3. Materials and Methods
3.1. General Remarks
All experiments were performed in an inert atmosphere using a standard glove box and Schlenk techniques. Toluene, hexane, and tetrahydrofuran (THF) were purchased from Thermo Fisher Scientific Korea and distilled from benzophenone ketyl. The hexane (HPLC grade) used for the polymerization reactions was purchased from Thermo Fisher Scientific Korea (Seoul, Korea) and purified over a Na/K alloy. The ethylene gas was purified by contact with molecular sieves and copper for more than 12 h under a pressure of 48 bar. Silica (SYLOPOL-2410) was obtained from GRACE and used as received (Yeosu, South Korea). The 1H NMR (600 MHz) and 13C NMR (150 MHz) spectra were recorded on a JEOL (Tokyo, Japan) ECZ 600 instrument. Gel permeation chromatography (GPC) was performed in 1,2,4-trichlorobenzene at 160 °C using an Agilent PL-GPC 220 system equipped with an RI detector and two columns (PLgel mixed-B 7.5 × 300 mm from Varian (Polymer Lab (Salop, UK)).
3.2. Synthesis of 1
A solution of nBuLi in hexane (8.44 g, 2.5 M, 30.4 mmol) was added dropwise to a solution of thiophene-fused cyclopentadiene compound 2,4,5-Me3C7H3S (5.00 g, 30.4 mmol) in hexane (100 mL) at −78 °C. The resulting solution was slowly warmed to room temperature and then stirred overnight at room temperature to precipitate the corresponding thiophene-fused cyclopentadienyl–Li as a white solid, which was isolated by filtration (4.66 g, 90%). A solution of Me2SiCl2 (1.14 g, 8.81 mmol) in toluene (7.0 mL) was added to the prepared Li compound (1.00 g, 5.88 mmol) dispersed in toluene (5.0 mL) at −30 °C. The solution was slowly warmed to room temperature and stirred overnight at room temperature. After the removal of the solvent using a vacuum line, the product was extracted with hexane (20 mL) via filtration over Celite. The solvent was removed using a vacuum line to obtain the desired compound as a yellowish-brown liquid (1.39 g, 92%). 1H NMR (C6D6): δ 6.49 (q, 4J = 1.2 Hz, 1H), 3.23 (s, 1H), 2.28 (s, 3H, CH3), 1.97 (s, 3H, CH3), 1.92 (q, 4J = 1.2 Hz, 3H, CH3), 0.21 (s, 3H, SiCH3), and 0.02 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 152.5, 141.6, 136.8, 135.6, 131.0, 116.6, 48.8, 16.0, 14.8, 11.7, −0.4, and −0.8 ppm. HRMS(EI): m/z calcd. ((M+) C12H17ClSSi) 256.0509. Found: 256.0509.
3.3. Synthesis of 2
Compound 2 was synthesized using the same conditions and procedure as those for 1, with (Me4C7S)−Li+ (1.08 g, 5.88 mmol). A yellowish-brown liquid compound was obtained in 92% yield (1.46 g). 1H NMR (C6D6): δ 3.20 (s, 1H), 2.15 (s, 3H, CH3), 2.05 (s, 3H, CH3), 2.00 (s, 3H, CH3), 1.99 (s, 3H, CH3), 0.27 (s, 3H, SiCH3), and 0.03 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 151.6, 136.8, 134.6, 134.0, 131.8, 125.4, 48.3, 14.6, 13.6, 12.2, 12.1, −0.2, and −0.9 ppm. HRMS(EI): m/z calcd. ((M+) C13H17ClSSi) 270.0663. Found: 270.0665.
3.4. Synthesis of 3
Compound 3 was synthesized using the same conditions and procedure as those for 1, with (Me3C7H2S)−Li+ (0.92 g, 5.88 mmol). A yellowish-brown liquid compound was obtained in 92% yield (1.31 g). 1H NMR (C6D6): δ 7.00 (d, J = 4.8 Hz, 1H), 6.80 (d, J = 4.8 Hz, 1H), 3.21 (s, 1H), 1.94 (s, 3H, CH3), 1.89 (s, 3H, CH3), 0.16 (s, 3H, SiCH3), and −0.01 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 152.8, 138.2, 137.7, 130.9, 127.0, 117.8, 48.4, 14.8, 11.7, −0.5, and −0.8 ppm. HRMS(EI): m/z calcd. ((M+) C11H15ClSSi) 242.0352. Found: 242.0352.
3.5. Synthesis of 4
A solution of nBuLi in hexane (8.34 g, 2.5 M, 30.1 mmol) was added slowly to a solution of fluorene (5.00 g, 30.1 mmol) in toluene (50 mL) at room temperature. The resulting solution was heated to 80 °C and then stirred for 5 h to precipitate fluorenyl–Li as orange solids, which were isolated by filtration (4.66 g, 90%). The prepared fluorenyl–Li (0.248 g, 1.56 mmol) was added to a solution of 1 (0.400 g, 1.56 mmol) in THF (6.0 mL) at −30 °C, and the mixture was slowly warmed to room temperature. The solution was stirred overnight, and the solvent was removed using a vacuum line. The product was extracted with hexane (20 mL) and collected by filtration over Celite. After removing the solvent using a vacuum line, the product was purified by silica gel column chromatography and eluted with hexane and toluene (30:1, v/v). A yellowish-brown, oily compound was obtained (0.181 g, 30%). 1H NMR (C6D6): δ 7.80 (d, J = 7.6 Hz, 1H), 7.77 (d, J = 7.6 Hz, 1H), 7.74 (d, J = 7.6 Hz, 1H), 7.34 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.26 (t, J = 7.6 Hz, 1H), 7.24 (t, J = 7.6 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 6.62 (s, 1H), 4.47 (s, 1H), 3.60 (s, 1H), 2.34 (s, 3H, CH3), 2.00 (s, 3H, CH3), 1.76 (s, 3H, CH3), −0.28 (s, 3H, SiCH3), and −0.49 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 151.9, 145.5, 141.4, 141.3, 140.7, 138.2, 137.6, 130.1, 126.0, 125.9, 124.7, 124.5, 120.5, 120.4, 116.9, 46.1, 40.8, 16.1, 15.0, 11.9, −6.7, and −7.2 ppm. HRMS(EI): m/z calcd. ((M+) C25H26SSi) 386.1527. Found: 386.1527.
