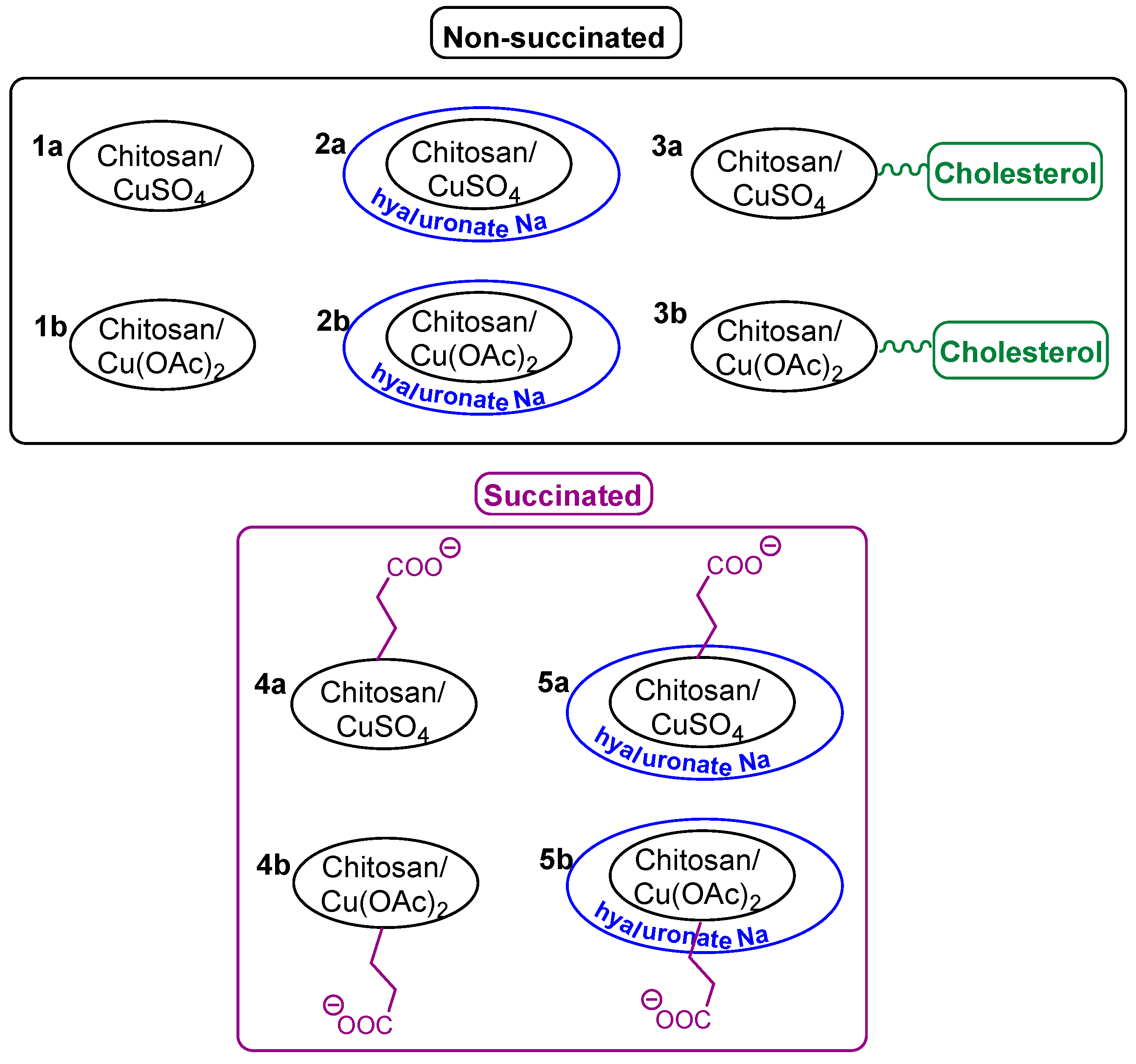
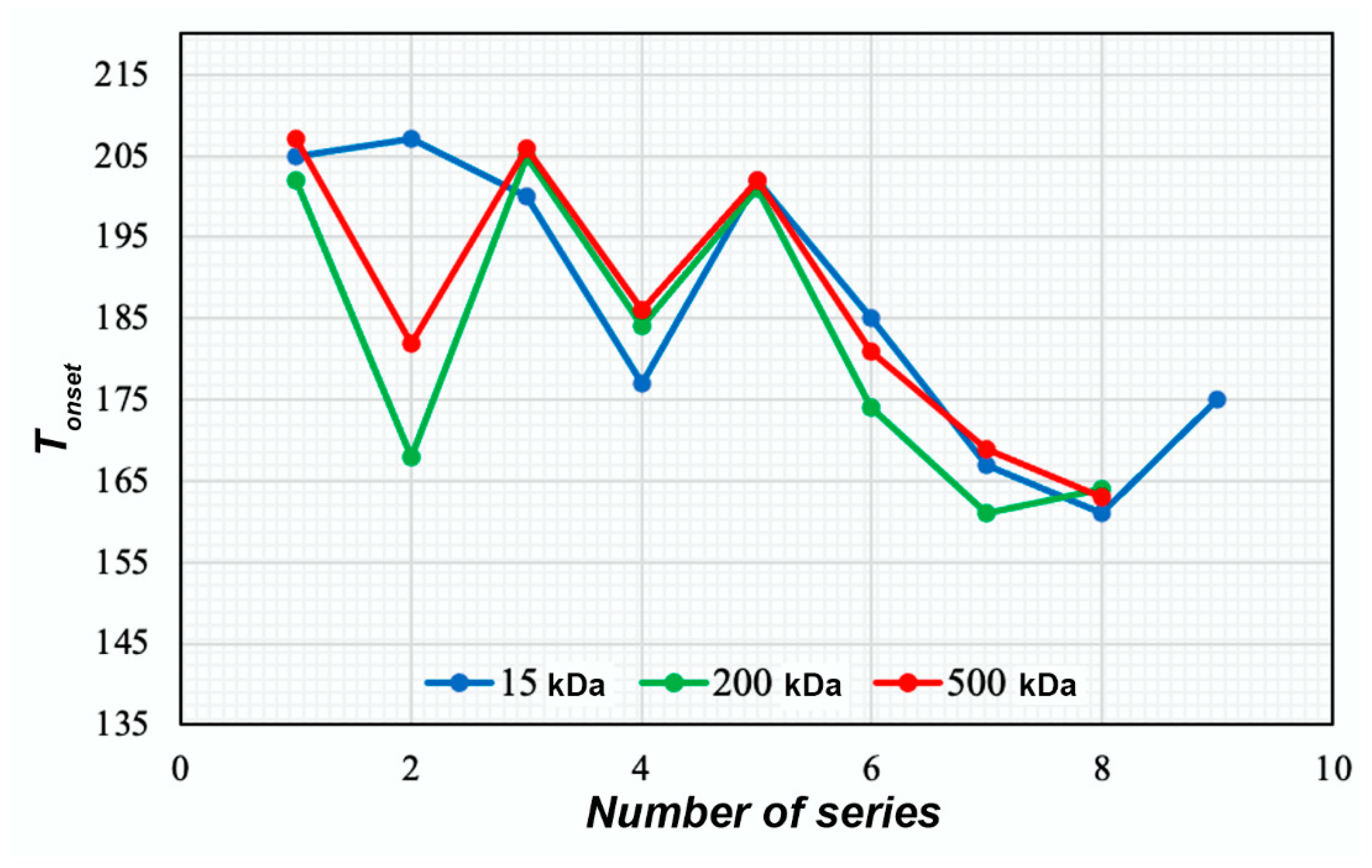
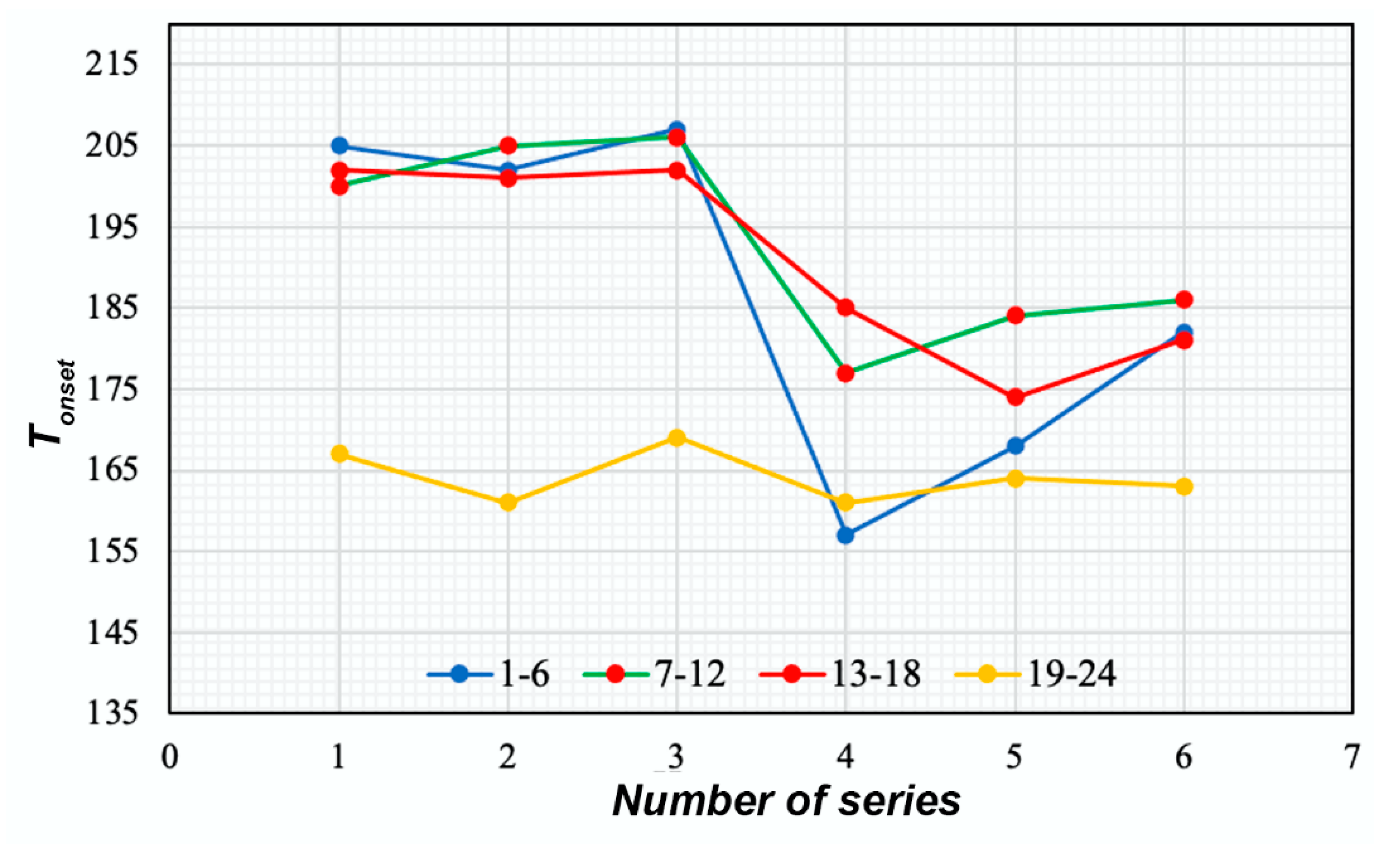
| Composition of the Sample | No. | Size (nm) | Ζeta-Potential (mV) | Copper(II) Content (%) | Tonset (°C) | |
|---|---|---|---|---|---|---|
| Non-succinated | CS12+CuSO4 | 1 | 307 | 13.98 ± 0.32 | 17.4 | 205 |
| CS200+CuSO4 | 2 | 266 | 14.16 ± 0.13 | 18.1 | 202 | |
| CS500+CuSO4 | 3 | 286 | 14.07 ± 0.14 | 17.7 | 207 | |
| CS12+CuOAc | 4 | - | - | 13.3 | 157 | |
| CS200+CuOAc | 5 | - | - | 13.8 | 168 | |
| CS500+CuOAc | 6 | - | - | 13.6 | 182 | |
| CS12+CuSO4+NaHA | 7 | 329 | 7.08 ± 0.10 | 13.2 | 200 | |
| CS200+CuSO4+NaHA | 8 | 271 | 7.03 ± 1.12 | 13.0 | 205 | |
| CS500+CuSO4+NaHA | 9 | 288 | 7.37 ± 0.07 | 13.0 | 206 | |
| CS12+CuOAc+NaHA | 10 | - | - | 8.3 | 177 | |
| CS200+CuOAc+NaHA | 11 | - | - | 8.5 | 184 | |
| CS500+CuOAc+NaHA | 12 | - | - | 8.7 | 186 | |
| CS12Chol+CuSO4 | 13 | 99 | 8.22 ± 0.17 | 15.7 | 202 | |
| CS200Chol+CuSO4 | 14 | 108 | 8.55 ± 0.15 | 15.7 | 201 | |
| CS500Chol+CuSO4 | 15 | 93 | 8.37 ± 0.21 | 15.5 | 202 | |
| CS12Chol+CuOAc | 16 | - | - | 10.9 | 185 | |
| CS200Chol+CuOAc | 17 | - | - | 10.4 | 174 | |
| CS500Chol+CuOAc | 18 | - | - | 10.6 | 181 | |
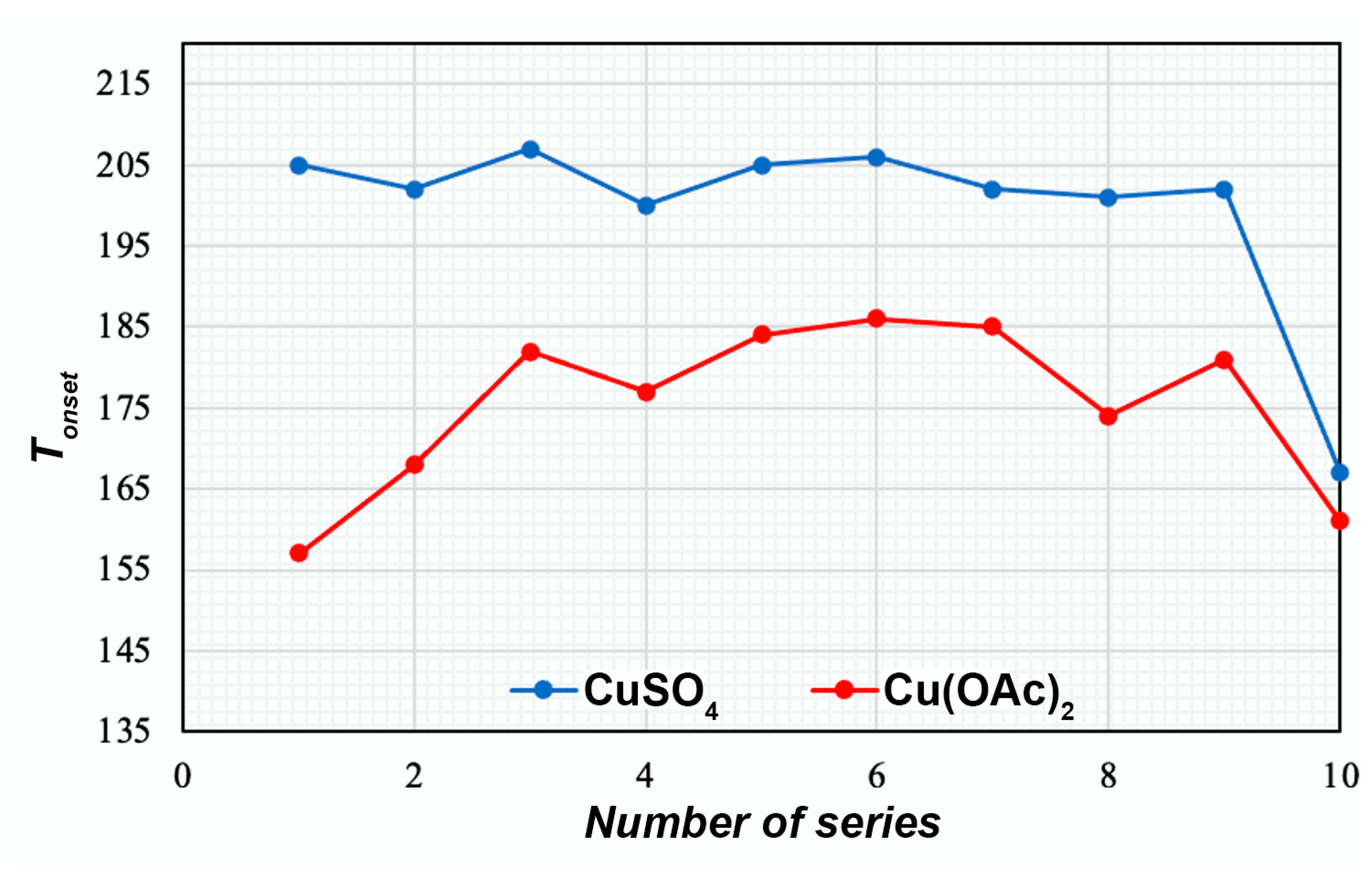
| Succinated | CS12+CuSO4 | 19 | 287 | −4.14 ± 0.22 | 10.1 | 167 |
| CS200+CuSO4 | 20 | 271 | −4.62 ± 0.10 | 10.0 | 161 | |
| CS500+CuSO4 | 21 | 290 | −4.23 ± 0.17 | 10.0 | 169 | |
| CS12+CuOAc | 22 | - | - | 7.4 | 161 | |
| CS200+CuOAc | 23 | - | - | 7.6 | 164 | |
| CS500+CuOAc | 24 | - | - | 7.7 | 163 | |
| CS12+CuSO4+NaHA | 25 | 315 | −21.06 ± 0.11 | 8.3 | 175 | |
| CS200+CuSO4+NaHA | 26 | 276 | −21.18 ± 0.15 | 8.4 | - | |
| CS500+CuSO4+NaHA | 27 | 283 | −20.88 ± 0.18 | 8.2 | - | |
| CS12+CuOAc+NaHA | 28 | 315 | −21.12 ± 0.30 | 5.6 | - | |
| CS200+CuOAc+NaHA | 29 | 284 | −20.66 ± 0.14 | 5.8 | - | |
| CS500+CuOAc+NaHA | 30 | 303 | −20.74 ± 0.21 | 5.8 | - |
2. Results and Discussion
2.1. Preparation and Characterization of Chitosan/Copper(II) Composites
2.1.1. Preparation of Chitosan/Copper(II) Composites
2.1.2. Characterization of the Chitosan/Copper(II) Composites
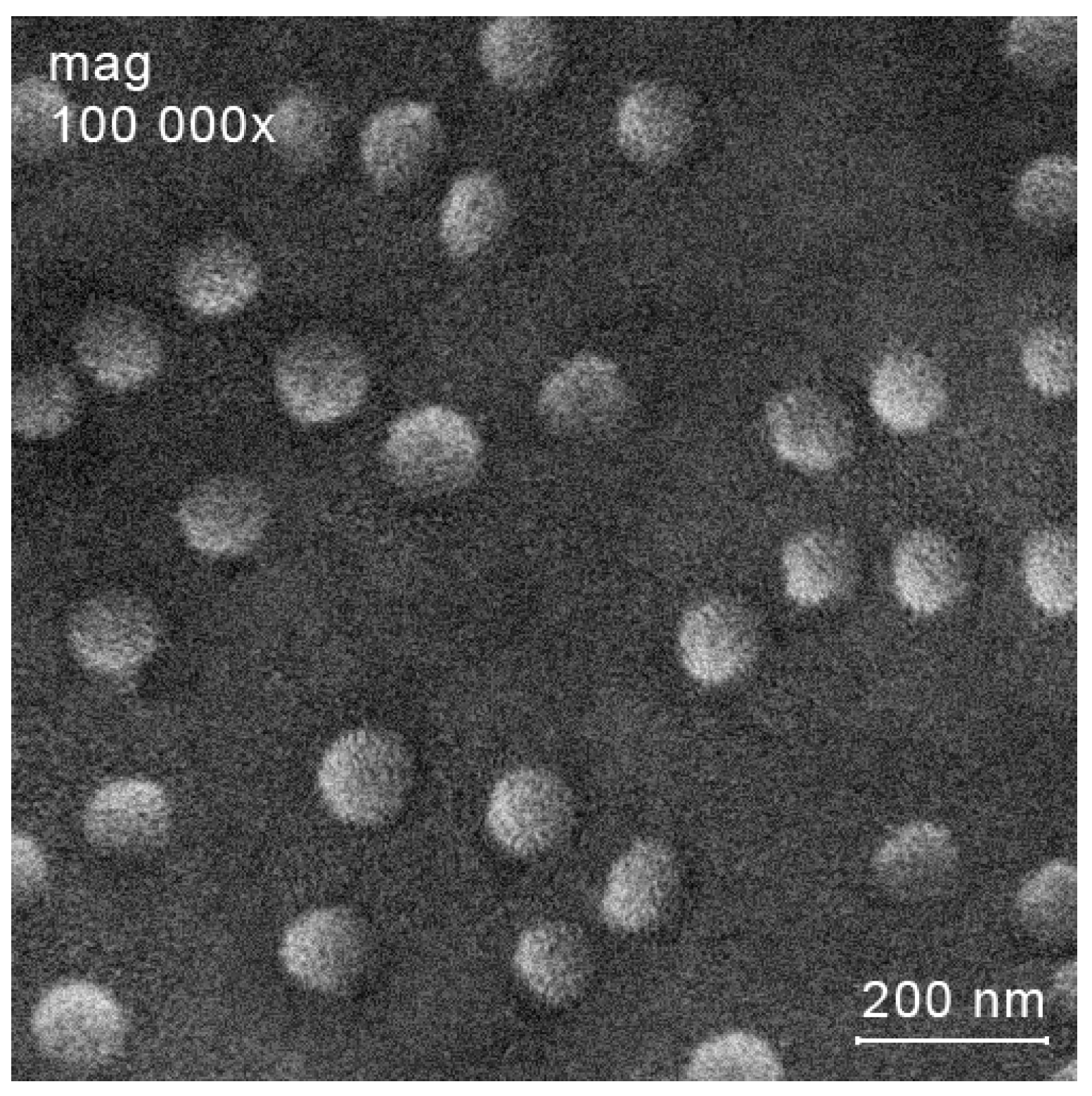
Form of the Samples
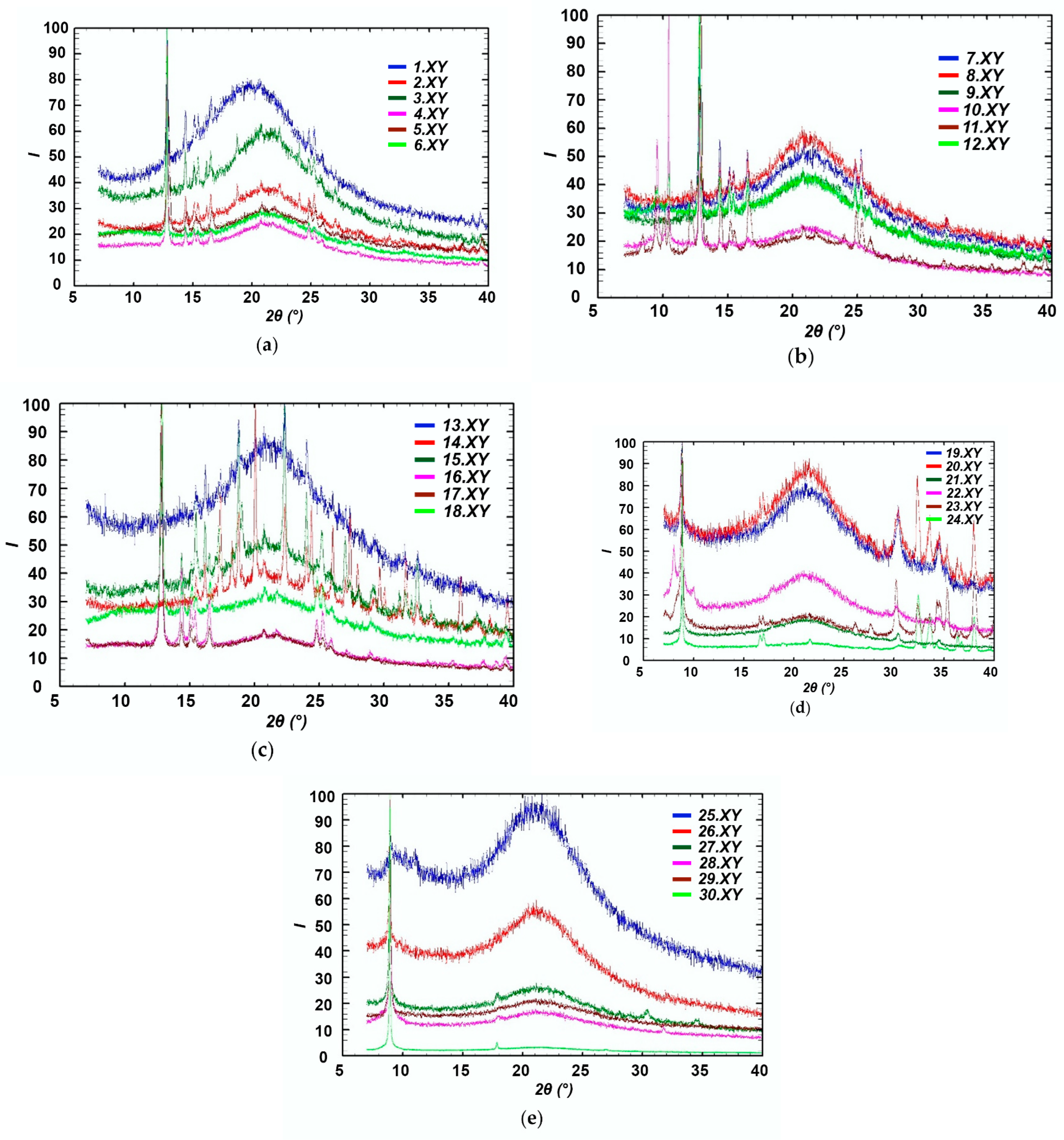
X-ray Diffraction Study
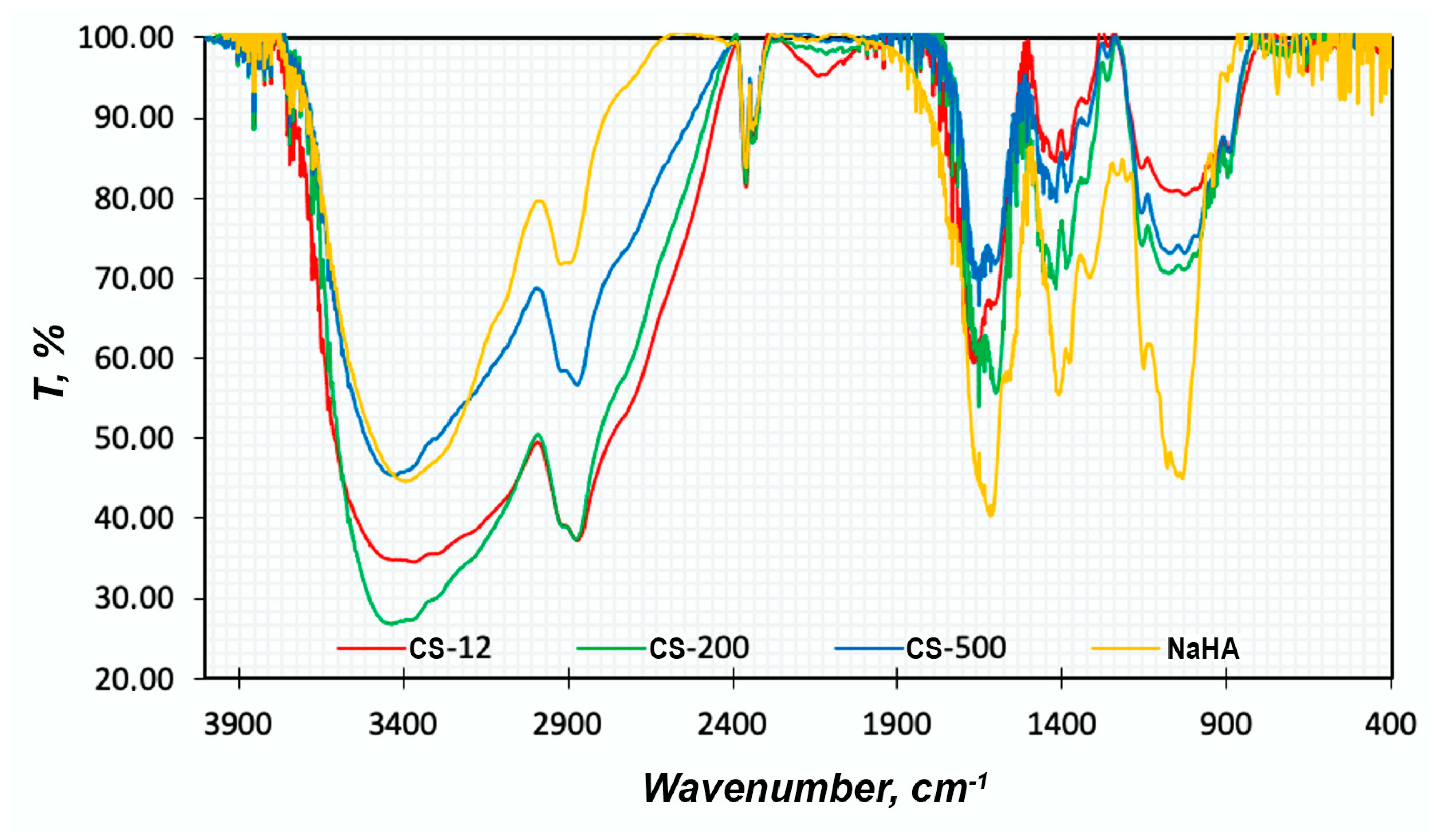
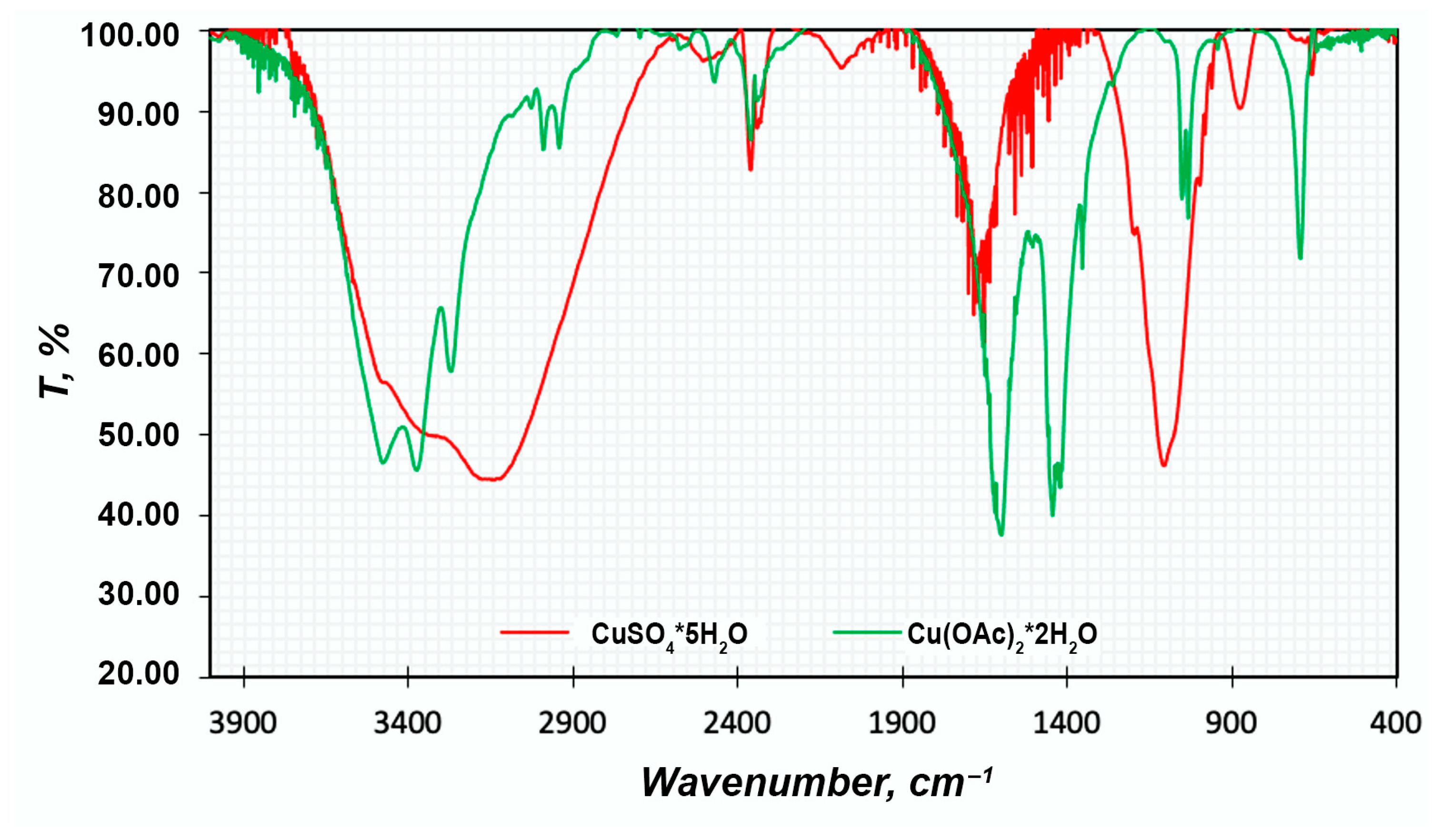
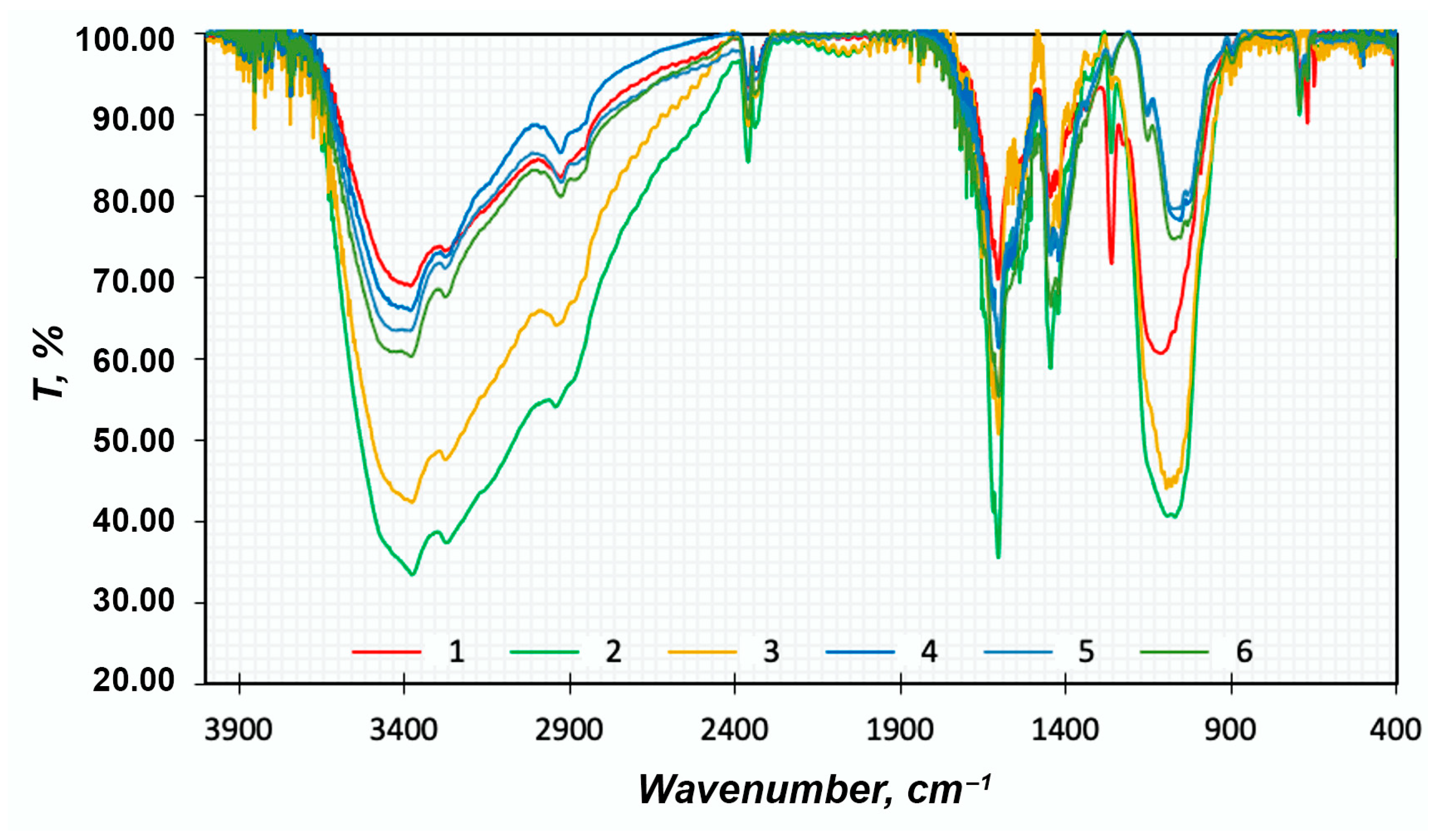
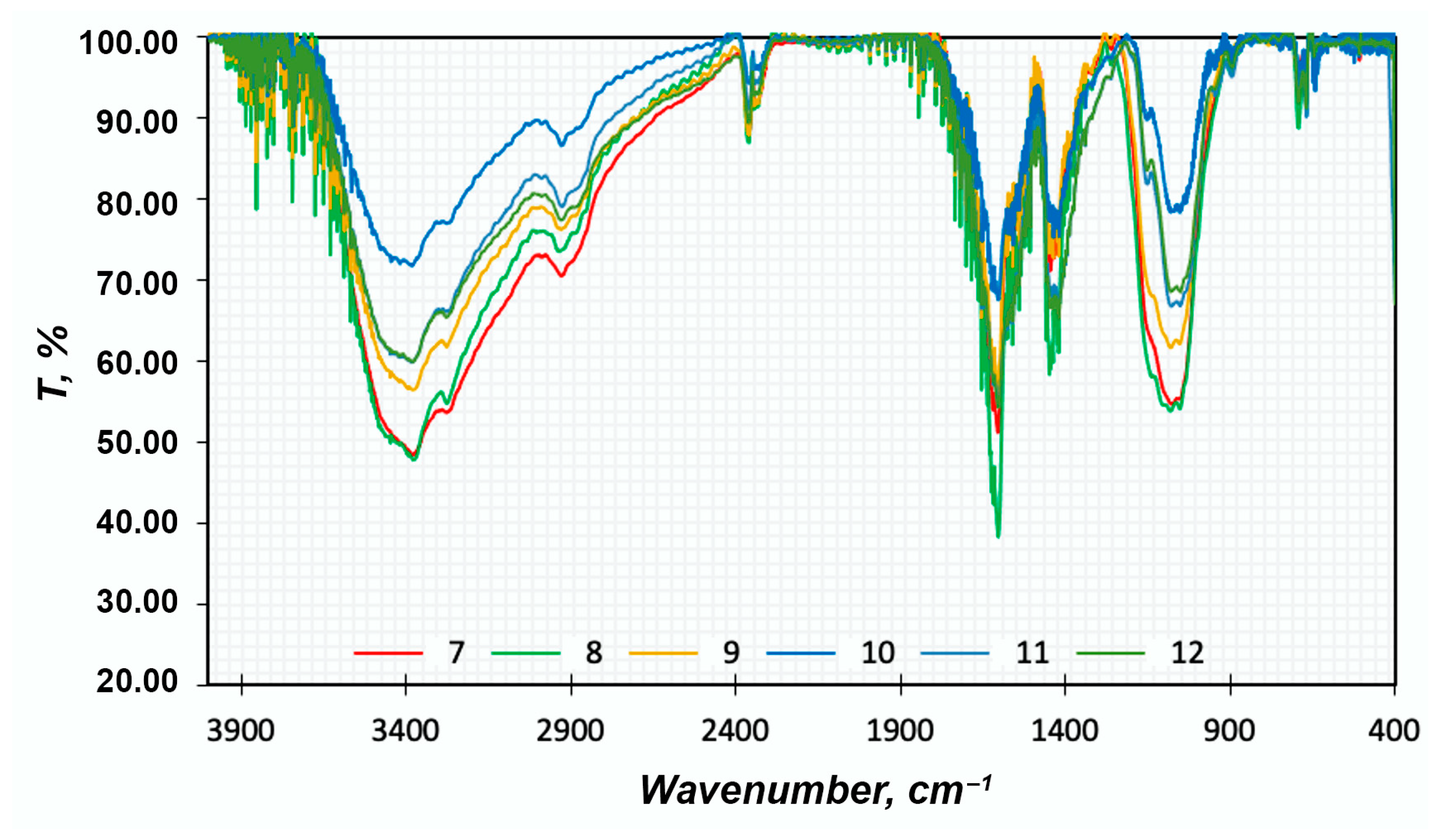
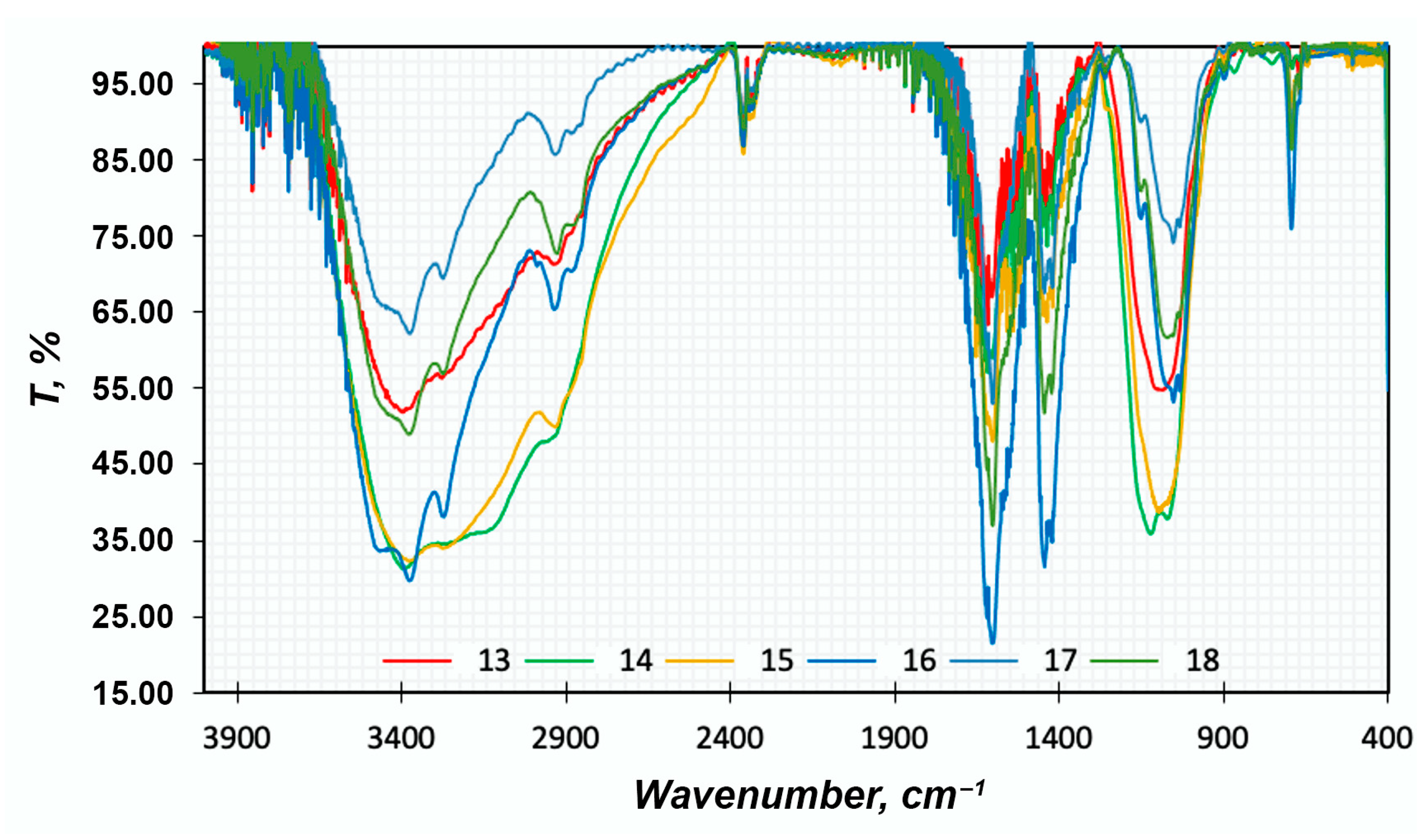
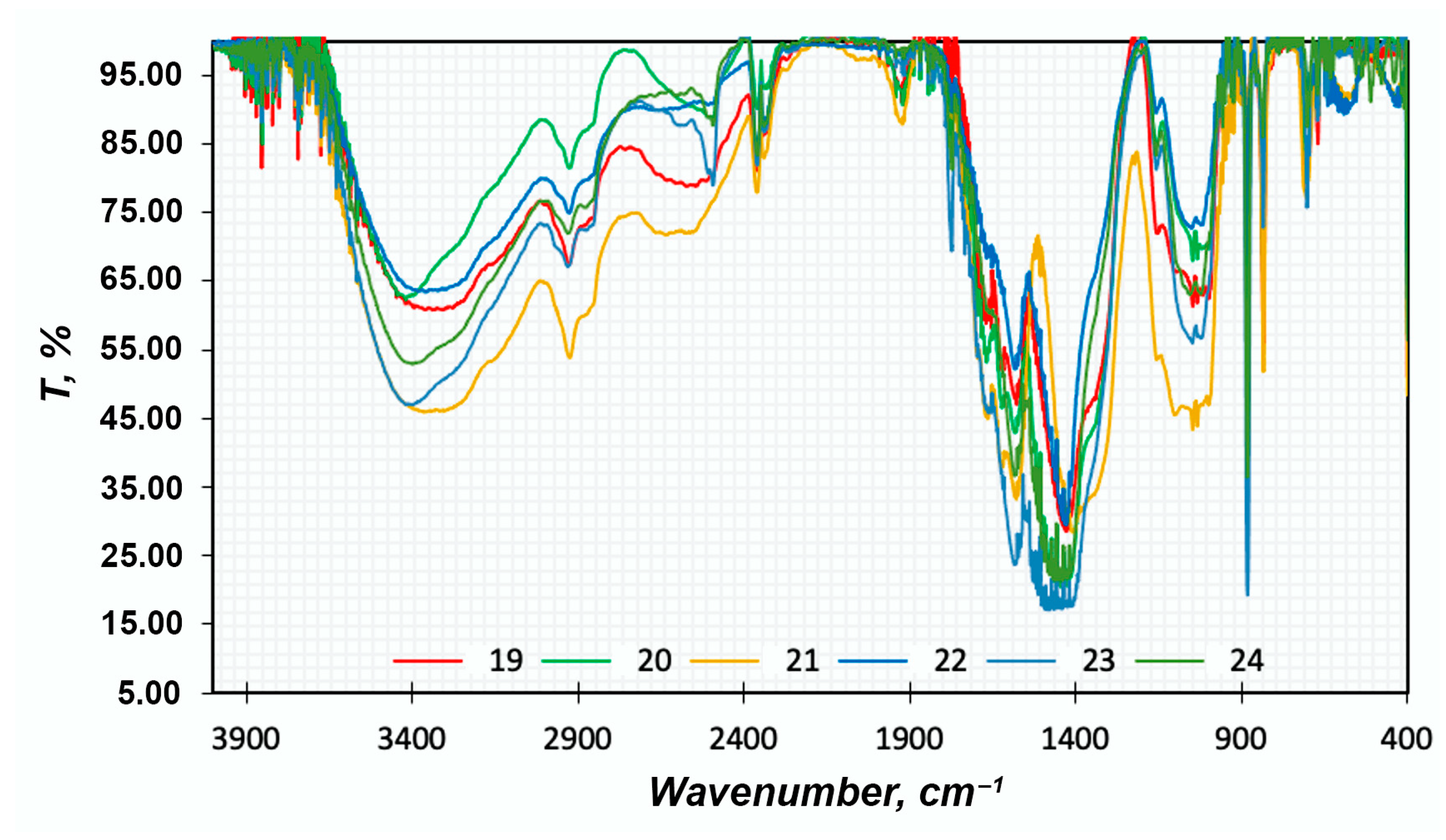
