
Recent Advances in Catalysis for Methanation of CO2 from Biogas


Abstract
:1. Introduction
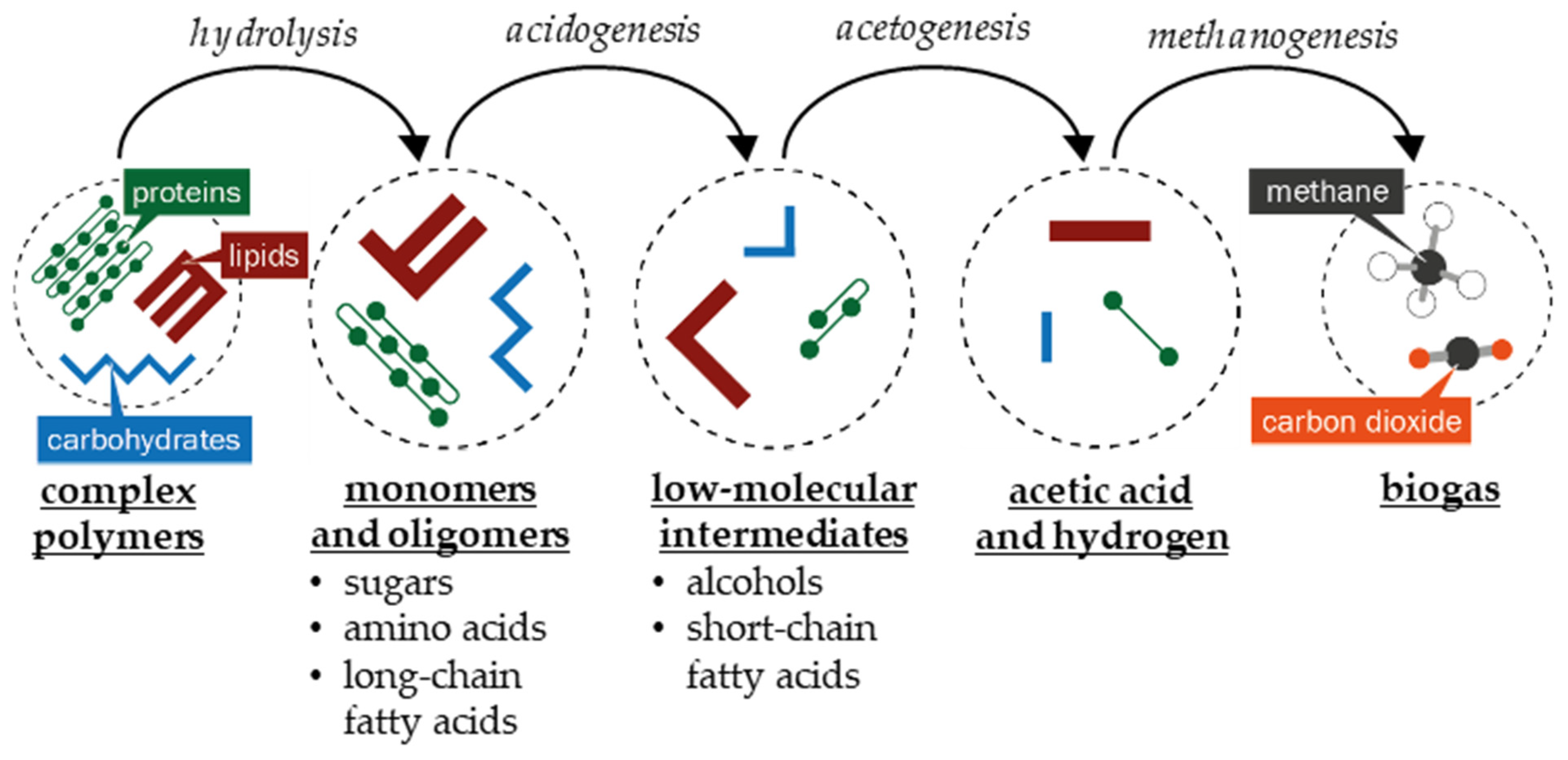
2. Biogas Composition
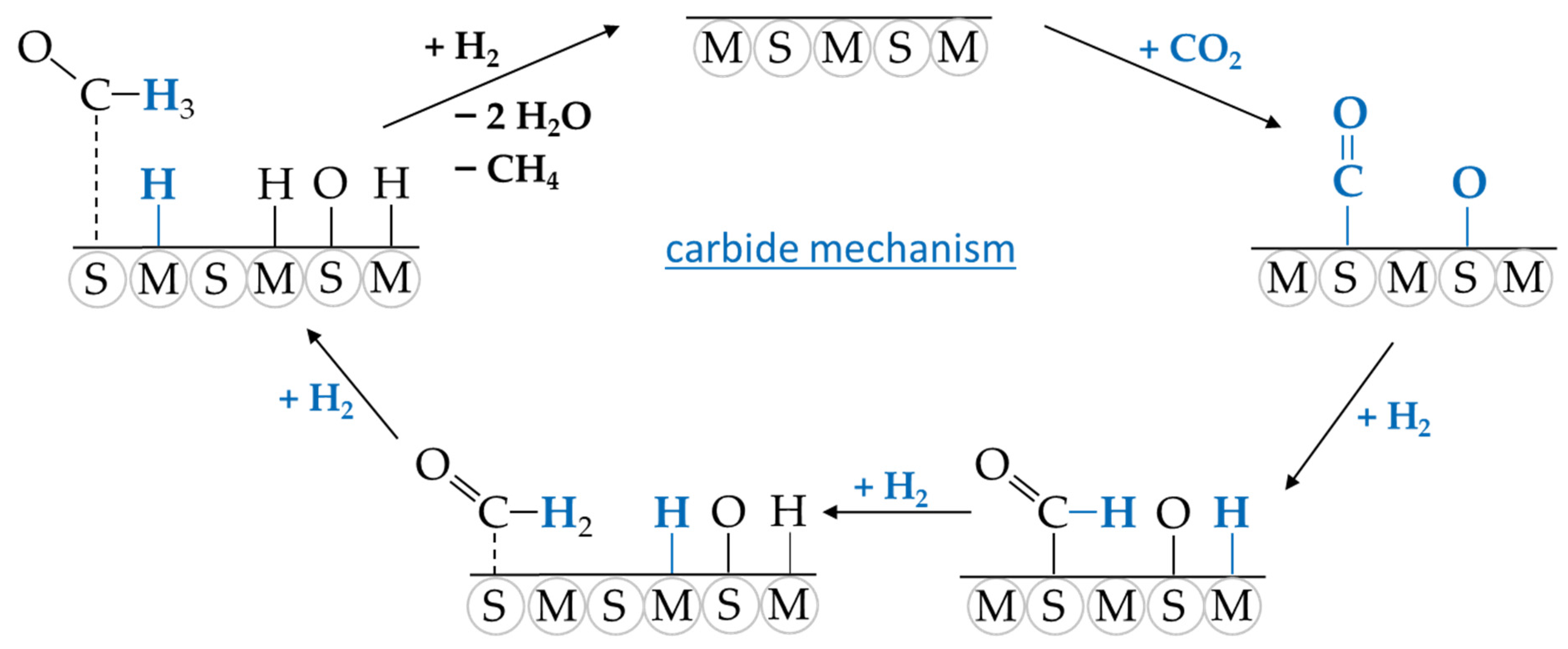
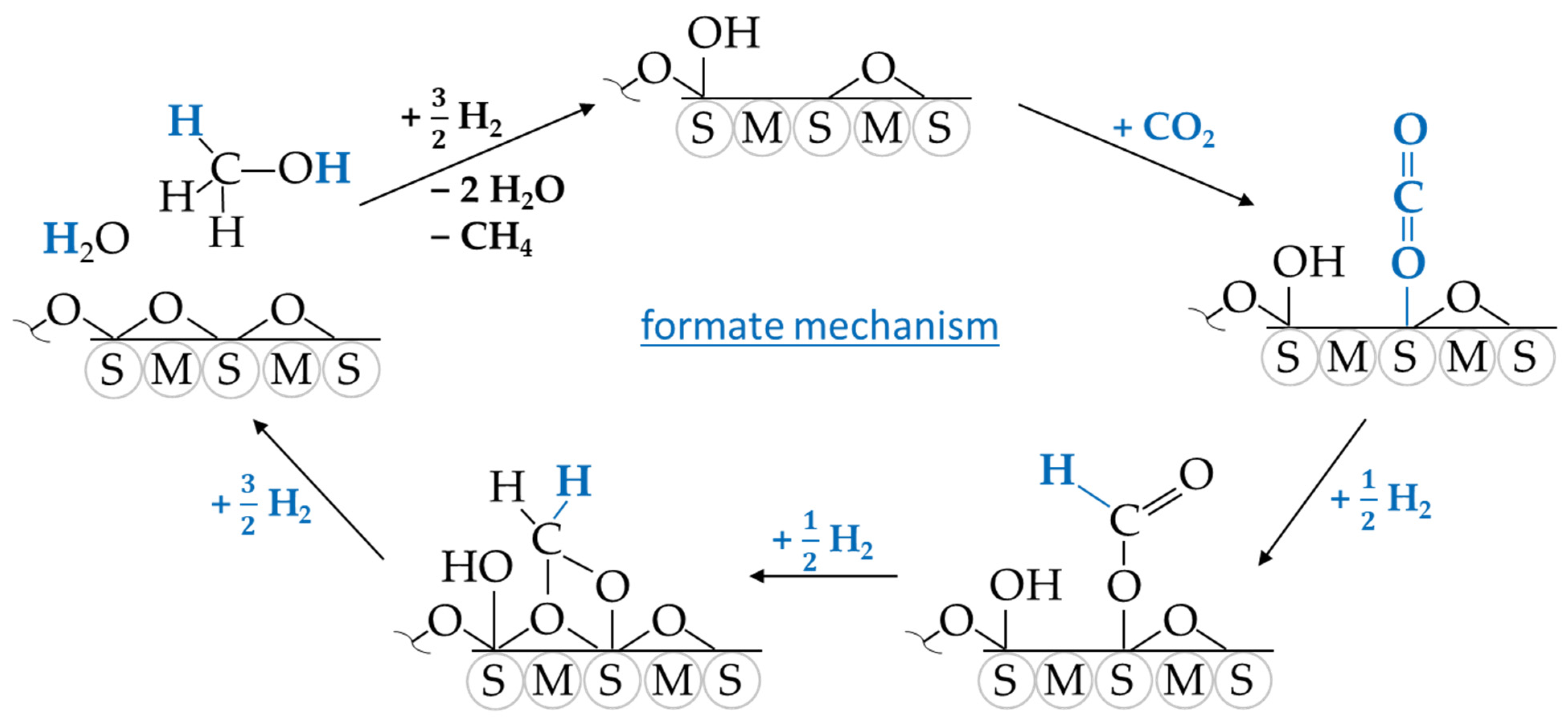
3. CO2 Methanation
4. Catalysts for Biogas Methanation
4.1. Active Metal
4.2. Support Material
4.3. Promoters
4.4. Deactivation of Catalysts
4.5. Increasing Sulfur Resistance
4.5.1. Active Metal
4.5.2. Support
4.5.3. Promoters
4.6. Catalyst Regeneration
5. Review Criteria and Catalyst Nomenclature
6. Influence of CH4 in the Feed Gas
6.1. Influence of CH4 on Ni/Al2O3
6.2. Influence of CH4 on Ni/CeO2
6.3. Influence of CH4 on Ni/CeO2-ZrO2
6.4. Further Studies