3.6. Synthesis of 5
Compound 5 was synthesized under the same conditions and procedure as 4, using 2 (0.423 g, 1.56 mmol). A yellowish-brown, oily compound was obtained by silica gel column chromatography with hexane and toluene (30:1, v/v) as eluents (0.269 g, 43%). 1H NMR (C6D6): δ 7.79 (d, J = 7.6 Hz, 1H), 7.76 (d, J = 7.6 Hz, 2H), 7.33 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.26 (t, J = 7.6 Hz, 1H), 7.23 (td, 3J = 7.6 Hz, 4J = 1.4 Hz, 1H), 7.13 (td, 3J = 7.6 Hz, 4J = 1.4 Hz, 1H), 4.47 (s, 1H), 3.55 (s, 1H), 2.22 (s, 3H, CH3), 2.14 (s, 3H, CH3), 2.07 (s, 3H, CH3), 1.74 (s, 3H, CH3), −0.23 (s, 3H, SiCH3), and −0.44 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 151.0, 145.6, 145.6, 141.4, 138.1, 136.0, 133.7, 131.0, 126.7, 126.5, 126.0, 125.8, 125.6, 124.7, 124.6, 120.5, 120.3, 45.6, 40.8, 14.7, 13.7, 12.4, 12.2, −6.5, and −6.9 ppm. HRMS(EI): m/z calcd. ((M+) C26H28SSi) 400.1684. Found: 400.1681.
3.7. Synthesis of 6
Compound 6 was synthesized using the same conditions and procedure as those for 4, with 3 (0.379 g, 1.56 mmol). A yellowish-brown, oily compound was obtained by silica gel column chromatography with hexane and toluene (30:1, v/v) as eluents (0.174 g, 30%). 1H NMR (C6D6): δ 7.78 (d, J = 7.6 Hz, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.25 (t, J = 7.6 Hz, 1H), 7.23 (td, 3J = 7.6 Hz, 4J = 1.4 Hz, 1H), 7.15 (td, 3J = 7.6 Hz, 4J = 1.4 Hz, 1H), 6.97 (d, J = 4.8 Hz, 1H), 6.86 (d, J = 4.8 Hz, 1H), 4.34 (s, 1H), 3.51 (s, 1H), 1.92 (s, 3H, CH3), 1.68 (s, 3H, CH3), and −0.39 (s, 3H, SiCH3), −0.56 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 152.3, 145.5, 145.4, 141.4, 141.3, 140.2, 139.2, 130.0, 126.7, 126.5, 126.2, 126.0, 125.9, 124.7, 124.6, 120.5, 120.4, 118.1, 45.7, 40.7, 15.0, 11.8, −6.7, and −7.2 ppm. HRMS(EI): m/z calcd. ((M+) C24H24SSi) 372.1368. Found: 372.1371.
3.8. Synthesis of 7
A solution of nBuLi in hexane (4.98 g, 2.5 M, 18.0 mmol) was added dropwise to a solution of 2,7-di-tert-butylfluorene (5.00 g, 18.0 mmol) in toluene (50 mL) at room temperature. The resulting solution was heated to 80 °C and then stirred for 5 h to precipitate 2,7-di-tert-butylfluorenyl-Li as a yellow solid, which was isolated by filtration (4.32 g, 85%). Compound 7 was prepared using the same conditions and procedures as those for 4, with the prepared 2,7-di-tert-butylfluorenyl-Li (0.444 g, 1.56 mmol). A yellowish-brown oily compound was obtained via silica gel column chromatography with hexane and toluene (30:1, v/v) as eluents (0.210 g, 27%). 1H NMR (C6D6): δ 8.02 (s, 1H), 7.83 (d, J = 8.3 Hz, 1H), 7.80 (d, J = 8.3 Hz, 1H), 7.54 (s, 1H), 7.43 (dd, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 7.34 (dd, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 6.64 (q, J = 1.2 Hz, 1H), 4.47 (s, 1H), 3.74 (s, 1H), 2.35 (s, 3H, CH3), 2.02 (s, 3H, CH3), 1.86 (s, 3H, CH3), 1.39 (s, 9H, tBu), 1.32 (s, 9H, tBu), −0.15 (s, 3H, SiCH3), and −0.40 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 152.0, 149.3, 149.1, 145.8, 145.7, 140.8, 139.1, 139.0, 138.2, 137.7, 130.2, 123.2, 123.1, 124.9, 121.6, 121.5, 119.9, 119.8, 117.0, 46.4, 40.6, 35.1, 34.9, 31.9, 31.9, 16.1, 15.1, 11.9, −6.3, and −6.9 ppm. HRMS(EI): m/z calcd. ((M+) C33H42SSi) 498.2774. Found: 498.2776. The yield was improved by reversing the reaction sequence. Thus, tert-butyl methyl ether (0.889 g, 3.3 eq/Li+) was added to a solution of 2,7-di(tert-butyl)fluorenyl–Li (0.766 g, 2.70 mmol) in hexane (27 mL) tert-butyl methyl ether (0.889 g, 3.3 eq/Li+). After cooling to −78 °C, a solution of Me2SiCl2 (0.522 g, 4.04 mmol) in hexane (7.5 mL) was added. After overnight stirring at room temperature, the solvent was removed under vacuum. The product was extracted using hexane (15 mL). The extract was collected by filtration over Celite. The solvent was removed using a vacuum line to obtain 2,7-di(tert-butyl)fluorenyl-Si(Me)Cl (0.90 g, 90%). The prepared 2,7-di(tert-butyl)fluorenyl-Si(Me)Cl was dissolved in THF (10 mL). After cooling to −30 °C, thiophene-fused cyclopentadienyl–Li compound (2,4,5-Me3C7H2S)Li (0.413 g, 2.43 mmol) was added. The solution was stirred overnight, and the solvent was removed using a vacuum line. The product was extracted with hexane (15 mL) and collected by filtration over Celite. After removing the solvent using a vacuum line, the product was purified by silica gel column chromatography with hexane and toluene (30:1, v/v) as eluents. A yellowish-brown, oily compound was obtained in 81% yield (0.98 g).