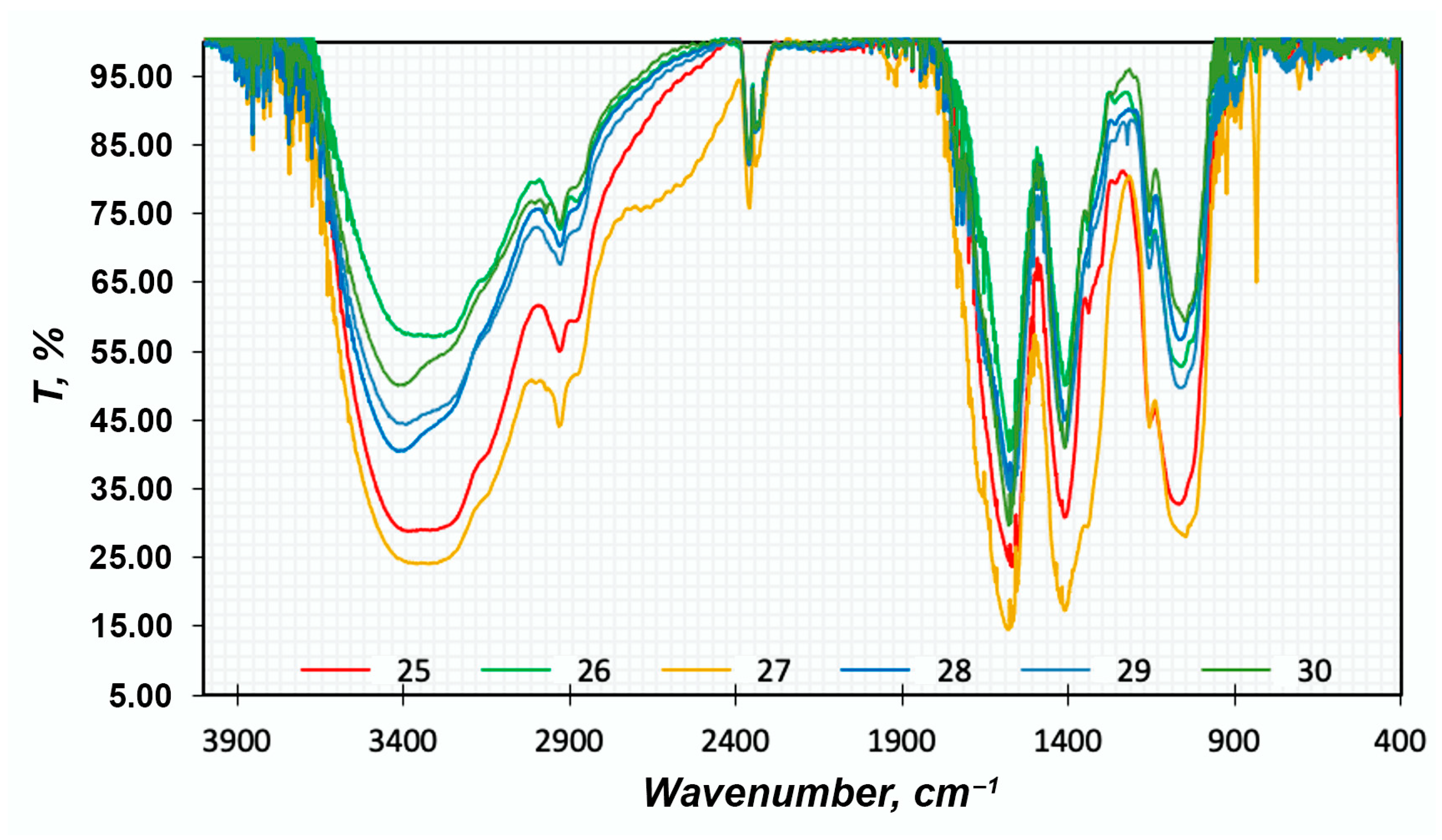
FTIR
TGA
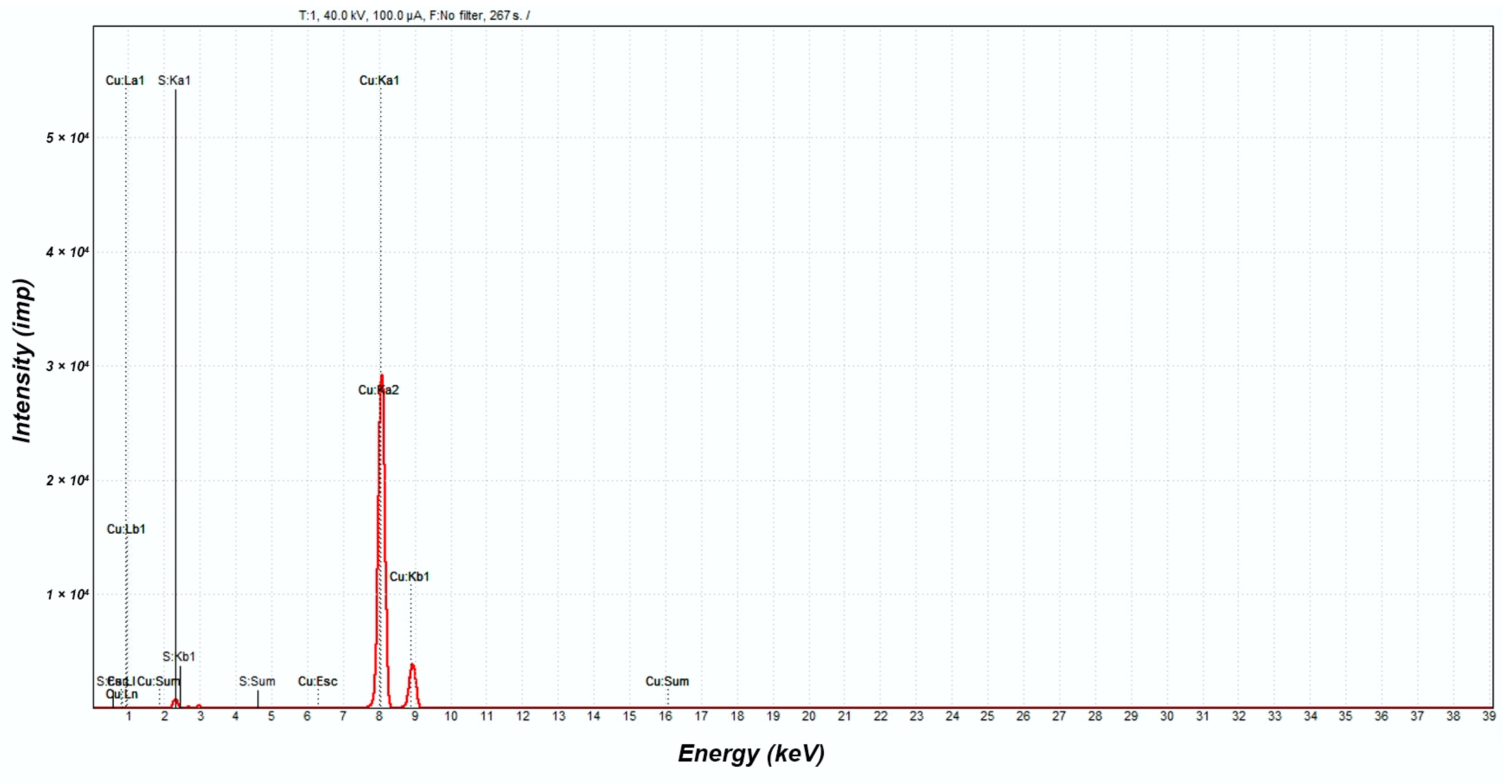
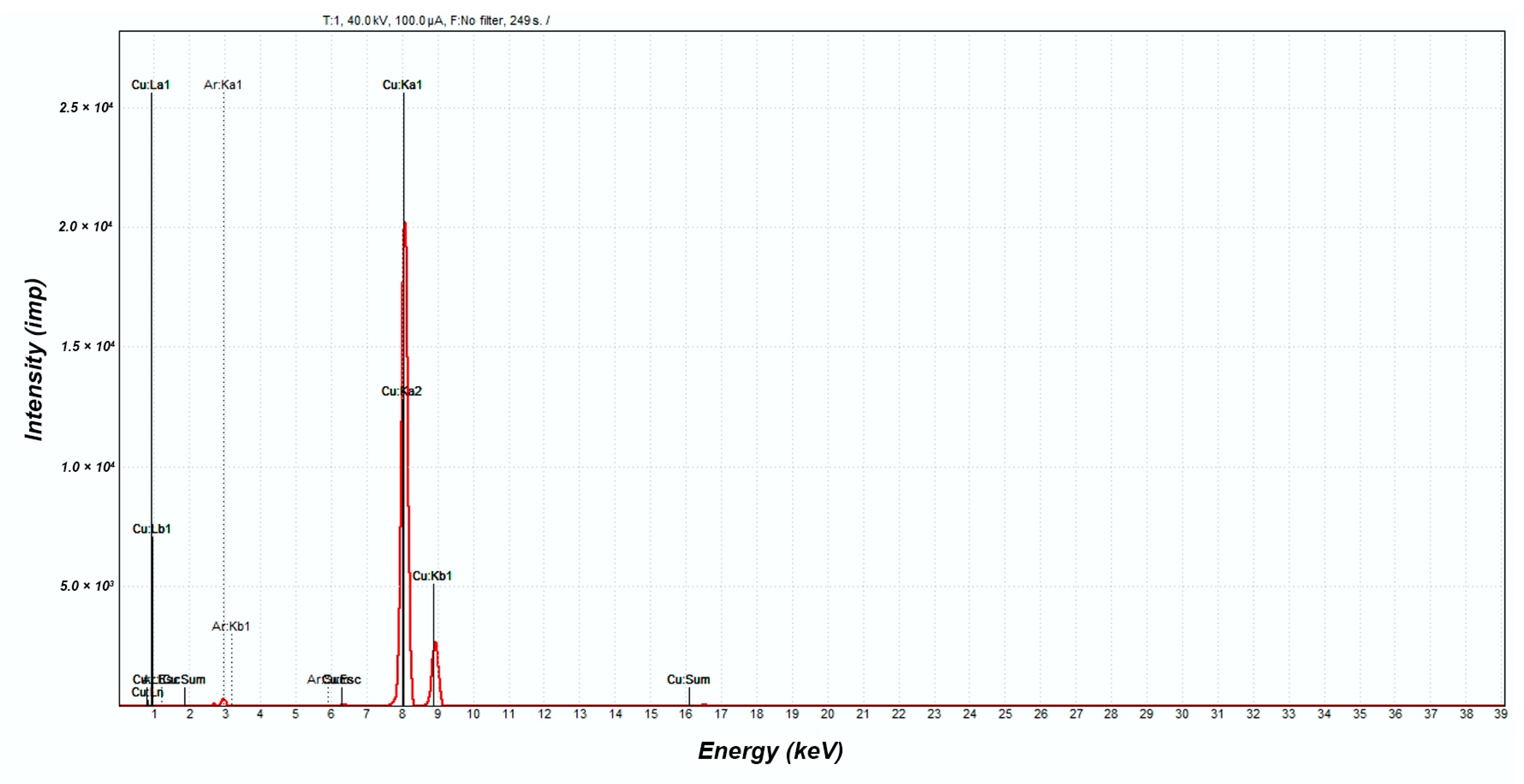
X-ray Fluorescence Analysis
2.2. Catalytic Studies
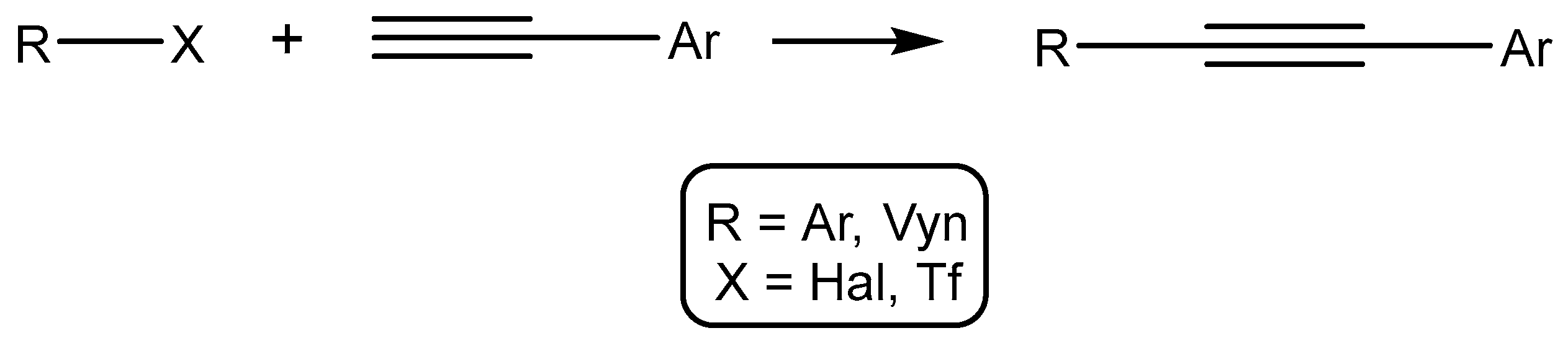
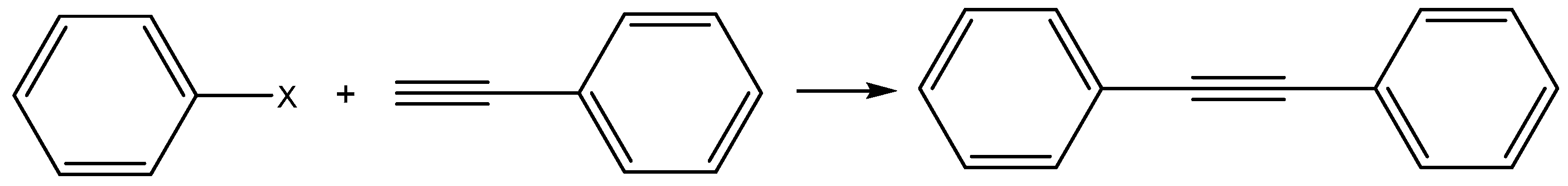
2.2.1. Catalytic Studies of the Sonogashira Reaction
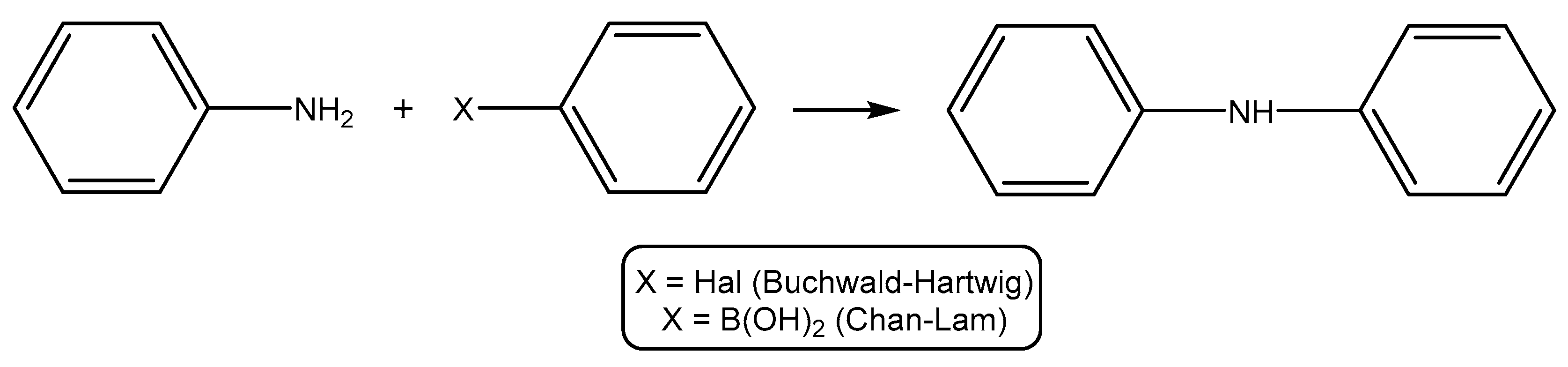
2.2.2. Catalytic Studies of the Buchwald–Hartwig and Chan–Lam Reactions
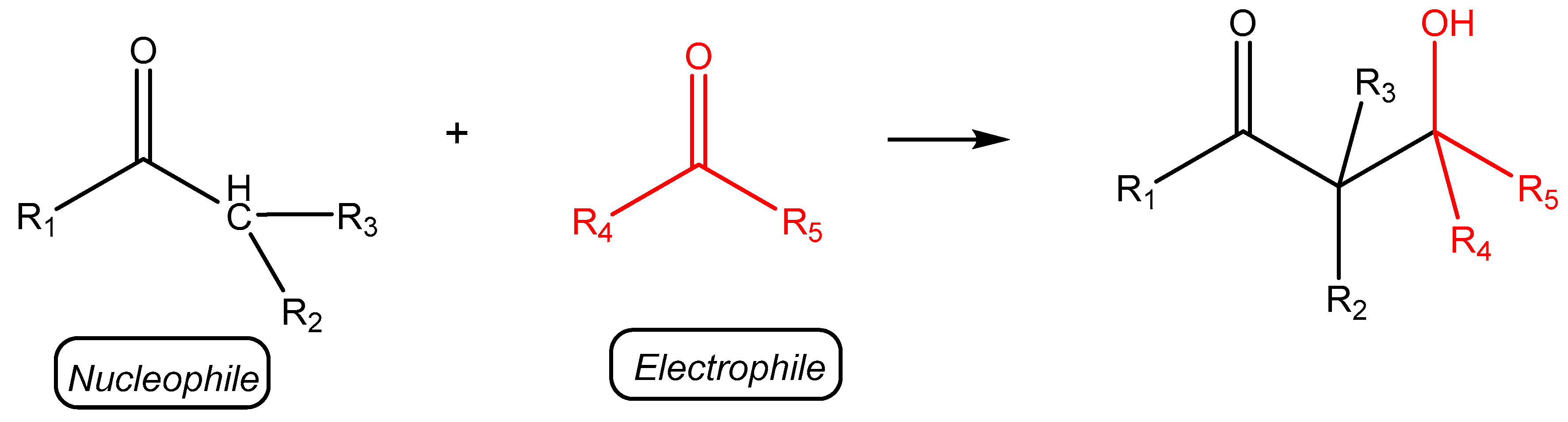
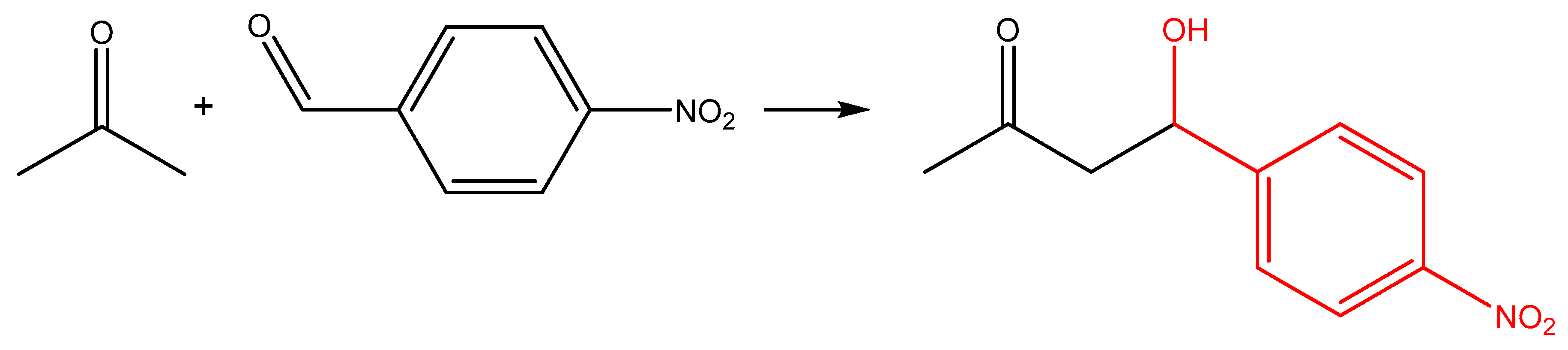
2.2.3. Catalytic Studies of the Aldol Reaction
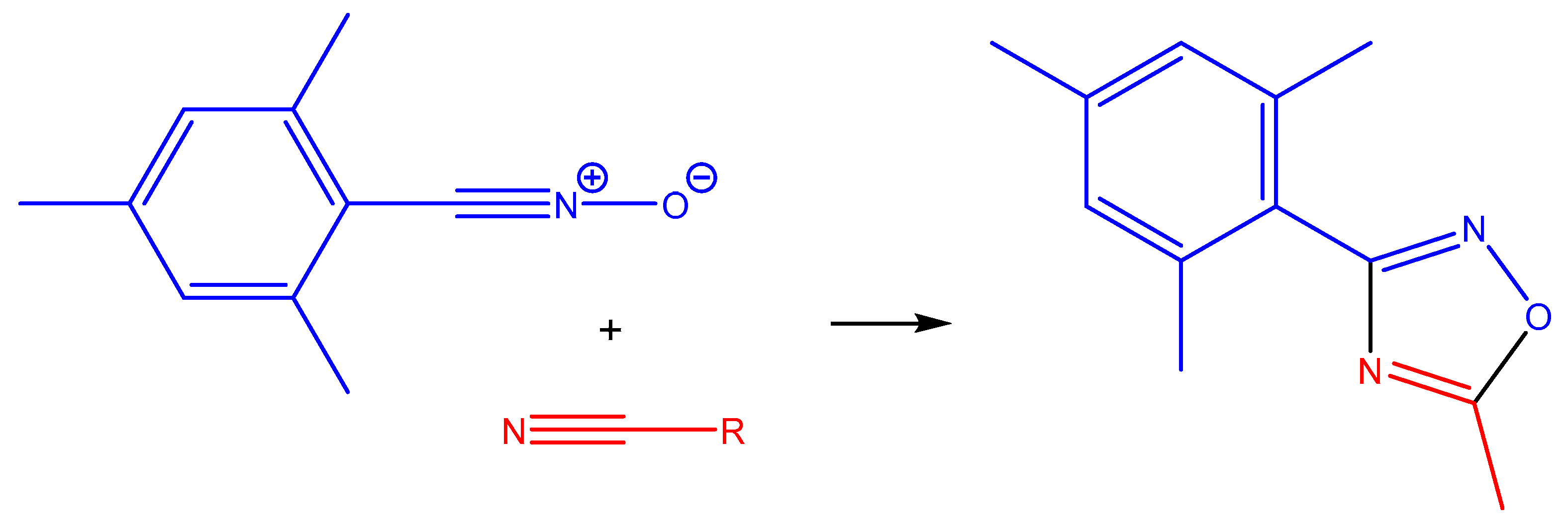
2.2.4. Catalytic Studies of the 1,3-Dipolar Cycloaddition of Nitrile Oxides to Nitriles
2.2.5. Final Remarks on Catalytic Studies
3. Materials and Methods
3.1. Materials
3.2. Preparation and Characterization of Chitosan/Copper(II) Composites
3.3. Catalytic Experiments
3.3.1. Sonogashira Reaction
3.3.2. Buchwald–Hartwig and Chan–Lam Reactions
3.3.3. Aldol Reaction
1,3-Dipolar Cycloaddition Reaction
3.4. Instrumentation
4. Conclusions
- (i)
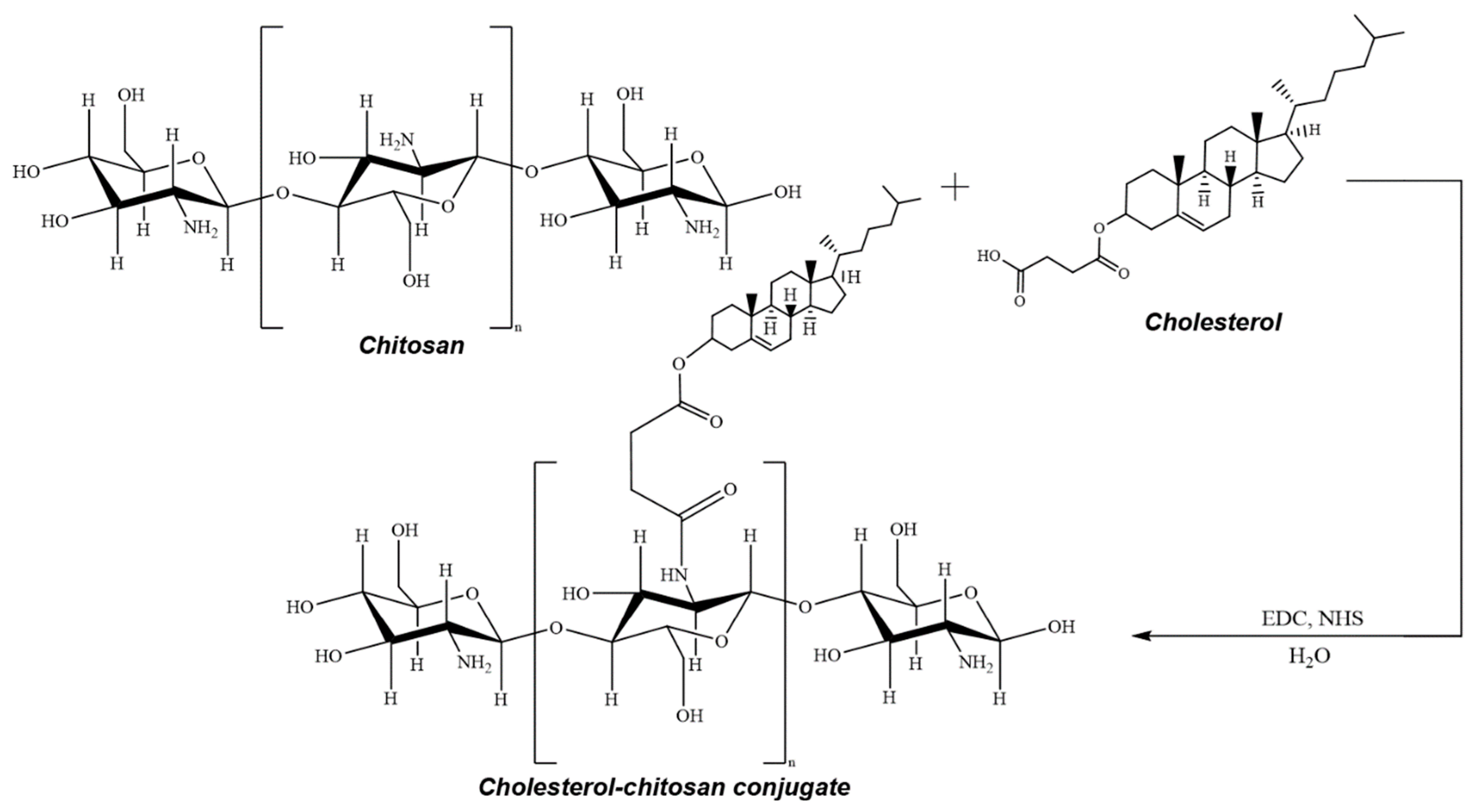
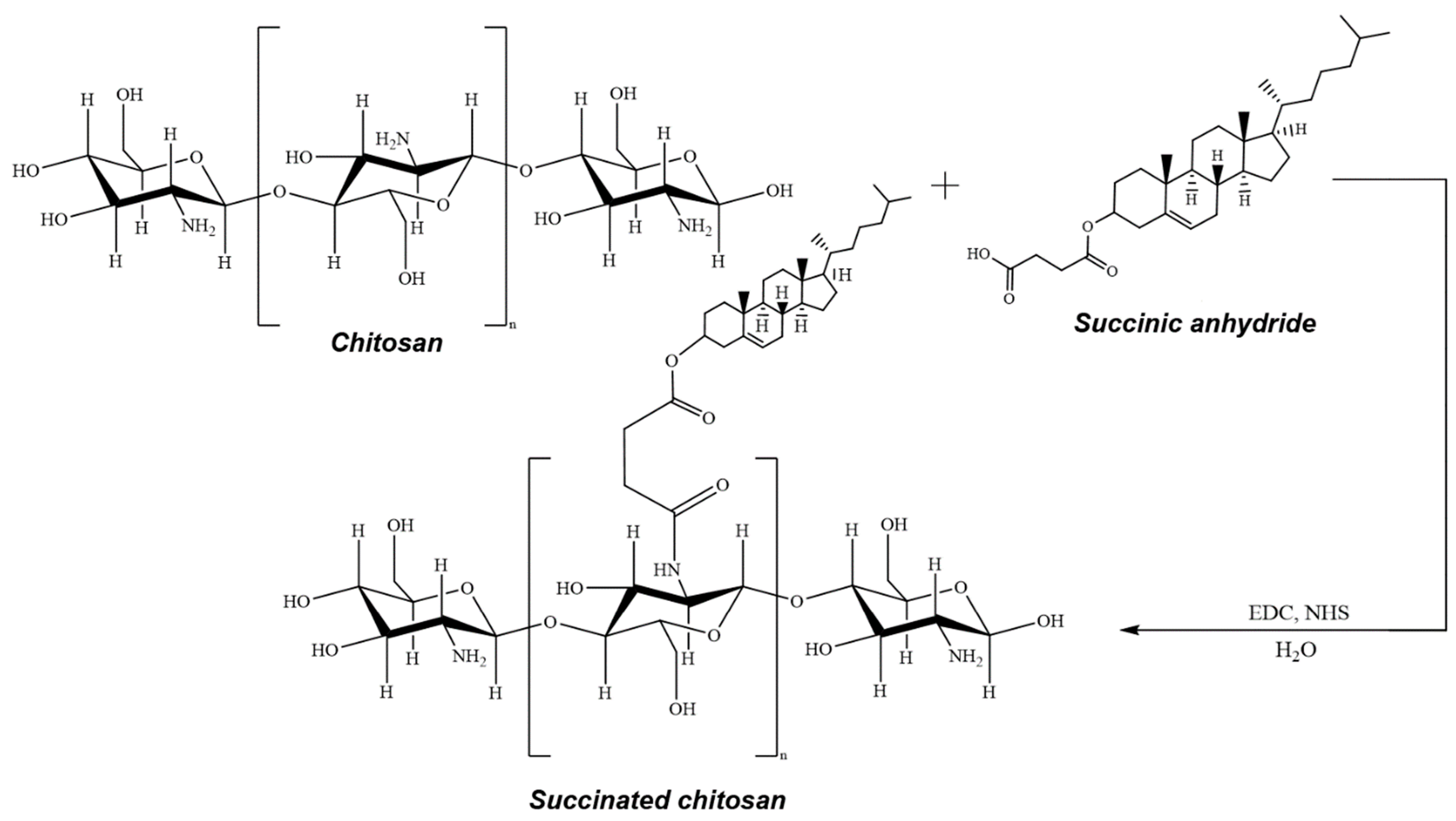
- Indeed, treatment of chitosan (or its cholesterol conjugate) with copper(II) sulfate or acetate, followed by coating with a layer of sodium hyaluronate or succination (if necessary), makes it possible to obtain a wide range of structurally similar systems (Figure 1, Table 1). In addition, we were able to obtain some of these systems in the form of nanoparticles (mainly copper(II) sulfate-based systems), and the second part is a coarse-grained powder (mainly copper(II) acetate-based systems). The molecular weight of the used chitosan practically does not affect the characteristics of the systems obtained;
- (ii)
- Cholesterol-containing systems have proven to be highly efficient catalysts for cross-couplings (Sonogashira, Buchwald–Hartwig, and Chan–Lam); succinated systems coated with a layer of hyaluronic acid are catalysts for the aldol reaction; systems containing inorganic copper(II) salt nanoparticles are capable of catalyzing the nitrile-oxide-to-nitrile 1,3-dipolar cycloaddition;
- (iii)
- The elaborated catalytic systems efficiently catalyze the mentioned reactions in the greenest solvent available, i.e., water, under aerobic conditions. The studied catalytic reactions proceed selectively, and the isolation of the product does not require column chromatography. The product is separated from the catalyst by simple filtration or centrifugation.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kolcsár, V.J.; Szőllősi, G. Chitosan as a chiral ligand and organocatalyst: Preparation conditions–property–catalytic performance relationships. Catal. Sci. Technol. 2021, 11, 7652–7666. [Google Scholar] [CrossRef]
