
Preparation Strategy Using Pre-Nucleation Coupled with In Situ Reduction for a High-Performance Catalyst towards Selective Hydrogen Production from Formic Acid

 , and
, and
Abstract
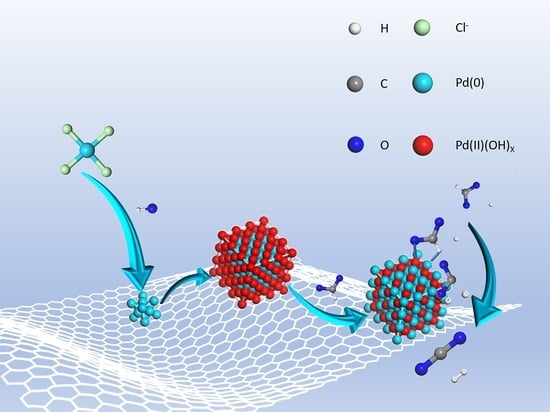
:
1. Introduction
2. Results and Discussion
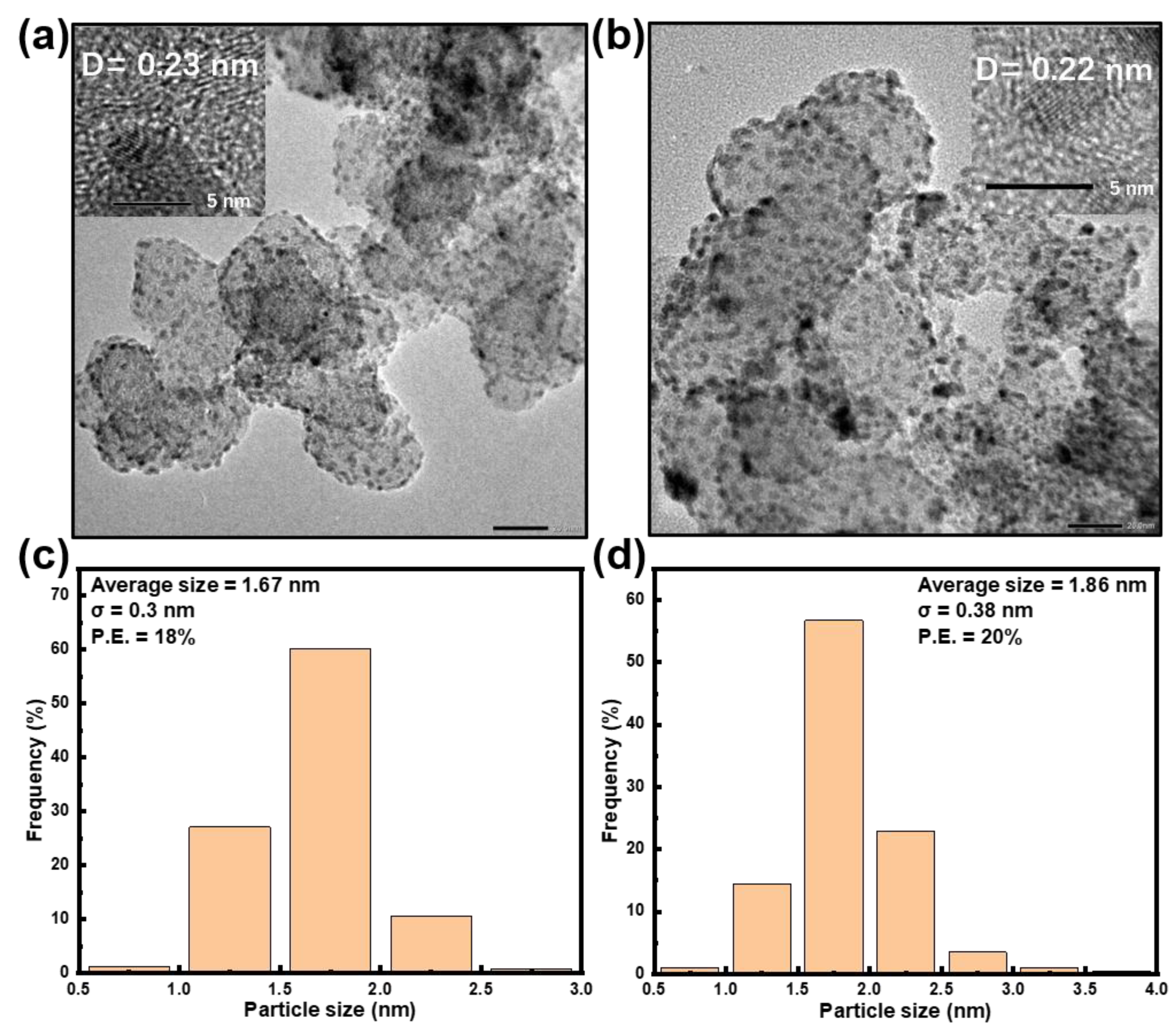
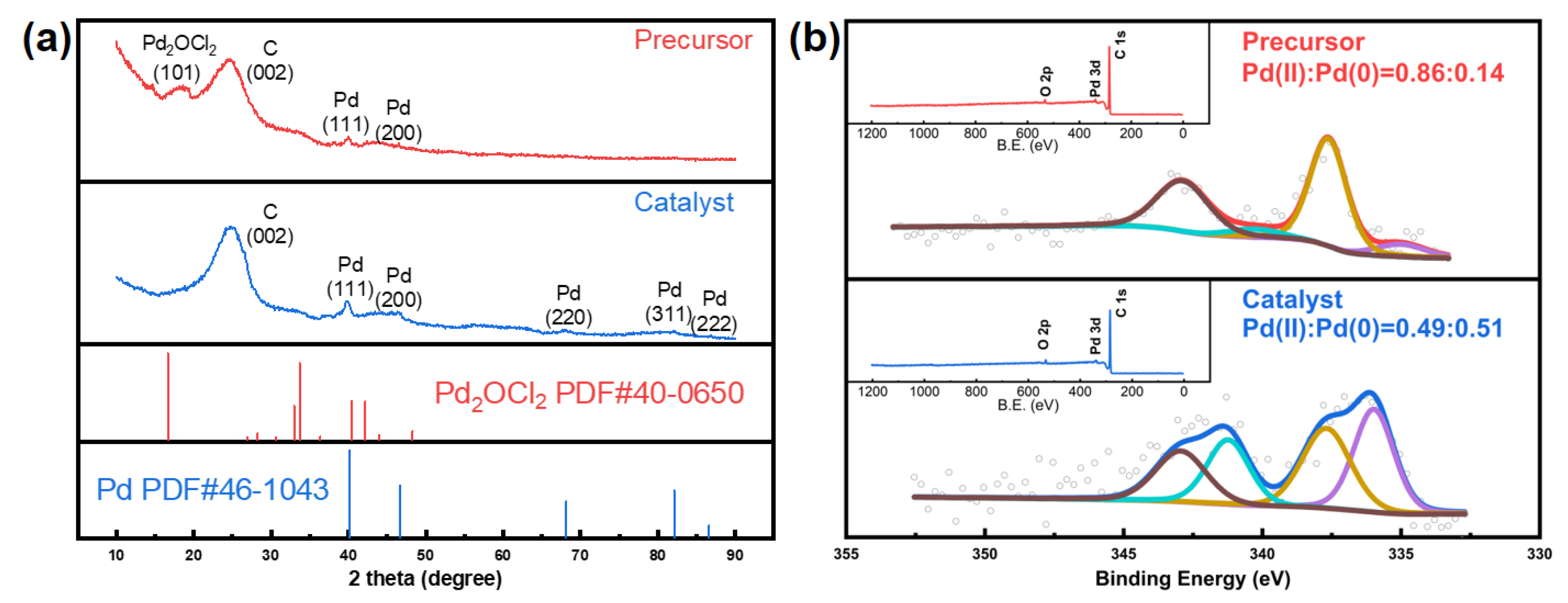
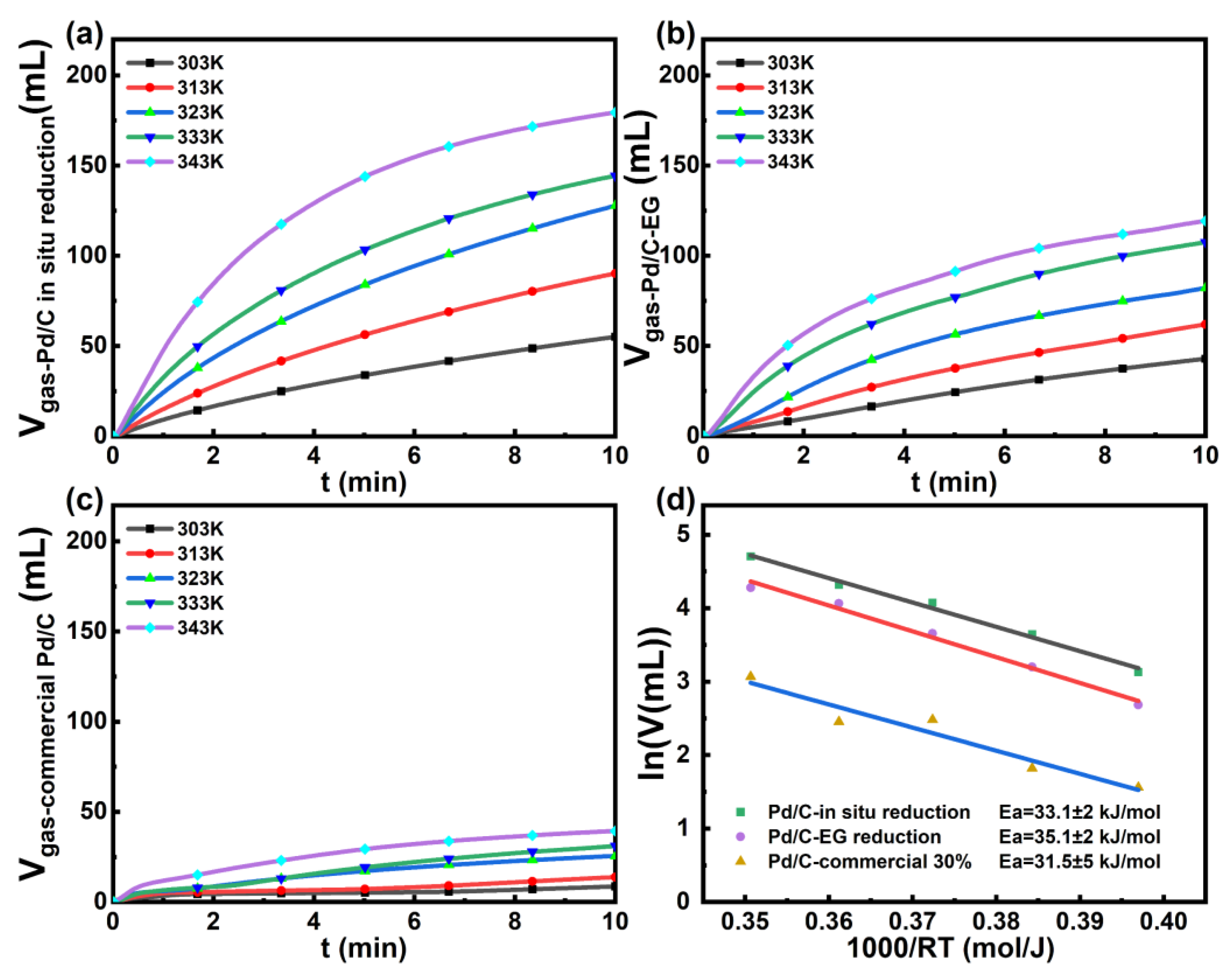
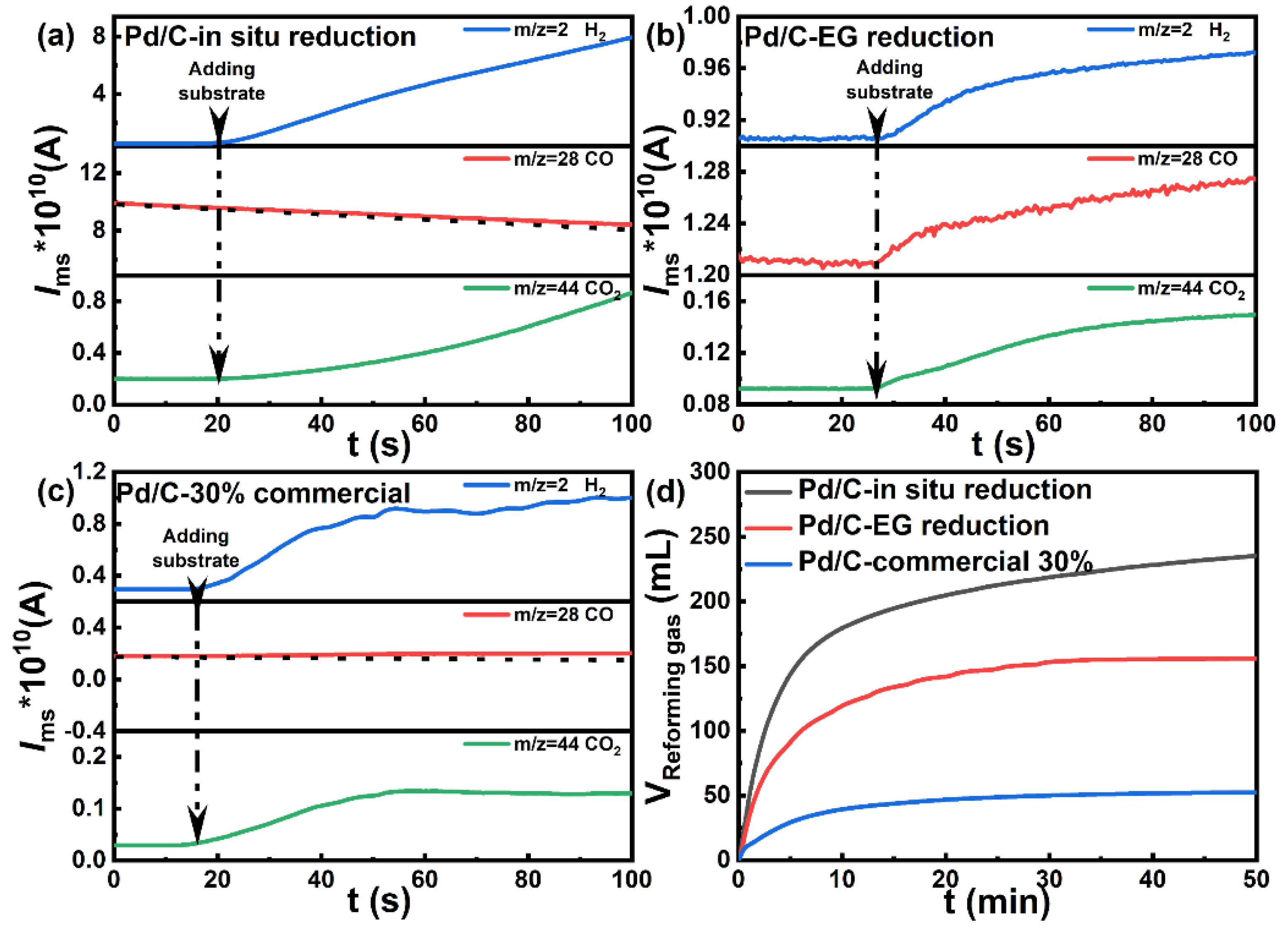
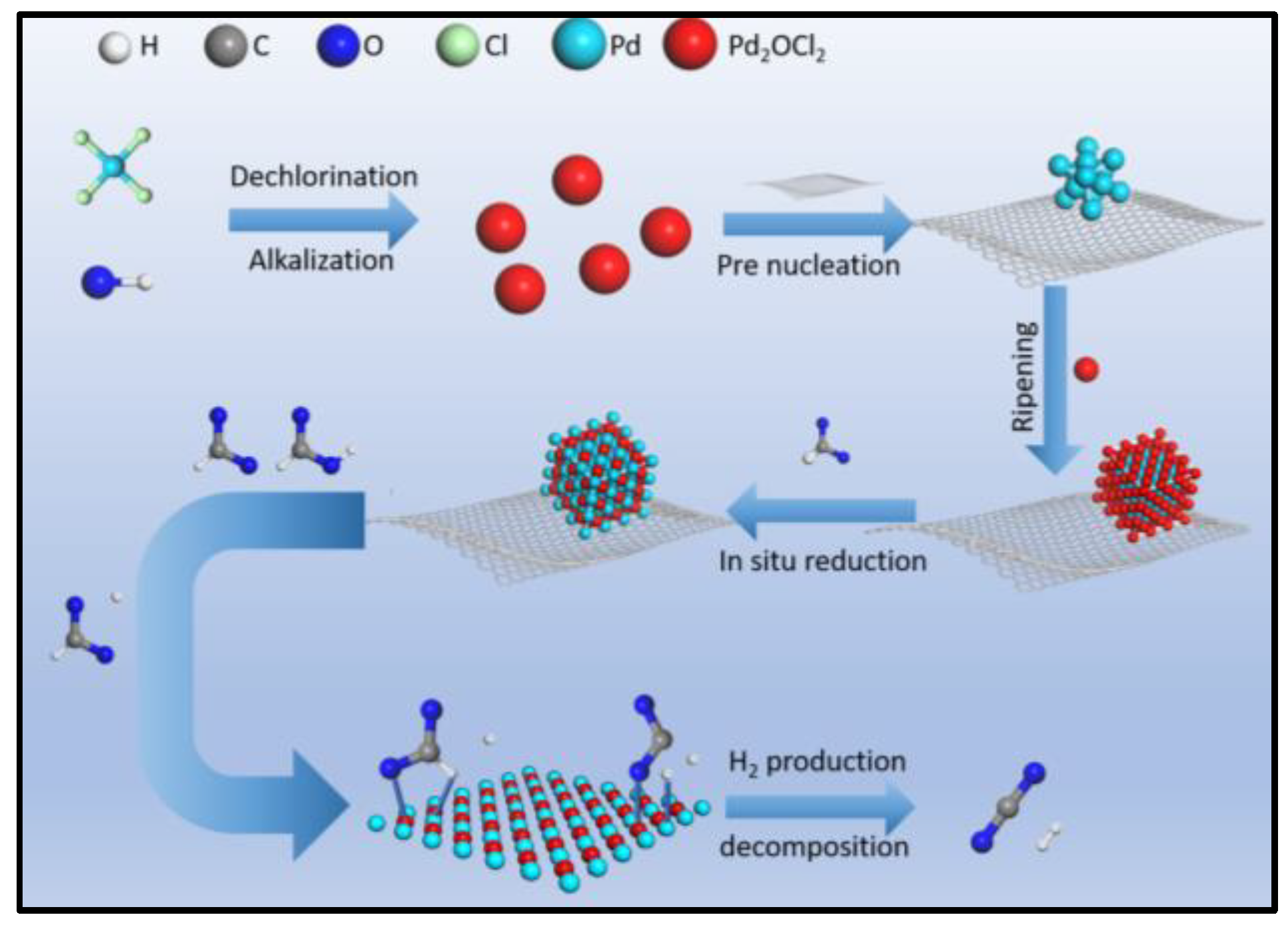
Preparation and Characterization
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Preparation of the Catalyst
3.3. Test for Formic Acid Decomposition
3.4. In Situ Mass Spectra (ISMS) Test
3.5. Catalyst Characterization
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, K.; Li, Y.; Wang, Y.M.; Ge, J.J.; Liu, C.P.; Xing, W. Enhanced electrocatalytic performance for the hydrogen evolution reaction through surface enrichment of platinum nanoclusters alloying with ruthenium in situ embedded in carbon. Energy Environ. Sci. 2018, 11, 1232–1239. [Google Scholar] [CrossRef]
- Lubitz, W.; Tumas, W. Hydrogen: An Overview. Chem. Rev. 2007, 107, 3900–3903. [Google Scholar] [CrossRef] [PubMed]
