
Oxidative N-Dealkylation of N,N-Dimethylanilines by Non-Heme Manganese Catalysts


Abstract
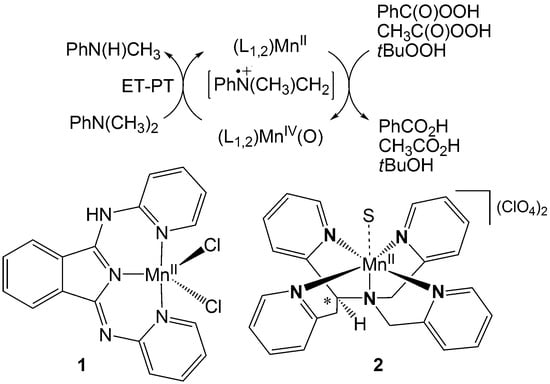
:
1. Introduction
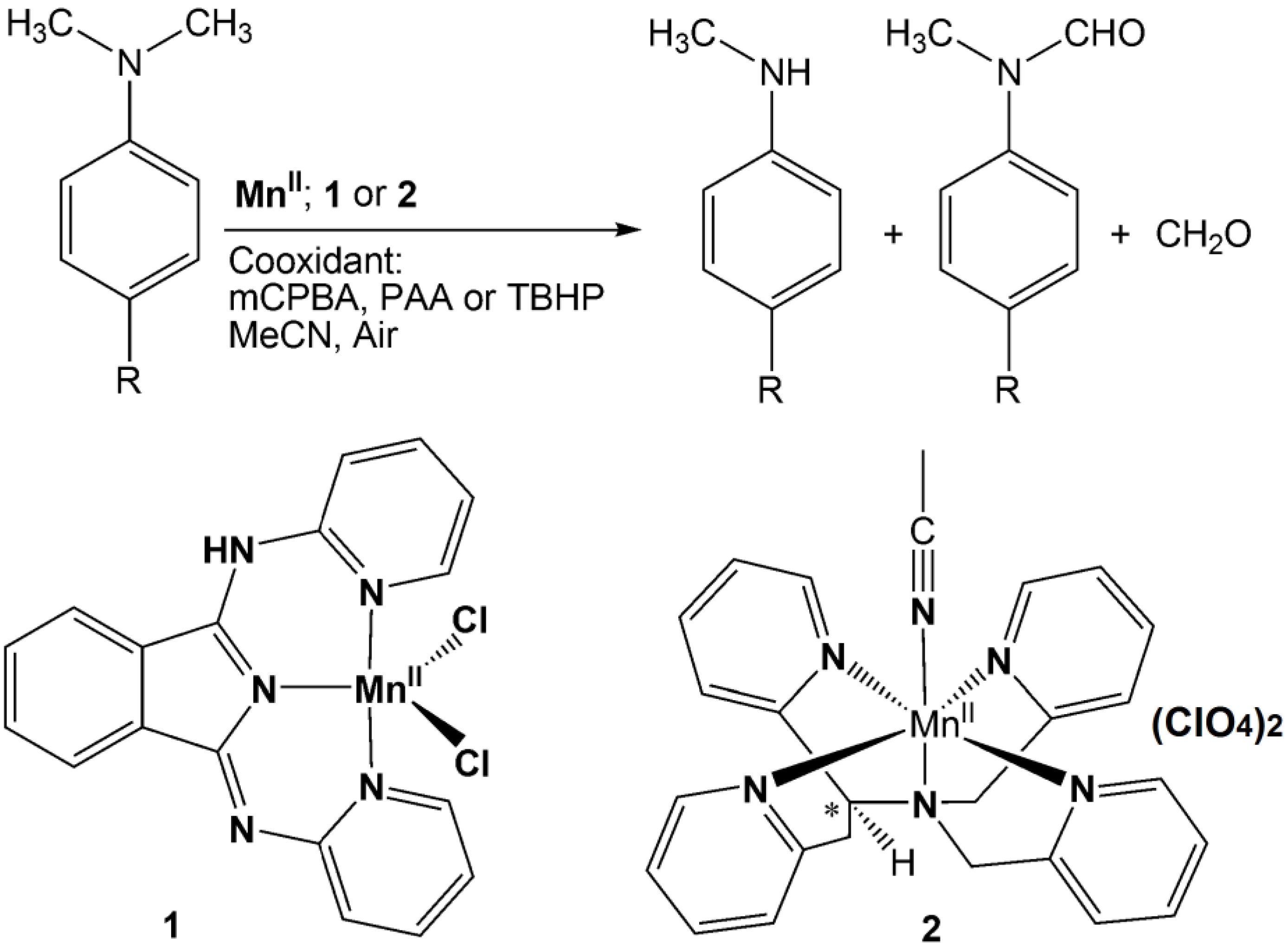
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Methods
3.2. Description of the Catalytic Oxidation of N,N-Dimethylaniline under Air (Ar)
3.3. Reaction of Oxomanganese(IV) Compex with N,N-Dimethylaniline under Air
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guengerich, F.P. Enzymatic oxidation of xenobiotic chemicals. Crit. Rev. Mol. Biol. 1990, 25, 97–153. [Google Scholar]
- Hawkins, B.K.; Dawson, J.H. Intrasubstituent isotope effect studies of oxidative N-demethylations catalyzed by secondary amine monooxygenase. Comparison to cytochrome P-450. J. Am. Chem. Soc. 1992, 114, 3547–3549. [Google Scholar] [CrossRef]
- Guengerich, F.P. Reactions and significance of cytochrome P-450 enzymes. J. Biol. Chem. 1991, 266, 10019–10022. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Macdonald, L.T. Chemical mechanisms of catalysis by cytochromes P-450: A unified view. Acc. Chem. Res. 1984, 17, 9–16. [Google Scholar] [CrossRef]
- Porter, T.D.; Coon, M.J. Cytochrome P-450. Multiplicity of isoforms, substrates, and catalytic and regulatory mechanisms. J. Biol. Chem. 1991, 266, 13469–13472. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, P.F. Mechanisms of cytochrome P450 and peroxides-catalyzed xenobiotic metabolism. FASEB J. 1992, 6, 686–694. [Google Scholar] [CrossRef]
- Hanzlik, R.P.; Ling, K.-H.J. Active site dynamics of xylene hydroxylation by cytochrome P-450 as revealed by kinetic deuterium isotope effects. J. Am. Chem. Soc. 1993, 115, 9363–9370. [Google Scholar] [CrossRef]
- Newcomb, M.; Shen, R.; Choi, S.-Y.; Toy, P.H.; Hollenberg, P.F.; Vaz, A.D.M.; Coon, J. Cytochrome P450-Catalyzed Hydroxylation of Mechanistic Probes that Distinguish between Radicals and Cations. Evidence for Cationic but Not for Radical Intermediates. J. Am. Chem. Soc. 2000, 122, 2677–2686. [Google Scholar]