6.5. Discussion on the Effect of Methane in the Feed
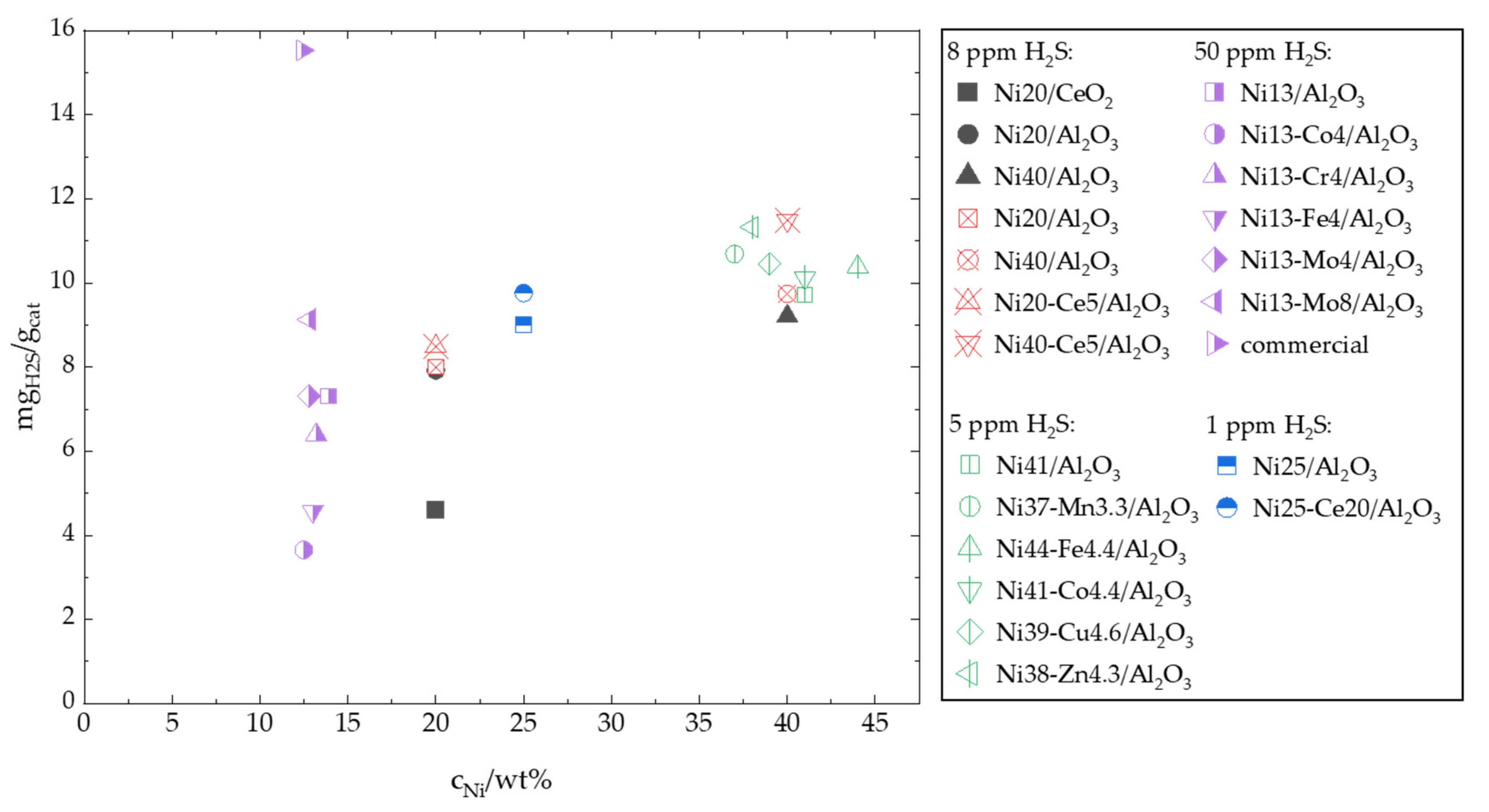
7. Influence of H2S in the Feed Gas
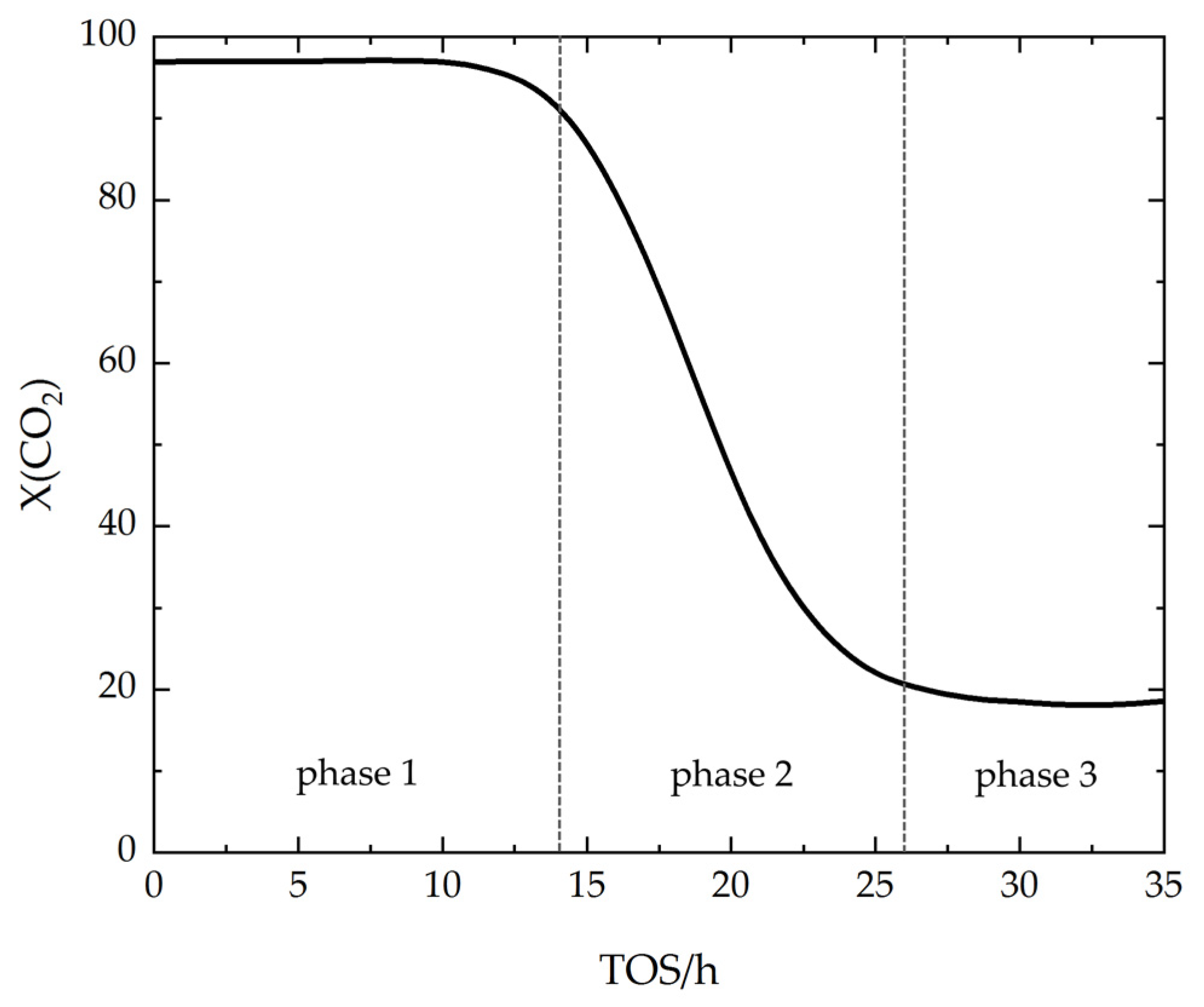
- Phase 1 usually starts with a constant and high CO2 conversion, which slightly decreases after a certain time on stream with H2S in the feed gas. The decline in CO2 conversion indicates that the catalyst can no longer reach equilibrium conversion due to H2S poisoning [41]. During H2S poisoning, deactivation of the catalyst particles proceeds along the catalyst bed with time on stream. CO2 conversion is an integral parameter across the entire catalyst bed, which only begins to decrease as the poisoning approaches the rear end of the catalyst bed [41,50].
