
Crystal Contact Engineering for Enhanced Cross-Linking Efficiency of HheG Crystals

Abstract
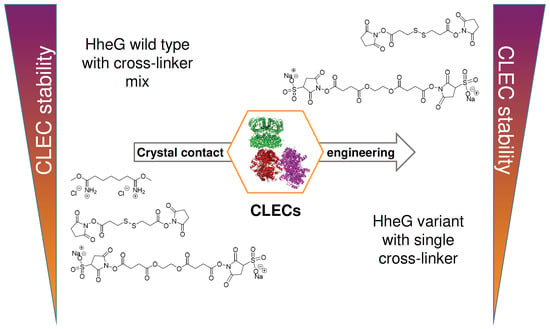
:
1. Introduction
2. Results and Discussion
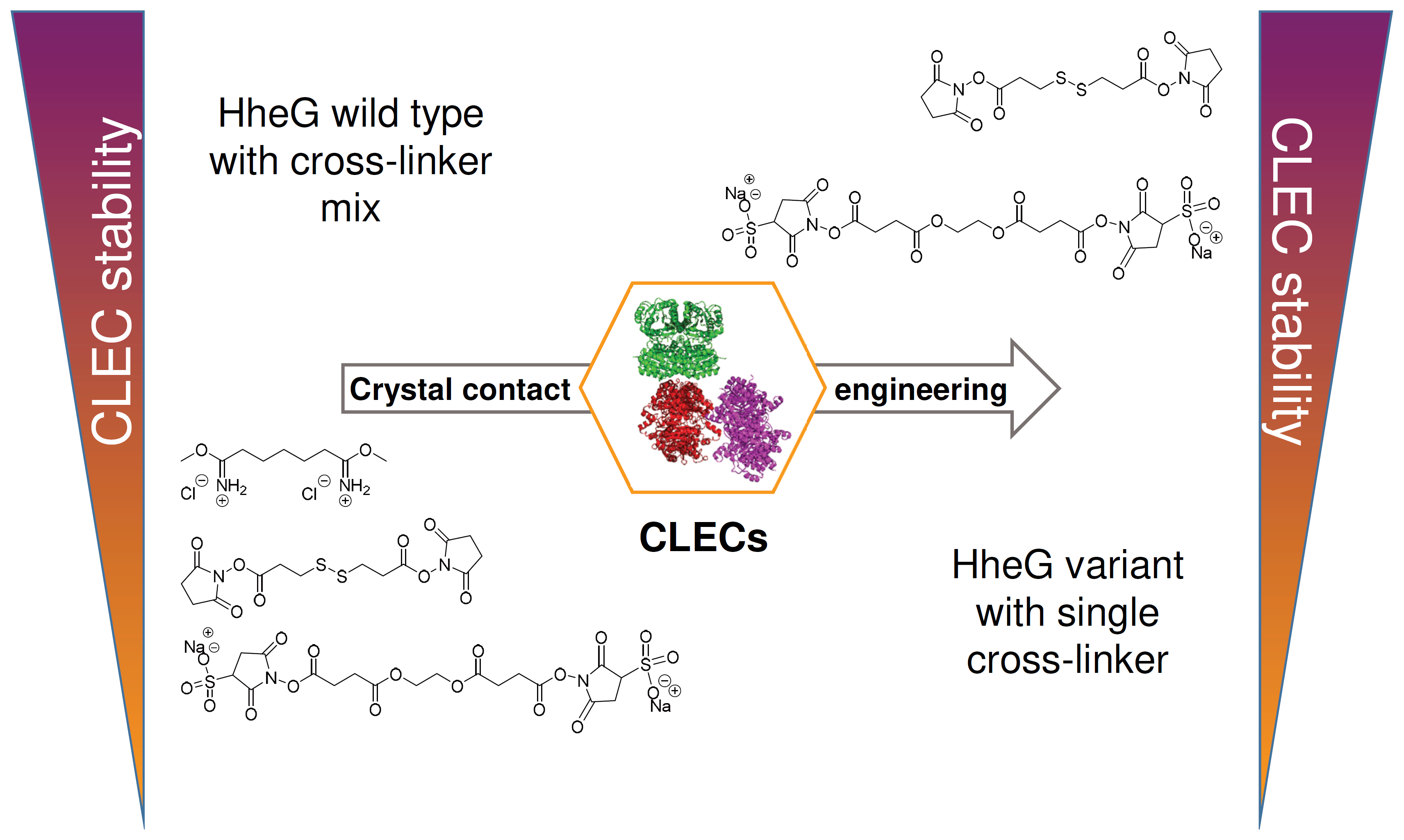
2.1. Crystal Contact Engineering
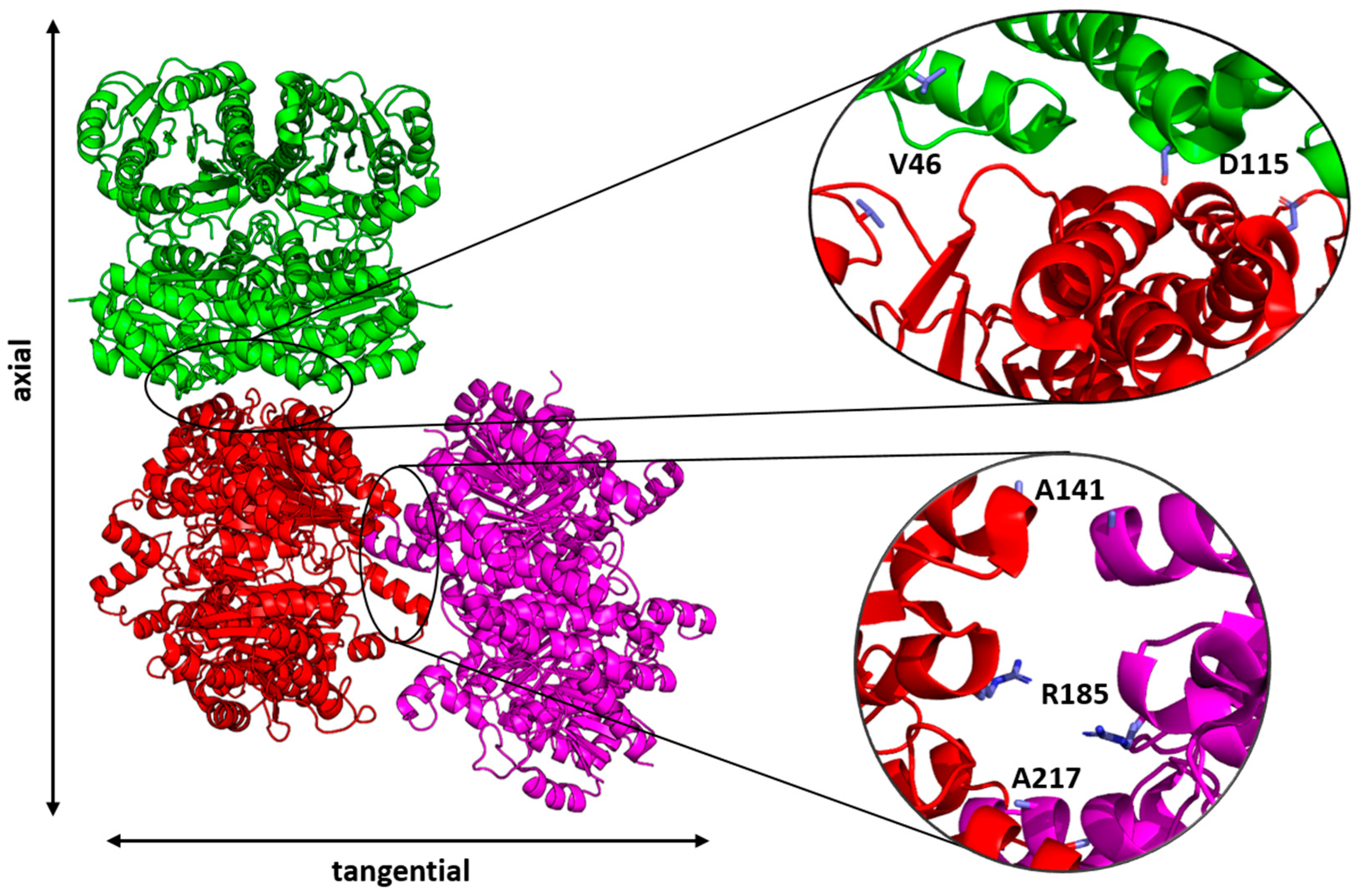
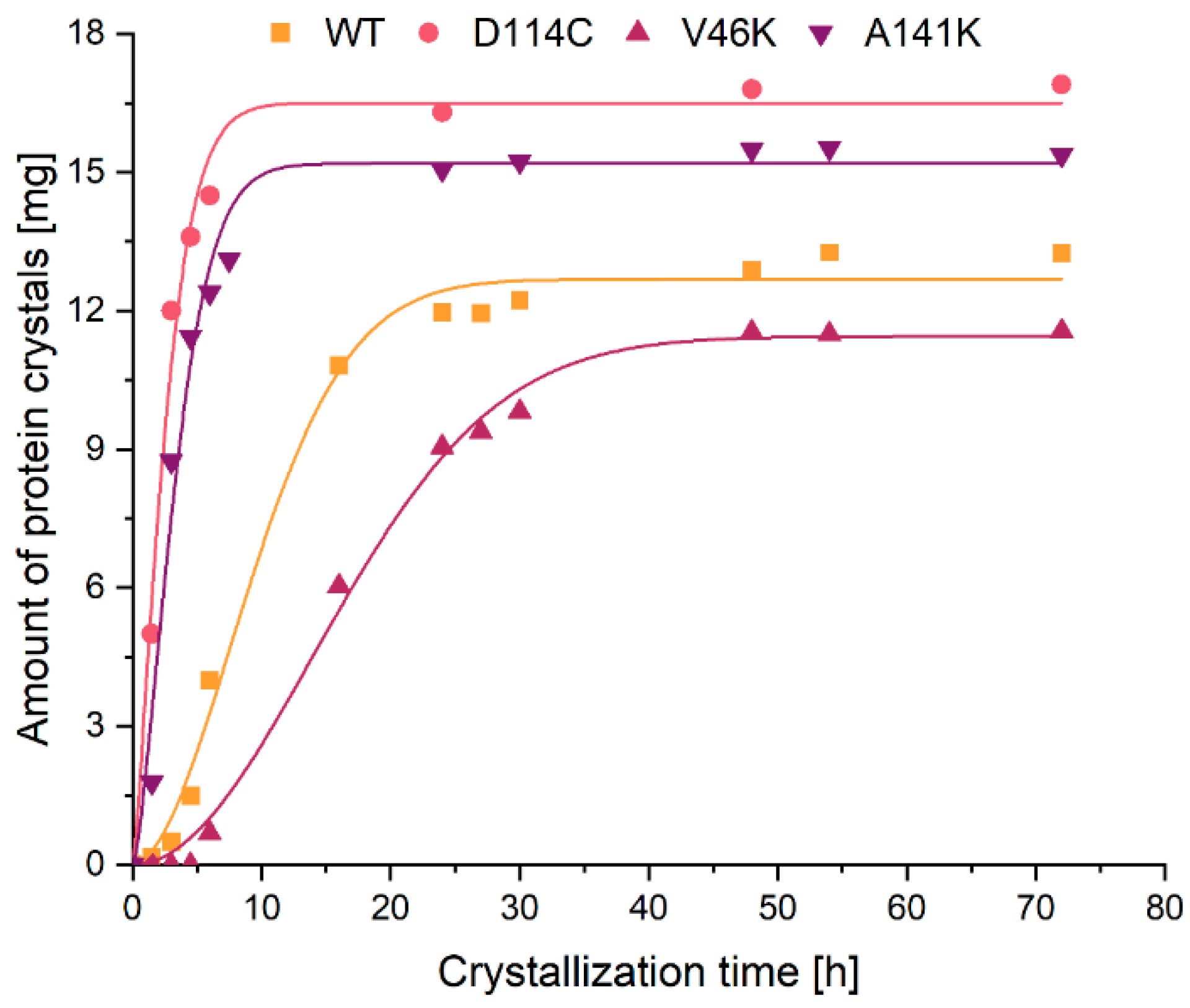
2.2. Crystallization
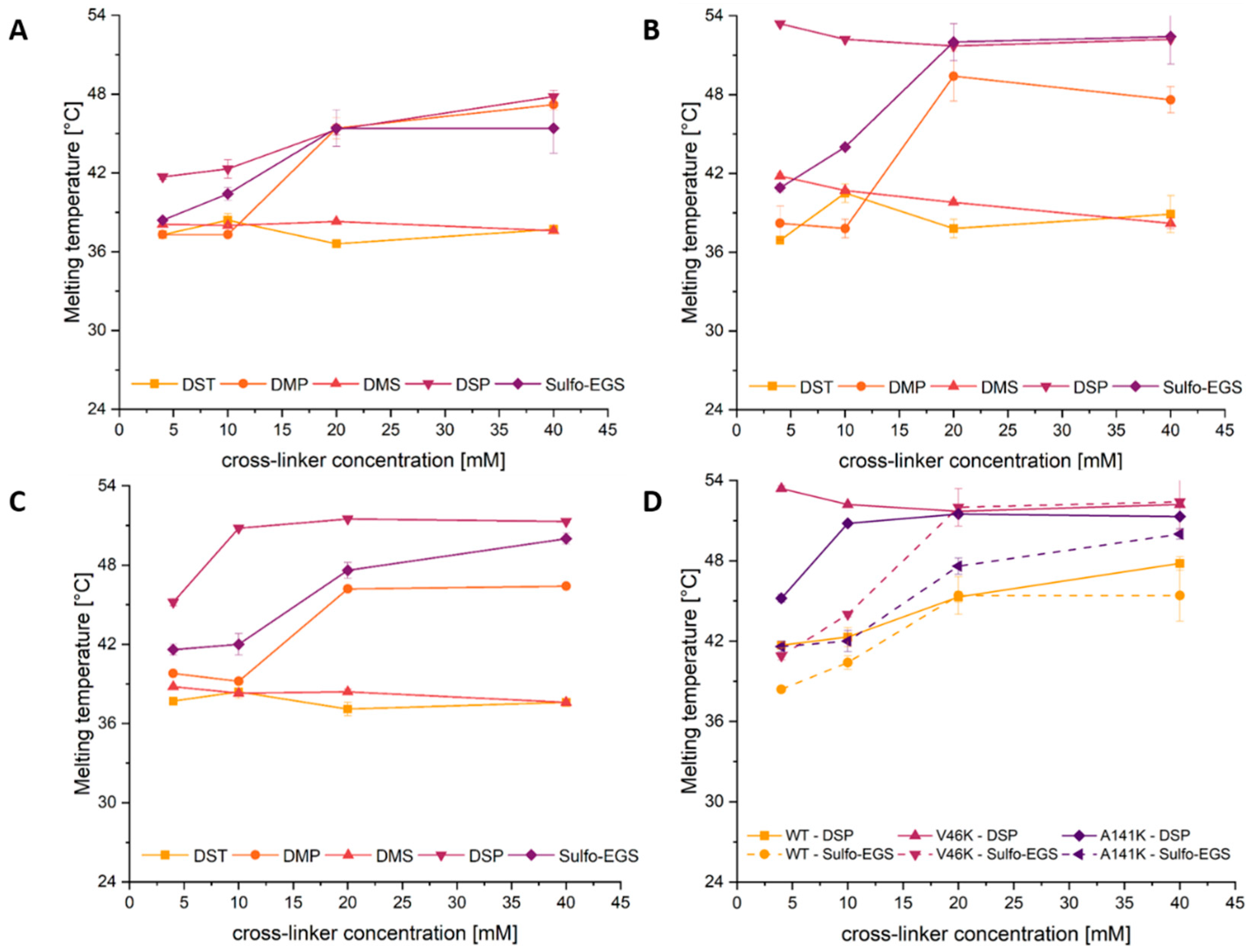
2.3. Cross-Linking
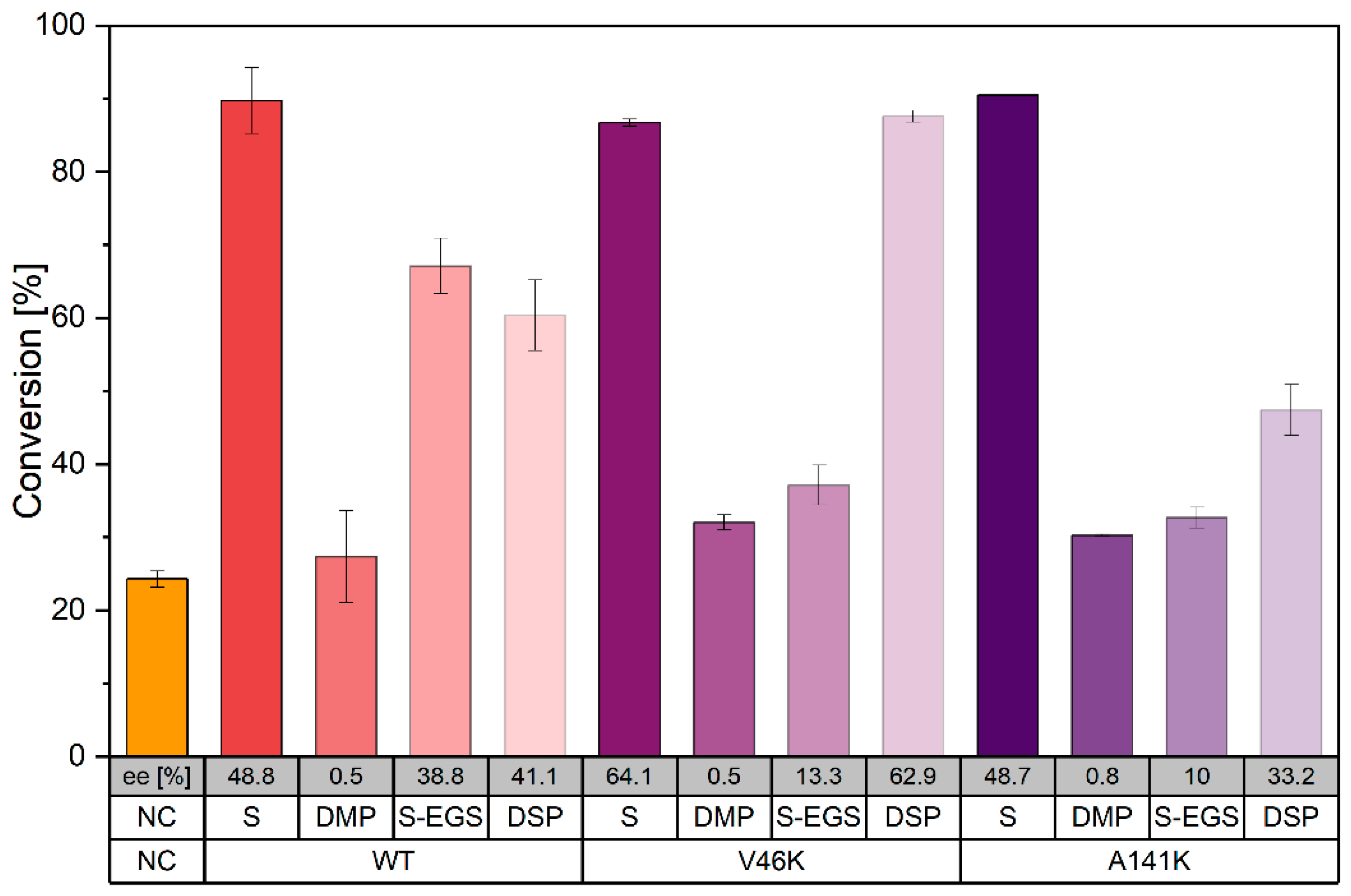
2.4. CLEC Activity
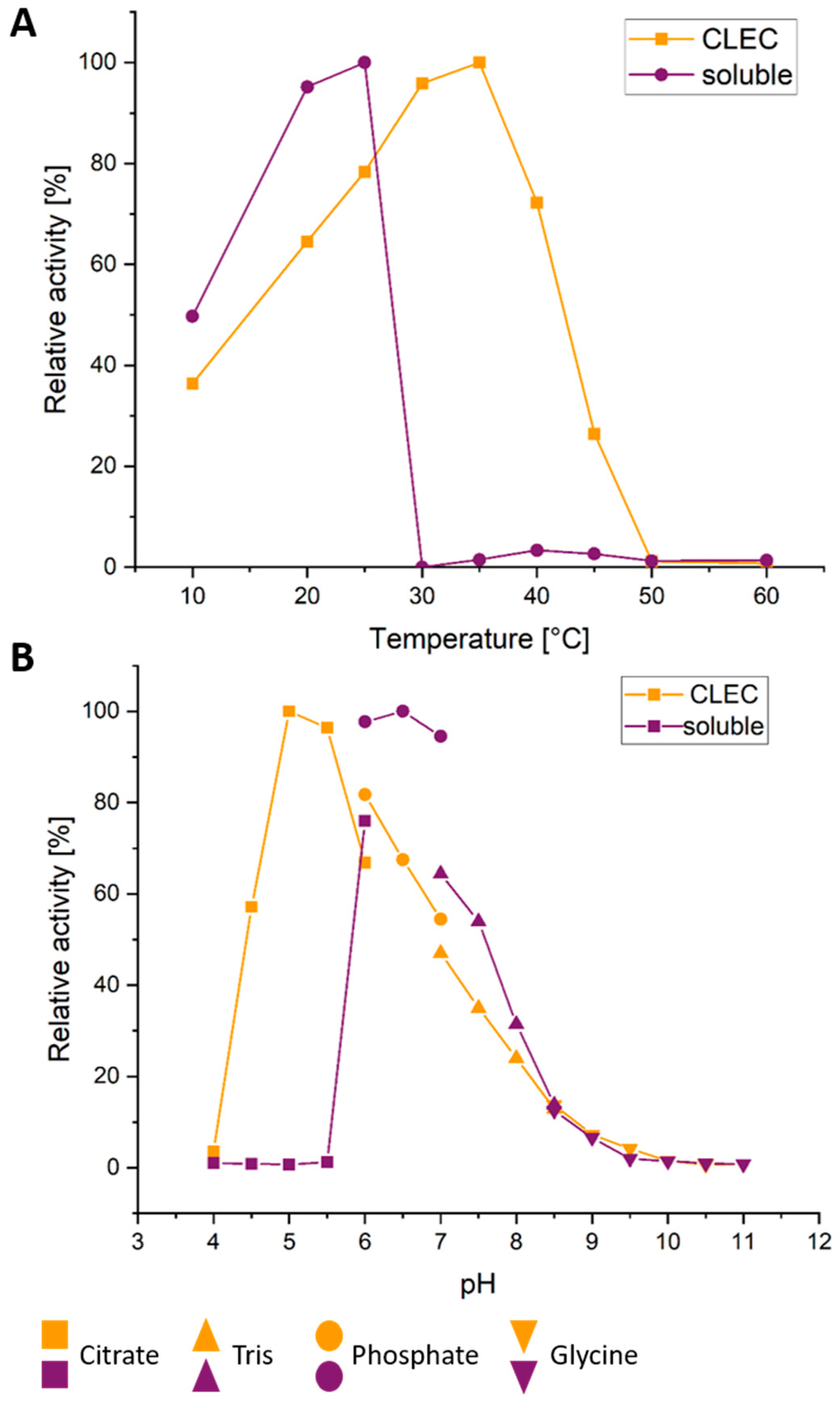
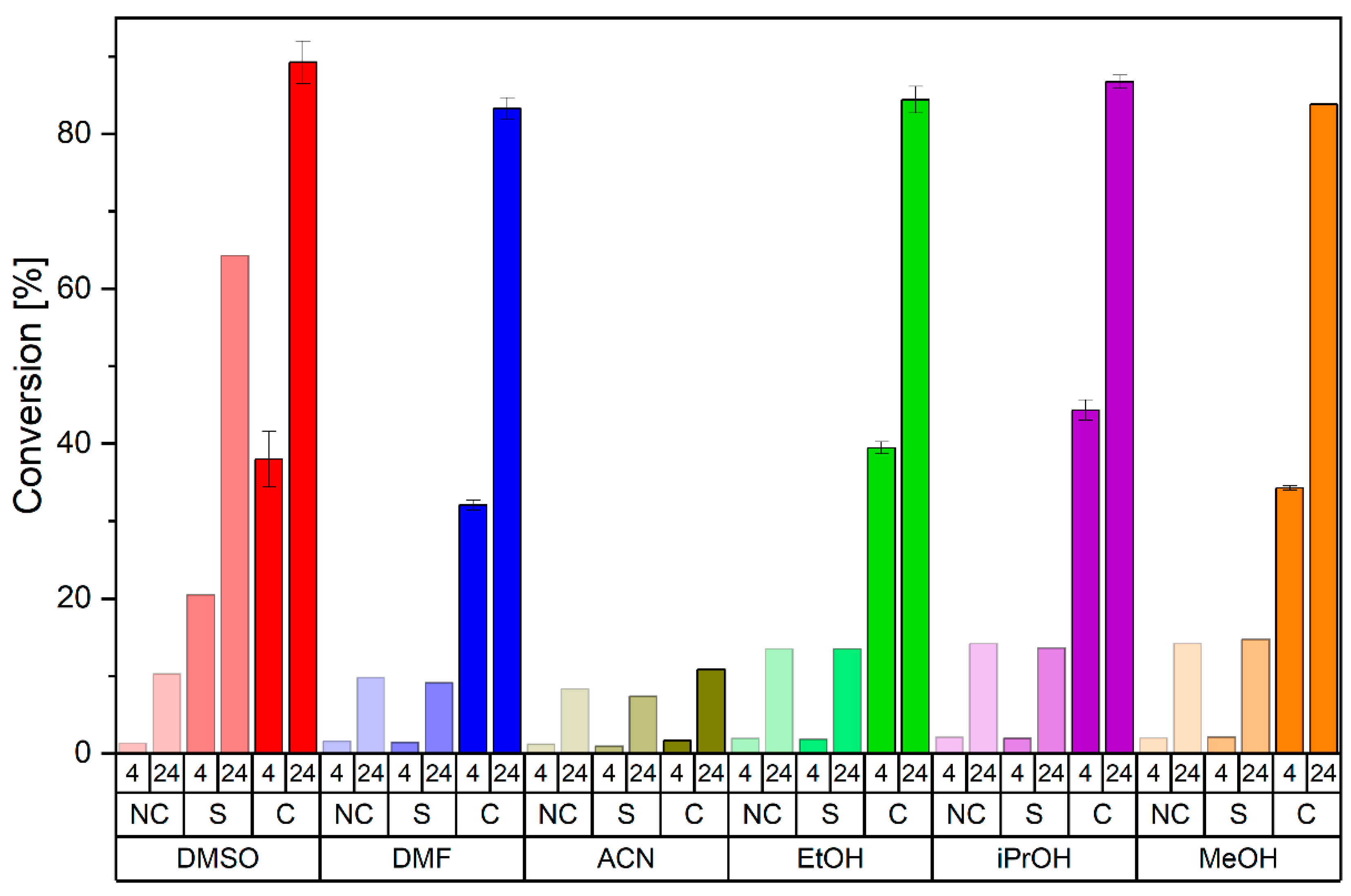
2.5. Further CLEC Characterization
3. Materials and Methods
3.1. Chemicals
3.2. Bacterial Strains and Plasmids
3.3. HheG Engineering
3.4. Protein Production and Purification
3.5. Activity and Enantioselectivity Determination
3.6. Crystallization and Cross-Linking
3.7. Temperature, pH and Solvent Activity Profiles
3.8. Thermal Shift Assay
3.9. Thermal Inactivation
3.10. Determination of Half-Life Times
3.11. Reusability
3.12. Particle Size Distribution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cao, L.; van Langen, L.; Sheldon, R.A. Immobilised Enzymes: Carrier-Bound or Carrier-Free? Curr. Opin. Biotechnol. 2003, 14, 387–394. [Google Scholar] [CrossRef]
- Liao, Q.; Du, X.; Jiang, W.; Tong, Y.; Zhao, Z.; Fang, R.; Feng, J.; Tang, L. Cross-Linked Enzyme Aggregates (CLEAs) of Halohydrin Dehalogenase from Agrobacterium radiobacter AD1: Preparation, Characterization and Application as a Biocatalyst. J. Biotechnol. 2018, 272–273, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Clair, N.S.; Wang, Y.-F.; Margolin, A.L. Cofactor-Bound Cross-Linked Enzyme Crystals (CLEC) of Alcohol Dehydrogenase. Angew. Chem. Int. Ed. 2000, 39, 380–383. [Google Scholar] [CrossRef]
- Persichetti, R.A.; Clair, N.L.S.; Griffith, J.P.; Navia, M.A.; Margolin, A.L. Cross-Linked Enzyme Crystals (CLECs) of Thermolysin in the Synthesis of Peptides. J. Am. Chem. Soc. 1995, 117, 2732–2737. [Google Scholar] [CrossRef]