- Toy, P.H.; Newcomb, M.; Hollenberg, P.F. Hypersensitive Mechanistic Probe Studies of Cytochrome P450-Catalyzed Hydroxylation Reactions. Impications for the Cationic Pathway. J. Am. Chem. Soc. 1998, 120, 7719–7729. [Google Scholar]
- Li, X.-X.; Wang, Y.; Zheng, Q.-C.; Zhang, H.-X. detoxification of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MTPT) by cytochrome P450 enzymes: A theoretical investigation. J. Inorg. Biochem. 2016, 154, 21–28. [Google Scholar] [CrossRef]
- Macdonald, L.T.; Zirvi, K.; Burka, L.T.; Peyman, P.; Guengerich, F.P. Mechanism of cytochrome P-450 inhibition by cyclopropylamines. J. Am. Chem. Soc. 1982, 104, 2050–2052. [Google Scholar] [CrossRef]
- Stearns, R.A.; Ortiz de Montillano, P.R. Cytochrome P-450 catalyzed oxidation of quadriclane. Evidence for a radical cation intermediate. J. Am. Chem. Soc. 1985, 107, 4081–4082. [Google Scholar] [CrossRef]
- Augusto, O.; Beilan, H.S.; Ortiz de Montillano, P.R. The catalytic mechanism of cytochrome P-450. Spin-trapping evidence for one-electron substrate oxidation. J. Biol. Chem. 1982, 257, 11288–11295. [Google Scholar] [CrossRef] [PubMed]
- Shono, T.; Toda, T.; Oshino, N. Electron transfer from nitrogen in microsomal oxidation of amine and amide. Simulation of microsomal oxidation by anodic oxidation. J. Am. Chem. Soc. 1982, 104, 2639–2641. [Google Scholar] [CrossRef]
- Hall, L.R.; Hanzlik, R.P. Kinetic deuterium isotope effects on the N-demethylation of tertiary amides by cytochrome P-450. J. Biol. Chem. 1990, 265, 12349–12355. [Google Scholar] [CrossRef]
- Guengerich, P.F.; Yun, C.-H.; Macdonald, T.L. Evidence for a 1-electron oxidation mechanism in N-dealkylation of N,N-dialkylanilines by cytochrome P450 2B1. Kinetic hydrogen isotope effects, linear free energy relationships, comparisons with horseradish peroxidase, and studies with oxygen surrogates. J. Biol. Chem. 1996, 271, 27321–27329. [Google Scholar] [CrossRef] [Green Version]
- Karki, S.B.; Dinnocenzo, J.P.; Jones, J.P.; Korzekwa, K.R. Mechanism of Oxidative Amine Dealkylation of Substituted N,N-Dimethylanilines by Cytochrome P-450: Application of Isotope Effect Profiles. J. Am. Chem. Soc. 1995, 117, 3657–3664. [Google Scholar] [CrossRef]