3.9. Synthesis of 8
A solution of nBuLi in hexane (5.90 g, 2.5 M, 21.3 mmol) was added dropwise to a solution of 2,7-dichlorofluorene (5.00 g, 21.3 mmol) in toluene (50 mL) at −78 °C. The resulting solution was stirred overnight to precipitate 2,7-dichlorofluorenyl–Li as a light-yellow solid, which was isolated by filtration (4.51 g, 88%). Compound 8 was prepared using the same conditions and procedures as those for 4, with the prepared 2,7-dichlorofluorenyl–Li (0.376 g, 1.56 mmol). A yellowish-brown, oily compound was obtained by silica gel column chromatography with hexane and toluene (30:1, v/v) as eluents (0.412 g, 58%). 1H NMR (C6D6): δ 7.58 (s, 1H), 7.28 (d, J = 8.3 Hz, 1H), 7.25 (s, 1H), 7.24 (d, J = 8.3 Hz, 1H), 7.22 (dd, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 7.17 (dd, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 6.52 (s, 1H), 3.91 (s, 1H), 3.21 (s, 1H), 2.37 (s, 3H, CH3), 1.92 (s, 3H, CH3), 1.62 (s, 3H, CH3), −0.33 (s, 3H, SiCH3), and −0.36 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 151.9, 147.3, 147.1, 141.0, 138.7, 138.5, 137.8, 137.1, 132.6, 132.5, 130.4, 126.4, 126.2, 124.9, 124.9, 121.2, 121.1, 117.0, 45.9, 40.9, 16.1, 14.7, 11.8, −5.4, and −5.9 ppm. HRMS(EI): m/z calcd. ((M+) C25H24Cl2SSi) 454.0741. Found: 454.0745.
3.10. Synthesis of 9
A solution of nBuLi in hexane (1.11 g, 2.5 M, 4.02 mmol) was added dropwise to a solution of 4 (0.777 g, 2.01 mmol) in THF (3.5 mL) at −30 °C. After slowly warming to room temperature, the solution was stirred at room temperature for 1 h. After cooling to −30 °C again, MeMgBr (1.41 mL, 1.37 M solution in THF–toluene, 4.62 mmol) and ZrCl4·(THF)2 (0.743 g, 1.97 mmol) were added successively. After the solution was stirred overnight at room temperature, the solvent was removed using a vacuum line. The product was extracted with toluene (30 mL) and collected by filtration over Celite. The solvent was removed using a vacuum line to obtain a yellowish-brown solid, which was redissolved in toluene (4 mL). Yellow solids were deposited when the solution was stored in a freezer at −30 °C (0.906 g, 91%). 1H NMR (C6D6): δ 7.95 (d, J = 8.3 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.73 (d, J = 8.3 Hz, 1H), 7.45 (d, J = 8.3 Hz, 1H), 7.34 (t, J = 8.3 Hz, 1H), 7.24 (t, J = 8.3 Hz, 1H), 7.00 (t, J = 8.3 Hz, 2H), 6.17 (q, 4J = 1.2 Hz, 1H), 2.07 (q, J = 1.2 Hz, 3H, CH3), 2.00 (s, 3H, CH3), 1.75 (s, 3H, CH3), 1.06 (s, 3H, SiCH3), 0.83 (s, 3H, SiCH3), −1.08 (s, 3H, ZrCH3), and −1.57 (s, 3H, ZrCH3) ppm. 13C{1H} NMR (C6D6): δ 145.2, 140.1, 132.4, 131.0, 129.1, 128.7, 127.4, 126.9, 126.7, 126.4, 126.1, 125.4, 124.8, 124.0, 123.5, 123.4, 117.1, 116.0, 77.0, 62.7, 40.8, 38.3, 16.2, 15.5, 12.2, 1.5, and 1.3 ppm.
3.11. Synthesis of 10
Compound 10 was synthesized using the same conditions and procedure as those for 9, with 5 (0.175 g, 0.437 mmol). A yellow solid was obtained in 73% yield (0.166 g). 1H NMR (C6D6): δ 7.95 (d, J = 8.3 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.74 (d, J = 8.3 Hz, 1H), 7.45 (d, J = 8.3 Hz, 1H), 7.34 (t, J = 8.3 Hz, 1H), 7.23 (t, J = 8.3 Hz, 1H), 7.01 (td, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 7.00 (td, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 2.06 (s, 3H, CH3), 1.96 (s, 3H, CH3), 1.87 (s, 3H, CH3), 1.76 (s, 3H, CH3), 1.08 (s, 3H, SiCH3), 0.84 (s, 3H, SiCH3), −1.08 (s, 3H, ZrCH3), and −1.60 (s, 3H, ZrCH3) ppm. 13C{1H} NMR (C6D6): δ 140.1, 137.1, 132.6, 131.1, 129.5, 127.7, 127.3, 126.8, 126.7, 126.6, 126.5, 125.4, 124.8, 124.4, 123.9, 123.4, 123.4, 117.6, 76.7, 62.8, 39.7, 38.5, 15.3, 13.5, 12.4, 11.9, 1.7, and 1.2 ppm.