- In phase 2, the CO2 conversion decreases rapidly within a short time. This phase represents the poisoning of the remaining active catalyst particles at the rear end of the catalyst bed [41].
- Finally, in phase 3, CO2 conversion reaches a minimum, which remains constant over time on stream. The constant low CO2 conversion indicates saturation of sulfur on the catalyst surface. Due to the formation of strong sulfur-metal bonds, the active sites are blocked and no longer available for CO2 adsorption [87]. Hence, CO2 adsorption is only possible on the catalyst support. Since the active metal is crucial to activate H2 for CH4 formation, CO2 methanation activity is lost and only CO is formed as a product.
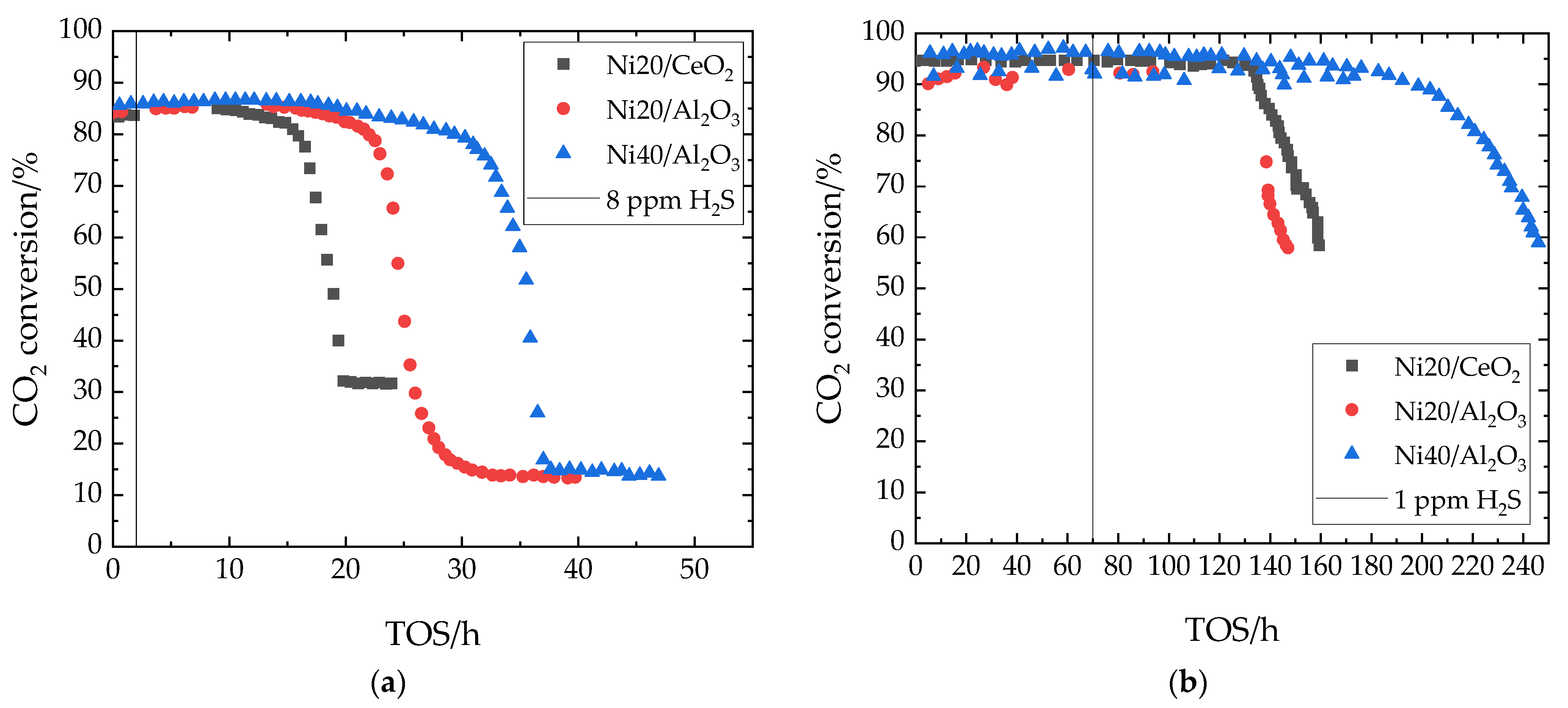
7.1. Sulfur Tolerance of Unpromoted Ni Catalysts
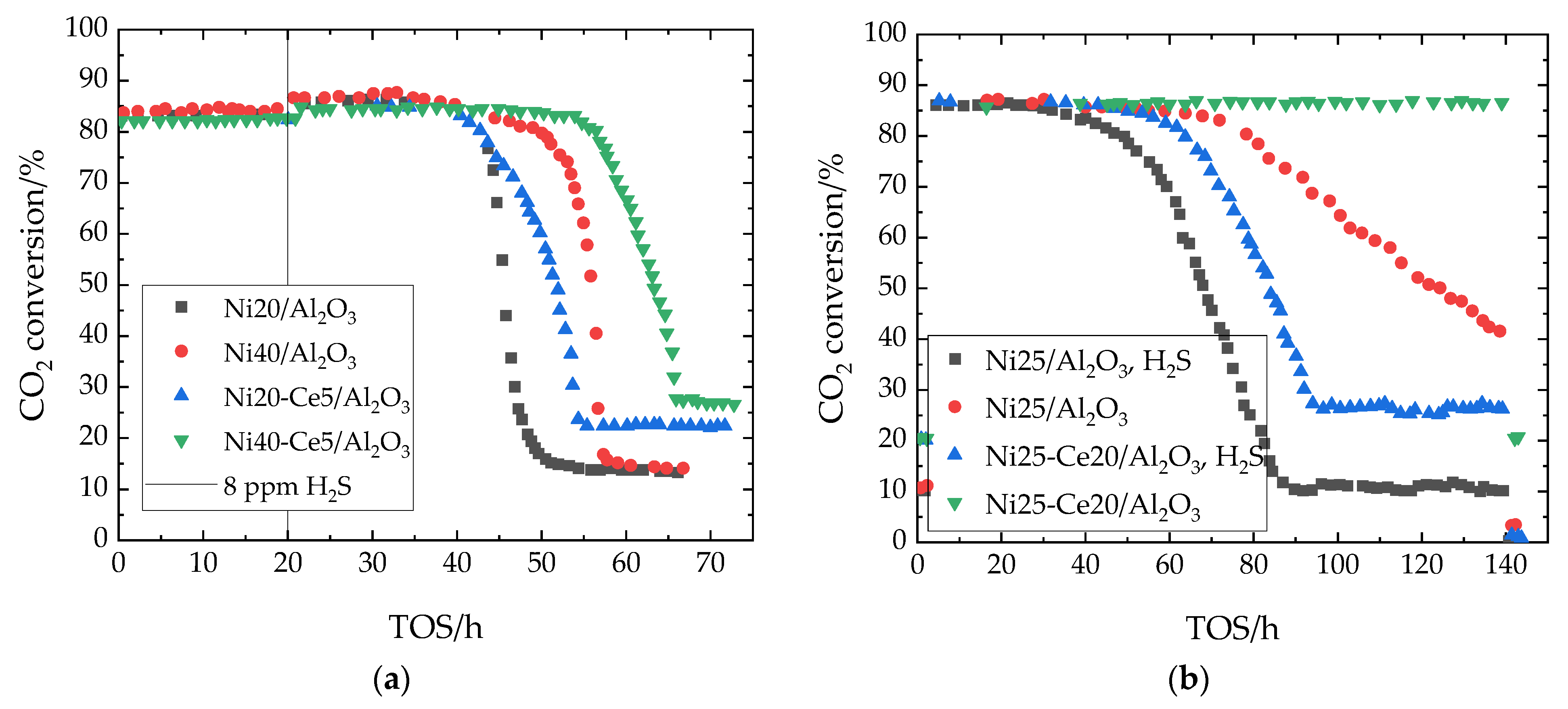