- Midilli, A.; Ay, M.; Dincer, I.; Rosen, M.A. On hydrogen and hydrogen energy strategies I: Current status and needs. Renew. Sust. Energ. Rev. 2005, 9, 255–271. [Google Scholar] [CrossRef]
- Eberle, U.; Felderhoff, M.; Schueth, F. Chemical and Physical Solutions for Hydrogen Storage. Angew. Chem. Int. Ed. 2009, 48, 6608–6630. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef] [PubMed]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Enthaler, S.; von Langermann, J.; Schmidt, T. Carbon dioxide and formic acid-the couple for environmental-friendly hydrogen storage? Energy Environ. Sci. 2010, 3, 1207–1217. [Google Scholar] [CrossRef]
- Yang, L.; Li, G.; Ge, J.; Liu, C.; Jin, Z.; Wang, G.; Xing, W. TePbPt alloy nanotube as electrocatalyst with enhanced performance towards methanol oxidation reaction. J. Mater. Chem. A 2018, 6, 16798–16803. [Google Scholar] [CrossRef]
- Wang, P.; Steinmann, S.N.; Fu, G.; Michel, C.; Sautet, P. Key Role of Anionic Doping for H2 Production from Formic Acid on Pd(111). ACS Catal. 2017, 7, 1955–1959. [Google Scholar] [CrossRef] [Green Version]
