
Lignin Depolymerization in the Presence of Base, Hydrogenation Catalysts, and Ethanol


 ,
,
Abstract
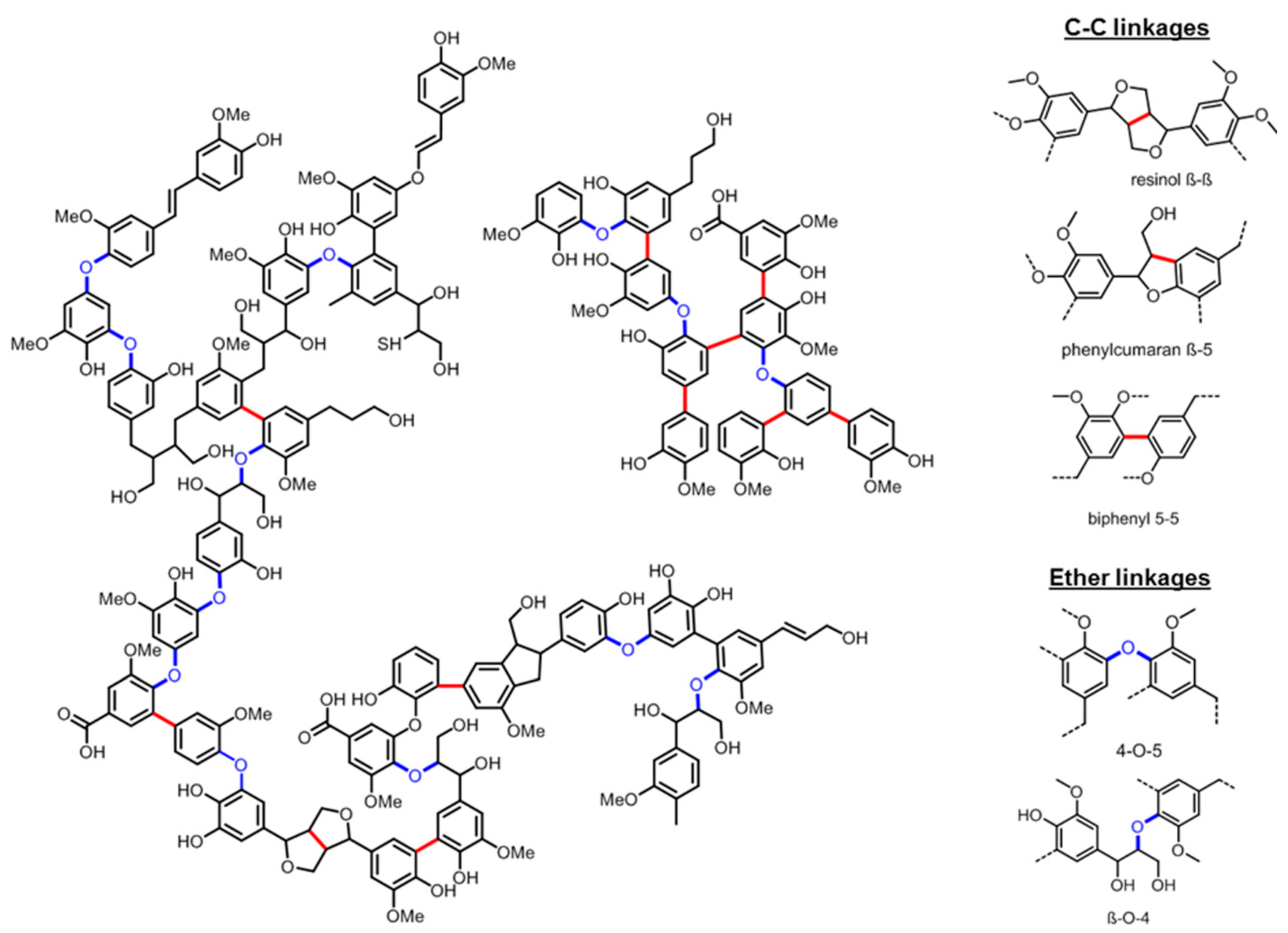
:1. Introduction
2. Results and Discussions
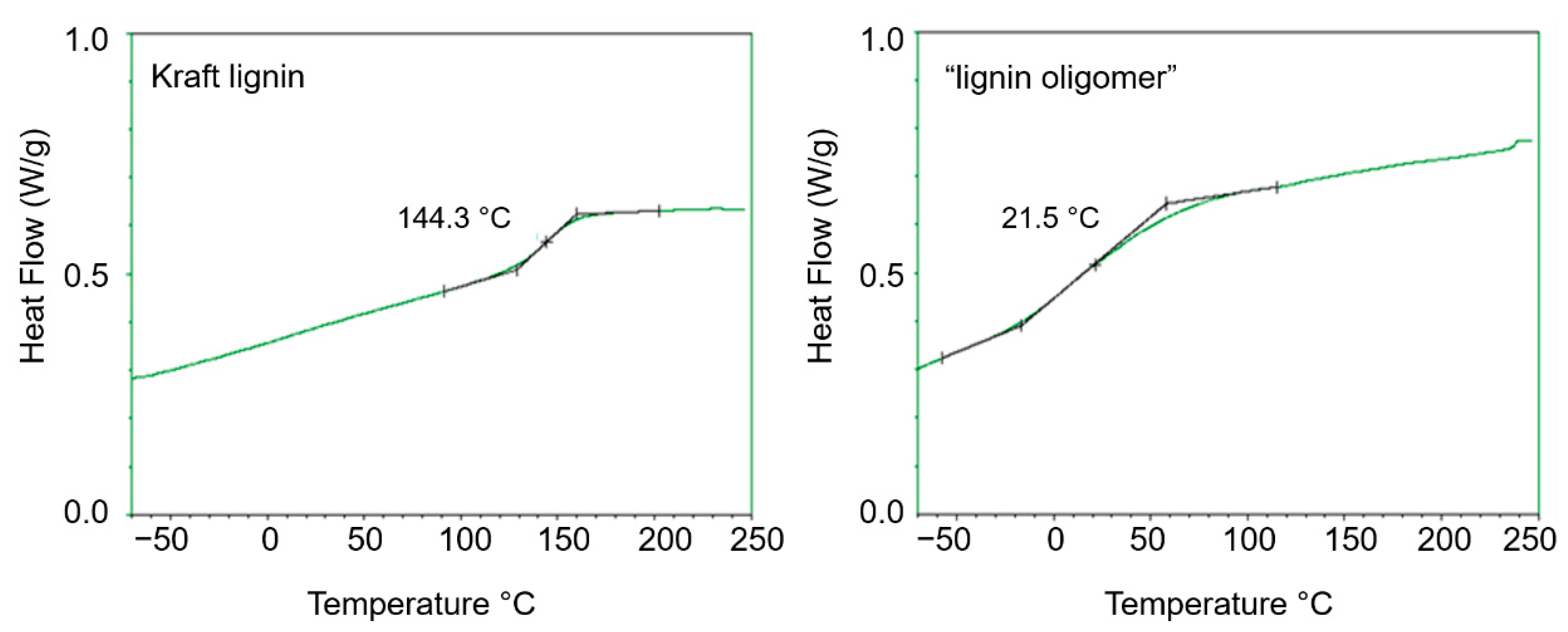
2.1. Kraft Lignin Depolymerization in Alkaline Media in the Presence of Ru/C Catalyst and Ethanol
2.2. Catalyst Screening
2.3. Effect of Alcohol Addition
2.4. Effect of Gas Nature and Pressure
2.5. Effect of Base Concentration
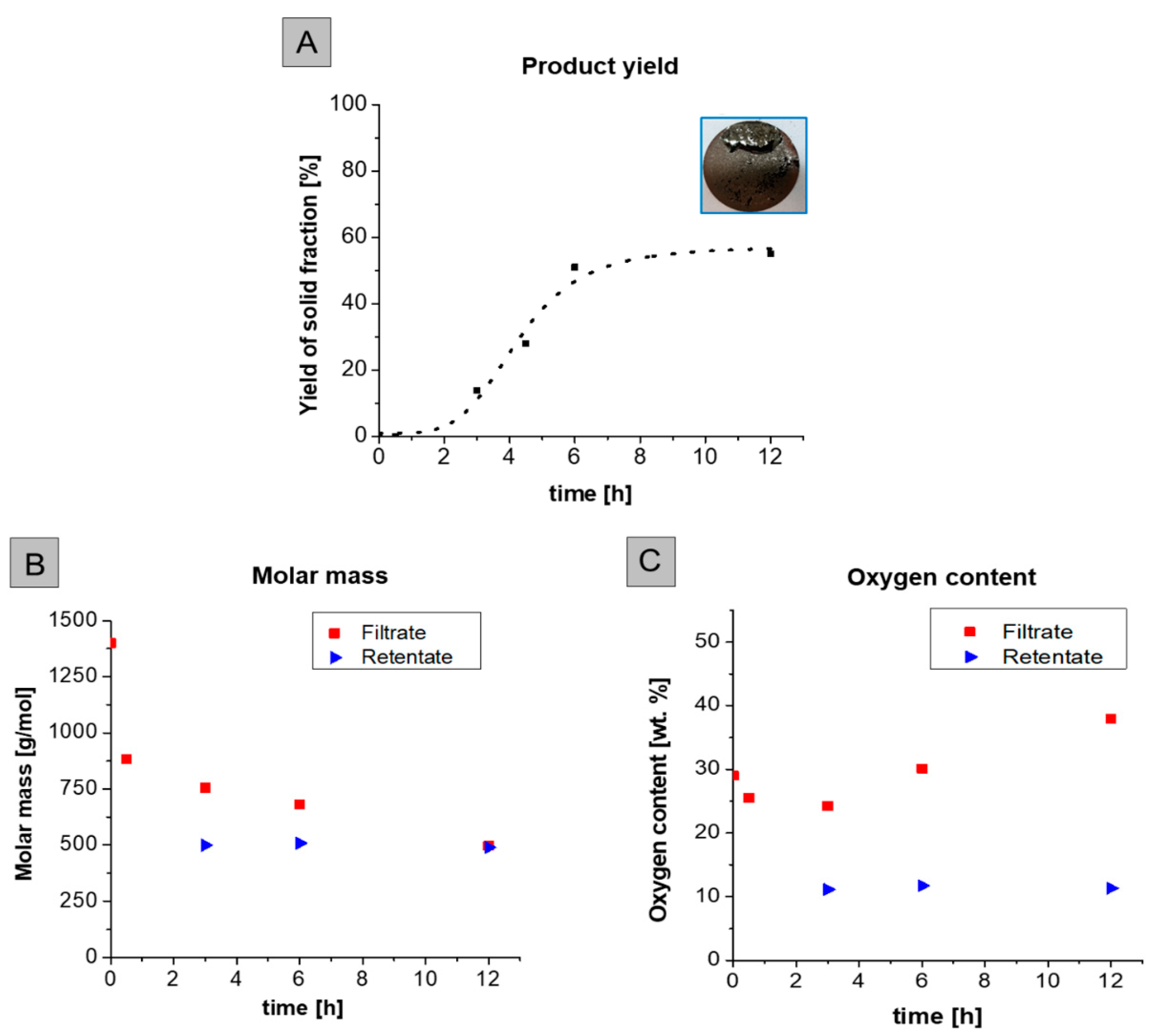
2.6. Effect of Reaction Time
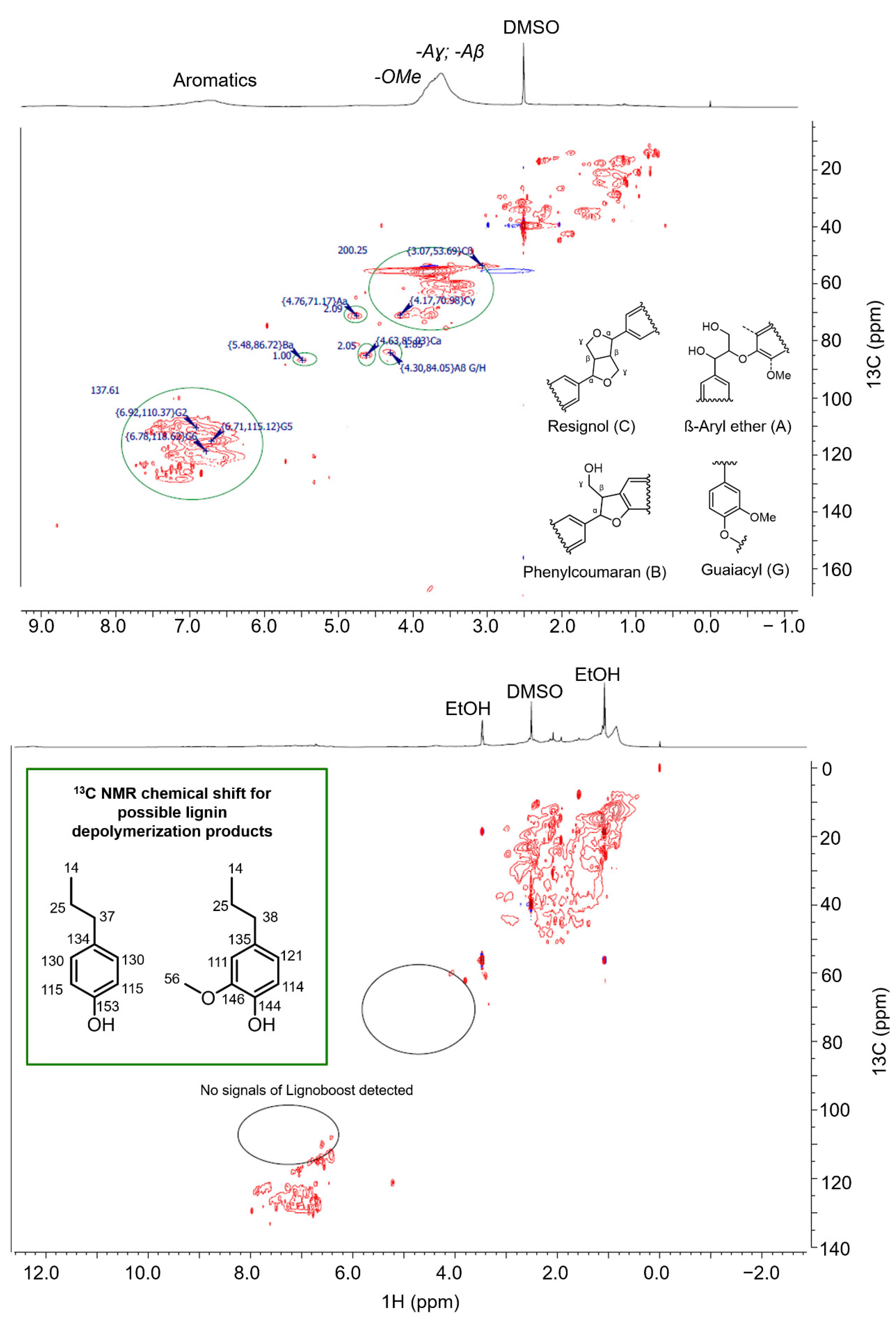
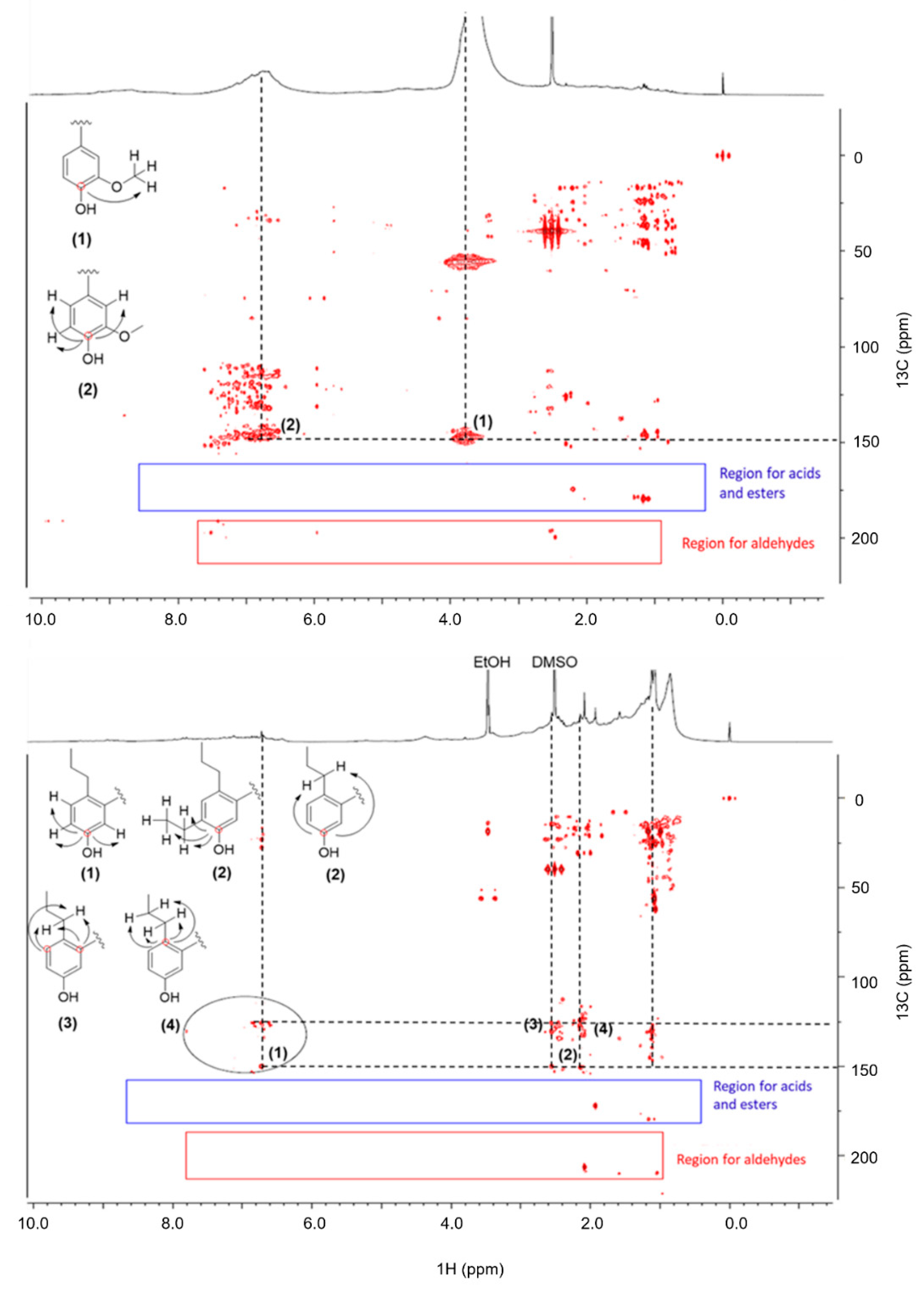
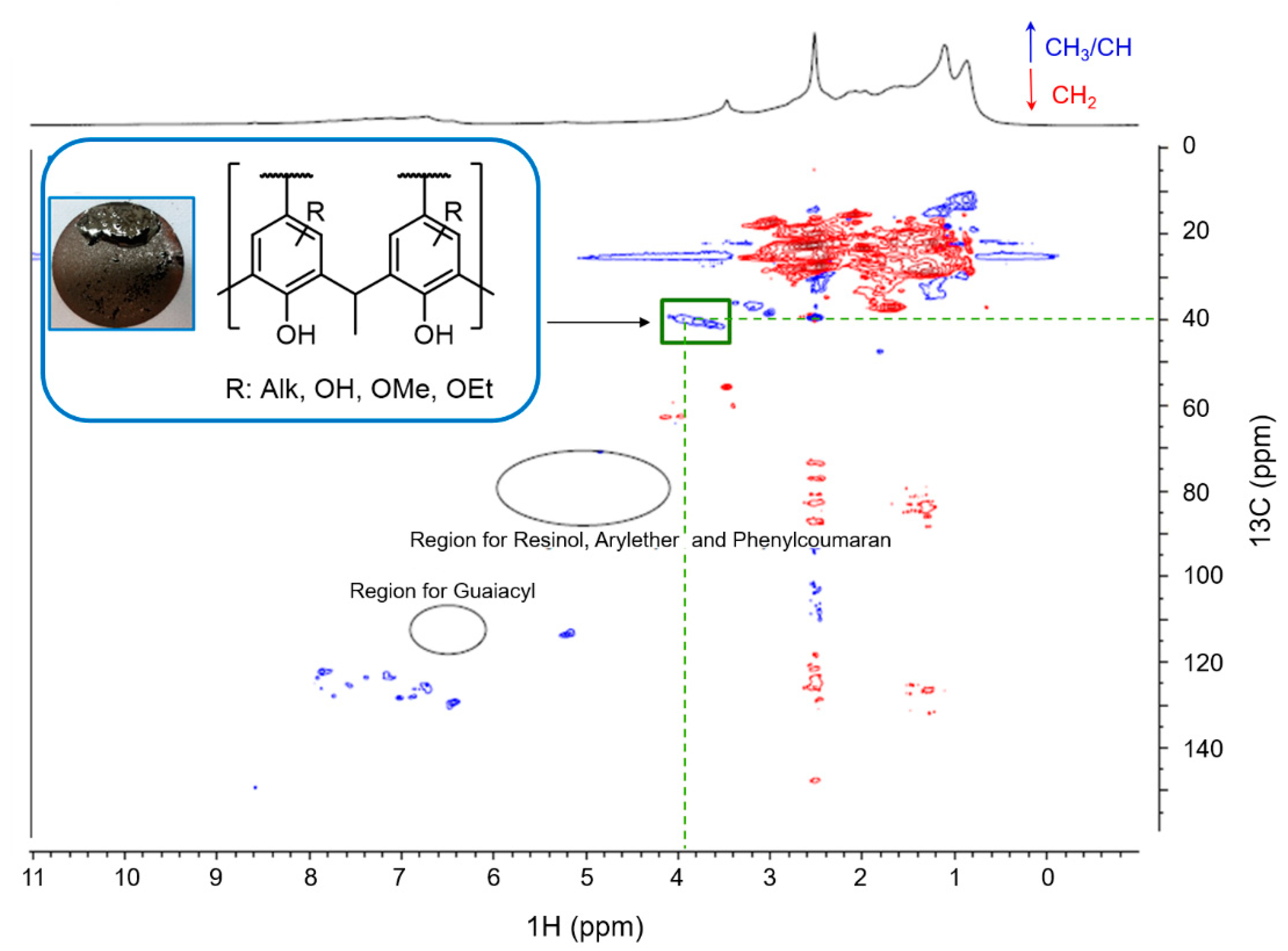
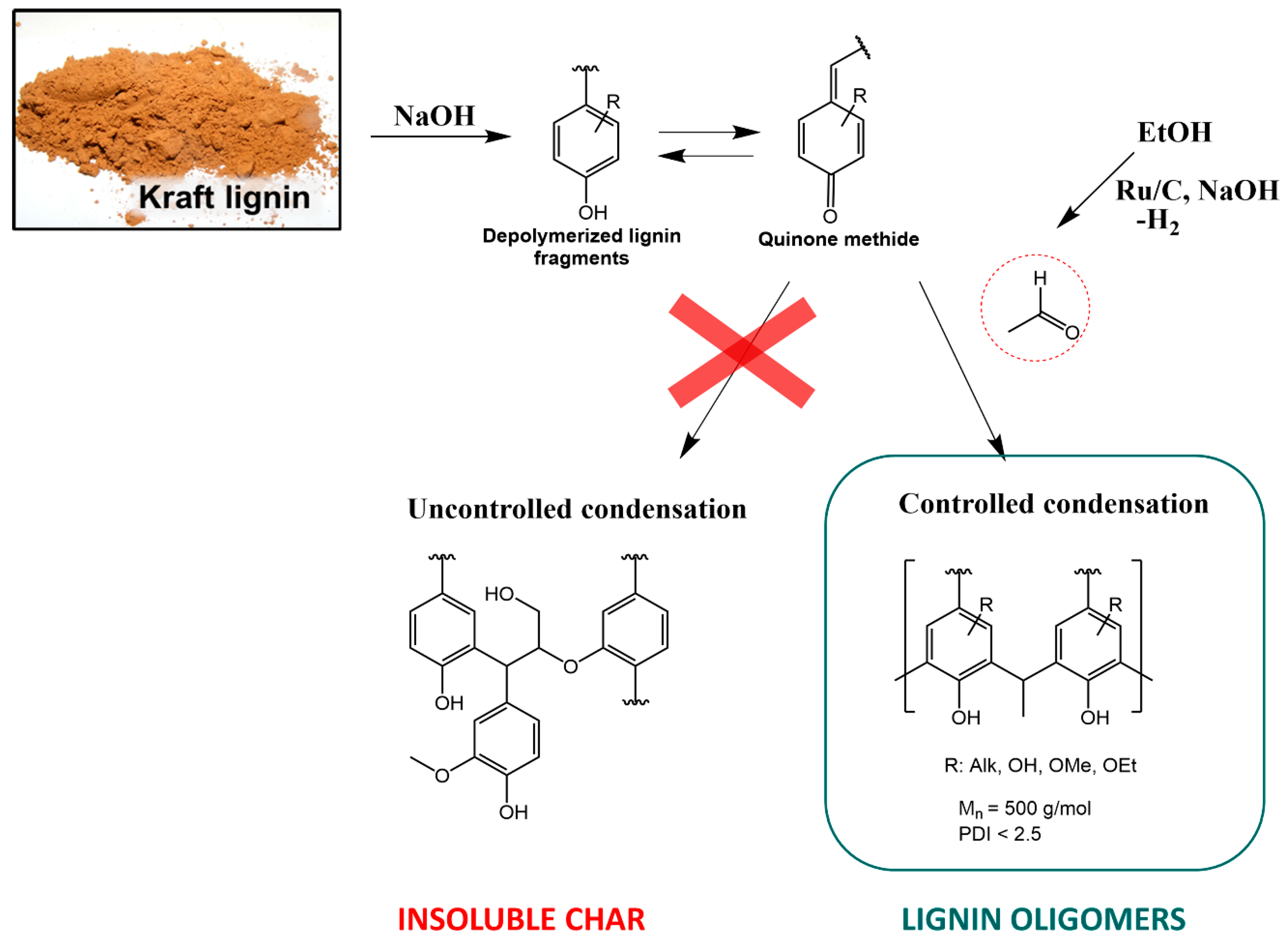
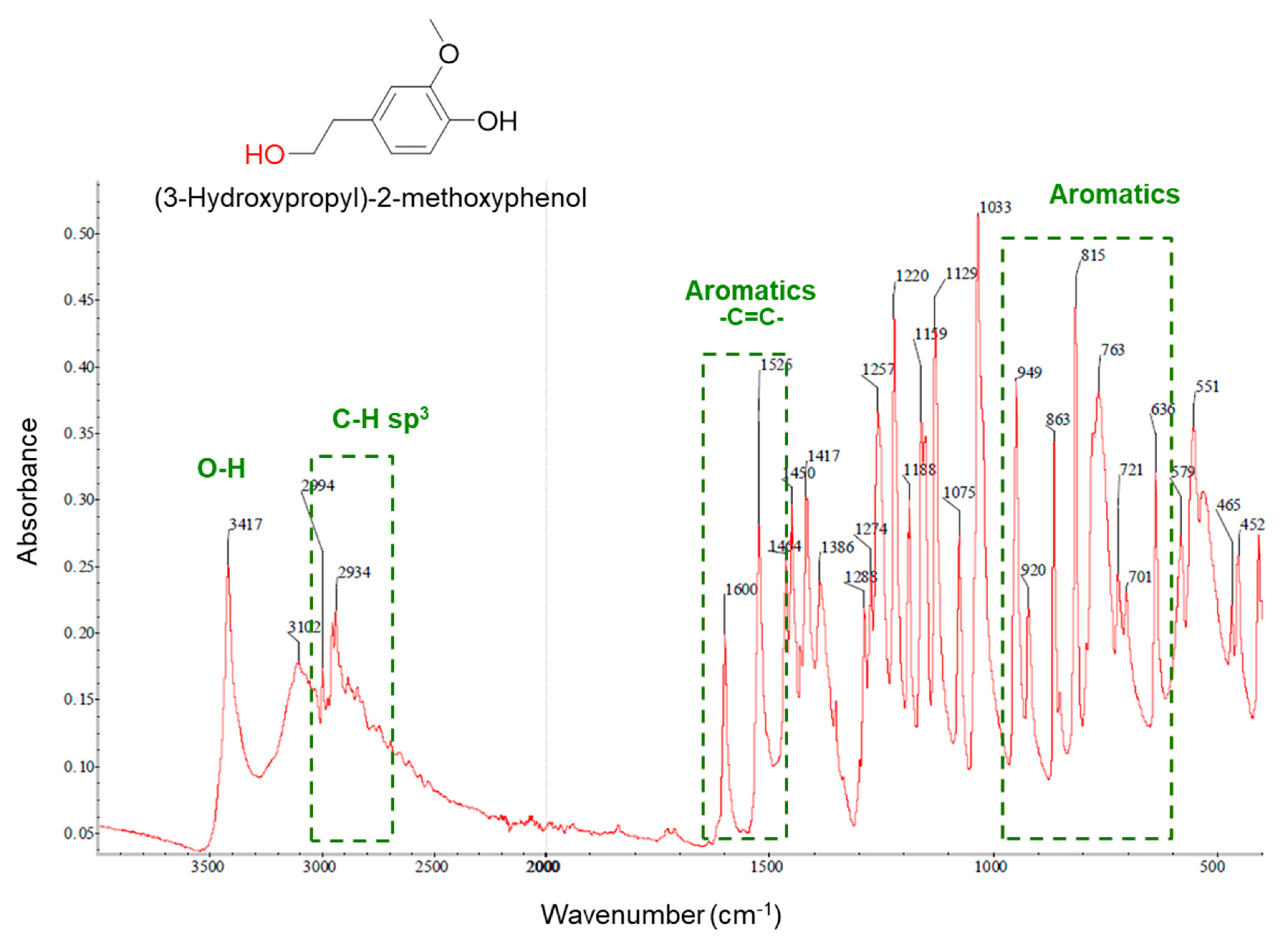
2.7. Elucidation of Reaction Mechanism
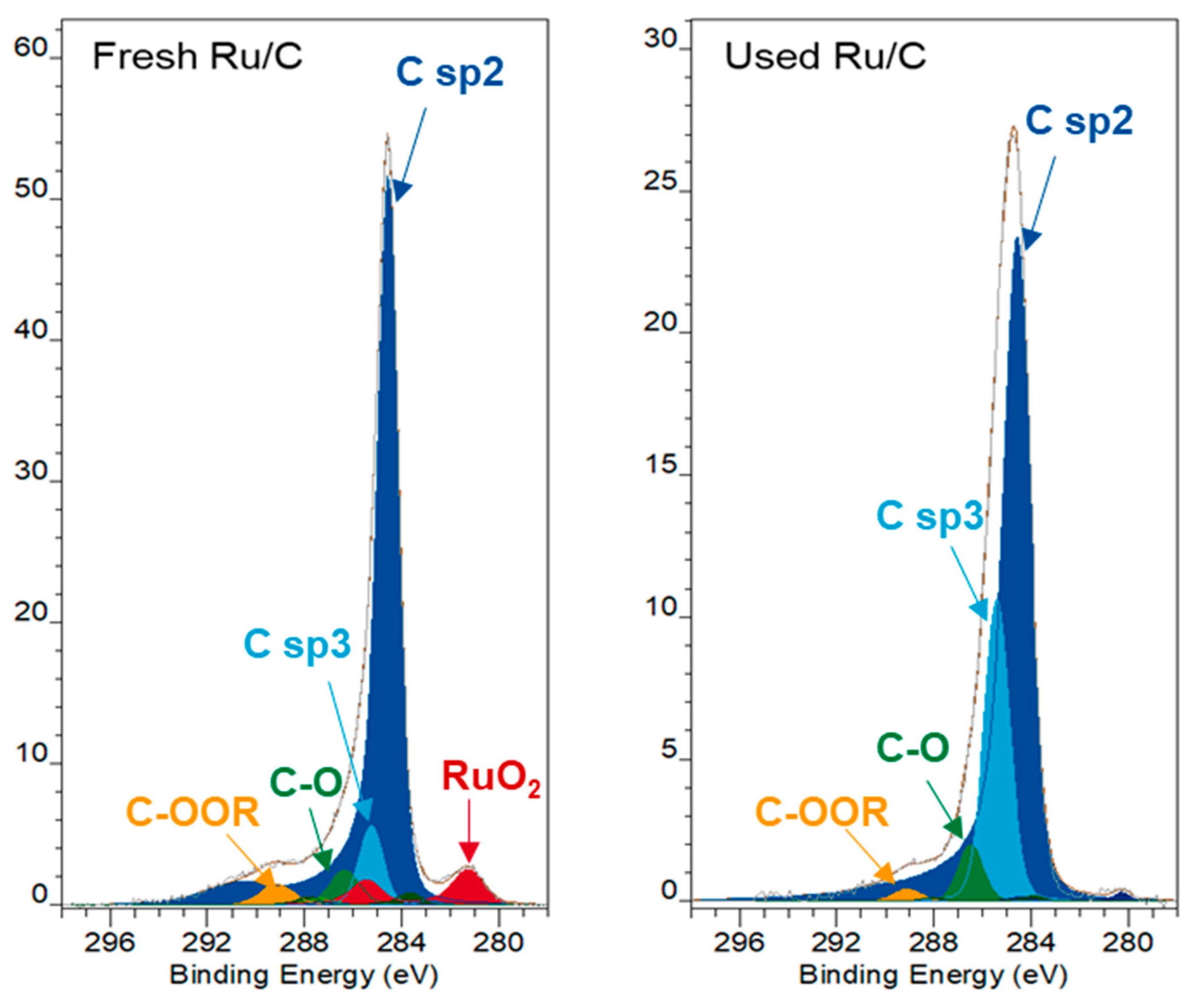
2.8. Recycling and Characterization of Spent Catalyst
3. Materials and Methods
3.1. General
3.2. Catalytic Depolymerization Process
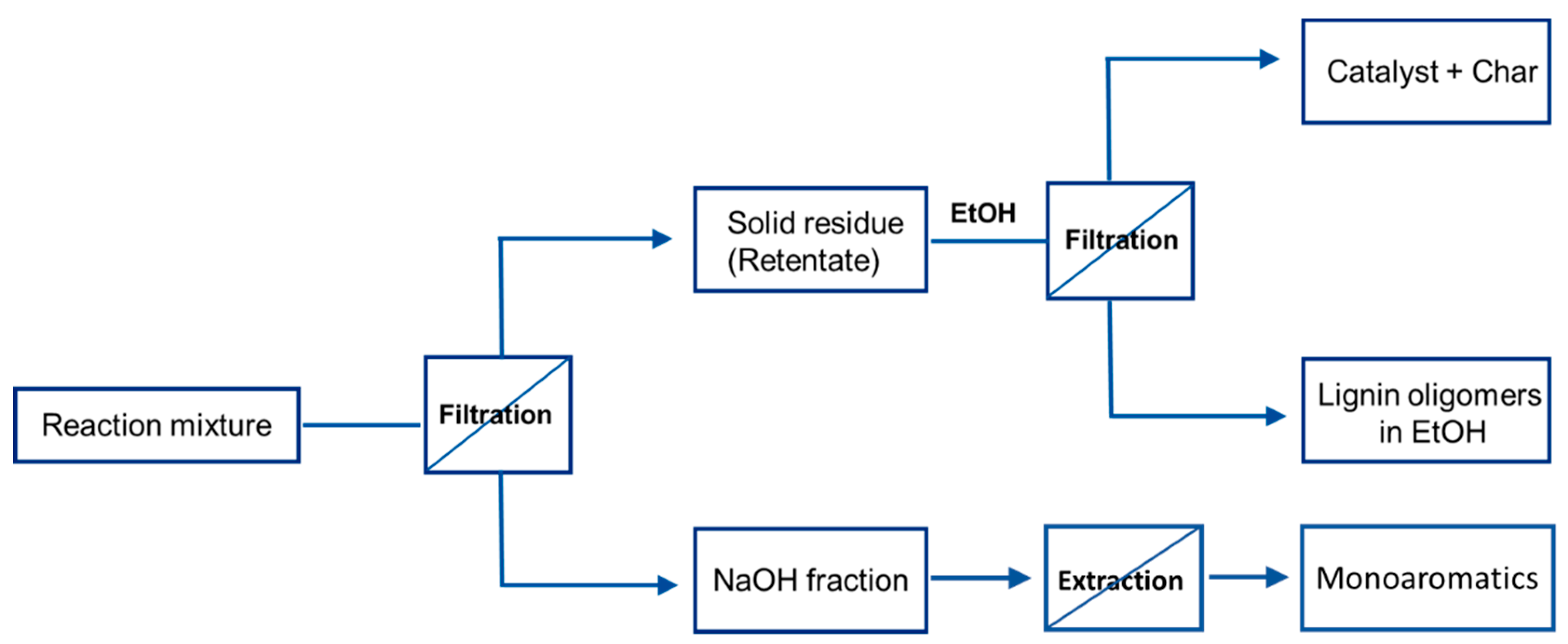
3.3. Product Work-Up
3.3.1. Recycling
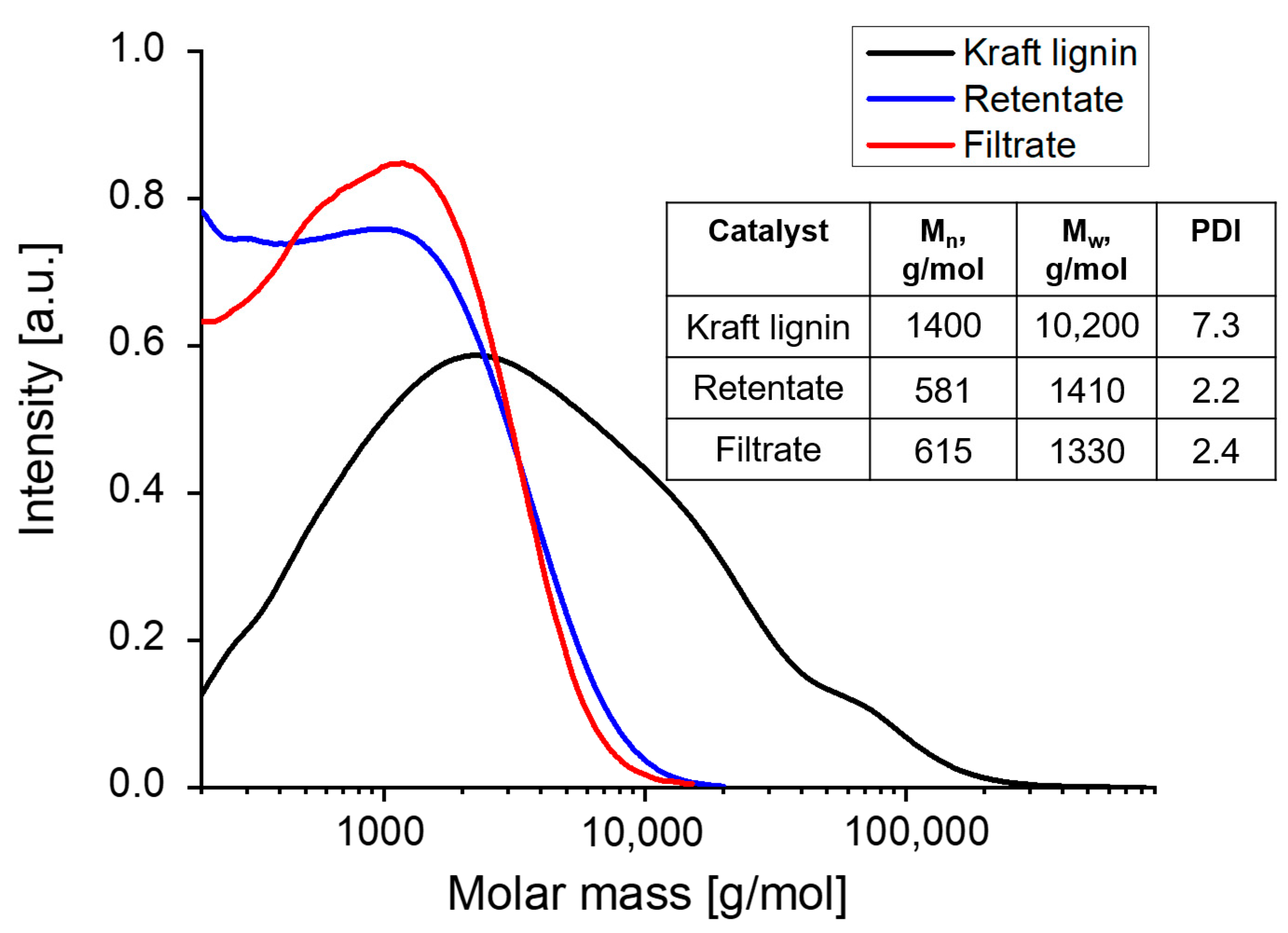
3.3.2. GPC Analysis
3.3.3. Elemental Analysis
3.3.4. GC-MS
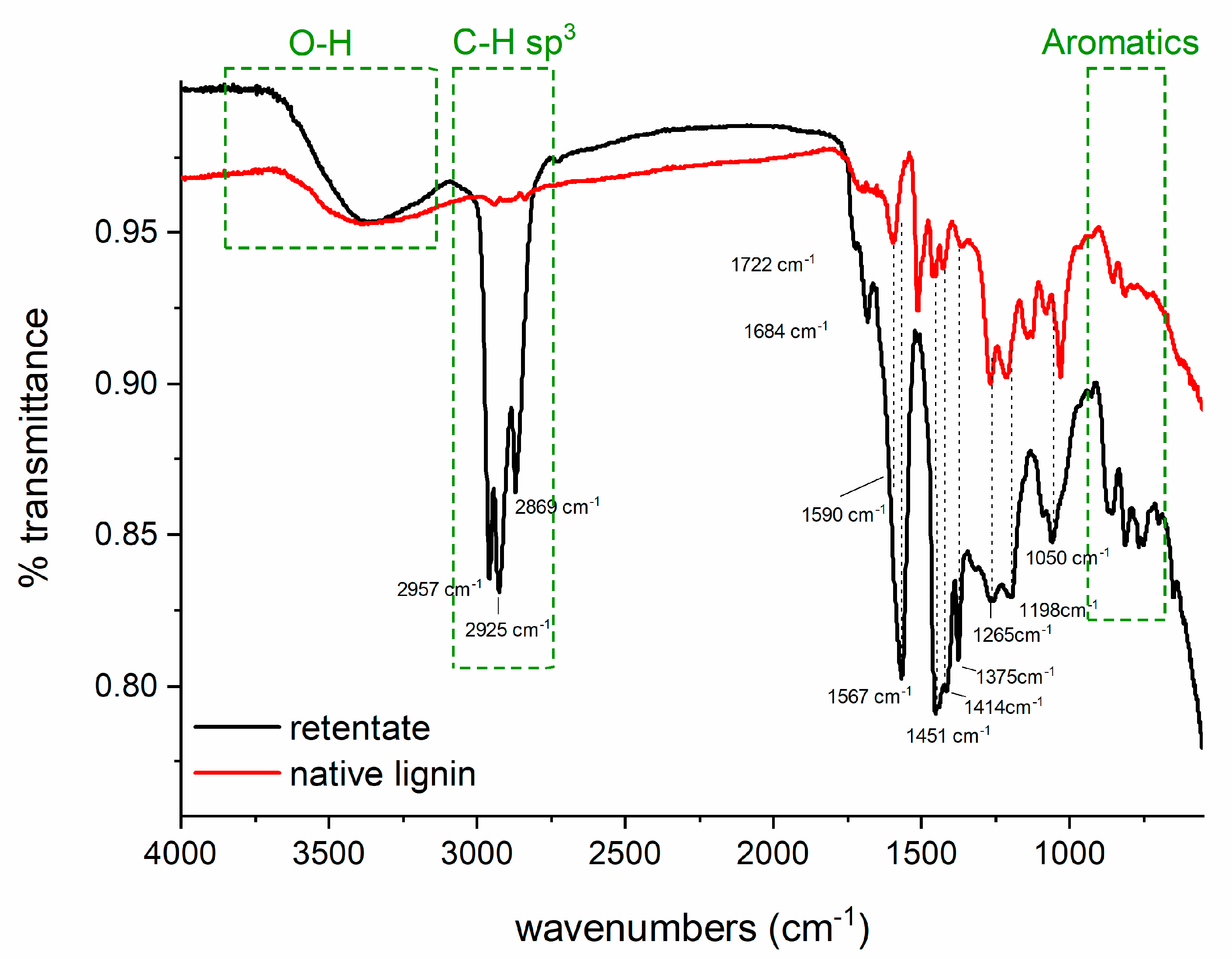
3.3.5. FTIR Analysis
3.3.6. NMR
3.3.7. XPS
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A




References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kärkäs, M.D.; Matsuura, B.S.; Monos, T.M.; Magallanes, G.; Stephenson, C.R.J. Transition-metal catalyzed valoriza-tion of lignin: The key to a sustainable car-bon-neutral future. Org. Biomol. Chem. 2016, 14, 1853–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.P.; Kim, C.S. Lignin Depolymerization and Conversion: A Re-view of Thermochemical Methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Berlin, A.; Balakshin, M. Industrial Lignins. In Bioenergy Research: Advances and Applications; Elsevier: Amsterdam, The Netherlands, 2014; pp. 315–336. [Google Scholar]
- Tomani, P. The Lignoboost Process. Cellul. Chem. Technol. 2010, 44, 53–58. [Google Scholar]