7.2. Sulfur Tolerance of Ni/Al2O3 with Ce Promoter
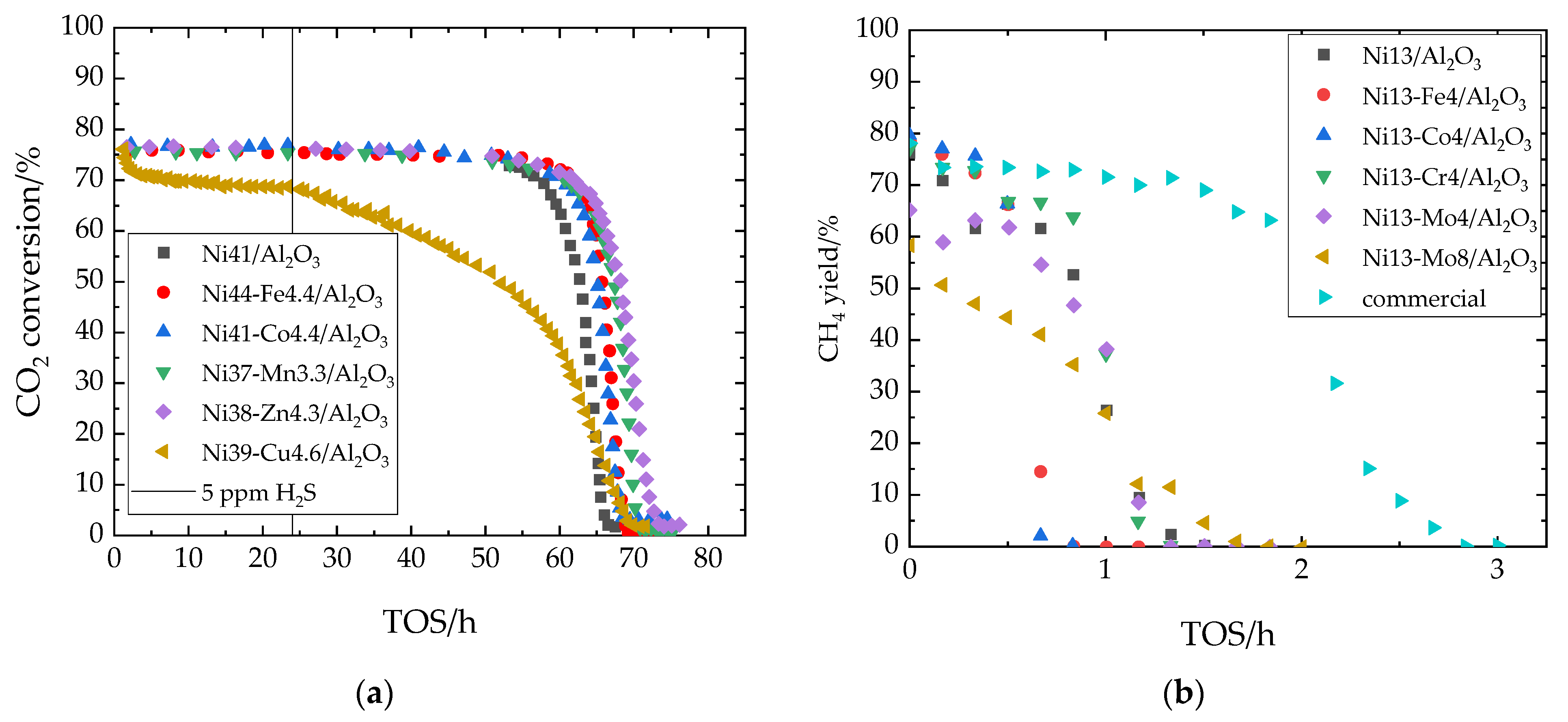
7.3. Sulfur Tolerance of Ni/Al2O3 with Transition Metal Promoters
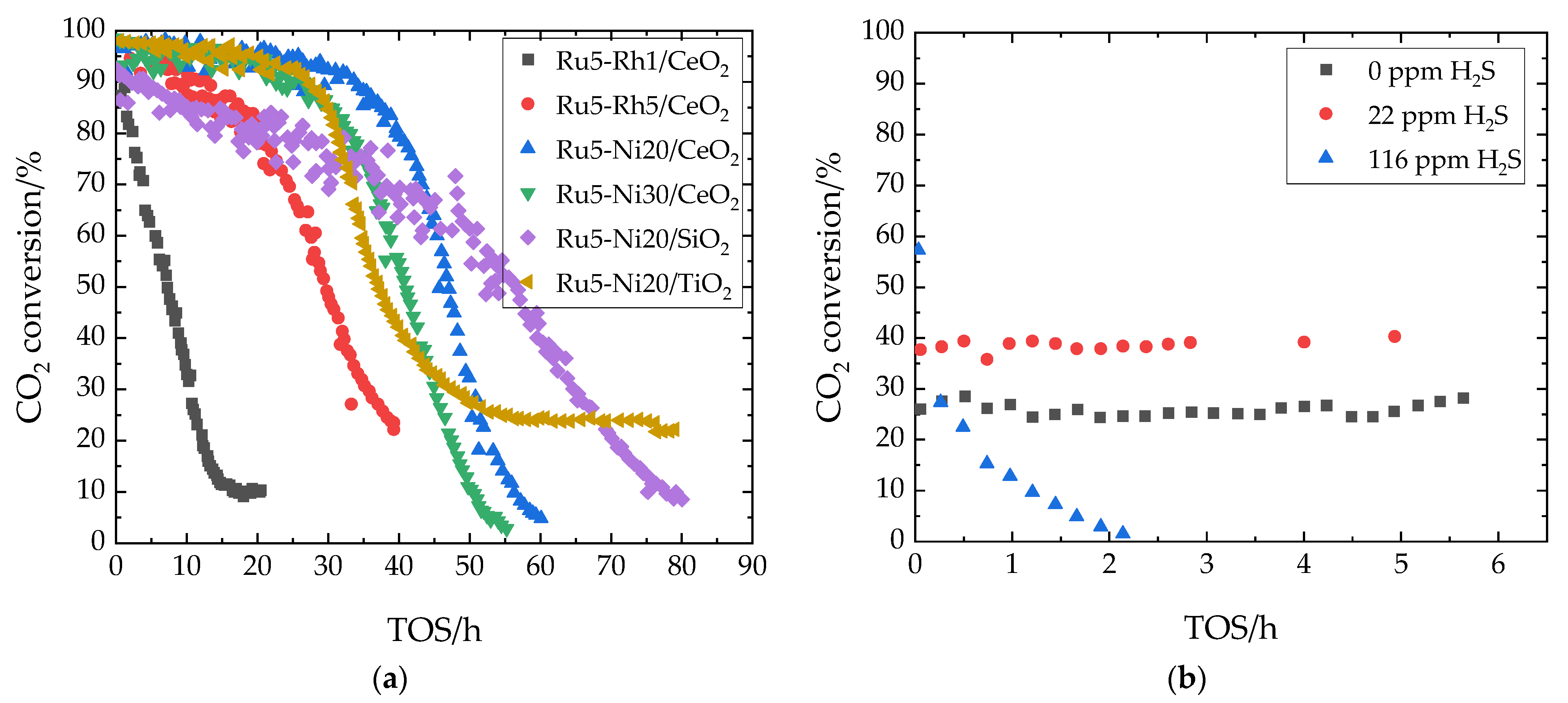
7.4. Sulfur Tolerance of Unpromoted and Promoted Noble Metals on Different Supports
7.5. Discussion on the Effect of H2S in the Feed
8. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
References
- Pastor-Pérez, L.; Patel, V.; Le Saché, E.; Reina, T.R. CO2 methanation in the presence of methane: Catalysts design and effect of methane concentration in the reaction mixture. J. Energy Inst. 2020, 93, 415–424. [Google Scholar] [CrossRef]
- Umweltbundesamt. Climate Protection Plan 2050 of the Federal Government (German, Klimaschutzplan 2050 der Bundesregierung: Diskussionsbeitrag des Umweltbundesamtes); Umweltbundesamt: Dessau-Roßlau, Germany, 2016.