- Noritomi, H.; Koyama, K.; Kato, S.; Nagahama, K. Increased Thermostability of Cross-Linked Enzyme Crystals of Subtilisin in Organic Solvents. Biotechnol. Tech. 1998, 12, 467–469. [Google Scholar] [CrossRef]
- Ayala, M.; Horjales, E.; Pickard, M.A.; Vazquez-Duhalt, R. Cross-Linked Crystals of Chloroperoxidase. Biochem. Biophys. Res. Commun. 2002, 295, 828–831. [Google Scholar] [CrossRef]
- Lalonde, J.J.; Govardhan, C.; Khalaf, N.; Martinez, A.G.; Visuri, K.; Margolin, A.L. Cross-Linked Crystals of Candida rugosa Lipase: Highly Efficient Catalysts for the Resolution of Chiral Esters. J. Am. Chem. Soc. 1995, 117, 6845–6852. [Google Scholar] [CrossRef]
- Staar, M.; Henke, S.; Blankenfeldt, W.; Schallmey, A. Biocatalytically Active and Stable Cross-Linked Enzyme Crystals of Halohydrin Dehalogenase HheG by Protein Engineering. ChemCatChem 2022, 14, e202200145. [Google Scholar] [CrossRef]
- Roy, J.J.; Abraham, T.E. Continuous Biotransformation of Pyrogallol to Purpurogallin Using Cross-Linked Enzyme Crystals of Laccase as Catalyst in a Packed-Bed Reactor. J. Chem. Technol. Biotechnol. 2006, 81, 1836–1839. [Google Scholar] [CrossRef]
- Fernández-Penas, R.; Verdugo-Escamilla, C.; Martínez-Rodríguez, S.; Gavira, J.A. Production of Cross-Linked Lipase Crystals at a Preparative Scale. Cryst. Growth Des. 2021, 21, 1698–1707. [Google Scholar] [CrossRef]
- Conejero-Muriel, M.; Rodríguez-Ruiz, I.; Martínez-Rodríguez, S.; Llobera, A.; Gavira, J.A. McCLEC, a Robust and Stable Enzymatic Based Microreactor Platform. Lab Chip 2015, 15, 4083–4089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.D.; Jia, S.R. Optimization Protocols and Improved Strategies of Cross-Linked Enzyme Aggregates Technology: Current Development and Future Challenges. Crit. Rev. Biotechnol. 