3.12. Synthesis of 11
Compound 11 was synthesized using the same conditions and procedure as those for 9, with 6 (0.233 g, 0.626 mmol). A yellow solid was obtained in 60% yield (0.181 g). 1H NMR (C6D6): δ 7.92 (d, J = 8.3 Hz, 1H), 7.83 (d, J = 8.3 Hz, 1H), 7.70 (d, J = 8.3 Hz, 1H), 7.45 (d, J = 8.3 Hz, 1H), 7.34 (t, J = 8.3 Hz, 1H), 7.23 (t, J = 8.3 Hz, 1H), 7.00 (td, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 6.99 (td, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 6.73 (d, J = 5.4 Hz, 1H), 6.44 (d, J = 5.4 Hz, 1H), 1.97 (s, 3H, CH3), 1.74 (s, 3H, CH3), 1.04 (s, 3H, SiCH3), 0.82 (s, 3H, SiCH3), −1.10 (s, 3H, ZrCH3), and −1.65 (s, 3H, ZrCH3) ppm. 13C{1H} NMR (C6D6): δ 139.5, 132.4, 131.2, 130.5, 129.9, 129.4, 129.3, 127.4, 126.8, 126.6, 126.2, 125.4, 124.8, 124.0, 123.5, 123.5, 118.3, 117.5, 76.9, 62.9, 41.9, 38.6, 15.5, 12.1, 1.6, and 1.3 ppm.
3.13. Synthesis of 12
Compound 12 was synthesized using the same conditions and procedure as those for 9, with 7 (0.295 g, 0.591 mmol). A yellow solid was obtained in 24% yield (0.086 g). The solubility of this compound in C6D6 was too low to record the 13C NMR spectrum. 1H NMR (C6D6): δ 7.96, (d, J = 8.3 Hz, 1H), 7.87 (d, J = 8.3 Hz, 1H), 7.76 (s, 1H), 7.58 (s, 1H), 7.50 (dd, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 7.43 (dd, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 6.22 (q, 4J = 1.2 Hz, 1H), 2.12 (q, 4J = 1.2 Hz, 3H, CH3), 2.03 (s, 3H, CH3), 1.87 (s, 3H, CH3), 1.35 (s, 9H, tBu), 1.30 (s, 9H, tBu), 1.16 (s, 3H, SiCH3), 0.98 (s, 3H, SiCH3), −1.09 (s, 3H, ZrCH3), and −1.59 (s, 3H, ZrCH3) ppm.
3.14. Synthesis of 13
Compound 13 was synthesized using the same conditions and procedure as those for 9, with 8 (0.200 g, 0.440 mmol). A yellow solid was obtained in 74% yield (0.183 g). 1H NMR (C6D6): δ 7.75 (s, 1H), 7.53 (s, 1H), 7.52 (d, J = 8.3 Hz, 1H), 7.41 (d, J = 8.3 Hz, 1H), 7.31 (dd, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 7.21 (dd, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H), 6.09 (q, 4J = 1.2 Hz, 1H), 2.05 (q, 4J = 1.2 Hz, 3H, CH3), 1.93 (s, 3H, CH3), 1.72 (s, 3H, CH3), 0.89 (s, 3H, SiCH3), 0.67 (s, 3H, SiCH3), −1.06 (s, 3H, ZrCH3), and −1.56 (s, 3H, ZrCH3) ppm. 13C{1H} NMR (C6D6): δ 125.9, 140.3, 133.2, 133.0, 133.0, 131.8, 129.7, 129.3, 126.5, 125.8, 125.0, 124.3, 124.2, 124.1, 123.6, 123.2, 117.7, 115.8, 77.2, 63.7, 41.4, 38.8, 16.1, 15.3, 12.1, 0.9, and 0.8 ppm.
3.15. Synthesis of 14
A solution of nBuLi in hexane (3.17 g, 2.5 M, 1.14 mmol) was added dropwise to a solution of 2-methyl-4-(4-
tert-butylphenyl)indenene (3.00 g, 1.14 mmol) in hexane (30 mL), and the resulting solution was stirred overnight. A light-yellow solid precipitated was isolated by filtration (2.54 g, 83%). Compound
14 was synthesized using the same conditions and procedures as those for
4, with the prepared 2-methyl-4-(4-
tert-butylphenyl)indenyl–Li (0.420 g, 1.56 mmol). A yellowish-brown, oily compound was obtained by silica gel column chromatography with hexane and toluene (30:1,
v/
v) as eluents (0.445 g, 59%). The product was obtained as a mixture of two diastereomers in a ratio of 1.0:0.10. Signals for minor isomers were not recorded (see
Figure S14).
1H NMR (C
6D
6): δ 7.64 (d,
J = 8.3 Hz, 2H), 7.42 (d,
J = 7.6 Hz, 1H), 7.41 (d,
J = 8.3 Hz, 2H), 7.28 (d,
J = 7.6 Hz, 1H), 7.14 (t,
J = 7.6 Hz, 1H), 7.00 (s, 1H), 6.62 (s, 1H), 6.96 (s, 1H), 6.62 (s, 1H), 3.99 (s, 1H), 3.49 (s, 1H), 2.33 (s, 3H, CH
3), 2.14 (s, 3H, CH
3), 2.02 (s, 3H, CH
3), 1.89 (s, 3H, CH
3), 1.31 (s, 9H, tBu), −0.19 (s, 3H, SiCH
3), and −0.26 (s, 3H, SiCH
3) ppm.
13C{
1H} NMR (C
6D
6): δ 152.1, 149.7, 147.5, 146.1, 143.7, 140.7, 139.3, 138.3, 137.7, 134.7, 130.2, 129.2, 126.7, 126.0, 125.7, 123.6, 122.5, 116.8, 47.6, 46.4, 34.6, 31.6, 18.0, 16.0, 15.1, 11.8, −6.3, and −6.8 ppm.
3.16. Synthesis of 15
Compound
15 was synthesized using the same conditions and procedure as those for
9, with
14 (0.207 g, 0.428 mmol). A yellow solid was obtained in 67% yield (0.178 g), as a mixture of two isomers in a 1.0:0.54 ratio (refer to
Figure S15a), which was used for polymerization without separation of the isomers. Solids (0.074 g) were deposited in a toluene solution containing 0.250 g of the isomer mixture and were stored in a freezer at −30 °C. The
1H NMR spectrum of the deposited solids indicated that, mainly, the minor isomer in the crude mixture was deposited, along with a small portion of the major isomer, in a 1.0:0.13 ratio (refer to
Figure S15b). Contaminant THF signals (0.5 eq) were detected even after crystallization.