- Billig, E.; Decker, M.; Benzinger, W.; Ketelsen, F.; Pfeifer, P.; Peters, R.; Stolten, D.; Thrän, D. Non-fossil CO2 recycling—The technical potential for the present and future utilization for fuels in Germany. J. CO2 Util. 2019, 30, 130–141. [Google Scholar] [CrossRef]
- Adnan, A.I.; Ong, M.Y.; Nomanbhay, S.; Chew, K.W.; Show, O.L. Technologies for Biogas Upgrading to Biomethane: A Review. Bioengineering 2019, 6, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.H.; Tan, C.S. A Review: CO2 Utilization. Aerosol Air Qual. Res. 2014, 14, 480–499. [Google Scholar] [CrossRef] [Green Version]
- Shahbaz, M.; AlNouss, A.; Ghiat, I.; Mckay, G.; Mackey, H.; Elkhalifa, S.; Al-Ansari, T. A comprehensive review of biomass based thermochemical conversion technologies integrated with CO2 capture and utilisation within BECCS networks. Resour. Conserv. Recycl. 2021, 173, 105734. [Google Scholar] [CrossRef]
- Gabrielli, P.; Gazzani, M.; Mazzotti, M. The Role of Carbon Capture and Utilization, Carbon Capture and Storage, and Biomass to Enable a Net-Zero-CO2 Emissions Chemical Industry. Ind. Eng. Chem. Res. 2020, 59, 7033–7045. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Li, C.E.; Prajitno, H.; Yoo, J.; Patel, J.; Yang, Y.X.; Lim, S. Recent trend in thermal catalytic low temperature CO2 methanation: A critical review. Catal. Today 2021, 368, 2–19. [Google Scholar] [CrossRef]
- Ghaib, K.; Ben-Fares, F.Z. Power-to-Methane: A state-of-the-art review. Renew. Sust. Energ. Rev. 2018, 81, 433–446. [Google Scholar] [CrossRef]
- Dannesboe, C.; Hansen, J.B.; Johannsen, I. Catalytic methanation of CO2 in biogas: Experimental results from a reactor at full scale. React. Chem. Eng. 2020, 5, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Ryckebosch, E.; Drouillon, M.; Vervaeren, H. Techniques for transformation of biogas to biomethane. Biomass Bioenergy 2011, 35, 1633–1645. [Google Scholar] [CrossRef]
- Villadsen, S.N.B.; Fosbøl, P.L.; Angelidaki, I.; Woodley, J.M.; Nielsen, L.P.; Møller, P. The Potential of Biogas; the Solution to Energy Storage. ChemSusChem 2019, 12, 2147–2153. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Cherevotan, A.; Peter, S.C. Thermochemical CO2 Hydrogenation to Single Carbon Products: Scientific and Technological Challenges. ACS Energy Lett. 2018, 3, 1938–1966. [Google Scholar] [CrossRef] [Green Version]
- Calbry-Muzyka, A.S.; Schildhauer, T.J. Direct Methanation of Biogas—Technical Challenges and Recent Progress. Front. Energy Res. 2020, 8, 356. [Google Scholar] [CrossRef]
- Kuo, J.; Dow, J. Biogas production from anaerobic digestion of food waste and relevant air quality implications. J. Air Waste Manag. Assoc. 2017, 67, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.A. Catalyst deactivation: Common causes. In Proceedings of the Nitrogen + Syngas 2013 International Conference, Berlin, Germany, 5–8 March 2013; pp. 213–232. [Google Scholar]
- Bartholomew, C.H. Mechanisms of Nickel Catalyst Poisoning. Stud. Surf. Sci. Catal. 1987, 34, 81–104. [Google Scholar]
- Coppola, G.; Papurello, D. Biogas Cleaning: Activated Carbon Regeneration for H2S Removal. Clean Technol. 2018, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Barelli, L.; Bidini, G.; de Arespacochaga, N.; Pérez, L.; Sisani, E. Biogas use in high temperature fuel cells: Enhancement of KOH-KI activated carbon performance toward H2S removal. Int. J. Hydrog. Energy 2017, 42, 10341–10353. [Google Scholar] [CrossRef]
- Guilera, J.; Soto, R.; Alarcón, A.; Andreu, T. Satisfactory catalyst stability in SNG production using real biogas despite sulfur poisoning evidences at different reactor zones. Fuel 2021, 306, 121682. [Google Scholar] [CrossRef]
- Balat, M.; Balat, H. Biogas as a Renewable Energy Source—A Review. Energ. Source Part A 2009, 31, 1280–1293. [Google Scholar] [CrossRef]
- Weiland, P. Biogas production: Current state and perspectives. Appl. Microbiol. Biotechnol. 2010, 85, 849–860. [Google Scholar] [CrossRef]
- Weinrich, S.; Nelles, M. Basics of Anaerobic Digestion: Biochemical Conversion and Process Modelling. DBFZ Rep. 2021, 40, 10–80. [Google Scholar]
- Kwaśny, J.; Balcerzak, W. Sorbents Used for Biogas Desulfurization in the Adsorption Process. Pol. J. Environ. Stud. 2016, 25, 37–43. [Google Scholar] [CrossRef]
- Ramsay, I.R.; Pullammanappallil, P.C. Protein degradation during anaerobic wastewater treatment: Derivation of stoichiometry. Biodegradation 2001, 12, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Köppel, W.; Schreck, H.; Lubenau, U.; Erler, R. Monitoring Biogas II; 2014; Available online: https://www.dvgw.de/medien/dvgw/forschung/berichte/g1_03_10.pdf (accessed on 8 March 2022).
- Bragança, I.; Sánchez-Soberón, F.; Pantuzza, G.F.; Alves, A.; Ratola, N. Impurities in biogas: Analytical strategies, occurrence, effects and removal technologies. Biomass Bioenergy 2020, 143, 105878. [Google Scholar] [CrossRef]
- Yang, L.C.; Ge, X.M.; Wan, C.X.; Yu, F.; Li, Y.B. Progress and perspectives in converting biogas to transportation fuels. Renew. Sust. Energ. Rev. 2014, 40, 1133–1152. [Google Scholar] [CrossRef]
- Schweigkofler, M.; Niessner, R. Determination of Siloxanes and VOC in Landfill Gas and Sewage Gas by Canister Sampling and GC-MS/AES Analysis. Environ. Sci. Technol. 1999, 33, 3680–3685. [Google Scholar] [CrossRef]
- Cabrera-Codony, A.; Montes-Morán, M.A.; Sánchez-Polo, M.; Martín, M.J.; Gonzalez-Olmos, R. Biogas upgrading: Optimal activated carbon properties for siloxane removal. Environ. Sci. Technol. 2014, 48, 7187–7195. [Google Scholar] [CrossRef]
- Li, W.H.; Wang, H.Z.; Jiang, X.; Zhu, J.; Liu, Z.M.; Guo, X.W.; Song, C.S. A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. RSC Adv. 2018, 8, 7651–7669. [Google Scholar] [CrossRef] [Green Version]
- Tsiotsias, A.I.; Charisiou, N.D.; Yentekakis, I.V.; Goula, M.A. The Role of Alkali and Alkaline Earth Metals in the CO2 Methanation Reaction and the Combined Capture and Methanation of CO2. Catalysts 2020, 10, 812. [Google Scholar] [CrossRef]
- Strucks, P.; Failing, L.; Kaluza, S. A Short Review on Ni-Catalyzed Methanation of CO2: Reaction Mechanism, Catalyst Deactivation, Dynamic Operation. Chem. Ing. Tech. 2021, 93, 1526–1536. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported Catalysts for CO2 Methanation: A Review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Wang, F.; He, S.; Chen, H.; Wang, B.; Zheng, L.R.; Wei, M.; Evans, D.G.; Duan, X. Active Site Dependent Reaction Mechanism over Ru/CeO2 Catalyst toward CO2 Methanation. J. Am. Chem. Soc. 2016, 138, 6298–6305. [Google Scholar] [CrossRef] [PubMed]