- El Kadib, A. Chitosan as a Sustainable Organocatalyst: A Concise Overview. ChemSusChem 2015, 8, 217–244. [Google Scholar] [CrossRef]
- Aranaz, I.; Alcántara, A.R.; Civera, M.C.; Arias, C.; Elorza, B.; Heras Caballero, A.; Acosta, N. Chitosan: An Overview of Its Properties and Applications. Polymers 2021, 13, 3256. [Google Scholar] [CrossRef]
- Kritchenkov, A.S.; Egorov, A.R.; Artemjev, A.A.; Kritchenkov, I.S.; Volkova, O.V.; Kiprushkina, E.I.; Zabodalova, L.A.; Suchkova, E.P.; Yagafarov, N.Z.; Tskhovrebov, A.G.; et al. Novel heterocyclic chitosan derivatives and their derived nanoparticles: Catalytic and antibacterial properties. Int. J. Biol. Macromol. 2020, 149, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Kritchenkov, A.S.; Kletskov, A.V.; Egorov, A.R.; Kurasova, M.N.; Tskhovrebov, A.G.; Khrustalev, V.N. Ultrasound and click chemistry lead to a new chitin chelator. Its Pd(II) complex is a recyclable catalyst for the Sonogashira reaction in water. Carbohydr. Polym. 2021, 252, 117167. [Google Scholar] [CrossRef] [PubMed]
- Kritchenkov, A.S.; Egorov, A.R.; Dysin, A.P.; Volkova, O.V.; Zabodalova, L.A.; Suchkova, E.P.; Kurliuk, A.V.; Shakola, T.V. Ultrasound-assisted Cu(I)-catalyzed azide-alkyne click cycloaddition as polymer-analogous transformation in chitosan chemistry. High antibacterial and transfection activity of novel triazol betaine chitosan derivatives and their nanoparticles. Int. J. Biol. Macromol. 2019, 137, 592–603. [Google Scholar] [CrossRef]
- Kritchenkov, A.S.; Egorov, A.R.; Abramovich, R.A.; Kurliuk, A.V.; Shakola, T.V.; Kultyshkina, E.K.; Meza, M.J.B.; Pavlova, A.V.; Suchkova, E.P.; Thuy, G.L.; et al. Water-soluble triazole chitin derivative and its based nanoparticles: Synthesis, characterization, catalytic and antibacterial properties. Carbohydr. Polym. 2021, 257, 117593. [Google Scholar] [CrossRef]
- Egorov, A.R.; Khubiev, O.; Rubanik, V.V.; Rubanik, V.V.; Lobanov, N.N.; Savilov, S.V.; Kirichuk, A.A.; Kritchenkov, I.S.; Tskhovrebov, A.G.; Kritchenkov, A.S. The first selenium containing chitin and chitosan derivatives: Combined synthetic, catalytic and biological studies. Int. J. Biol. Macromol. 2022, 209, 2175–2187. [Google Scholar] [CrossRef]