- Manchester, J.I.; Dinnocenzo, J.P.; Higgins, L.A.; Jones, J.P. A New Mechanistic Probe for Cytochrome P450: An Application of Isotope Effect Profiles. J. Am. Chem. Soc. 1997, 119, 5069–5070. [Google Scholar] [CrossRef]
- Griffin, B.W.; Ting, P.L. Mechanism of N-demethylation of aminopyrine by hydrogen peroxide catalyzed by horseradish peroxidase, metmyoglobin, and protohemin. Biochemistry 1978, 17, 2206–2211. [Google Scholar] [CrossRef]
- Van der Zee, J.; Duling, R.; Mason, R.P.; Eling, T.E. The oxidation of N-substituted aromatic amines by horseradish peroxidase. J. Biol. Chem. 1989, 264, 19828–19836. [Google Scholar] [CrossRef]
- Miwa, G.T.; Walsh, J.S.; Kedderis, G.L.; Hollenberg, P.F. The use of intramolecular isotope effects to distinguish between deprotonation and hydrogen atom abstraction mechanisms in cytochrome P-450- and peroxidase-catalyzed N-demethylation reactions. J. Biol. Chem. 1983, 258, 14445–14449. [Google Scholar] [CrossRef] [PubMed]
- Hagel, J.M.; Facchini, P.J. Biochemistry and occurance of O-demethylation in plant metabolism. Front. Physiol. 2010, 1, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.R.L.; Mortimer, D.N. Model systems for cytochrome P450-dependent monooxygenases. Part 5. Amine oxidation. Part 17. Oxidative N-dealkylation of tertiary amines by metalloporphyrin-catalysed model systems for cytochrome P450 monooxygenases. J. Chem. Soc., Perkin Trans. 1986, 2, 1743–1749. [Google Scholar] [CrossRef]
- Murata, S.; Miura, M.; Nomura, M. Oxidation of 3- or 4-substituted N,N-dimethylanilines with molecular oxygen in the presence of either ferric chloride or [Fe(salen)]OAc. J. Org. Chem. 1989, 54, 4700–4702. [Google Scholar] [CrossRef]
- Murata, S.; Miura, M.; Nomura, M. Iron-catalysed oxidation of N,N-dimethylaniline with molecular oxygen. J. Chem. Soc. Chem. Commun. 1989, 2, 116–118. [Google Scholar] [CrossRef]