- Younas, M.; Loong Kong, L.; Bashir, M.J.K.; Nadeem, H.; Shehzad, A.; Sethupathi, S. Recent Advancements, Fundamental Challenges, and Opportunities in Catalytic Methanation of CO2. Energy Fuels 2016, 30, 8815–8831. [Google Scholar] [CrossRef]
- Wei, W.; Jinlong, G. Methanation of carbon dioxide: An overview. Front. Chem. Sci. Eng. 2011, 5, 2–10. [Google Scholar] [CrossRef]
- Rachow, F. Process Optimization for the Methanation of CO2 (German, Prozessoptimierung für die Methanisierung von CO2: Vom Labor zum Technikum). Ph.D. Dissertation, BTU Cottbus-Senftenberg, Cottbus, Germany, 2017. [Google Scholar]
- Stangeland, K.; Kalai, D.; Li, H.L.; Yu, Z.X. CO2 Methanation: The Effect of Catalysts and Reaction Conditions. Energy Procedia 2017, 105, 2022–2027. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, J.; Chu, M.; Yue, J.R.; Cui, Y.B.; Xu, G.W. Cooperation Between Active Metal and Basic Support in Ni-Based Catalyst for Low-Temperature CO2 Methanation. Catal. Lett. 2020, 150, 1418–1426. [Google Scholar] [CrossRef]
- Wolf, M.; Wong, L.H.; Schüler, C.; Hinrichsen, O. CO2 methanation on transition-metal-promoted Ni-Al catalysts: Sulfur poisoning and the role of CO2 adsorption capacity for catalyst activity. J. CO2 Util. 2020, 36, 276–287. [Google Scholar] [CrossRef]
- Gao, J.J.; Liu, Q.; Gu, F.N.; Liu, B.; Zhong, Z.Y.; Su, F.B. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar] [CrossRef]
- Swalus, C.; Jacquemin, M.; Poleunis, C.; Bertrand, P.; Ruiz, P. CO2 methanation on Rh/γ-Al2O3 catalyst at low temperature: “In situ” supply of hydrogen by Ni/activated carbon catalyst. Appl. Catal. B 2012, 125, 41–50. [Google Scholar] [CrossRef]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrog. Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Boggula, R.R.; Fischer, D.; Casaretto, R.; Born, J. Methanation potential: Suitable catalyst and optimized process conditions for upgrading biogas to reach gas grid requirements. Biomass Bioenergy 2020, 133, 105447. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Vannice, M.A. The Catalytic Synthesis of Hydrocarbons from Carbon Monoxide and Hydrogen. Catal. Rev. Sci. Eng. 1976, 14, 153–191. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Yentekakis, I.V.; Goula, M.A. Bimetallic Ni-Based Catalysts for CO2 Methanation: A Review. Nanomaterials 2020, 11, 28. [Google Scholar] [CrossRef]
- Walker, S.B.; Sun, D.; Kidon, D.; Siddiqui, A.; Kuner, A.; Fowler, M.; Simakov, D.S.A. Upgrading biogas produced at dairy farms into renewable natural gas by methanation. Int. J. Energy Res. 2018, 42, 1714–1728. [Google Scholar] [CrossRef]
- Wolf, M.; Schüler, C.; Hinrichsen, O. Sulfur poisoning of co-precipitated Ni–Al catalysts for the methanation of CO2. J. CO2 Util. 2019, 32, 80–91. [Google Scholar] [CrossRef]
- Mills, A.G.A.; Steffgen, F.W. Catalytic Methanation. Catal. Rev. 1974, 8, 159–210. [Google Scholar] [CrossRef]
- Ewald, S.; Kolbeck, M.; Kratky, T.; Wolf, M.; Hinrichsen, O. On the deactivation of Ni-Al catalysts in CO2 methanation. Appl. Catal. A-Gen. 2019, 570, 376–386. [Google Scholar] [CrossRef]
- Liu, C.J.; Ye, J.Y.; Jiang, J.J.; Pan, Y.X. Progresses in the Preparation of Coke Resistant Ni-based Catalyst for Steam and CO2 Reforming of Methane. ChemCatChem 2011, 3, 529–541. [Google Scholar] [CrossRef]
- Zhou, L.; Li, L.; Wei, N.; Li, J.; Basset, J.M. Effect of NiAl2O4 Formation on Ni/Al2O3 Stability during Dry Reforming of Methane. ChemCatChem 2015, 7, 2508–2516. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Pérez, L.; Saché, E.L.; Jones, C.; Gu, S.; Arellano-Garcia, H.; Reina, T.R. Synthetic natural gas production from CO2 over Ni-x/CeO2-ZrO2 (x = Fe, Co) catalysts: Influence of promoters and space velocity. Catal. Today 2018, 317, 108–113. [Google Scholar] [CrossRef]
- Atzori, L.; Rombi, E.; Meloni, D.; Sini, M.F.; Monaci, R.; Cutrufello, M.G. CO and CO2 Co-Methanation on Ni/CeO2-ZrO2 Soft-Templated Catalysts. Catalysts 2019, 9, 415. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Razzaq, R.; Li, C.; Muhmmad, Y.; Zhang, S. Catalytic Methanation of Carbon Dioxide by Active Oxygen Material CexZr1−xO2 Supported Ni-Co Bimetallic Nanocatalysts. AIChE J. 2013, 59, 2567–2576. [Google Scholar] [CrossRef]
- Sakpal, T.; Lefferts, L. Structure-dependent activity of CeO2 supported Ru catalysts for CO2 methanation. J. Catal. 2018, 367, 171–180. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, H.M.; Park, J.N. Bifunctional Mechanism of CO2 Methanation on Pd-MgO/SiO2 Catalyst: Independent Roles of MgO and Pd on CO2 Methanation. J. Phys. Chem. C 2010, 114, 7128–7131. [Google Scholar] [CrossRef]
- Gac, W.; Zawadzki, W.; Rotko, M.; Słowik, G.; Greluk, M. CO2 Methanation in the Presence of Ce-Promoted Alumina Supported Nickel Catalysts: H2S Deactivation Studies. Top. Catal. 2019, 62, 524–534. [Google Scholar] [CrossRef] [Green Version]
- Chein, R.Y.; Wang, C.C. Experimental Study on CO2 Methanation over Ni/Al2O3, Ru/Al2O3, and Ru-Ni/Al2O3 Catalysts. Catalysts 2020, 10, 1112. [Google Scholar] [CrossRef]
- Alarcón, A.; Guilera, J.; Soto, R.; Andreu, T. Higher tolerance to sulfur poisoning in CO2 methanation by the presence of CeO2. Appl. Catal. B 2020, 263, 118346. [Google Scholar] [CrossRef]
- Argyle, M.; Bartholomew, C. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Mateos, D.; Barrio, V.L.; Requies, J.M.; Cambra, J.F. A study of deactivation by H2S and regeneration of a Ni catalyst supported on Al2O3, during methanation of CO2. Effect of the promoters Co, Cr, Fe and Mo. RSC Adv. 2020, 10, 16551–16564. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Hrbek, J. Interaction of Sulfur with Well-Defined Metal and Oxide Surfaces: Unraveling the Mysteries behind Catalyst Poisoning and Desulfurization. Acc. Chem. Res. 1999, 32, 719–728. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R.; Pedersen, K. Sulfur poisoning of Boudouard and methanation reactions on nickel catalysts. J. Catal. 1979, 59, 395–404. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, C.; Li, Y.; Song, C.; Bolin, T.B. Sulfur poisoning mechanism of steam reforming catalysts: An X-ray absorption near edge structure (XANES) spectroscopy study. Phys. Chem. Chem. Phys. 2010, 12, 5502. [Google Scholar] [CrossRef] [PubMed]
- Freund, H.-J.; Goodman, D.W. Handbook of Heterogeneous Catalysis, 2nd ed.; Weitkamp, J., Ed.; Wiley VCH: Weinheim, Germany, 2008. [Google Scholar]