- Kritchenkov, A.S.; Kurachenkov, A.I.; Egorov, A.R.; Yagafarov, N.Z.; Fortalnova, E.A.; Lobanov, N.N.; Dysin, A.P.; Khomik, A.S.; Khrustalev, V.N. Novel zinc(II)/chitosan-based composite: Ultrasound-assisted synthesis, catalytic and antibacterial activity. Mendeleev Commun. 2020, 30, 642–644. [Google Scholar] [CrossRef]
- Dalpozzo, R.; Mancuso, R. Copper-Catalyzed Synthesis of Coumarins. A Mini-Review. Catalysts 2021, 11, 1382. [Google Scholar] [CrossRef]
- Manna, S. Copper-Catalyzed Diastereo- and Enantioselective Borylative Cyclization. Catalysts 2022, 12, 734. [Google Scholar] [CrossRef]
- Weidlich, T.; Špryncová, M.; Čegan, A. Copper-Catalyzed Reactions of Aryl Halides with N-Nucleophiles and Their Possible Application for Degradation of Halogenated Aromatic Contaminants. Catalysts 2022, 12, 911. [Google Scholar] [CrossRef]
- Kujur, S.; Verma, S.; Pathak, D.D. Development of a Graphene Oxide-Supported N-Heterocyclic Carbene Copper(I) Complex as a Heterogeneous Catalyst for the Selective N-Monoalkylation of Amines. Catalysts 2022, 12, 1458. [Google Scholar] [CrossRef]
- Zelin, J.; Meyer, C.I.; Duarte, H.A.; Marchi, A. A Stable and Reusable Supported Copper Catalyst for the Selective Liquid-Phase Hydrogenation of 5-Hydroxymethylfurfural to 2,5-Bis(hydroxymethyl)furan. Catalysts 2022, 12, 1476. [Google Scholar] [CrossRef]
- Tarahomi, M.; Alinezhad, H.; Maleki, B. Immobilizing Pd nanoparticles on the ternary hybrid system of graphene oxide, Fe3O4 nanoparticles, and PAMAM dendrimer as an efficient support for catalyzing sonogashira coupling reaction. Appl. Organomet. Chem. 2019, 33, e5203. [Google Scholar] [CrossRef]
- Laffafchi, F.; Tajbakhsh, M.; Sarrafi, Y.; Maleki, B.; Ghani, M. Cu-Modified Magnetic Creatine as an Efficient Catalyst for Regioselective Preparation of 1,2,3-Triazoles Derivatives. Polycycl. Aromat. Compd. 2022. [Google Scholar] [CrossRef]
- Chemler, S.R. Copper catalysis in organic synthesis. Beilstein J. Org. Chem. 2015, 11, 2252–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavadetscher, J.; Hoffmann, S.; Lilienkampf, A.; Mackay, L.; Yusop, R.M.; Rider, S.A.; Mullins, J.J.; Bradley, M. Copper Catalysis in Living Systems and In Situ Drug Synthesis. Angew. Chem. Int. Ed. 2016, 55, 15662–15666. [Google Scholar] [CrossRef]
- Shiri, P. An overview on the copper-promoted synthesis of five-membered heterocyclic systems. Appl. Organomet. Chem. 2020, 34, e5600. [Google Scholar] [CrossRef]