2015, 35, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Jegan Roy, J.; Emilia Abraham, T. Strategies in Making Cross-Linked Enzyme Crystals. Chem. Rev. 2004, 104, 3705–3722. [Google Scholar] [CrossRef] [PubMed]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in Aqueous Solution, Reaction with Proteins, and Application to Enzyme Crosslinking. BioTechniques 2004, 37, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, T.; Endo, Y. Effects of Glutaraldehyde Exposure on Human Health. J. Occup. Health 2006, 48, 75–87. [Google Scholar] [CrossRef]
- Leung, H.-W. Ecotoxicology of Glutaraldehyde: Review of Environmental Fate and Effects Studies. Ecotoxicol. Environ. Saf. 2001, 49, 26–39. [Google Scholar] [CrossRef]
- Roy, J.J.; Abraham, T.E. Preparation and Characterization of Cross-Linked Enzyme Crystals of Laccase. J. Mol. Catal. B Enzym. 2006, 38, 31–36. [Google Scholar] [CrossRef]
- Mattson, G.; Conklin, E.; Desai, S.; Nielander, G.; Savage, M.D.; Morgensen, S. A Practical Approach to Crosslinking. Mol. Biol. Rep. 1993, 17, 167–183. [Google Scholar] [CrossRef]
- Staros, J.V. N-Hydroxysulfosuccinimide Active Esters: Bis(N-Hydroxysulfosuccinimide) Esters of Two Dicarboxylic Acids Are Hydrophilic, Membrane-Impermeant, Protein Cross-Linkers. Biochemistry 1982, 21, 3950–3955. [Google Scholar] [CrossRef]
- Staros, J.V. Membrane-Impermeant Crosslinking Reagents: Probes of the Structure and Dynamics of Membrane Proteins. Acc. Chem. Res. 1988, 21, 435–441. [Google Scholar] [CrossRef]
- Auclair, J.R.; Boggio, K.J.; Petsko, G.A.; Ringe, D.; Agar, J.N. Strategies for Stabilizing Superoxide Dismutase (SOD1), the Protein Destabilized in the Most Common Form of Familial Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2010, 107, 21394–21399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabarek, Z.; Gergely, J. Zero-Length Crosslinking Procedure with the Use of Active Esters. Anal. Biochem. 1990, 185, 131–135. [Google Scholar] [CrossRef]