- Narog, D.; Lechowicz, U.; Pietryga, T.; Sobkowiak, A. Iron(II, III)-catalyzed oxidative N-dealkylation of amines with dioxygen. J. Mol. Catal. A Chem. 2004, 212, 25–33. [Google Scholar] [CrossRef]
- Lakk-Bogáth, D.; Kripli, B.; Meena, B.I.; Speier, G.; Kaizer, J. Catalytic and stoichiometric oxidation of N,N-dimethylanilines mediated by nonheme oxoiron(IV) complex with tetrapyridyl ligand. Polyhedron 2019, 169, 169–175. [Google Scholar] [CrossRef]
- Meth-Cohn, O.; Stanforth, S.P. Comprehensive Organic Synthesis; Trost, B., Fleming, I., Eds.; Pergamon: Oxford, UK, 1991; Volume 4, p. 777. [Google Scholar]
- Downie, I.M.; Earle, M.J.; Heaney, H.; Shuhaibar, K.F. Vilsmeier formylation and glyoxylation reactions of nucleophilic aromatic compounds using pyrophosphoryl chloride. Tetrahedron 1993, 49, 4015–4034. [Google Scholar] [CrossRef]
- Barbieri, A.; De Gennaro, M.; Di Stefano, S.; Lanzalunga, O.; Lapi, A.; Mazzonna, M.; Olivoa, G.; Ticconia, B. Isotope effect profiles in the N-demethylation of N,N-dimethylanilines: A key to determine the pKa of nonheme Fe(III)-OH complexes. Chem. Commun. 2015, 51, 5032–5035. [Google Scholar]
- Nehru, K.; Seo, M.S.; Kim, J.; Nam, W. Oxidative N-Dealkylation Reactions by Oxoiron(IV) Complexes of Nonheme and Heme Ligands. Inorg. Chem. 2007, 46, 293–298. [Google Scholar] [CrossRef]
- Yoon, H.; Morimoto, Y.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Electron-transfer properties of a nonheme manganese(IV)-oxo complex acting as a stronger one-electron oxidant than the iron(IV)-oxo analogue. Chem. Commun. 2012, 48, 11187–11189. [Google Scholar] [CrossRef] [PubMed]
- Kaizer, J.; Csay, T.; Kővári, P.; Speier, G.; Párkányi, L. Catalase mimics of a manganese(II) complex: The effect of axial ligands and pH. J. Mol. Catal. Chem. 2008, 280, 203–209. [Google Scholar] [CrossRef]
- Kripli, B.; Garda, Z.; Sólyom, B.; Tircsó, G.; Kaizer, J. Formation, stability and catalase-like activity of mononuclear manganese(II) and oxomanganese(IV) complexes in protic and aprotic solvents. New J. Chem. 2020, 44, 5545–5555. [Google Scholar] [CrossRef]