- Shiri, P.; Aboonajmi, J. A systematic review on silica-, carbon-, and magnetic materials-supported copper species as efficient heterogeneous nanocatalysts in "click" reactions. Beilstein J. Org. Chem. 2020, 16, 551–586. [Google Scholar] [CrossRef]
- Ueda, S.; Nagasawa, H. Synthesis of 2-Arylbenzoxazoles by Copper-Catalyzed Intramolecular Oxidative C–O Coupling of Benzanilides. Angew. Chem. Int. Ed. 2008, 47, 6411–6413. [Google Scholar] [CrossRef]
- Jiao, L.-Y.; Zhang, Z.; Hong, Q.; Ning, Z.-H.; Liu, S.; Sun, M.; Hao, Q.; Xu, L.; Li, Z.; Ma, X.-X. Recyclable copper catalyst on chitosan for facile preparation of alkyl/aryl mixed phosphates via deaminated esterification between diphenylphosphoryl azides and aliphatic alcohols. Mol. Catal. 2020, 494, 111120. [Google Scholar] [CrossRef]
- Shen, C.; Xu, J.; Yu, W.; Zhang, P. A highly active and easily recoverable chitosan@copper catalyst for the C–S coupling and its application in the synthesis of zolimidine. Green Chem. 2014, 16, 3007–3012. [Google Scholar] [CrossRef]
- Baig, R.B.N.; Varma, R.S. Copper on chitosan: A recyclable heterogeneous catalyst for azide–alkyne cycloaddition reactions in water. Green Chem. 2013, 15, 1839–1843. [Google Scholar] [CrossRef]
- Williams, A.; Ibrahim, I.T. Carbodiimide chemistry: Recent advances. Chem. Rev. 1981, 81, 589–636. [Google Scholar] [CrossRef]
- Cammarata, C.R.; Hughes, M.E.; Ofner, C.M., III. Carbodiimide Induced Cross-Linking, Ligand Addition, and Degradation in Gelatin. Mol. Pharm. 2015, 12, 783–793. [Google Scholar] [CrossRef] [Green Version]
- Bashir, S.; Teo, Y.Y.; Ramesh, S.; Ramesh, K.; Khan, A.A. N-succinyl chitosan preparation, characterization, properties and biomedical applications: A state of the art review. Rev. Chem. Eng. 2015, 31, 563–597. [Google Scholar] [CrossRef]
- Yanat, M.; Schroën, K. Preparation methods and applications of chitosan nanoparticles; with an outlook toward reinforcement of biodegradable packaging. React. Funct. Polym. 2021, 161, 104849. [Google Scholar] [CrossRef]
- Eremin, D.B.; Ananikov, V.P. Understanding active species in catalytic transformations: From molecular catalysis to nanoparticles, leaching, “Cocktails” of catalysts and dynamic systems. Coord. Chem. Rev. 2017, 346, 2–19. [Google Scholar] [CrossRef]
- Wang, D.; Gao, S. Sonogashira coupling in natural product synthesis. Org. Chem. Front. 2014, 1, 556–566. [Google Scholar] [CrossRef]
- Nagarapu, L.; Karnakanti, S.; Bantu, R. Total synthesis of sapinofuranone A from d-ribose. Tetrahedron 2012, 68, 5829–5832. [Google Scholar] [CrossRef]