- Brinkley, M. A Brief Survey of Methods for Preparing Protein Conjugates with Dyes, Haptens and Crosslinking Reagents. Bioconjugate Chem. 1992, 3, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Hetrick, E.M.; Sperry, D.C.; Nguyen, H.K.; Strege, M.A. Characterization of a Novel Cross-Linked Lipase: Impact of Cross-Linking on Solubility and Release from Drug Product. Mol. Pharm. 2014, 11, 1189–1200. [Google Scholar] [CrossRef]
- Kubiak, M.; Kampen, I.; Schilde, C. Structure-Based Modeling of the Mechanical Behavior of Cross-Linked Enzyme Crystals. Crystals 2022, 12, 441. [Google Scholar] [CrossRef]
- Rehman, S.; Bhatti, H.N.; Bilal, M.; Asgher, M. Cross-Linked Enzyme Aggregates (CLEAs) of Pencilluim notatum Lipase Enzyme with Improved Activity, Stability and Reusability Characteristics. Int. J. Biol. Macromol. 2016, 91, 1161–1169. [Google Scholar] [CrossRef]
- Schallmey, A.; Schallmey, M. Recent Advances on Halohydrin Dehalogenases—From Enzyme Identification to Novel Biocatalytic Applications. Appl. Microbiol. Biotechnol. 2016, 100, 7827–7839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasnaoui-Dijoux, G.; Majerić Elenkov, M.; Spelberg, J.H.L.; Hauer, B.; Janssen, D.B. Catalytic Promiscuity of Halohydrin Dehalogenase and Its Application in Enantioselective Epoxide Ring Opening. ChemBioChem 2008, 9, 1048–1051. [Google Scholar] [CrossRef]
- Wan, N.; Tian, J.; Zhou, X.; Wang, H.; Cui, B.; Han, W.; Chen, Y. Regioselective Ring-Opening of Styrene Oxide Derivatives Using Halohydrin Dehalogenase for Synthesis of 4-Aryloxazolidinones. Adv. Synth. Catal. 2019, 361, 4651–4655. [Google Scholar] [CrossRef]
- Majerić Elenkov, M.; Tang, L.; Meetsma, A.; Hauer, B.; Janssen, D.B. Formation of Enantiopure 5-Substituted Oxazolidinones through Enzyme-Catalysed Kinetic Resolution of Epoxides. Org. Lett. 2008, 10, 2417–2420. [Google Scholar] [CrossRef]