- Kaizer, J.; Kripli, B.; Speier, G.; Párkányi, L. Synthesis, structure, and catalase-like activity of a novel manganese(II) complex: Dichloro[1,3-bis(2′-benzimidazolylimino)isoindoline]manganese(II). Polyhedron 2009, 28, 933–936. [Google Scholar] [CrossRef]
- Kaizer, J.; Baráth, G.; Speier, G.; Réglier, M.; Giorgi, M. Synthesis, structure and catalase mimics of novel homoleptic manganese(II) complexes of 1,3-bis(2′-pyridylimino)isoindoline, Mn(4R-ind)2 (R = H, Me). Inorg. Chem. Commun. 2007, 10, 292–294. [Google Scholar] [CrossRef]
- Kaizer, J.; Baráth, G.; Csonka, R.; Speier, G.; Korecz, L.; Rockenbauer, A.; Párkányi, L. Catechol oxidase and phenoxazinone synthase activity of a manganese(II) isoindoline complex. J. Inorg. Biochem. 2008, 102, 773–780. [Google Scholar] [CrossRef]
- Pap, J.S.; Kripli, B.; Váradi, T.; Giorgi, M.; Kaizer, J.; Speier, G. Comparison of the SOD-like activity of hexacoordinate Mn(II), Fe(II) and Ni(II) complexes having isoindoline-based ligands. J. Inorg. Biochem. 2011, 105, 911–918. [Google Scholar] [CrossRef]
- Meena, B.I.; Lakk-Bogáth, D.; Keszei, S.; Kaizer, J. Bleach catalysis in aqueous medium by iron(III)-isoindoline complexes and hydrogen peroxide. Comptes Rendus. Chim. 2021, 24, 351–360. [Google Scholar] [CrossRef]
- Meena, B.I.; Lakk-Bogáth, D.; Kaizer, J. Effect of redox potential on manganese-mediated benzylalcohol and sulfideoxidation. Comptes Rendus. Chim. 2021, 24, 281–290. [Google Scholar] [CrossRef]
- Csonka, R.; Speier, G.; Kaizer, J. Isoindoline-derived ligands and applications. RSC Adv. 2015, 5, 18401–18419. [Google Scholar] [CrossRef]
- Nam, W. Dioxygen Activation by Metalloenzymes and Models. Acc. Chem. Res. 2007, 40, 465. [Google Scholar] [CrossRef] [Green Version]
- Nam, W. High-valent Iron(IV)-Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions. Acc. Chem. Res. 2007, 40, 522–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lee, Y.-M.; Guo, M.; Fukuzumi, S.; Nam, W. Unprecedented Reactivities of Highly Reactive Manganese (III)–Iodosylarene Porphyrins in Oxidation Reactions. J. Am. Chem. Soc. 2020, 142, 19879–19884. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Corona, T.; Ray, K.; Nam, W. Heme and Nonheme High-Valent Iron and Manganese Oxo Cores in Biological and Abiological Oxidation Reactions. ACS Cent. Sci. 2019, 5, 13–28. [Google Scholar] [CrossRef]