- Miller, M.W.; Johnson, C.R. Sonogashira Coupling of 2-Iodo-2-cycloalkenones: Synthesis of (+)- and (−)-Harveynone and (−)-Tricholomenyn A. J. Org. Chem. 1997, 62, 1582–1583. [Google Scholar] [CrossRef]
- Garlaschelli, L.; Magistrali, E.; Vidari, G.; Zuffardi, O. Tricholomenyns A and B, novel antimitotic acetylenic cyclohexenone derivatives from the fruiting bodies of Tricholoma acerbum. Tetrahedron Lett. 1995, 36, 5633–5636. [Google Scholar] [CrossRef]
- Osadchii, S.A.; Shul’ts, E.E.; Vasilevskii, S.F.; Polukhina, E.V.; Stepanov, A.A.; Tolstikov, G.A. Study of alkaloids of the Siberian and Altai flora 13. Synthesis of alkynyllappaconitines. Russ. Chem. Bull. 2007, 56, 356–360. [Google Scholar] [CrossRef]
- Tolstikova, G.T.; Bryzgalov, O.A.; Sorokina, V.I.; Osadchii, A.S.; Shults, E.E.; Dolgikh, P.M.; Khvostov, V.M. Solification with Hydrobromic Acid as a Factor Defining the Antiarrhythmic Effect of Lappaconitine Derivatives. Lett. Drug Des. Discov. 2009, 6, 475–477. [Google Scholar] [CrossRef]
- Magano, J.; Dunetz, J.R. Large-Scale Applications of Transition Metal-Catalyzed Couplings for the Synthesis of Pharmaceuticals. Chem. Rev. 2011, 111, 2177–2250. [Google Scholar] [CrossRef] [PubMed]
- Hervé, G.; Len, C. Heck and Sonogashira couplings in aqueous media—Application to unprotected nucleosides and nucleotides. Sustain. Chem. Process. 2015, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Mohjer, F.; Mofatehnia, P.; Rangraz, Y.; Heravi, M.M. Pd-free, Sonogashira cross-coupling reaction. An update. J. Organomet. Chem. 2021, 936, 121712. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Q.; Li, J.-H.; Dong, Z.-B. A Review on the Latest Progress of Chan-Lam Coupling Reaction. Adv. Synth. Catal. 2020, 362, 3311–3331. [Google Scholar] [CrossRef]
- Heravi, M.M.; Kheilkordi, Z.; Zadsirjan, V.; Heydari, M.; Malmir, M. Buchwald-Hartwig reaction: An overview. J. Organomet. Chem. 2018, 861, 17–104. [Google Scholar] [CrossRef]
- Yuan, C.; Zhang, L.; Zhao, Y. Cu(II)-Catalyzed C-N Coupling of (Hetero)aryl Halides and N-Nucleophiles Promoted by α-Benzoin Oxime. Molecules 2019, 24, 4177. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.-N.; Zheng, H.; Li, T.; Wei, W.-T. Recent Advances in Copper-Catalyzed C−N Bond Formation Involving N-Centered Radicals. ChemSusChem 2021, 14, 5340–5358. [Google Scholar] [CrossRef]
- Sahoo, H.; Mukherjee, S.; Grandhi, G.S.; Selvakumar, J.; Baidya, M. Copper Catalyzed C–N Cross-Coupling Reaction of Aryl Boronic Acids at Room Temperature through Chelation Assistance. J. Org. Chem. 2017, 82, 2764–2771. [Google Scholar] [CrossRef] [PubMed]