- Neuberg, S.; Pennemann, H.; Shanmugam, V.; Thiermann, R.; Zapf, R.; Gac, W.; Greluk, M.; Zawadzki, W.; Kolb, G. CO2 Methanation in Microstructured Reactors—Catalyst Development and Process Design. Chem. Eng. Technol. 2019, 42, 2076–2084. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Weatherbee, G.D.; Jarvi, G.A. Sulfur Poisoning of Nickel Methanation Catalysts: I. In situ Deactivation by H2S of Nickel and Nickel Bimetallics. J. Catal. 1979, 60, 257–269. [Google Scholar] [CrossRef]
- Yan, X.L.; Liu, Y.; Zhao, B.R.; Wang, Y.; Liu, C.J. Enhanced sulfur resistance of Ni/SiO2 catalyst for methanation via the plasma decomposition of nickel precursor. Phys. Chem. Chem. Phys. 2013, 15, 12132–12138. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, W.J.; Chu, W. Effect of glow discharge plasma treatment on the performance of Ni/SiO2 catalyst in CO2 methanation. J. Fuel Chem. Technol. 2013, 41, 96–101. [Google Scholar] [CrossRef]
- Yan, X.L.; Liu, Y.; Zhao, B.R.; Wang, Z.; Wang, Y.; Liu, C.J. Methanation over Ni/SiO2: Effect of the catalyst preparation methodologies. Int. J. Hydrog. Energy 2013, 38, 2283–2291. [Google Scholar] [CrossRef]
- Yuan, C.; Yao, N.; Wang, X.; Wang, J.; Lv, D.; Li, X. The SiO2 supported bimetallic Ni–Ru particles: A good sulfur-tolerant catalyst for methanation reaction. Chem. Eng. J. 2015, 260, 1–10. [Google Scholar] [CrossRef]
- Lee, K.; Song, C.; Janik, M.J. Density functional theory study of sulfur tolerance of CO adsorption and dissociation on Rh–Ni binary metals. Appl. Catal. A-Gen. 2010, 389, 122–130. [Google Scholar] [CrossRef]
- Rangan, M.; Yung, M.M.; Medlin, J.W. Experimental and computational investigations of sulfur-resistant bimetallic catalysts for reforming of biomass gasification products. J. Catal. 2011, 282, 249–257. [Google Scholar] [CrossRef]
- Shafiq, I.; Shafique, S.; Akhter, P.; Yang, W.; Hussain, M. Recent developments in alumina supported hydrodesulfurization catalysts for the production of sulfur-free refinery products: A technical review. Catal. Rev. 2022, 64, 1–86. [Google Scholar] [CrossRef]
- Wang, B.; Ding, G.; Shang, Y.; Lv, J.; Wang, H.; Wang, E.; Li, Z.; Ma, X.; Qin, S.; Sun, Q. Effects of MoO3 loading and calcination temperature on the activity of the sulphur-resistant methanation catalyst MoO3/γ-Al2O3. Appl. Catal. A-Gen. 2012, 431–432, 144–150. [Google Scholar] [CrossRef]
- Wang, B.; Yao, Y.; Jiang, M.; Li, Z.; Ma, X.; Qin, S.; Sun, Q. Effect of cobalt and its adding sequence on the catalytic performance of MoO3/Al2O3 toward sulfur-resistant methanation. J. Energy Chem. 2014, 23, 35–42. [Google Scholar] [CrossRef]
- Zaman, S.; Smith, K.J. A Review of Molybdenum Catalysts for Synthesis Gas Conversion to Alcohols: Catalysts, Mechanisms and Kinetics. Catal. Rev. Sci. Eng. 2012, 54, 41–132. [Google Scholar] [CrossRef]
- Kim, M.Y.; Ha, S.B.; Koh, D.J.; Byun, C.; Park, E.D. CO methanation over supported Mo catalysts in the presence of H2S. Catal. Commun. 2013, 35, 68–71. [Google Scholar] [CrossRef]
- Struis, R.P.W.J.; Schildhauer, T.J.; Czekaj, I.; Janousch, M.; Biollaz, S.M.A.; Ludwig, C. Sulphur poisoning of Ni catalysts in the SNG production from biomass: A TPO/XPS/XAS study. Appl. Catal. A-Gen. 2009, 362, 121–128. [Google Scholar] [CrossRef]
- Mullins, D.R. The surface chemistry of cerium oxide. Surf. Sci. Rep. 2015, 70, 42–85. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Gao, J.; Zhang, M.; Li, H.; Gu, F.; Xu, G.; Zhong, Z.; Su, F. Highly active and stable Ni/γ-Al2O3 catalysts selectively deposited with CeO2 for CO methanation. RSC Adv. 2014, 4, 16094–16103. [Google Scholar] [CrossRef]
- Guilera, J.; del Valle, J.; Alarcón, A.; Díaz, J.A.; Andreu, T. Metal-oxide promoted Ni/Al2O3 as CO2 methanation micro-size catalysts. J. CO2 Util. 2019, 30, 11–17. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Charojrochkul, S.; Kim-Lohsoontorn, P.; Assabumrungrat, S. Role and advantages of H2S in catalytic steam reforming over nanoscale CeO2-based catalysts. J. Catal. 2010, 276, 6–15. [Google Scholar] [CrossRef]
- Fitzharris, W.D.; Katzer, J.R.; Manogue, W.H. Sulfur Deactivation of Nickel Methanation Catalyst. J. Catal. 1982, 76, 369–384. [Google Scholar] [CrossRef]
- Hinnemann, B.; Nørskov, J.K.; Topsøe, H. A density functional study of the chemical differences between Type I and Type II MoS2-based structures in hydrotreating catalysts. J. Phys. Chem. 2005, 109, 2245–2253. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wang, B.; Yao, Y.; Wang, H.; Li, Z.; Ma, X.; Qin, S.; Sun, Q. The role of the distribution of Ce species on MoO3/CeO2–Al2O3 catalysts in sulfur-resistant methanation. Catal. Commun. 2013, 35, 32–35. [Google Scholar] [CrossRef]
- Diaz, A.; Odriozola, J.A.; Montes, M. Influence of alkali additives on activity and toxicity of H2S and tiophene over a Ni/SiO2 catalyst. Appl. Catal. A-Gen. 1998, 166, 163–172. [Google Scholar] [CrossRef]
- Alexander, A.M.; Hargreaves, J.S.J. Alternative catalytic materials: Carbides, nitrides, phosphides and amorphous boron alloys. Chem. Soc. Rev. 2010, 39, 4388–4401. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Agrawal, P.K.; Katzer, J.R. Sulfur Poisoning of Metals. Adv. Catal. 1982, 135–242. [Google Scholar] [CrossRef]
- Aguinaga, A.; Montes, M. Regeneration of a nickel/silica catalyst poisoned by thiophene. Appl. Catal. A-Gen. 1992, 90, 131–144. [Google Scholar] [CrossRef]
- Zhang, X.; Witte, J.; Schildhauer, T.; Bauer, C. Life cycle assessment of power-to-gas with biogas as the carbon source. Sustain. Energy Fuels 2020, 4, 1427–1436. [Google Scholar] [CrossRef]
- Zhuang, Y.; Simakov, D.S.A. Single-Pass Conversion of CO2/CH4 Mixtures over the Low-Loading Ru/γ-Al2O3 for Direct Biogas Upgrading into Renewable Natural Gas. Energy Fuels 2021, 35, 10062–10074. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Stangeland, K.; Kalai, D.Y.; Li, H.; Yu, Z. Active and stable Ni based catalysts and processes for biogas upgrading: The effect of temperature and initial methane concentration on CO2 methanation. Appl. Energy 2018, 227, 206–212. [Google Scholar] [CrossRef]
- Han, D.; Kim, Y.; Byun, H.; Cho, W.; Baek, Y. CO2 Methanation of Biogas Over 20 wt% Ni-Mg-Al Catalyst: On the Effect of N2, CH4, and O2 on CO2 conversion Rate. Catalysts 2020, 10, 1201. [Google Scholar] [CrossRef]
- Gac, W.; Zawadzki, W.; Rotko, M.; Greluk, M.; Słowik, G.; Pennemann, H.; Neuberg, S.; Zapf, R.; Kolb, G. Direct Conversion of Carbon Dioxide to Methane over Ceria- and Alumina-Supported Nickel Catalysts for Biogas Valorization. ChemPlusChem 2021, 86, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Jürgensen, L.; Ehimen, E.A.; Born, J.; Holm-Nielsen, J.B. Dynamic biogas upgrading based on the Sabatier process: Thermodynamic and dynamic process simulation. Bioresour. Technol. 2015, 178, 323–329. [Google Scholar] [CrossRef]