- Lancefield, C.S.; Wienk, H.L.J.; Boelens, R.; Weckhuysen, B.M.; Bruijnincx, P.C.A. Identification of a diagnostic structural motif reveals a new reaction intermediate and condensation pathway in kraft lignin formation. Chem. Sci. 2018, 9, 6348–6360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crestini, C.; Lange, H.; Sette, M.; Argyropoulos, D.S. On the structure of softwood kraft lignin. Green Chem. 2017, 19, 4104–4121. [Google Scholar] [CrossRef]
- Hunter-Cevera, K.R.; Neubert, M.G.; Olson, R.J.; Solow, A.R.; Shalapyonok, A.; Sosik, H.M. Physiological and eco-logical drivers of early spring blooms of a coastal phytoplankter. Science 2016, 354, 326–329. [Google Scholar] [CrossRef] [Green Version]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin bio-synthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Guo, T.; Liu, Y.; Kühn, F.E.; Wang, C.; Zhao, Z.K.; Xiao, J.; Li, C.; Zhang, T. Sustainable Production of Benzylamines from Lignin. Angew. Chem. Int. Ed. 2021, 60, 20666–20671. [Google Scholar] [CrossRef]
- Elangovan, S.; Afanasenko, A.; Haupenthal, J.; Sun, Z.; Liu, Y.; Hirsch, A.; Barta, K. From Wood to Tetrahydro-2-benzazepines in Three Waste-Free Steps Modular Synthesis of Biologically Active Lig-nin-Derived Scaffolds Enhanced Reader. ACS Cent. Sci. 2019, 5, 1707–1716. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Cao, D.; Qiu, Z.; Li, C.-J. Palladium-Catalyzed Formal Cross-Coupling of Diaryl Ethers with Amines: Slicing the 4-O -5 Linkage in Lignin Models. Angew. Chem. Int. Ed. 2018, 57, 3752–3757. [Google Scholar] [CrossRef] [PubMed]
- Katahira, R.; Mittal, A.; McKinney, K.; Chen, X.; Tucker, M.P.; Johnson, D.K.; Beckham, G.T. Base-Catalyzed Depolymerization of Biorefinery Lignins. ACS Sustain. Chem. Eng. 2016, 4, 1474–1486. [Google Scholar] [CrossRef]
- Du, X.; Li, J.; Lindström, M.E. Modification of industrial softwood kraft lignin using Mannich reaction with and without phenolation pretreatment. Ind. Crops Prod. 2014, 52, 729–735. [Google Scholar] [CrossRef]