- Majerić Elenkov, M.; Hoeffken, H.W.; Tang, L.; Hauer, B.; Janssen, D.B. Enzyme-Catalyzed Nucleophilic Ring Opening of Epoxides for the Preparation of Enantiopure Tertiary Alcohols. Adv. Synth. Catal. 2007, 349, 2279–2285. [Google Scholar] [CrossRef]
- Lutje Spelberg, J.H.; van Hylckama Vlieg, J.E.T.; Bosma, T.; Kellogg, R.M.; Janssen, D.B. A Tandem Enzyme Reaction to Produce Optically Active Halohydrins, Epoxides and Diols. Tetrahedron Asymmetry 1999, 10, 2863–2870. [Google Scholar] [CrossRef]
- Majerić Elenkov, M.; Primožič, I.; Hrenar, T.; Smolko, A.; Dokli, I.; Salopek-Sondi, B.; Tang, L. Catalytic Activity of Halohydrin Dehalogenases towards Spiroepoxides. Org. Biomol. Chem. 2012, 10, 5063–5072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-R.; Wan, N.-W.; Ma, J.-M.; Cui, B.-D.; Han, W.-Y.; Chen, Y.-Z. Enzymatic Kinetic Resolution of Bulky Spiro-Epoxyoxindoles via Halohydrin Dehalogenase-Catalyzed Enantio- and Regioselective Azidolysis. ACS Catal. 2021, 11, 9066–9072. [Google Scholar] [CrossRef]
- Xu, Q.; Huang, K.-S.; Wang, Y.-F.; Wang, H.-H.; Cui, B.-D.; Han, W.-Y.; Chen, Y.-Z.; Wan, N.-W. Stereodivergent Synthesis of Epoxides and Oxazolidinones via the Halohydrin Dehalogenase-Catalyzed Desymmetrization Strategy. ACS Catal. 2022, 12, 6285–6293. [Google Scholar] [CrossRef]
- Koopmeiners, J.; Diederich, C.; Solarczek, J.; Voß, H.; Mayer, J.; Blankenfeldt, W.; Schallmey, A. HheG, a Halohydrin Dehalogenase with Activity on Cyclic Epoxides. ACS Catal. 2017, 7, 6877–6886. [Google Scholar] [CrossRef]
- Calderini, E.; Wessel, J.; Süss, P.; Schrepfer, P.; Wardenga, R.; Schallmey, A. Selective Ring-Opening of Di-Substituted Epoxides Catalysed by Halohydrin Dehalogenases. ChemCatChem 2019, 11, 2099–2106. [Google Scholar] [CrossRef]