- Szailer, T.; Novák, É.; Oszkó, A.; Erdőhelyi, A. Effect of H2S on the hydrogenation of carbon dioxide over supported Rh catalysts. Top. Catal. 2007, 46, 79–86. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Unit | Agricultural Waste 1 | Residual Waste 2 | Bio Waste 3 | Sewage Sludge 4 |
|---|---|---|---|---|---|
| CH4 | vol% | 52–57 | 59–64 | 58–61 | 63–65 |
| CO2 | vol% | 36–48 | 33–38 | 39–41 | 35–37 |
| O2 | vol% | <2 | <0.5 | <0.6 | <0.2 |
| N2 | vol% | 0–4.5 | 0–2.2 | 1.4–1.6 | 0.2 |
| H2S | mg/m3 | 10–560 | 5–320 | 390–860 | 5–20 |
| R-SH | mg/m3 | 0–5 | 1–8 | 3–5 | 1–2 |
| NH3 | mg/m3 | 0–12 | 2–15 | <2 | <1 |
| H2 | ppmv | 10–500 | 50–390 | 100–190 | 120–350 |
| BTEX | mg/m3 | <2 | <90 | <3 | <39 |
| Si | mg/m3 | <2 | <2 | <2 | 4–15 |
| Promoter | Effect | Source |
|---|---|---|
| Fe |
| [1,48] |
| Co |
| [1,48] |
| Ru |
| [48] |
| Mg |
| [2,44,45,59] |
| Mn |
| [1] |
| Ce |
| [42,60,61,62] |
| Zr |
| [39] |
| Mo |
| [63] |
| Mechanism | Type | Description |
|---|---|---|
| Poisoning | Chemical | Species adsorb strongly on catalytic sites and block them for reaction |
| Fouling | Mechanical | Physical deposition of species on catalytic surface and in pores |
| Thermal degradation and sintering | Thermal Thermal/chemical | Thermally induced loss of catalytic surface area, support area, and active phase support reactions |
| Vapor formation | Chemical | Loss of catalyst compounds by reaction with gas |
| Vapor-solid and solid-solid reactions | Chemical | Reaction of catalyst compounds among each other or with gas leading to deactivation |
| Attrition/crushing | Mechanical | Loss of active material due to abrasion and crushing |
| Reaction Conditions | Catalyst | T/°C | CH4/CO2 | H2/CO2/CH4/N2/vol% 1 | X (CO2) | S (CH4) | Ref. |
|---|---|---|---|---|---|---|---|
| Table 5 | Ni20/Al2O3 | 350 | 0/100 50/50 67/33 | 57/14/00/29 57/14/14/14 57/14/29/00 | 71.5% 70.8% 70.6% | 99.7% 99.5% 99.4% | [97] |
| Table 5 | Ni20-Ru0.5/Al2O3 | 350 | 0/100 67/33 | 57/14/00/29 57/14/29/00 | 82% 2 81% 2 | >99% >99% | [97] |
| Table 6 | Ni20-Mg3/Al2O3 | 400 | 0/100 40/60 50/50 65/35 | 80/20/00/00 71/18/12/00 67/17/17/00 58/15/27/00 | 74% 2 67% 64% 54% | 96% 2 97% 2 97% 2 97% 2 | [98] |
| Table 7 3 | Ni20/Al2O3 | 350 | 0/100 50/50 | 62/15/00/23 5 62/15/15/08 5 | 88.5% 82% 2 | 100% 79% 2 | [99] |
| Table 7 3 | Ni40/Al2O3 | 350 | 0/100 50/50 | 62/15/00/23 5 62/15/15/08 5 | 91.4% 81% 2 | 100% 79% 2 | [99] |
| Table 7 3 | Ni20/CeO2 | 350 | 0/100 50/50 | 62/15/00/23 5 62/15/15/08 5 | 91.7% 81% 2 | 100% 75% 2 | [99] |
| Table 8 4 | Ni20/Al2O3 | 350 | 50/50 | 62/15/15/08 5 | 93.7% | 100% | [99] |
| Table 8 4 | Ni40/Al2O3 | 350 | 50/50 | 62/15/15/08 5 | 95.7% | 100% | [99] |
| Table 8 4 | Ni20/CeO2 | 350 | 50/50 | 62/15/15/08 5 | 94.6% | 100% | [99] |
| Table 9 | Ni15-Co3/ CeO2-ZrO2 | 350 | 0/100 21/79 36/64 47/53 | 80/20/00/00 76/19/05/00 72/18/10/00 68/17/15/00 | 71% 2 72% 74% 78% | 98% 2 97% 98% 99% | [1] |
| Catalysts | Ni20/Al2O3 Ni20-Ru0.5/Al2O3 |
| Activation conditions | 600 °C, 6 h, H2/N2 |
| T | 350 °C |
| p | 1 bar |
| H2/CO2 | 4 |
| WHSV | 56,000 mL gcat−1 h−1 |
| CH4,in | 0, 14, 29 vol% |
| Catalyst | Ni20-Mg3/Al2O3 |
| Activation conditions | 700 °C, 4 h, H2/N2 |
| T | 400 °C |
| p | not specified |
| H2/CO2 | 4 |
| GHSV | 30,000 h−1 |
| CH4,in | 0, 12, 17, 27 vol% |
| Catalysts | Ni20/Al2O3 Ni40/Al2O3 |
| Activation conditions | 600 °C, 2 h, H2 |
| T | 350 °C, 600 °C |
| p | 2 bar |
| H2/CO2 | 4 |
| WHSV | 30 L gcat−1 h−1 |
| CH4,in | 0, 15.4 vol% |
| Catalysts | Ni20/Al2O3 Ni40/Al2O3 |
| Activation conditions | 500 °C, 2 h, H2 |
| T | 350 °C |
| p | 12.5 bar |
| H2/CO2 | 4 |
| WHSV | 45 L gcat−1 h−1 |
| CH4,in | 15.4 vol% |
| Catalyst | Ni15-Co3/CeO2-ZrO2 |
| Activation conditions | 450 °C, 1 h, H2/N2 |
| T | 200–450 °C |
| p | 1 bar |
| H2/CO2 | 4 |
| WHSV | 12,000 mL g−1 h−1 |
| CH4,in | 0, 5, 10, 15 vol% |
| Reduction T | 400 °C, 600 °C |
| T | 350–600 °C |
| p | 1–3.8 bar |
| WHSV | 90,000–420,000 mL g−1 h−1 |
| X(CO2), 0 vol% CH4 | X(CO2), 14 vol% CH4 | X(CO2), 29 vol% CH4 | |
|---|---|---|---|
| 1 bar | 85.0% | 82.9% | 81.5% |
| 2 bar | 88.3% | 86.6% | 85.6% |
| 12.5 bar | 94.0% | 93.2% | 92.7% |
| Catalysts | Ni20/CeO2 Ni20/Al2O3 Ni40/Al2O3 |
| Activation conditions | 600 °C, 2 h, H2 |
| T | 450 °C |
| p | 2 bar |
| H2/CO2 | 4 |
| WHSV | not specified |
| H2Sin | 8 ppm |
| Catalysts | Ni20/CeO2 Ni20/Al2O3 Ni40/Al2O3 |
| Activation conditions | 500 °C, 2 h, H2 |
| T | 350 °C |
| p | 12.5 bar |
| H2/CO2 | 4 |
| WHSV | 45 L gcat−1 h−1 |
| H2Sin | 1 ppm |
| CH4,in | 15.4 vol% |
| Catalysts | Ni20/Al2O3 Ni40/Al2O3 Ni20-Ce5/Al2O3 Ni40-Ce5/Al2O3 |
| Activation conditions | 600 °C, 2 h, H2 |
| T | 475 °C |
| p | 1.9 bar |
| H2/CO2 | 4 |
| WHSV | 4.6 LCO2 h−1 g−1 |
| H2Sin | 8 ppm |
| Catalysts | Ni25/Al2O3 Ni25-Ce20/Al2O3 |
| Activation conditions | 500 °C, 3 h, H2 |
| T | 250 °C, 500 °C |
| p | 5 bar |
| H2/CO2 | 4 |
| GHSV | 40,000 mL g−1 h−1 |
| H2Sin | 1–5 ppm |
| Catalysts | Ni41/Al2O3 Ni37-Mn3.3/Al2O3 Ni44-Fe4.4/Al2O3 Ni41-Co4.4/Al2O3 Ni39-Cu4.6/Al2O3 Ni38-Zn4.3/Al2O3 |
| Activation conditions | 450 °C, 4 h, H2/Ar |
| T | 400 °C |
| p | 1 bar |
| H2/CO2 | 4 |
| GHSV | not specified |
| H2Sin | 5 ppm |
| Catalysts | Ni13/Al2O3 Ni13-Co4/Al2O3 Ni13-Cr4/Al2O3 Ni13-Fe4/Al2O3 Ni13-Mo4/Al2O3 Ni13-Mo8/Al2O3 |
| Activation conditions | 400 °C, 4 h, H2/N2 |
| T | 300–500 °C |
| p | 10 bar |
| H2/CO2 | 4 |
| WHSV | 33.5 gfeed gcat−1 h−1 |
| H2Sin | 50 ppm |
| Catalysts | Ru5/CeO2 Ru5-Rh1/CeO2 Ru5-Rh5/CeO2 Ru5-Ni20/CeO2 Ru5-Ni30/CeO2 Ru5-Ni20/SiO2 Ru5-Ni20/TiO2 |
| Activation conditions | 500 °C, 2 h, H2 |
| T | 375 °C |
| p | 12.5 bar |
| H2/CO2 | 4 |
| WHSV | 40 L gcat−1 h−1 |
| H2Sin | 1 ppm |
| Catalysts | Rh/TiO2 Ru/TiO2 Pd/TiO2 Rh/Al2O3 Rh/SiO2 Rh/CeO2 Rh/ZrO2 Rh/MgO |
| Activation conditions | 400 °C, 1 h |
| T | 275 °C |
| p | not specified |
| H2/CO2 | 4 |
| GHSV | not specified |
| H2Sin | 22 ppm, 116 ppm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nieß, S.; Armbruster, U.; Dietrich, S.; Klemm, M. Recent Advances in Catalysis for Methanation of CO2 from Biogas. Catalysts 2022, 12, 374. https://doi.org/10.3390/catal12040374
Nieß S, Armbruster U, Dietrich S, Klemm M. Recent Advances in Catalysis for Methanation of CO2 from Biogas. Catalysts. 2022; 12(4):374. https://doi.org/10.3390/catal12040374
Chicago/Turabian StyleNieß, Selina, Udo Armbruster, Sebastian Dietrich, and Marco Klemm. 2022. "Recent Advances in Catalysis for Methanation of CO2 from Biogas" Catalysts 12, no. 4: 374. https://doi.org/10.3390/catal12040374