- Deuss, P.J.; Scott, M.; Tran, F.; Westwood, N.J.; de Vries, J.G.; Barta, K. Aromatic monomers by in situ conversion of reactive intermediates in the acid-catalyzed depolymerization of lignin. J. Am. Chem. Soc. 2015, 137, 7456–7467. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Tymchyshyn, M.; Xu, C. Reductive Depolymerization of Kraft and Organosolv Lignin in Supercritical Acetone for Chemicals and Materials. ChemCatChem 2016, 8, 1968–1976. [Google Scholar] [CrossRef]
- Yu, X.; Wei, Z.; Lu, Z.; Pei, H.; Wang, H. Activation of lignin by selective oxidation: An emerging strategy for boost-ing lignin depolymerization to aromatics. Bioresour. Technol. 2019, 291, 121885. [Google Scholar] [CrossRef]
- Hanson, S.K.; Baker, R.T. Knocking on wood: Base metal complexes as catalysts for selective oxidation of lignin models and extracts. Acc. Chem. Res. 2015, 48, 2037–2048. [Google Scholar] [CrossRef]
- Adam, M.; Ocone, R.; Mohammad, J.; Berruti, F.; Briens, C. Kinetic Investigations of Kraft Lignin Pyrolysis. Ind. Eng. Chem. Res. 2013, 52, 8645–8654. [Google Scholar] [CrossRef]
- Jiang, G.; Nowakowski, D.J.; Bridgwater, A.V. Effect of the Temperature on the Composition of Lignin Pyrolysis Products. Energy Fuels 2010, 24, 4470–4475. [Google Scholar] [CrossRef]
- Kawamoto, H. Lignin pyrolysis reactions. J. Wood Sci. 2017, 63, 117–132. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.; Zhang, T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright Side of Lignin Depolymerization: Toward New Plat-form Chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schutyser, W.; Renders, T.; Van Den Bosch, S.; Koelewijn, S.-F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Dutta, T.; Walter, E.D.; Isern, N.G.; Cort, J.R.; Simmons, B.A.; Singh, S. Chemoselective Methylation of Phenolic Hydroxyl Group Prevents Quinone Methide Formation and Repolymerization During Lignin Depolymerization. ACS Sustain. Chem. Eng. 2017, 5, 3913–3919. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kim, C.S. Recent Efforts to Prevent Undesirable Reactions from Fractionation to Depolymerization of Lignin: Toward Maximizing the Value from Lignin. Front. Energy Res. 2018, 6, 170. [Google Scholar] [CrossRef] [Green Version]