- Tedsree, K.; Li, T.; Jones, S.; Chan, C.W.A.; Yu, K.M.K.; Bagot, P.A.J.; Marquis, E.A.; Smith, G.D.W.; Tsang, S.C.E. Hydrogen production from formic acid decomposition at room temperature using a Ag-Pd core-shell nanocatalyst. Nat. Nanotechnol. 2011, 6, 302–307. [Google Scholar] [CrossRef]
- Li, J.; Chen, W.; Zhao, H.; Zheng, X.; Wu, L.; Pan, H.; Zhu, J.; Chen, Y.; Lu, J. Size-dependent catalytic activity over carbon-supported palladium nanoparticles in dehydrogenation of formic acid. J. Catal. 2017, 352, 371–381. [Google Scholar] [CrossRef]
- Zhang, S.; Jiang, B.; Jiang, K.; Cai, W.B. Surfactant-Free Synthesis of Carbon-Supported Palladium Nanoparticles and Size-Dependent Hydrogen Production from Formic Acid-Formate Solution. ACS Appl. Mater. Interfaces 2017, 9, 24678–24687. [Google Scholar] [CrossRef]
- Lv, Q.; Meng, Q.; Liu, W.; Sun, N.; Jiang, K.; Ma, L.; Peng, Z.; Cai, W.; Liu, C.; Ge, J.; et al. Pd–PdO Interface as Active Site for HCOOH Selective Dehydrogenation at Ambient Condition. J. Phys. Chem. C 2018, 122, 2081–2088. [Google Scholar] [CrossRef]
- Wang, X.; Meng, Q.L.; Gao, L.Q.; Liu, J.; Ge, J.J.; Liu, C.P.; Xing, W. Metal organic framework derived nitrogen-doped carbon anchored palladium nanoparticles for ambient temperature formic acid decomposition. Int. J. Hydrog. Energy 2019, 44, 28402–28408. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Li, X.F.; Wei, Z.Z.; Mao, S.J.; Deng, J.; Cao, Y.L.; Wang, Y. Efficient synthesis of ultrafine Pd nanoparticles on an activated N-doping carbon for the decomposition of formic acid. Catal. Commun. 2018, 108, 55–58. [Google Scholar] [CrossRef]
- Wei, J.; Wang, H.Z.; Zhang, Q.H.; Li, Y.G. One-pot Hydrothermal Synthesis of N-Doped Carbon Quantum Dots Using the Waste of Shrimp for Hydrogen Evolution from Formic Acid. Chem. Lett. 2015, 44, 241–243. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, D. Hydrogen production from formic acid dehydrogenation over a Pd supported on N-doped mesoporous carbon catalyst: A role of nitrogen dopant. Appl. Catal. A-Gen. 2020, 608, 117887. [Google Scholar] [CrossRef]
- Golub, F.S.; Beloshapkin, S.; Gusel’nikov, A.V.; Bolotov, V.A.; Parmon, V.N.; Bulushev, D.A. Boosting Hydrogen Production from Formic Acid over Pd Catalysts by Deposition of N-Containing Precursors on the Carbon Support. Energies 2019, 12, 3885. [Google Scholar] [CrossRef] [Green Version]
- Xing, Z.H.; Guo, Z.L.; Chen, X.Y.; Zhang, P.; Yang, W.S. Optimizing the activity of Pd based catalysts towards room-temperature formic acid decomposition by Au alloying. Catal. Sci. Technol. 2019, 9, 588–592. [Google Scholar] [CrossRef]
- Li, S.J.; Zhou, Y.T.; Kang, X.; Liu, D.X.; Gu, L.; Zhang, Q.H.; Yan, J.M.; Jiang, Q. A Simple and Effective Principle for a Rational Design of Heterogeneous Catalysts for Dehydrogenation of Formic Acid. Adv. Mater. 2019, 31, 1806781. [Google Scholar] [CrossRef]
- Hong, W.; Kitta, M.; Tsumori, N.; Himeda, Y.; Autrey, T.; Xu, Q. Immobilization of highly active bimetallic PdAu nanoparticles onto nanocarbons for dehydrogenation of formic acid. J. Mater. Chem. A 2019, 7, 18835–18839. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Yan, J.-M.; Ping, Y.; Wang, H.-L.; Zheng, W.-T.; Jiang, Q. An Efficient CoAuPd/C Catalyst for Hydrogen Generation from Formic Acid at Room Temperature. Angew. Chem. Int. Ed. 2013, 52, 4406–4409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Metin, O.; Su, D.; Sun, S. Monodisperse AgPd Alloy Nanoparticles and Their Superior Catalysis for the Dehydrogenation of Formic Acid. Angew. Chem. Int. Ed. 2013, 52, 3681–3684. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, Y.; Xing, W.; Liu, C.; Liao, J.; Lu, T. High-quality hydrogen from the catalyzed decomposition of formic acid by Pd–Au/C and Pd–Ag/Cw. Chem. Commun. 2008, 3540, 3542. [Google Scholar]