- Solarczek, J.; Kaspar, F.; Bauer, P.; Schallmey, A. G-Type Halohydrin Dehalogenases Catalyze Ring Opening Reactions of Cyclic Epoxides with Diverse Anionic Nucleophiles. Chem. Eur. J. 2022, e202202343. [Google Scholar] [CrossRef]
- Solarczek, J.; Klünemann, T.; Brandt, F.; Schrepfer, P.; Wolter, M.; Jacob, C.R.; Blankenfeldt, W.; Schallmey, A. Position 123 of Halohydrin Dehalogenase HheG Plays an Important Role in Stability, Activity, and Enantioselectivity. Sci. Rep. 2019, 9, 5106. [Google Scholar] [CrossRef] [Green Version]
- Kubiak, M.; Staar, M.; Kampen, I.; Schallmey, A.; Schilde, C. The Depth-Dependent Mechanical Behavior of Anisotropic Native and Cross-Linked HheG Enzyme Crystals. Crystals 2021, 11, 718. [Google Scholar] [CrossRef]
- Dasgupta, S.; Iyer, G.H.; Bryant, S.H.; Lawrence, C.E.; Bell, J.A. Extent and Nature of Contacts between Protein Molecules in Crystal Lattices and between Subunits of Protein Oligomers. Proteins 1997, 28, 494–514. [Google Scholar] [CrossRef]
- Iyer, G.H.; Dasgupta, S.; Bell, J.A. Ionic Strength and Intermolecular Contacts in Protein Crystals. J. Cryst. Growth 2000, 217, 429–440. [Google Scholar] [CrossRef]
- Han, K.-K.; Richard, C.; Delacourte, A. Chemical Cross-Links of Proteins by Using Bifunctional Reagents. Int. J. Biochem. 1984, 16, 129–145. [Google Scholar] [CrossRef]
- Derewenda, Z.S. Rational Protein Crystallization by Mutational Surface Engineering. Structure 2004, 12, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Abdul Wahab, M.K.H.; El-Enshasy, H.A.; Bakar, F.D.A.; Murad, A.M.A.; Jahim, J.M.; Illias, R.M. Improvement of Cross-Linking and Stability on Cross-Linked Enzyme Aggregate (CLEA)-Xylanase by Protein Surface Engineering. Process Biochem. 2019, 86, 40–49. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Chayen, N.E. Comparative Studies of Protein Crystallization by Vapour-Diffusion and Microbatch Techniques. Acta Cryst. D 1998, 54, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Avrami, M. Kinetics of Phase Change. I General Theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Moreno-Cencerrado, A.; Iturri, J.; Toca-Herrera, J.L. In-Situ 2D Bacterial Crystal Growth as a Function of Protein Concentration: An Atomic Force Microscopy Study. Microsc. Res. Tech. 2018, 81, 1095–1104. [Google Scholar] [CrossRef]
- Khayati, G.R.; Nourafkan, E.; Karimi, G.; Moradgholi, J. Synthesis of Cuprous Oxide Nanoparticles by Mechanochemical Oxidation of Copper in High Planetary Energy Ball Mill. Adv. Powder Technol. 2013, 24, 301–305. [Google Scholar] [CrossRef]
- Sinz, A. Chemical Cross-Linking and Mass Spectrometry for Mapping Three-Dimensional Structures of Proteins and Protein Complexes. J. Mass Spectrom. 2003, 38, 1225–1237. [Google Scholar] [CrossRef]
- Lomant, A.J.; Fairbanks, G. Chemical Probes of Extended Biological Structures: Synthesis and Properties of the Cleavable Protein Cross-Linking Reagent [35S]Dithiobis(Succinimidyl Propionate). J. Mol. Biol. 1976, 104, 243–261. [Google Scholar] [CrossRef] [PubMed]
- Margolin, A.L.; Navia, M.A. Protein Crystals as Novel Catalytic Materials. Angew. Chem. Int. Ed. 2001, 40, 2204–2222. [Google Scholar] [CrossRef]
- Kasvinsky, P.J.; Madsen, N.B. Activity of Glycogen Phosphorylase in the Crystalline State. J. Biol. Chem. 1976, 251, 6852–6859. [Google Scholar] [CrossRef] [PubMed]
- Cseri, L.; Razali, M.; Pogany, P.; Szekely, G. Chapter 3.15—Organic Solvents in Sustainable Synthesis and Engineering. In Green Chemistry; Török, B., Dransfield, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 513–553. ISBN 978-0-12-809270-5. [Google Scholar]