- Klein, I.; Marcum, C.; Kenttämaa, H.; Abu-Omar, M.M. Mechanistic investigation of the Zn/Pd/C catalyzed cleavage and hydrodeoxygenation of lignin. Green Chem. 2016, 18, 2399–2405. [Google Scholar] [CrossRef]
- Zakzeski, J.; Jongerius, A.L.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Catalytic lignin valorization process for the pro-duction of aromatic chemicals and hydrogen. ChemSusChem 2012, 5, 1602–1609. [Google Scholar] [CrossRef]
- Shuai, L.; Sitison, J.; Sadula, S.; Ding, J.; Thies, M.C.; Saha, B. Selective C–C Bond Cleavage of Methylene-Linked Lignin Models and Kraft Lignin. ACS Catal. 2018, 8, 6507–6512. [Google Scholar] [CrossRef]
- Lv, W.; Si, Z.; Tian, Z.; Wang, C.; Zhang, Q.; Xu, Y.; Wang, T.; Ma, L. Synergistic Effect of EtOAc/H2O Biphasic Solvent and Ru/C Catalyst for Cornstalk Hydrol-ysis Residue Depolymerization. ACS Sustain. Chem. Eng. 2017, 5, 2981–2993. [Google Scholar] [CrossRef]
- Shen, X.-J.; Huang, P.-L.; Wen, J.-L.; Sun, R.-C. A facile method for char elimination during base-catalyzed depolymerization and hydrogenolysis of lignin. Fuel Process. Technol. 2017, 167, 491–501. [Google Scholar] [CrossRef]
- Long, J.; Xu, Y.; Wang, T.; Yuan, Z.; Shu, R.; Zhang, Q.; Ma, L. Efficient base-catalyzed decomposition and in situ hydrogenolysis process for lignin depolymerization and char elimination. Appl. Energy 2015, 141, 70–79. [Google Scholar] [CrossRef]
- Hossain, A.; Phung, T.K.; Rahaman, M.S.; Tulaphol, S.; Jasinski, J.B.; Sathitsuksanoh, N. Catalytic cleavage of the β-O-4 aryl ether bonds of lignin model compounds by Ru/C catalyst. Appl. Catal. A Gen. 2019, 582, 117100. [Google Scholar] [CrossRef]
- Wang, X.; Rinaldi, R. Solvent effects on the hydrogenolysis of diphenyl ether with Raney nickel and their implications for the conversion of lignin. ChemSusChem 2012, 5, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Barta, K.; Matson, T.D.; Fettig, M.L.; Scott, S.L.; Iretskii, A.V.; Ford, P.C. Catalytic disassembly of an organosolv lignin via hydrogen transfer from supercritical methanol. Green Chem. 2010, 12, 1640. [Google Scholar] [CrossRef] [Green Version]