- Mahrwald, R. Modern Aldol Reactions; Wiley-VCH Verlag GmbH & Co: Weinheim, Germany, 2004; p. 679. [Google Scholar]
- Mukaiyama, T. The Directed Aldol Reaction. In Organic Reactions; Wiley: Hoboken, NJ, USA, 1982; pp. 203–331. [Google Scholar] [CrossRef]
- Heathcock, C.H. The Aldol Reaction: Acid and General Base Catalysis. In Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon: Oxford, UK, 1991; pp. 133–179. [Google Scholar]
- Mestres, R. A green look at the aldol reaction. Green Chem. 2004, 6, 583–603. [Google Scholar] [CrossRef]
- Li, J.J.; Johnson, D.S.; Sliskovic, D.R.; Roth, B.D. Contemporary Drug Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2004; p. 221. [Google Scholar]
- Braun, M.; Devant, R.D. (R)- and (S)-2-acetoxy-1,1,2-triphenylethanol—Effective synthetic equivalents of a chiral acetate enolate. Tetrahedron Lett. 1984, 25, 5031–5034. [Google Scholar] [CrossRef]
- Schetter, B.; Mahrwald, R. Modern aldol methods for the total synthesis of polyketides. Angew. Chem. Int. Ed. 2006, 45, 7506–7525. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Mandal, S.; Ghosh, S.K.; Ghosh, A.; Saha, R.; Banerjee, S.; Saha, B. Review of the aldol reaction. Synth. Commun. 2016, 46, 1327–1342. [Google Scholar] [CrossRef]
- Bargujar, S.; Ratnani, S. Aldol condensation: Green perspectives. J. Iran. Chem. Soc. 2022, 19, 2171–2190. [Google Scholar] [CrossRef]
- Breugst, M.; Reissig, H.-U. The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himo, F.; Demko, Z.P.; Noodleman, L.; Sharpless, K.B. Why Is Tetrazole Formation by Addition of Azide to Organic Nitriles Catalyzed by Zinc(II) Salts? J. Am. Chem. Soc. 2003, 125, 9983–9987. [Google Scholar] [CrossRef]
- Sommer, M.G.; Rechkemmer, Y.; Suntrup, L.; Hohloch, S.; van der Meer, M.; van Slageren, J.; Sarkar, B. Structural snapshots in the copper(ii) induced azide–nitrile cycloaddition: Effects of peripheral ligand substituents on the formation of unsupported μ1,1-azido vs. μ1,4-tetrazolato bridged complexes. Dalton Trans. 2016, 45, 17770–17781. [Google Scholar] [CrossRef]
- Kuznetsov, M.L.; Kukushkin, V.Y.; Haukka, M.; Pombeiro, A.J.L. 1,3-Dipolar cycloaddition of nitrile oxides to free and Pt-bound nitriles: A theoretical study of the activation effect, reactivity and mechanism. Inorg. Chim. Acta 2003, 356, 85–94. [Google Scholar] [CrossRef]
- Nitrile Oxides, Nitrones and Nitronates in Organic Synthesis: Novel Strategies in Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2008; p. 768.