- Qin, Y.-L.; Wang, J.; Meng, F.-Z.; Wang, L.-M.; Zhang, X.-B. Efficient PdNi and PdNi@Pd-catalyzed hydrogen generation via formic acid decomposition at room temperature. Chem. Commun. 2013, 49, 10028–10030. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Feng, L.; Hu, C.; Liu, C.; Xing, W. High-quality hydrogen generated from formic acid triggered by in situ prepared Pd/C catalyst for fuel cells. Catal. Sci. Technol. 2015, 5, 2581–2584. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Yan, J.-M.; Wang, H.-L.; Ping, Y.; Jiang, Q. Pd/C Synthesized with Citric Acid: An Efficient Catalyst for Hydrogen Generation from Formic Acid/Sodium Formate. Sci. Rep. 2012, 2, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Tsumori, N.; Kitta, M.; Xu, Q. Fast Dehydrogenation of Formic Acid over Palladium Nanoparticles Immobilized in Nitrogen-Doped Hierarchically Porous Carbon. ACS Catal. 2018, 8, 12041–12045. [Google Scholar] [CrossRef]
- Jiang, K.; Xu, K.; Zou, S.; Cai, W.-B. B-Doped Pd Catalyst: Boosting Room-Temperature Hydrogen Production from Formic Acid-Formate Solutions. J. Am. Chem. Soc. 2014, 136, 4861–4864. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.M.; Wang, Z.L.; Gu, L.; Li, S.J.; Wang, H.L.; Zheng, W.T.; Jiang, Q. AuPd-MnOx/MOF-Graphene: An Efficient Catalyst for Hydrogen Production from Formic Acid at Room Temperature. Adv. Energy Mater. 2015, 5, 1500107. [Google Scholar] [CrossRef]
- Bulut, A.; Yurderi, M.; Karatas, Y.; Say, Z.; Kivrak, H.; Kaya, M.; Gulcan, M.; Ozensoy, E.; Zahmakiran, M. MnOx-Promoted PdAg Alloy Nanoparticles for the Additive-Free Dehydrogenation of Formic Acid at Room Temperature. Acs Catal. 2015, 5, 6099–6110. [Google Scholar] [CrossRef]
- Zhu, Q.L.; Tsumori, N.; Xu, Q. Sodium hydroxide-assisted growth of uniform Pd nanoparticles on nanoporous carbon MSC-30 for efficient and complete dehydrogenation of formic acid under ambient conditions. Chem. Sci. 2014, 5, 195–199. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | MSP (molH2/(gPd·h)) | Ea (kJ/mol) | T (K) |
|---|---|---|---|
| this work | 8.94 | 33.1 | 303 |
| Pd/C [29] | 1.5 | \ | 303 |
| Pd-B [29] | 3.5 | \ | 303 |
| Pd@CN [28] | \ | 46.9 | 303 |
| Ag@Pd/C [10] | 0.164 | 30 | 293 |
| Pd1Au1/C [21] | \ | 34.4 | 333 |