- Macala, G.S.; Matson, T.D.; Johnson, C.L.; Lewis, R.S.; Iretskii, A.V.; Ford, P.C. Hydrogen Transfer from Supercritical Methanol over a Solid Base Catalyst: A Model for Lignin Depolymerization. ChemSusChem 2009, 2, 215–217. [Google Scholar] [CrossRef]
- Matson, T.D.; Barta, K.; Iretskii, A.V.; Ford, P.C. One-pot catalytic conversion of cellulose and of woody biomass solids to liquid fuels. J. Am. Chem. Soc. 2011, 133, 14090–14097. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Catalytic depolymerization of lignin in supercritical ethanol. ChemSusChem 2014, 7, 2276–2288. [Google Scholar] [CrossRef]
- Huang, X.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics. Green Chem. 2015, 17, 4941–4950. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-Y.; Park, J.; Hwang, H.; Kim, J.K.; Song, I.K.; Choi, J.W. Catalytic de-polymerization of lignin macromolecule to alkylated phenols over various metal cata-lysts in supercritical tert-butanol. J. Anal. Appl. Pyrolysis 2015, 113, 99–106. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Park, J.; Kim, U.-J.; Choi, J.W. Conversion of Lignin to Phenol-Rich Oil Fraction under Supercritical Alcohols in the Presence of Metal Catalysts. Energy Fuels 2015, 29, 5154–5163. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Yagi, T.; Shinohara, S.; Fukunaga, T.; Nakasaka, Y.; Tago, T.; Masuda, T. Production of phenols from lignin via depolymerization and catalytic cracking. Fuel Process. Technol. 2013, 108, 69–75. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, Y.; Wu, K. Role of Co-Solvents in Biomass Conversion Reactions Using Sub/Supercritical Water. In Near-Critical and Supercritical Water and Their Applications for Biorefineries; Fang, Z., Xu, C., Eds.; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar]
- Sebhat, W. Valorisation de la Lignine par Catalyse Hétérogène en Condition Sous-Critique en Milieux Aqueux et Eau/Alcool. PhD. Thesis, Université Claude Bernard—Lyon I, Lyon, France, 2016. [Google Scholar]
- Saisu, M.; Sato, T.; Watanabe, M.; Adschiri, T.; Arai, K. Conversion of Lignin with Supercritical Water−Phenol Mixtures. Energy Fuels 2003, 17, 922–928. [Google Scholar] [CrossRef]