1H NMR (C
6D
6) of major isomer: δ 7.88 (d,
J = 8.3 Hz, 2H), 7.64 (d,
J = 8.3 Hz, 1H), 7.41 (d,
J = 8.3 Hz, 2H), 7.25 (d,
J = 8.3 Hz, 1H), 7.24 (s, 1H), 6.87 (dd,
J = 9.0 Hz and 6.9 Hz, 1H), 6.26 (s, 1H), 2.14 (s, 3H, CH
3), 2.09 (s, 3H, CH
3), 1.98 (s, 3H, CH
3), 1.83 (s, 3H, CH
3), 1.25 (s, 9H, tBu), 1.00 (s, 3H, SiCH
3), 0.65 (s, 3H, SiCH
3), 0.12 (s, 3H, ZrCH
3), and −1.38 (s, 3H, ZrCH
3) ppm.
1H NMR (C
6D
6) of minor isomer: δ 7.85 (d,
J = 8.3 Hz, 2H), 7.36 (d,
J = 8.3 Hz, 1H), 7.35 (d,
J = 8.3 Hz, 2H), 7.25 (d,
J = 8.3 Hz, 1H), 7.24 (s, 1H), 6.90 (dd,
J = 9.0 Hz and 6.9 Hz, 1H), 6.38 (s, 1H), 2.20 (s, 3H, CH
3), 2.19 (s, 3H, CH
3), 2.06 (s, 3H, CH
3), 1.76 (s, 3H, CH
3), 1.24 (s, 9H, tBu), 0.80 (s, 3H, SiCH
3), 0.77 (s, 3H, SiCH
3), −0.43 (s, 3H, ZrCH
3), and −0.91 (s, 3H, ZrCH
3) ppm.
3.17. Synthesis of 16
Compound 16 was synthesized using the same conditions and procedures as those for 4, with tert-BuN(H)Li (0.126 g, 1.59 mmol). A yellowish-brown liquid was obtained (0.423 g, 91%). 1H NMR (C6D6): δ 6.61 (q, 4J = 1.2 Hz, 1H), 3.22 (s, 1H), 2.36 (s, 3H, CH3), 2.04 (s, 3H, CH3), 2.04 (s, 3H, CH3), 1.12 (s, 9H, tBu), 0.63 (s, 1H, NH), 0.12 (s, 3H, SiCH3), and −0.03 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 151.3, 139.7, 138.3, 138.1, 129.0, 116.5, 50.3, 49.6, 33.9, 16.1, 15.2, 11.9, 0.0, and −1.6 ppm. HRMS(EI): m/z calcd. ((M+) C16H27NSSi) 293.1633. Found: 293.1631.
3.18. Synthesis of 17
A measured quantity of nBuLi (0.60 mL, 2.5 M in hexane, 1.51 mmol) was added dropwise at −78 °C to 16 (0.221 g, 0.754 mmol) dissolved in THF (2.0 g). After stirring overnight at room temperature, the solution was cooled to −78 °C. Subsequently, MeMgCl (0.52 mL, 3.1 M in THF, 1.58 mmol) and TiCl4·DME (0.211 g, 0.739 mmol) were added. The resulting solution was stirred overnight at room temperature. After the volatiles were removed using a vacuum line, the product was extracted with hexane (13 mL). The extract was collected by filtration over Celite. The solvent was removed using a vacuum line to obtain a yellowish-brown solid (0.133 g, 47%). 1H NMR (C6D6): δ 6.39 (q, 4J = 1.8 Hz, 1H), 2.20 (s, 3H, CH3), 2.19 (q, 4J = 1.8 Hz, 3H, CH3), 1.89 (s, 3H, CH3), 1.53 (s, 9H, tBu), 0.66 (s, 3H, CH3), 0.56 (s, 3H, CH3), 0.46 (s, 3H, CH3), and 0.25 (s, 3H, CH3) ppm. 13C{1H} NMR (C6D6): δ 147.2, 141.4, 137.2, 136.8, 122.1, 116.4, 88.2, 57.9, 55.8, 52.8, 34.7, 16.4, 15.6, 12.6, 4.6, and 4.2 ppm.