- McDonald, A.R.; Que, L., Jr. High-valent nonheme iron-oxo complexes: Synthesis, structure, and spectroscopy. Coord. Chem. Rev. 2013, 257, 414–428. [Google Scholar] [CrossRef]
- Lakk-Bogáth, D.; Csonka, R.; Speier, G.; Reglier, M.; Simaan, A.J.; Naubron, J.V.; Giorgi, M.; Lazar, K.; Kaizer, J. Oxoiron(IV) Complex Derived from Chiral Pentadentate Ligand asN4Py. Inorg. Chem. 2016, 55, 10090–10093. [Google Scholar] [CrossRef]
- Kaizer, J.; Klinker, E.J.; Oh, N.Y.; Rohde, J.U.; Song, W.J.; Stubna, A.; Kim, J.; Munck, E.; Nam, W.; Que, L., Jr. Nonheme FeIVO Complexes That Can Oxidize the C-H Bonds of Cyclohexane at Room Temperature. J. Am. Chem. Soc. 2004, 126, 472–473. [Google Scholar] [CrossRef]
- Klinker, E.J.; Kaizer, J.; Brennessel, W.W.; Woodrum, N.L.; Cramer, C.J.; Que, L., Jr. Structures of Nonheme Oxoiron(IV) Complexes from X-ray Crystallography, NMR Spectroscopy, and DFT Calculations. Angew. Chem. Int. Ed. 2005, 44, 3690–3694. [Google Scholar]
- Meunier, B.; Sorokin, A. Oxidation of Pollutants Catalyzed by Metallophthalocyanines. Acc. Chem. Res. 1997, 30, 470–476. [Google Scholar] [CrossRef]
- Mangematin, S.; Sorokin, A.B. Synthesis and catalytic properties of a novel phthalocyanine covalently grafted onto silica. J. Porphyr. Phthalocyanines 2001, 5, 674–680. [Google Scholar] [CrossRef]
- Sorokin, A.; Séris, J.-L.; Meunier, B. Efficient oxidative dechlorination and aromatic ring cleavage of chlorinated phenols catalyzed by iron sulfophthalocyanine. Science 1995, 268, 1163–1166. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.; Meunier, B. Oxidative Degradation of Polychlorinated Phenols Catalyzed by Metallosulfophthalocyanines. Chem. Eur. J. 1996, 2, 1308–1317. [Google Scholar] [CrossRef]
- Sorokin, B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8191. [Google Scholar] [CrossRef]