- Roberts, V.M.; Stein, V.; Reiner, T.; Lemonidou, A.; Li, X.; Lercher, J.A. To-wards quantitative catalytic lignin depolymerization. Chemistry 2011, 17, 5939–5948. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Umetsu, M.; Takami, S.; Adschiri, T. Disassembly of lignin and chemical recovery—rapid depolymerization of lignin without char formation in wa-ter–phenol mixtures. Fuel Process. Technol. 2004, 85, 803–813. [Google Scholar] [CrossRef]
- Lee, H.; Jae, J.; Ha, J.-M.; Suh, D.J. Hydro- and solvothermolysis of kraft lignin for maximizing production of monomeric aromatic chemicals. Bioresour. Technol. 2016, 203, 142–149. [Google Scholar] [CrossRef]
- Kang, S.; Li, X.; Fan, J.; Chang, J. Hydrothermal conversion of lignin: A review. Renew. Sustain. Energy Rev. 2013, 27, 546–558. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhou, Z.; Alma, M.H.; Sun, D.; Zhang, W.; Jiang, J. Direct Liquefaction of Alkali Lignin in Methanol and Water Mixture for the Production of Oligomeric Phenols and Aromatic Ethers. J. Biobased Mater. Bioenergy 2016, 10, 76–80. [Google Scholar] [CrossRef]
- Cheng, S.; Wilks, C.; Yuan, Z.; Leitch, M.; Xu, C. Hydrothermal degradation of alkali lignin to bio-phenolic compounds in sub/supercritical ethanol and water–ethanol co-solvent. Polym. Degrad. Stab. 2012, 97, 839–848. [Google Scholar] [CrossRef]
- Belkheiri, T.; Andersson, S.-I.; Mattsson, C.; Olausson, L.; Theliander, H.; Vamling, L. Hydrothermal liquefaction of kraft lignin in sub-critical water: The influence of the sodium and potassium fraction. Biomass Convers. Biorefin. 2018, 8, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Belkheiri, T.; Vamling, L.; Nguyen, T.; Maschietti, M.; Olausson, L.; Andersson, S.-I.; Amand, L.-E.; Theliander, H. Kraft lignin depolymerization in near-critical water: Effect of changing co-solvent. Cellul. Chem. Technol. 2014, 48, 813–818. [Google Scholar]