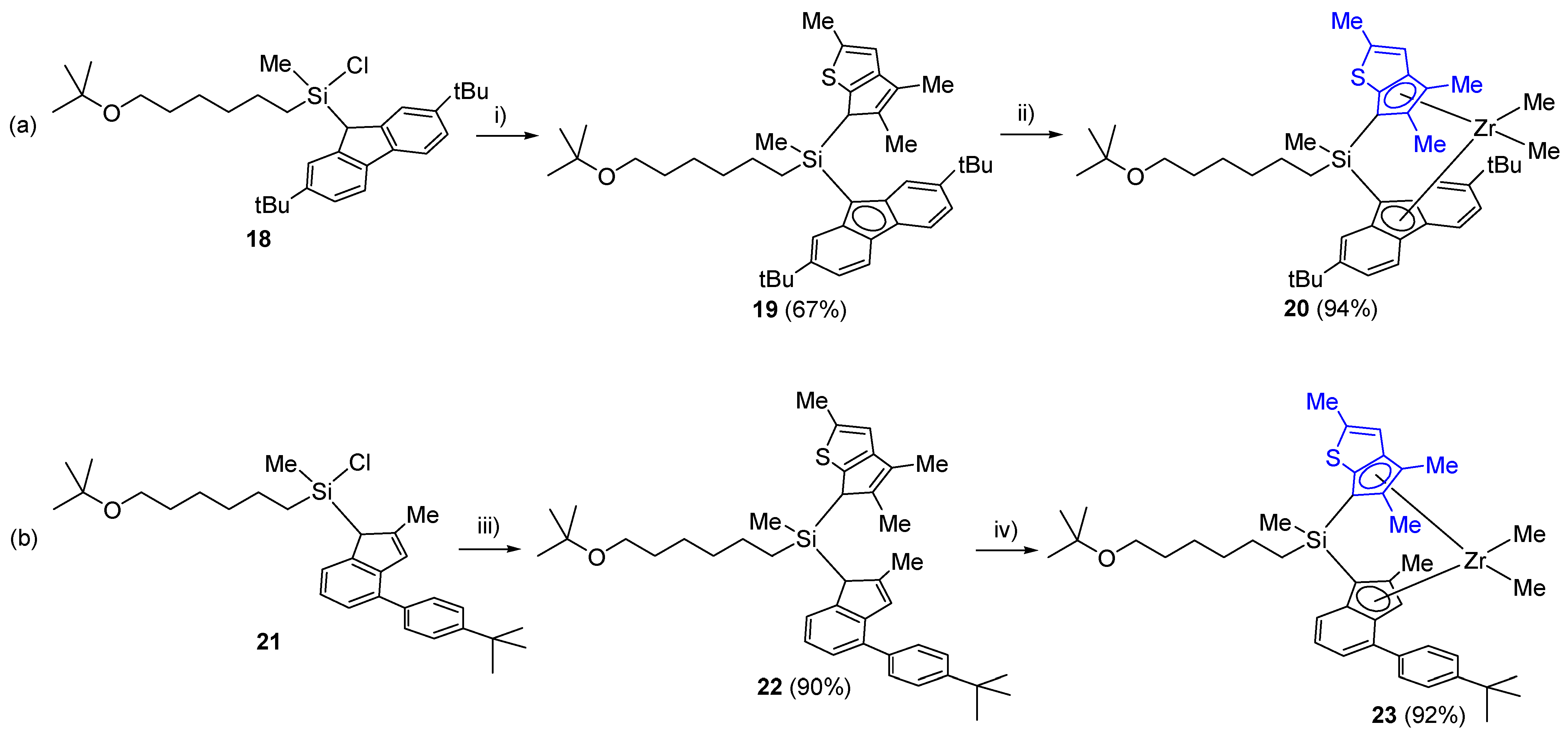
3.19. Synthesis of 18
A measured amount of tert-butyl methyl ether (0.886 g, 3 eq/Li+) was added to a solution of 2,7-di-tert-butylfluorenyl–Li (0.853 g, 3.00 mmol) in hexane (30 mL). After cooling to −78 °C, a solution of tBuO(CH2)6(Me)SiCl2 (0.814 g, 3.00 mmol) in hexane (7.5 mL) was added. After stirring overnight at room temperature, the solvent was removed under vacuum. The product was extracted using hexane (15 mL). The extract was collected by filtration over Celite. The solvent was removed using a vacuum line to obtain the desired compound (1.51 g, 98%). 1H NMR (C6D6): δ 7.89 (s, 1H), 7.81 (s, 1H), 7.77 (s, 1H), 7.76 (s, 1H), 7.42 (t, J = 2.07 Hz, 1H), 7.41 (t, J = 2.07 Hz, 1H), 1.50 (m, 2H), 1.39 and 1.37 (s, 18H, tBu), 1.27–1.02 (m, 6H), 1.13 (s, 9H, OtBu) 0.62–0.42 (m, 2H), and 0.09 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 149.4, 143.5, 139.0, 123.8, 122.3, 119.8, 72.0, 61.5, 43.1, 35.0, 33.2, 31.9, 30.9, 27.8, 26.3, 23.2, 15.7, and −1.5 ppm. HRMS(EI): m/z calcd. ((M+) C32H49ClOSi) 512.3240. Found: 512.3241.
3.20. Synthesis of 19
Thiophene-fused cyclopentadiene 2,4,5-Me
3C
7H
3S (0.325 g, 2.00 mmol) in THF (2.0 mL) was added to a Schlenk flask containing KH (0.104 g, 2.6 mmol) and THF (2.0 mL). After stirring overnight at room temperature, the solution was filtered over Celite to remove the remaining KH, owing to its addition in excess. To the filtrate were added CuCN (1.8 mg, 1 mol%) and a solution of
18 (1.02 g, 2.00 mmol) in THF (15 mL) cooled at −30 °C. The solution was stirred overnight, and the solvent was removed using a vacuum line. The product was extracted with hexane (30 mL) and collected by filtration over Celite. After removing the solvent using a vacuum line, the product was purified by silica gel column chromatography with hexane and diethyl ether (30:1,
v/
v) as eluents. A yellowish-brown, oily compound was obtained (0.854 g, 67%). The
1H NMR spectrum of the product was too complicated for complete analysis because of the mixture of the four diastereomers (
Figure S19). HRMS(EI):
m/
z calcd. ((M+) C
42H
60OSSi) 640.4138. Found: 640.4134.
3.21. Synthesis of 20
A solution of nBuLi in hexane (0.300 g, 2.5 M, 1.08 mmol) was added dropwise to a solution of 19 (0.347 g, 0.541 mmol) in THF (2 mL) at −30 °C, and the mixture was slowly warmed to room temperature. After the solution was stirred for 1 h at room temperature, the solution was cooled to −30 °C. MeMgBr (0.91 mL, 1.37 M solution in THF-toluene, 1.24 mmol). Next, ZrCl4·(THF)2 (0.200 g, 0.530 mmol) were successively added, and the solution was slowly warmed to room temperature. After stirring overnight, the solvent was removed under vacuum. The product was extracted with hexane (25 mL) and collected by filtration over Celite. The solvent was removed using a vacuum line to obtain a yellow solid (0.379 g, 94%). 1H NMR (C6D6): δ 7.97 and 7.95 (d, J = 1.4 Hz, 1H), 7.87 (d, J = 8.3 Hz, 1H), 7.79 and 7.78 (s, 1H), 7.64 and 7.62 (s, 1H), 7.51 and 7.50 (dd, 3J = 3.4 Hz, 4J = 1.4 Hz, 1H), 7.44 and 7.42 (dd, 3J = 8.7 Hz, 4J = 1.4 Hz, 1H), 6.23 (q, 4J = 1.4 Hz, 1H), 3.31 and 3.30 (q, J = 6.2 Hz, 2H), 2.13 (d, 4J = 1.4 Hz, 3H, SCCH3), 2.03 (s, 3H, CH3), 1.93 and 1.89 (s, 3H, CH3), 1.39, 1.36, 1.34, and 1.32 (s, 18H, tBu), 1.26 and 1.08 (s, 3H, Si–CH3), 1.15 and 1.14 (s, 9H, OtBu), −1.07 and −1.08 (s, 3H, ZrCH3), and −1.58 (s, 3H, ZrCH3) ppm. Anal. Calcd. (C44H64OSSiZr): C, 69.5; H, 8.48; and S, 4.31%. Found: C, 68.2; H, 8.04; and S, 4.08%.