- Bokach, N.A.; Kukushkin, V.Y. Coordination chemistry of dialkylcyanamides: Binding properties, synthesis of metal complexes, and ligand reactivity. Coord. Chem. Rev. 2013, 257, 2293–2316. [Google Scholar] [CrossRef]
- Jaleh, B.; Nasrollahzadeh, M.; Nasri, A.; Eslamipanah, M.; Moradi, A.; Nezafat, Z. Biopolymer-derived (nano)catalysts for hydrogen evolution via hydrolysis of hydrides and electrochemical and photocatalytic techniques: A review. Int. J. Biol. Macromol. 2021, 182, 1056–1090. [Google Scholar] [CrossRef]
- Razzaghi, M.; Homaei, A.; Vianello, F.; Azad, T.; Sharma, T.; Nadda, A.K.; Stevanato, R.; Bilal, M.; Iqbal, H.M.N. Industrial applications of immobilized nano-biocatalysts. Bioprocess Biosyst. Eng. 2022, 45, 237–256. [Google Scholar] [CrossRef]
- Dubashynskaya, N.V.; Golovkin, A.S.; Kudryavtsev, I.V.; Prikhodko, S.S.; Trulioff, A.S.; Bokatyi, A.N.; Poshina, D.N.; Raik, S.V.; Skorik, Y.A. Mucoadhesive cholesterol-chitosan self-assembled particles for topical ocular delivery of dexamethasone. Int. J. Biol. Macromol. 2020, 158, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Kritchenkov, A.S.; Luzyanin, K.V.; Bokach, N.A.; Kuznetsov, M.L.; Gurzhiy, V.V.; Kukushkin, V.Y. Selective Nucleophilic Oxygenation of Palladium-Bound Isocyanide Ligands: Route to Imine Complexes That Serve as Efficient Catalysts for Copper-/Phosphine-Free Sonogashira Reactions. Organometallics 2013, 32, 1979–1987. [Google Scholar] [CrossRef]
- Dombrovskii, A.V.; Tashchuk, K.G. Haloarylation of unsaturated compounds by aromatic diazo compounds. XVII. Arylation of α-chlorostyrene and preparation of tolans. Zh. Obshch. Khim. 1963, 33, 165–170. [Google Scholar]
- Astakhov, G.S.; Levitsky, M.M.; Bantreil, X.; Lamaty, F.; Khrustalev, V.N.; Zubavichus, Y.V.; Dorovatovskii, P.V.; Shubina, E.S.; Bilyachenko, A.N. Cu(II)-silsesquioxanes as efficient precatalysts for Chan-Evans-Lam coupling. J. Organomet. Chem. 2020, 906, 121022. [Google Scholar] [CrossRef]
- Kritchenkov, A.S.; Egorov, A.R.; Krytchankou, I.S.; Dubashynskaya, N.V.; Volkova, O.V.; Shakola, T.V.; Kurliuk, A.V.; Skorik, Y.A. Synthesis of novel 1H-tetrazole derivatives of chitosan via metal-catalyzed 1,3-dipolar cycloaddition. Catalytic and antibacterial properties of [3-(1H-tetrazole-5-yl)ethyl]chitosan and its nanoparticles. Int. J. Biol. Macromol. 2019, 132, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Bokach, N.A.; Khripoun, A.V.; Kukushkin, V.Y.; Haukka, M.; Pombeiro, A.J.L. A Route to 1,2,4-Oxadiazoles and Their Complexes via Platinum-Mediated 1,3-Dipolar Cycloaddition of Nitrile Oxides to Organonitriles. Inorg. Chem. 2003, 42, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Skorik, Y.A.; Kritchenkov, A.S.; Moskalenko, Y.E.; Golyshev, A.A.; Raik, S.V.; Whaley, A.K.; Vasina, L.V.; Sonin, D.L. Synthesis of N-succinyl- and N-glutaryl-chitosan derivatives and their antioxidant, antiplatelet, and anticoagulant activity. Carbohydr. Polym. 2017, 166, 166–172. [Google Scholar] [CrossRef] [PubMed]






















| Entry | Catalyst | Base/X | mol% | T, °C | Time, h | Yield,% |
|---|---|---|---|---|---|---|
| 1 | 13 | K3PO4/I | 20 | 100 | 10 | 54 |
| 2 | 14 | K3PO4/I | 20 | 100 | 10 | 56 |
| 3 | 15 | K3PO4/I | 20 | 100 | 10 | 50 |
| 4 | 16 | K3PO4/I | 20 | 100 | 10 | 18 |
| 5 | 17 | K3PO4/I | 20 | 100 | 10 | 16 |
| 6 | 18 | K3PO4/I | 20 | 100 | 10 | 16 |
| 7 | 13 | K2CO3/I | 20 | 100 | 10 | 58 |
| 8 | 14 | K2CO3/I | 20 | 100 | 10 | 60 |
| 9 | 15 | K2CO3/I | 20 | 100 | 10 | 60 |
| 10 | 16 | K2CO3/I | 20 | 100 | 10 | 22 |
| 11 | 17 | K2CO3/I | 20 | 100 | 10 | 22 |
| 12 | 18 | K2CO3/I | 20 | 100 | 10 | 20 |
| 13 | 13 | Cs2CO3/I | 20 | 100 | 10 | 59 |
| 14 | 14 | Cs2CO3/I | 20 | 100 | 10 | 57 |
| 15 | 15 | Cs2CO3/I | 20 | 100 | 10 | 57 |
| 16 | 16 | Cs2CO3/I | 20 | 100 | 10 | 20 |
| 17 | 17 | Cs2CO3/I | 20 | 100 | 10 | 20 |
| 18 | 18 | Cs2CO3/I | 20 | 100 | 10 | 23 |
| 19 | 13 | KF/I | 20 | 100 | 10 | 40 |
| 20 | 14 | KF/I | 20 | 100 | 10 | 44 |
| 21 | 15 | KF/I | 20 | 100 | 10 | 45 |
| 22 | 16 | KF/I | 20 | 100 | 10 | 17 |
| 23 | 17 | KF/I | 20 | 100 | 10 | 17 |
| 24 | 18 | KF/I | 20 | 100 | 10 | 15 |
| 25–30 | 13–18 | Et3N or Py/I | 20 | 100 | 10 | traces |
| 31 | 13 | LiOH/I | 20 | 100 | 10 | 100 |
| 32 | 14 | LiOH/I | 20 | 100 | 10 | 100 |
| 33 | 15 | LiOH/I | 20 | 100 | 10 | 100 |
| 34 | 16 | LiOH/I | 20 | 100 | 10 | 54 |
| 35 | 17 | LiOH/I | 20 | 100 | 10 | 53 |
| 36 | 18 | LiOH/I | 20 | 100 | 10 | 53 |
| 37 | 13 | LiOH/I | 20 | 90 | 3 | 100 |
| 38 | 14 | LiOH/I | 20 | 90 | 3 | 100 |
| 39 | 15 | LiOH/I | 20 | 90 | 3 | 100 |
| 40 | 16 | LiOH/I | 20 | 90 | 3 | 50 |
| 41 | 17 | LiOH/I | 20 | 90 | 3 | 52 |
| 42 | 18 | LiOH/I | 20 | 90 | 3 | 54 |
| 43 | 13 | LiOH/Br | 20 | 90 | 3 | 66 |
| 44 | 14 | LiOH/Br | 20 | 90 | 3 | 64 |
| 45 | 15 | LiOH/Br | 20 | 90 | 3 | 69 |
| 46 | 13 | LiOH/Cl | 20 | 90 | 3 | 31 |
| 47 | 14 | LiOH/Cl | 20 | 90 | 3 | 30 |
| 48 | 15 | LiOH/Cl | 20 | 90 | 3 | 26 |
| Entry | Catalyst | X | mol% | T (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 * | 13 | I | 20 | 120 | 24 | 18 |
| 2 * | 14 | I | 20 | 120 | 24 | 15 |
| 3 * | 15 | I | 20 | 120 | 24 | 16 |
| 4 * | 16 | I | 20 | 120 | 24 | traces |
| 5 * | 17 | I | 20 | 120 | 24 | traces |
| 6 * | 18 | I | 20 | 120 | 24 | traces |
| 7 * | 13 | B(OH)2 | 20 | 120 | 24 | 23 |
| 8 * | 14 | B(OH)2 | 20 | 120 | 24 | 22 |
| 9 * | 15 | B(OH)2 | 20 | 120 | 24 | 20 |
| 10 * | 16 | B(OH)2 | 20 | 120 | 24 | 5 |
| 11 * | 17 | B(OH)2 | 20 | 120 | 24 | 5 |
| 12 * | 18 | B(OH)2 | 20 | 120 | 24 | 5 |
| 13 | 13 + ZnI2 | I | 20 | 120 | 24 | 100 |
| 14 | 14 + ZnI2 | I | 20 | 120 | 24 | 100 |
| 15 | 15 + ZnI2 | I | 20 | 120 | 24 | 100 |
| 16 | 16 + ZnI2 | I | 20 | 120 | 24 | 43 |
| 17 | 17 + ZnI2 | I | 20 | 120 | 24 | 40 |
| 18 | 18 + ZnI2 | I | 20 | 120 | 24 | 40 |
| 19 | 13 + ZnI2 | B(OH)2 | 20 | 120 | 24 | 100 |
| 20 | 14 + ZnI2 | B(OH)2 | 20 | 120 | 24 | 100 |
| 21 | 15 + ZnI2 | B(OH)2 | 20 | 120 | 24 | 100 |
| 22 | 16 + ZnI2 | B(OH)2 | 20 | 120 | 24 | 48 |
| 23 | 17 + ZnI2 | B(OH)2 | 20 | 120 | 24 | 50 |
| 24 | 18 + ZnI2 | B(OH)2 | 20 | 120 | 24 | 45 |
| 25 | 13 + ZnI2 | I | 20 | 120 | 15 | 100 |
| 26 | 14 + ZnI2 | I | 20 | 120 | 15 | 100 |
| 27 | 15 + ZnI2 | I | 20 | 120 | 15 | 100 |
| 28 | 13 + ZnI2 | B(OH)2 | 20 | 120 | 15 | 100 |
| 19 | 14 + ZnI2 | B(OH)2 | 20 | 120 | 15 | 100 |
| 30 | 15 + ZnI2 | B(OH)2 | 20 | 120 | 15 | 100 |
| 31 | 13 + ZnI2 | I | 20 | 100 | 15 | 51 |
| 32 | 14 + ZnI2 | I | 20 | 100 | 15 | 54 |
| 33 | 15 + ZnI2 | I | 20 | 100 | 15 | 48 |
| 34 | 13 + ZnI2 | B(OH)2 | 20 | 100 | 15 | 55 |
| 35 | 14 + ZnI2 | B(OH)2 | 20 | 100 | 15 | 55 |
| 36 | 15 + ZnI2 | B(OH)2 | 20 | 100 | 15 | 59 |
| 37 | 13 + ZnI2 | I | 20 | 80 | 15 | 0 |
| 38 | 14 + ZnI2 | I | 20 | 80 | 15 | 0 |
| 39 | 15 + ZnI2 | I | 20 | 80 | 15 | 0 |
| 40 | 13 + ZnI2 | B(OH)2 | 20 | 80 | 15 | traces |
| 41 | 14 + ZnI2 | B(OH)2 | 20 | 80 | 15 | traces |
| 42 | 15 + ZnI2 | B(OH)2 | 20 | 80 | 15 | traces |
| 43 | 13 + ZnI2 | Br | 20 | 120 | 15 | 100 |
| Entry | Catalyst | mol% | T (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | 25 | 20 | 50 | 3 | 42 |
| 2 | 25 | 20 | 60 | 3 | 90 |
| 3 | 25 | 20 | 60 | 4 | 100 |
| 4 | 25 | 10 | 60 | 4 | 80 |
| 5 | 25 | 50 | 60 | 3 | 90 |
| 6 | 26 | 20 | 50 | 3 | 40 |
| 7 | 26 | 20 | 60 | 3 | 90 |
| 8 | 26 | 20 | 60 | 4 | 100 |
| 9 | 26 | 10 | 60 | 4 | 83 |
| 10 | 26 | 50 | 60 | 3 | 90 |
| 11 | 27 | 20 | 50 | 3 | 44 |
| 12 | 27 | 20 | 60 | 3 | 92 |
| 13 | 27 | 20 | 60 | 4 | 100 |
| 14 | 27 | 10 | 60 | 4 | 78 |
| 15 | 27 | 50 | 60 | 3 | 90 |
| 16 | 28 | 20 | 50 | 3 | 40 |
| 17 | 28 | 20 | 60 | 3 | 90 |
| 18 | 28 | 20 | 60 | 4 | 100 |
| 19 | 28 | 10 | 60 | 4 | 83 |
| 20 | 28 | 50 | 60 | 3 | 90 |
| 21 | 29 | 20 | 50 | 3 | 37 |
| 22 | 29 | 20 | 60 | 3 | 90 |
| 23 | 29 | 20 | 60 | 4 | 100 |
| 24 | 29 | 10 | 60 | 4 | 80 |
| 25 | 29 | 50 | 60 | 3 | 92 |
| 26 | 30 | 20 | 50 | 3 | 43 |
| 27 | 30 | 20 | 60 | 3 | 92 |
| 28 | 30 | 20 | 60 | 4 | 100 |
| 29 | 30 | 10 | 60 | 4 | 85 |
| 30 | 30 | 50 | 60 | 3 | 93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dysin, A.P.; Egorov, A.R.; Khubiev, O.; Golubev, R.; Kirichuk, A.A.; Khrustalev, V.N.; Lobanov, N.N.; Rubanik, V.V.; Tskhovrebov, A.G.; Kritchenkov, A.S. Novel Highly Efficient Green and Reusable Cu(II)/Chitosan-Based Catalysts for the Sonogashira, Buchwald, Aldol, and Dipolar Cycloaddition Reactions. Catalysts 2023, 13, 203. https://doi.org/10.3390/catal13010203
Dysin AP, Egorov AR, Khubiev O, Golubev R, Kirichuk AA, Khrustalev VN, Lobanov NN, Rubanik VV, Tskhovrebov AG, Kritchenkov AS. Novel Highly Efficient Green and Reusable Cu(II)/Chitosan-Based Catalysts for the Sonogashira, Buchwald, Aldol, and Dipolar Cycloaddition Reactions. Catalysts. 2023; 13(1):203. https://doi.org/10.3390/catal13010203
Chicago/Turabian StyleDysin, Artem P., Anton R. Egorov, Omar Khubiev, Roman Golubev, Anatoly A. Kirichuk, Victor N. Khrustalev, Nikolai N. Lobanov, Vasili V. Rubanik, Alexander G. Tskhovrebov, and Andreii S. Kritchenkov. 2023. "Novel Highly Efficient Green and Reusable Cu(II)/Chitosan-Based Catalysts for the Sonogashira, Buchwald, Aldol, and Dipolar Cycloaddition Reactions" Catalysts 13, no. 1: 203. https://doi.org/10.3390/catal13010203