- Arturi, K.R.; Strandgaard, M.; Nielsen, R.P.; Søgaard, E.G.; Maschietti, M. Hydrothermal liquefaction of lignin in near-critical water in a new batch reactor: In-fluence of phenol and temperature. J. Supercrit. Fluids 2017, 123, 28–39. [Google Scholar] [CrossRef]
- Kaiho, A.; Kogo, M.; Sakai, R.; Saito, K.; Watanabe, T. In situ trapping of enol intermediates with alcohol during acid-catalysed de-polymerisation of lignin in a nonpolar solvent. Green Chem. 2015, 17, 2780–2783. [Google Scholar] [CrossRef]
- Wang, X.; Wang, N.; Nguyen, T.T.; Qian, E.W. Catalytic Depolymerization of Lignin in Ionic Liquid Using a Continuous Flow Fixed-Bed Reaction System. Ind. Eng. Chem. Res. 2018, 57, 16995–17002. [Google Scholar] [CrossRef]
- Gabriëls, D.; Hernández, W.Y.; Sels, B.; van der Voort, P.; Verberckmoes, A. Review of catalytic systems and thermodynamics for the Guerbet condensation reaction and challenges for biomass valorization. Catal. Sci. Technol. 2015, 5, 3876–3902. [Google Scholar] [CrossRef] [Green Version]
- Panchenko, V.N.; Paukshtis, E.A.; Murzin, D.Y.; Simakova, I.L. Solid Base Assisted n -Pentanol Coupling over VIII Group Metals: Elucidation of the Guerbet Reaction Mechanism by DRIFTS. Ind. Eng. Chem. Res. 2017, 56, 13310–13321. [Google Scholar] [CrossRef]
- Granata, A.; Argyropoulos, D.S. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane,a Reagent for the Accurate Determination of the Uncondensed and Condensed Phenolic Moieties in Lignins. J. Agric. Food Chem. 1995, 43, 1538–1544. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
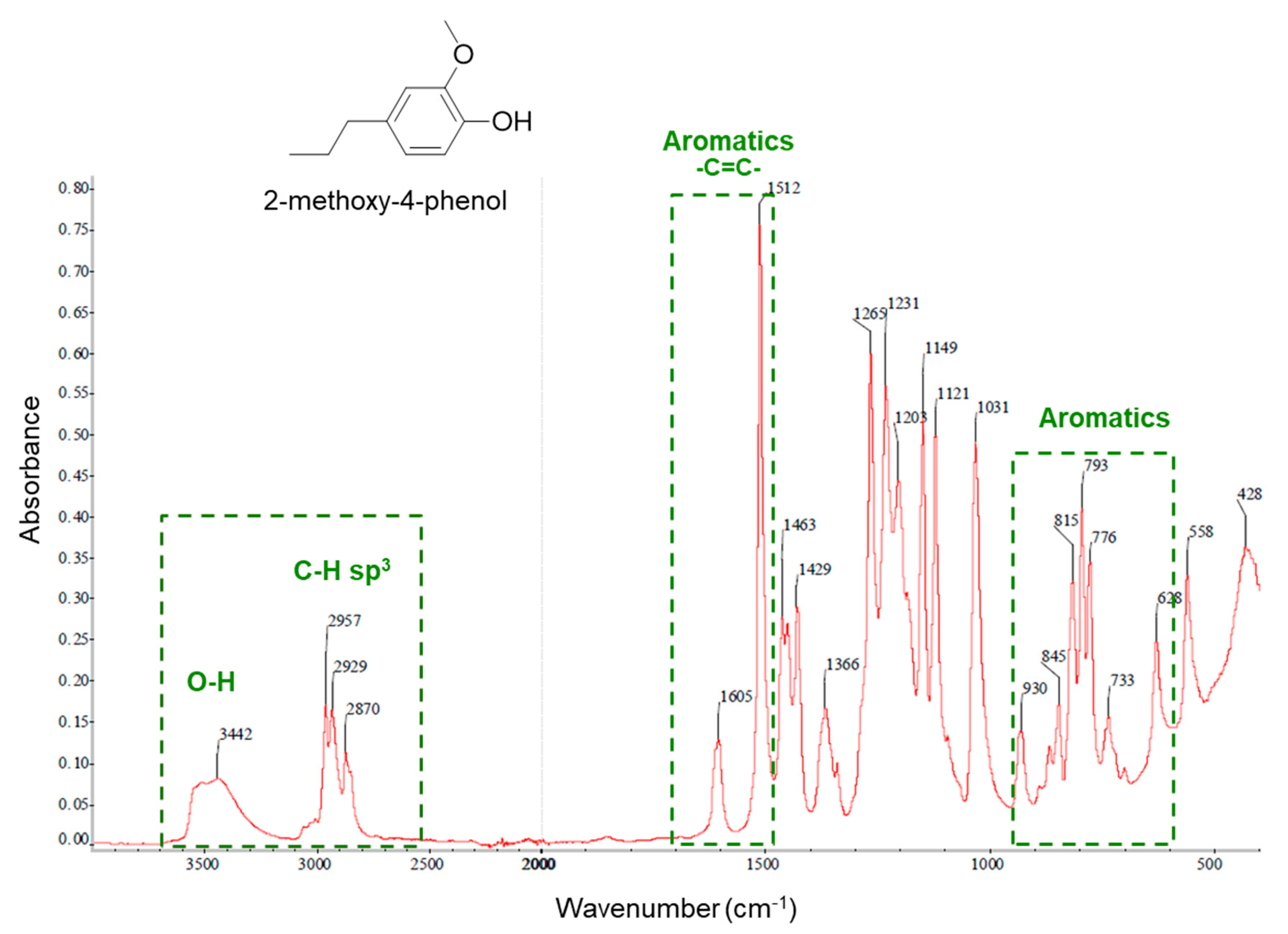{kind=link}
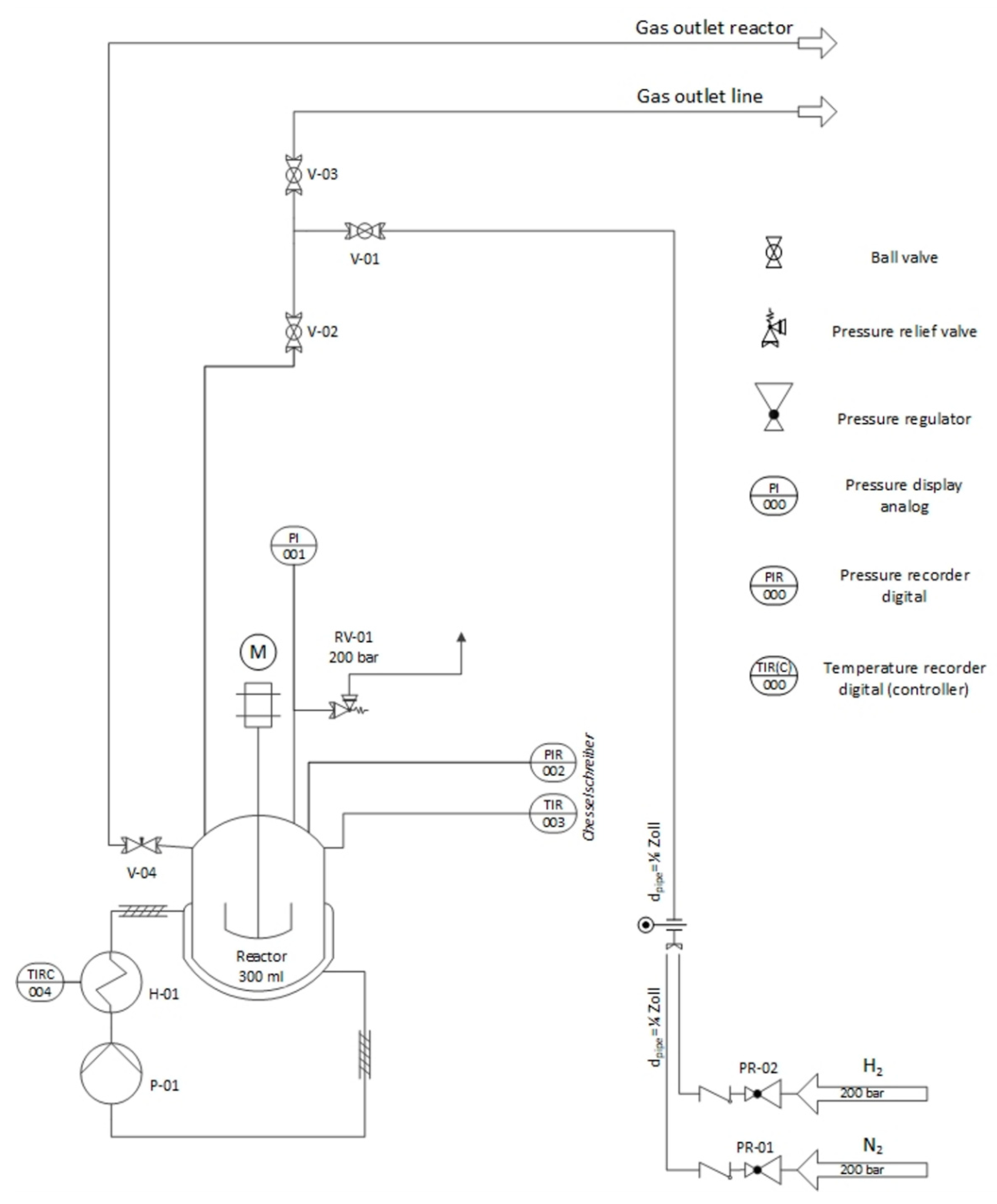{kind=link}
| Entry | Sample | C (wt.%) | O (wt.%) | H (wt.%) | O/C |
|---|---|---|---|---|---|
| 1 | Kraft lignin | 64.3 | 29.7 | 6.0 | 0.46 |
| 2 | Retentate | 78.7 | 13.3 | 8.0 | 0.17 |
| 3 | Filtrate | 57.5 | 35.7 | 6.8 | 0.62 |
| Entry | Catalyst | Cat. Amount, (wt.%) | Mn (g mol−1) | Mw (g mol−1) | PDI | Yield of Retentate (wt.%) |
|---|---|---|---|---|---|---|
| 1 | Ru/C | 0.01 | 539 ± 31 | 1247 ± 116 | 2.1 ± 0.1 | 49 ± 09 |
| 2 | Raney Ni | 0.01 | 385 | 632 | 1.6 | 34 |
| 3 | Ni NPs | 0.01 | 519 | 1120 | 2.2 | 48 |