| AuPd–MnOx/ZIF-8-rGO [30] | 1.65 | \ | 298 |
| PdAg-MnOx/N-SiO2 [31] | 1.05 | \ | 303 |
| Co0.30Au0.35Pd0.35/C [22] | 0.05 | \ | 298 |
| Pd/MSC-30 [32] | 3.59 | \ | 298 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, Q.; Yang, X.; Wang, X.; Xiao, M.; Li, K.; Jin, Z.; Ge, J.; Liu, C.; Xing, W. Preparation Strategy Using Pre-Nucleation Coupled with In Situ Reduction for a High-Performance Catalyst towards Selective Hydrogen Production from Formic Acid. Catalysts 2022, 12, 325. https://doi.org/10.3390/catal12030325
Meng Q, Yang X, Wang X, Xiao M, Li K, Jin Z, Ge J, Liu C, Xing W. Preparation Strategy Using Pre-Nucleation Coupled with In Situ Reduction for a High-Performance Catalyst towards Selective Hydrogen Production from Formic Acid. Catalysts. 2022; 12(3):325. https://doi.org/10.3390/catal12030325
Chicago/Turabian StyleMeng, Qinglei, Xiaolong Yang, Xian Wang, Meiling Xiao, Kui Li, Zhao Jin, Junjie Ge, Changpeng Liu, and Wei Xing. 2022. "Preparation Strategy Using Pre-Nucleation Coupled with In Situ Reduction for a High-Performance Catalyst towards Selective Hydrogen Production from Formic Acid" Catalysts 12, no. 3: 325. https://doi.org/10.3390/catal12030325