3.22. Synthesis of 21
Compound 21 was synthesized using the same conditions and procedure as those for 18, with 2-methyl-4-(4-tert-butylphenyl)indenyl-Li (0.805 g, 3.00 mmol). A yellow oily compound was obtained in 98% yield (1.46 g). 1H NMR (C6D6): δ 7.58 (d, J = 2.1 Hz, 1H), 7.56 (d, J = 2.1 Hz, 1H), 7.50 and 7.42 (d, J = 7.6 Hz, 1H), 7.41 (d, J = 2.1 Hz, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.19 and 7.16 (t, J = 7.6 Hz, 1H), 6.89 (s, 1H), 3.47 and 3.44 (s, 1H), 3.26 and 3.21 (t, J = 6.2 Hz, 2H, OCH2), 2.12 and 2.10 (s, 3H, CH3), 1.57–1.47 (m, 2H, CH2), 1.40–1.20 (m, 6H, CH2), 1.30 (s, 9H, tBu), 1.15 and 1.12 (s, 9H, OtBu), 0.84–0.39 (m, 2H, CH2), 0.28 and 0.02 (s, 3H, SiCH3) ppm. HRMS(EI): m/z calcd. ((M+) C31H45ClOSi) 496.2930. Found: 496.2928.
3.23. Synthesis of 22
Compound
22 was synthesized using the same conditions and procedure as those for
19, with
21 (0.955 g, 2.00 mmol). A yellowish-brown oil was obtained in 90% yield (1.11 g). The
1H NMR spectrum of the product was too complicated for complete analysis because of the mixture of four diastereomers (
Figure S21). HRMS(EI):
m/
z calcd. ((M+) C
41H
56OSSi) 624.3818. Found: 624.3821.
3.24. Synthesis of 23
Compound 23 was synthesized using the same conditions and procedure as those for 20, with 22 (0.396 g, 0.633 mmol). A yellow oil was obtained in 92% yield (0.427 g). It was isolated as a mixture of four diastereomers owing to the presence of three chiral centers. Signals of racemic-type and meso-type complexes in a 1.0:0.40 (or vice versa) ratio were further split into 1:0.9 and 1:1 ratios, respectively, due to the chiral center on the Si atom. Signals assigned to minor meso-type complexes are indicated in italics. 1H NMR (C6D6): δ 7.90–7.87 and 7.86–7.84 (m, 2H, 4-tBuPh-H), 7.73, 7.70, 7.39, and 7.37 (d, J = 8.7 Hz, 1H), 7.49, 7.42, 7.29, and 7.27 (d, J = 8.3 Hz, 1H), 7.42–7.38 (m, 2H, 4-tBuPh-H), 7.31 and 7.29 (s, 1H, indenyl-H), 6.96-6.89 (m, 1H), 6.37 and 6.25 (m, 1H, thiophene-H), 3.33–3.29 (m, 2H, OCH2), 2.26, 2.24, 2.15, and 2.14 (s, 3 H, CH3), 2.19, 2.18, 2.09, and 2.08 (d, 4J = 1.4 Hz, 3H, SCCH3), 2.07, 2.03, and 2.00 (s, 3H, CH3), 1.88, 1.86, 1.84, and 1.78 (s, 3 H, indenyl-CH3), 1.75–1.25 (m, 10H, CH2), 1.24, 1.23, and 1.23 (s, 9H, tBu), 1.16, 1.16, 1.16, and 1.15 (s, 9H, tBu), 1.08, 0.89, 0.84, and 0.73 (s, 3H, SiCH3), 0.18 and −0.37 (s, 3H, Zr–CH3), −0.84, −0.85, −1.31, and −1.32 (s, 3H, Zr–CH3). Anal. Calcd. (C43H60OSSiZr): C, 69.4; H, 8.13; S, 4.31%. Found: C, 68.2; H, 7.77; and S, 4.47%.
3.25. Synthesis of 24
A measured amount of tert-butyl methyl ether (0.716 g, 3.3 eq/Li+) was added to a suspension of 2,7-di-tert-butylfluorenyl–Li (0.700 g, 2.46 mmol) in hexane (15 mL). After cooling to −78 °C, a solution of Cl2Si(Me)-(CH2)6-(Me)SiCl2 (0.384 g, 1.23 mmol) in hexane (5 mL) was added. After stirring overnight at room temperature, the solvent was removed under vacuum. The product was extracted using hexane (15 mL). The extract was collected by filtration over the Celite. The solvent was removed using a vacuum line to obtain a white solid (0.97 g, 99%). 1H NMR (C6D6): δ 7.88 (s, 2H), 7.79 (s, 2H), 7.77 (s, 2H), 7.76 (s, 2H), 7.42 (t, J = 2.07 Hz, 2H), 7.41 (t, J = 2.07 Hz, 2H), 4.01 (s, 2H), 1.39 and 1.37 (s, 36H, tBu), 1.08–0.91 (m, 4H), 0.85–0.80 (m, 4H), 0.51–0.32 (m, 4H), and 0.11 (s, 6H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 149.4, 143.4, 138.9, 123.8, 122.2, 119.8, 43.1, 35.1, 32.7, 31.9, 23.0, 15.5, and −1.4 ppm.