| Entry | Alcohol | NaOH (aq) Content | Soluble Retentate | Char Formation |
|---|---|---|---|---|
| 1 | - | 90 wt.% | no | yes |
| 2 | 10 wt.% EtOH | 80 wt.% | yes (49 ± 09 wt.%) | no |
| 3 | 45 wt.% EtOH | 45 wt.% | yes | no |
| 5 | 10 wt.% MeOH | 80 wt.% | yes (18 wt.%) | no |
| 6 | 10 wt.% 2-Propanol | 80 wt.% | no | no |
| 7 | 10 wt.% t-BuOH | 80 wt.% | no | no |
| 8 | 10 wt.% n-Hexanol | 80 wt.% | no * | no |
| Entry | Gas | Pressure | Mn, g mol−1 | Mw, g mol−1 | PDI | Yield of Retentate, % |
|---|---|---|---|---|---|---|
| 1 | H2 | 30 bar (120 barg) | 539 ± 31 | 1247 ± 116 | 2.1 ± 0.1 | 49 ± 09 |
| 2 | N2 | 30 bar (120 barg) | 550 | 1290 | 2.3 | 70 |
| 3 | N2 | 1 bar (90 barg) | 678 | 1680 | 2.5 | 70 |
| 4 | H2 + N2 (1:1) | 30 bar (120 barg) | 542 | 1270 | 2.4 | 41 |
| Entry | Base | Lignin Content | Mn, g mol−1 | Mw, g mol−1 | PDI | O/C | Yield of Retentate, % |
|---|---|---|---|---|---|---|---|
| 1 | 1.0 M NaOH | 10 wt.% | 527 | 1170 | 2.2 | 0.17 | 59 |
| 2 | 0.3 M NaOH | 10 wt.% | 480 | 956 | 2.0 | 0.17 | 60 |
| Entry | Cycle | Mn, g mol−1 | Mw, g mol−1 | PDI | O/C | Yield of Retentate, % |
|---|---|---|---|---|---|---|
| 1 | I | 678 | 1680 | 2.5 | 0.18 | 70 |
| 2 | II | 504 | 1060 | 2.1 | 0.14 | 66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanenko, I.; Kurz, F.; Baumgarten, R.; Jevtovikj, I.; Lindner, J.-P.; Kundu, A.; Kindler, A.; Schunk, S.A. Lignin Depolymerization in the Presence of Base, Hydrogenation Catalysts, and Ethanol. Catalysts 2022, 12, 158. https://doi.org/10.3390/catal12020158
Romanenko I, Kurz F, Baumgarten R, Jevtovikj I, Lindner J-P, Kundu A, Kindler A, Schunk SA. Lignin Depolymerization in the Presence of Base, Hydrogenation Catalysts, and Ethanol. Catalysts. 2022; 12(2):158. https://doi.org/10.3390/catal12020158
Chicago/Turabian StyleRomanenko, Iuliia, Felix Kurz, Robert Baumgarten, Ivana Jevtovikj, Jean-Pierre Lindner, Arunabha Kundu, Alois Kindler, and Stephan Andreas Schunk. 2022. "Lignin Depolymerization in the Presence of Base, Hydrogenation Catalysts, and Ethanol" Catalysts 12, no. 2: 158. https://doi.org/10.3390/catal12020158