- Chen, J.; Lee, Y.-M.; Davis, K.M.; Wu, X.; Seo, M.S.; Cho, K.-B.; Yoon, H.; Park, Y.J.; Fukuzumi, S.; Pushkar, Y.N.; et al. A Mononuclear Non-Heme Manganese(IV)-Oxo Complex Binding Redox-Inactive Metal Ions. J. Am. Chem. Soc. 2013, 135, 6388–6391. [Google Scholar] [CrossRef] [PubMed]
- Leto, D.F.; Ingram, R.; Day, V.W.; Jackson, T.A. Spectroscopic properties and reactivity of a mononuclear oxomanganese(IV) complex. Chem. Commun. 2013, 49, 5378–5380. [Google Scholar]
- Leto, D.F.; Massie, A.A.; Rice, D.B.; Jackson, T.A. Spectroscopic and Computational Investigations of a Mononuclear Manganese(IV)-Oxo Complex Reveal Electronic Structure Contributions to Reactivity. J. Am. Chem. Soc. 2016, 138, 15413–15424. [Google Scholar] [CrossRef]
- Massie, A.A.; Denler, M.C.; Cardoso, L.T.; Walker, A.N.; Hossain, M.K.; Day, V.W.; Nordlander, E.; Jackson, T.A. Equatorial Ligand Perturbations Influence the Reactivity of Manganese(IV)-oxo Complexes. Angew. Chem. Int. Ed. 2017, 56, 4178–4182. [Google Scholar] [CrossRef] [Green Version]
- Denler, M.C.; Massie, A.A.; Singh, R.; Stewart-Jones, E.; Sinha, A.; Day, V.W.; Nordlander, E.; Jackson, T.A. MnIV-Oxo complex of a bis(benzimidazolyl)-containing N5 ligand reveals different reactivity trends for MnIV-oxo than FeIV-oxo species. Dalton Trans. 2019, 48, 5007–5021. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
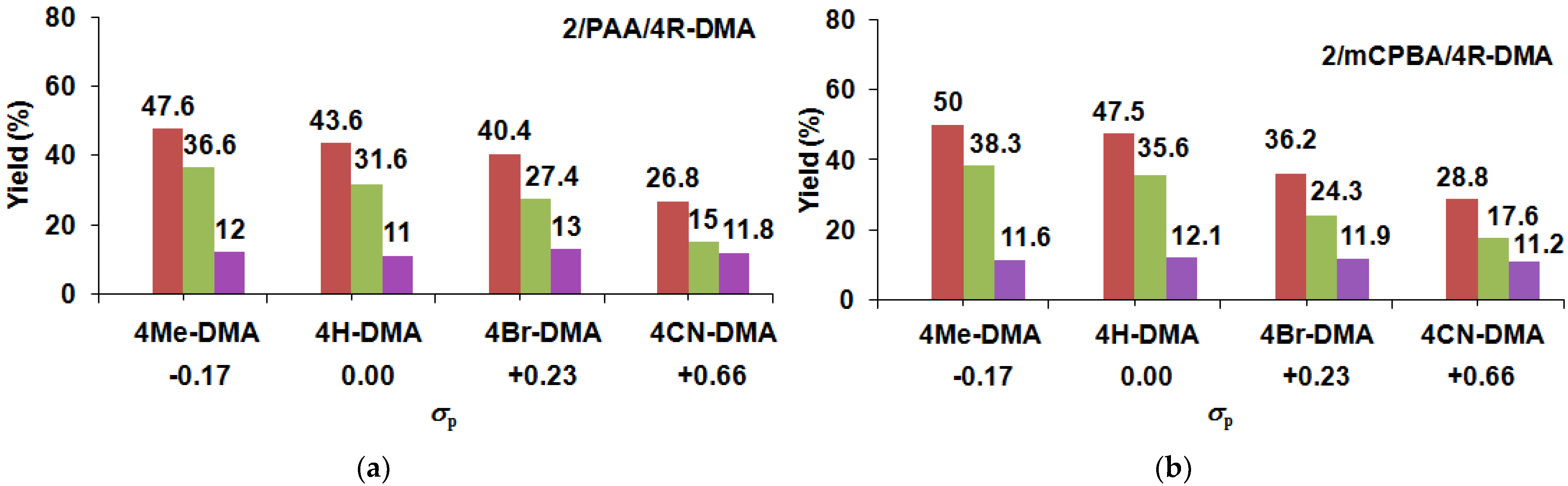
| Catalyst 1 | Co-Oxidant | Substrate 4R-DMA | Yield (%) 3 4R-MA | Yield (%) 3 4R-MFA | MA/MFA | ΣYield (%) 3 | TON 2 |
|---|---|---|---|---|---|---|---|
| Mn(ClO4)2 | PAA/Air | −H | 1.6 | 0.6 | 2.67 | 2.2 | 2.2 |
| Mn(OAc)2 | PAA/Air | −H | 9.9 | 4.3 | 2.30 | 14.2 | 14.2 |
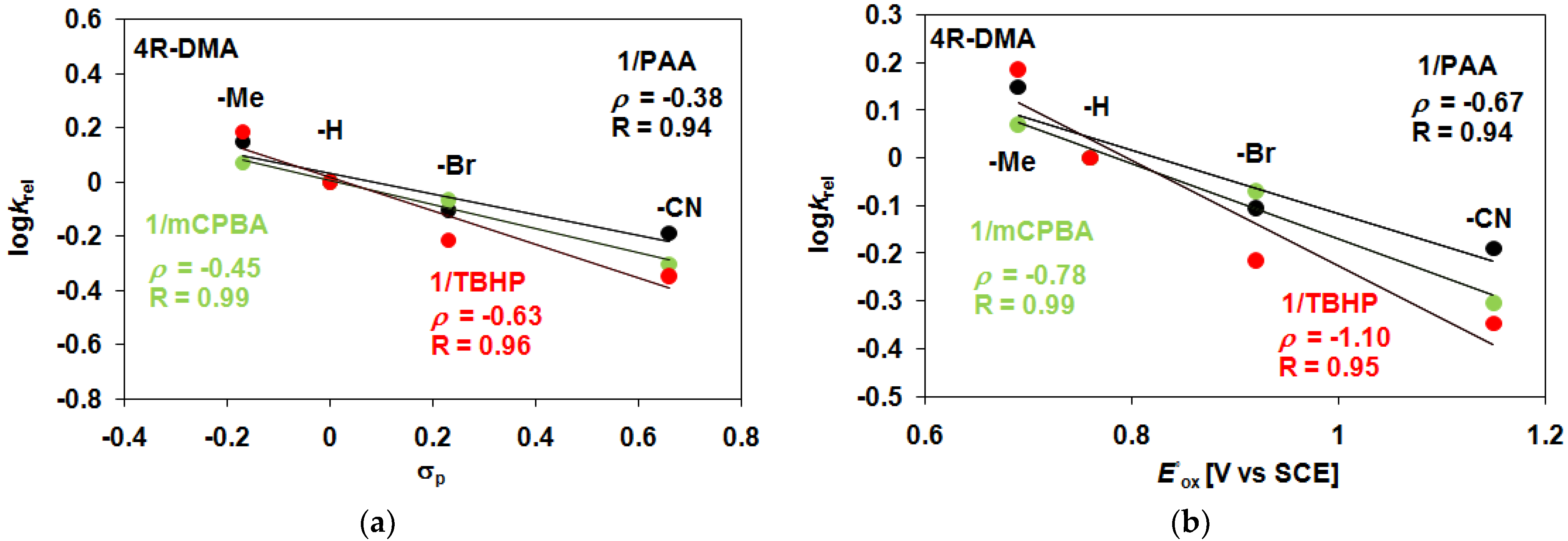
| 1 | PAA/Air | −H | 36.7 | 12.1 | 3.03 | 48.8 | 48.8 |
| 1 | PAA/Air | −Me | 48.3 | 11.7 | 4.13 | 60 | 60 |
| 1 | PAA/Air | −Br | 28.3 | 11.7 | 2.41 | 40 | 40 |
| 1 | PAA/Air | −CN | 23.3 | 11.1 | 2.09 | 34.4 | 34.4 |
| Mn(ClO4)2 | mCBPA/Air | −H | 1.9 | 0.86 | 2.21 | 2.76 | 2.76 |
| 1 | mCBPA/Air | −H | 40 | 11.2 | 3.57 | 51.2 | 51.2 |