3.26. Synthesis of 25
Thiophene-fused cyclopentadiene 2,4,5-Me3C7H3S (0.284 g, 1.7 mmol) in THF (2.0 mL) was added to a Schlenk flask containing KH (0.090 g, 2.2 mmol) and THF (2.0 mL). After stirring overnight at room temperature, the solution was filtered over Celite to remove the remaining KH, because of its addition in excess. To the filtrate were added CuCN (0.8 mg, 1 mol%) and a solution of 24 (0.69 g, 0.87 mmol) in THF (7 mL) cooled at −30 °C. The solution was stirred overnight, and the solvent was removed using a vacuum line. The product was extracted with hexane (20 mL) and collected by filtration over Celite. A yellowish-brown, oily compound was obtained (0.818 g, 90%). 1H NMR (C6D6): δ 7.95 (m, 2H), 7.82 (m, 4H), 7.57 (m, 2H), 7.41 (m, 4H), 6.62 (m, 2H), 4.34 (m, 2H), 3.72 (m, 2H), 2.35 (m, 6H), 2.35 (m, 6H), 2.03 (s, 6H), 1.95 (m, 6H), 1.38 (m, 36H, tBu), 1.27 (m, 6H), 0.50 (m, 6H), −0.05 (m, 3H, SiCH3), and −0.27 (m, 3H, SiCH3) ppm.
3.27. Synthesis of 26
A solution of nBuLi in hexane (0.621 g, 2.5 M, 2.28 mmol) was added dropwise to a solution of 25 (0.600 g, 0.570 mmol) in THF (6 mL) at −30 °C, and the mixture was slowly warmed to room temperature. After the solution was stirred for 1 h at room temperature, the solution was cooled to −30 °C. MeMgBr (1.92 mL, 1.37 M solution in THF–toluene, 2.62 mmol) and ZrCl4·(THF)2 (0.421 g, 1.12 mmol) were successively added, and the solution was slowly warmed to room temperature. After stirring overnight, the solvent was removed under vacuum. The product was extracted with hexane (30 mL) and collected by filtration over Celite. The solvent was removed using a vacuum line to obtain a yellow oily compound (0.456 g, 62%). 1H NMR (C6D6): δ 7.95 (m, 2H), 7.86 (m, 2H), 7.79 (m, 2H), 7.64 (m, 2H), 7.50 (m, 2H), 7.42 (m, 2H), 6.23 (m, 2H), 2.13 (m, 6H), 2.03 (m, 6H), 1.92 (s, 6H), 1.35 (m, 36H, tBu), 1.27 (m, 3H, SiCH3), 1.10 (m, 3H, SiCH3), 1.65 (m, 6H), 1.25 (m, 6H), −1.08 (m, 3H, ZrCH3), and −1.58 (m, 3H, ZrCH3) ppm.
3.28. Preparation of Supported Catalysts
Methylaluminoxane (MAO, 5.0 g, GRACE, 10 wt.% in toluene) was added to a solution of silica (GRACE, SYLOPOL-2410, 1.0 g) in toluene (10 mL) and the solution was stirred at 70 °C for 3 h. The solid was isolated by filtration and washed with toluene (10 mL). The resulting MAO–silica was re-dispersed in toluene (10 m), following which the prepared complex (150 μmol) was added. After stirring at 70 °C for 1 h, the solid was isolated by filtration and washed with toluene (20 mL). The residual solvents were completely removed using a vacuum line to obtain the supported catalyst.
3.29. Ethylene/1-Hexene Polymerization
A bomb reactor (450 mL) was evacuated at 120 °C for 4 h and then purged with nitrogen gas. Hexane (300 mL) containing Et3Al (0.20 mL) was added to the reactor, and the mixture was stirred for 1 h at 80 °C. Subsequently, the solution was removed using a cannula. The reactor was evacuated to remove any residual solvents. Next, the reactor was evacuated again at 120 °C for 4 h and recharged with nitrogen gas at atmospheric pressure. The reactor was then recharged with hexane (300 mL) containing 1-hexene (3.0 mL), and the temperature was set to 80 °C. Et3Al (0.2 mL) dissolved in hexane (3.0 mL) was injected and, subsequently, fine particles of the prepared supported catalyst (6.0 mg) were injected into the reactor using a syringe dispersed in hexane (3.0 mL). Ethylene gas was charged to 20 bar for a few seconds. The polymerization was performed for 30 min while maintaining the temperature at 80 °C and a constant pressure of 20 bar by feeding ethylene gas continuously. After the reactor was cooled to room temperature, the remaining ethylene gas was vented off and the generated polymer particles were collected by filtration.
3.30. X-ray Crystallography
Specimens of suitable quality and size were selected, mounted, and centered in the X-ray beam using a video camera. Reflection data were collected at 100 K on an APEX II CCD area diffractometer (Bruker) using graphite-monochromated Mo Kα radiation (λ = 0.7107 Ǻ). The hemisphere of the reflection data was collected as φ and ω scan frames at 0.5° per frame and an exposure time of 10 s per frame. The cell parameters were determined and refined using the SMART program. Data reduction was performed using the SAINT software. The data were corrected for Lorentz and polarization effects. Empirical absorption correction was applied using the SADABS program. The structure was solved by direct methods and refined by the full matrix least-squares method using the SHELXTL package and the olex2 program with anisotropic thermal parameters for all non-hydrogen atoms. Crystallographic data for 9 (CCDC# 2130400) that were used in all calculations are as follows: C26.88H29.64Cl0.12SSiZr, M = 508.33, triclinic, a = 9.00050(10), b = 10.1721(2), c = 13.1285(2) Å, α = 90.6600(8)°, β = 103.7878(8)°, γ = 95.4360(8), V = 1161.36(3) Å3, space group P-1, Z = 2, and 4421 unique (R(int) = 0.0262). The final wR2 was 0.0799 (I > 2σ(I)).


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}