| 1 | mCBPA/Air | −Me | 46 | 11 | 4.18 | 57 | 57 |
| 1 | mCBPA/Air | −Br | 34 | 11.8 | 2.88 | 45.8 | 45.8 |
| 1 | mCBPA/Air | −CN | 20 | 10 | 2 | 30 | 30 |
| Mn(ClO4)2 | TBHP/Air | −H | 0.62 | 0.09 | 6.6 | 0.71 | 0.71 |
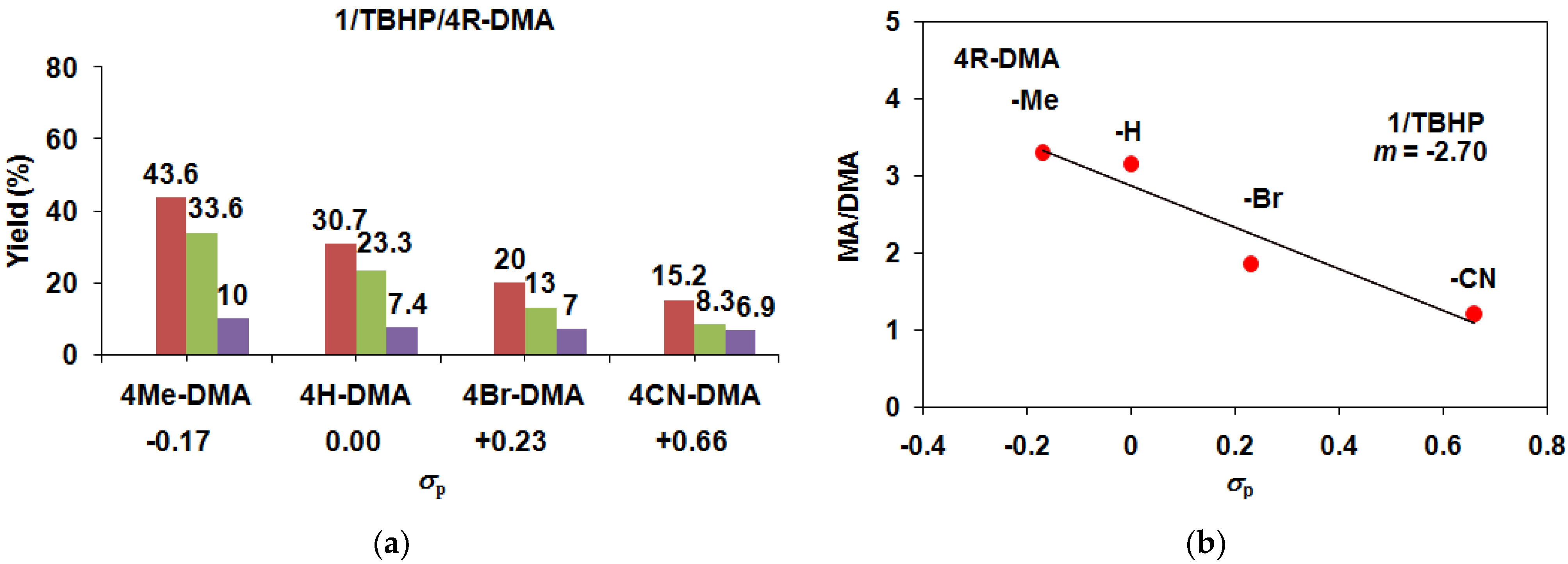
| 1 | TBHP/Air | −H | 23.3 | 7.4 | 3.15 | 30.7 | 30.7 |
| 1 | TBHP/Air | −Me | 33 | 10 | 3.30 | 43 | 43 |
| 1 | TBHP/Air | −Br | 13 | 7 | 1.86 | 20 | 20 |
| 1 | TBHP/Ar | −CN | 8.3 | 6.9 | 1.21 | 15.2 | 15.2 |
| Mn(ClO4)2 | PAA/Air | −H | 1.6 | 0.6 | 2.67 | 2.2 | 2.2 |
| Mn(OAc)2 | PAA/Air | −H | 9.9 | 4.3 | 2.30 | 14.2 | 14.2 |
| 2 | PAA/Air | −H | 31.6 | 12 | 2.64 | 43.6 | 43.6 |
| 2 | PAA/Ar | −H | 13.6 | - | - | 13.6 | 13.6 |
| 2 | PAA/Air | −Me | 36.6 | 11.0 | 3.33 | 47.6 | 47.6 |
| 2 | PAA/Air | −Br | 27.4 | 13.0 | 2.11 | 40.4 | 40.4 |
| 2 | PAA/Air | −CN | 15.0 | 11.8 | 1.27 | 26.8 | 26.8 |
| Mn(ClO4)2 | mCBPA/Air | −H | 1.9 | 0.86 | 2.21 | 2.79 | 2.8 |
| 2 | mCBPA/Air | −H | 35.6 | 12.1 | 2.94 | 47.7 | 47.7 |
| 2 | mCBPA/Ar | −H | 23.3 | - | - | 23.5 | 23.5 |
| 2 | mCBPA/Air | −Me | 38.3 | 11.6 | 3.30 | 50.0 | 50.0 |
| 2 | mCBPA/Air | −Br | 24.3 | 11.9 | 2.04 | 36.2 | 36.2 |
| 2 | mCBPA/Air | −CN | 17.6 | 11.2 | 1.58 | 28.8 | 28.8 |
| Mn(ClO4)2 | TBHP/Air | −H | 0.62 | 0.09 | 6.88 | 0.71 | 0.71 |
| 2 | TBHP/Air | −H | 15.8 | 6.8 | 2.32 | 22.6 | 22.6 |
| 2 | TBHP/Ar | −H | 9.9 | - | - | 9.9 | 9.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meena, B.I.; Lakk-Bogáth, D.; Török, P.; Kaizer, J. Oxidative N-Dealkylation of N,N-Dimethylanilines by Non-Heme Manganese Catalysts. Catalysts 2023, 13, 194. https://doi.org/10.3390/catal13010194
Meena BI, Lakk-Bogáth D, Török P, Kaizer J. Oxidative N-Dealkylation of N,N-Dimethylanilines by Non-Heme Manganese Catalysts. Catalysts. 2023; 13(1):194. https://doi.org/10.3390/catal13010194
Chicago/Turabian StyleMeena, Bashdar I., Dóra Lakk-Bogáth, Patrik Török, and József Kaizer. 2023. "Oxidative N-Dealkylation of N,N-Dimethylanilines by Non-Heme Manganese Catalysts" Catalysts 13, no. 1: 194. https://doi.org/10.3390/catal13010194