- Milčić, N.; Stepanić, V.; Crnolatac, I.; Findrik Blažević, Z.; Brkljača, Z.; Majerić Elenkov, M. Inhibitory Effect of DMSO on Halohydrin Dehalogenase: Experimental and Computational Insights into the Influence of an Organic Co-Solvent on the Structural and Catalytic Properties of a Biocatalyst. Chem. Eur. J. 2022, 28, e202201923. [Google Scholar] [CrossRef]
- Eyring, H.; Stearn, A.E. The Application of the Theory of Absolute Reacton Rates to Proteins. Chem. Rev. 1939, 24, 253–270. [Google Scholar] [CrossRef]
- Maurya, S.S.; Nadar, S.S.; Rathod, V.K. A Rapid Self-Assembled Hybrid Bio-Microflowers of Alpha–Amylase with Enhanced Activity. J. Biotechnol. 2020, 317, 27–33. [Google Scholar] [CrossRef]
- Taboada-Puig, R.; Junghanns, C.; Demarche, P.; Moreira, M.T.; Feijoo, G.; Lema, J.M.; Agathos, S.N. Combined Cross-Linked Enzyme Aggregates from Versatile Peroxidase and Glucose Oxidase: Production, Partial Characterization and Application for the Elimination of Endocrine Disruptors. Bioresour. Technol. 2011, 102, 6593–6599. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Yield [a] [mg/L] | Specific Activity [b] [U/mg] | Tm [°C] | eeP [c] [%] |
|---|---|---|---|---|
| WT | 204 | 2.32 ± 0.3 | 41 | 49.1 ± 0.1 |
| V46K | 103 | 0.95 ± 0.02 | 39 | 67.8 ± 0.3 |
| D115K | 192 | 1.73 ± 0.01 | 41 | 51.5 ± 0.1 |
| A141K | 82.8 | 2.52 ± 0.01 | 42.5 | 48.7 ± 0.1 |
| R185K | 197 | 2.26 ± 0.2 | 42.5 | 48.9 ± 0.2 |
| A217K | 301 | 2.39 ± 0.01 | 44 | 48.3 ± 0.1 |
| Mutagenic Primer | Sequence 5′–3′ |
|---|---|
| f_hheG_V46K | CAGCCGGTGATGGCACCATGAAAGGTGTTGAAGAAAGTTTTG |
| r_hheG_V46K | CAAAACTTTCTTCAACACCTTTCATGGTGCCATCACCGGCT |
| f_hheG_D115K | GCAAATTTCTGGATATGACCGATAAACAGTGGGCAAAAGTTAAAGCAACC |
| r_hheG_D115K | GGTTGCTTTAACTTTTGCCCACTGTTTATCGGTCATATCCAGAAATTTGC |
| f_hheG_A141K | GTTCTGCCTCCGATGGTTAAAGCCGGTGCAGGTCAGTG |
| r_hheG_A141K | CACTGACCTGCACCGGCTTTAACCATCGGAGGCAGAAC |
| f_hheG_R185K | GCAGTTGGTCTGGAACATGCAAAACATGGTGTTCAGGTTAATGC |
| r_hheG_R185K | GCATTAACCTGAACACCATGTTTTGCATGTTCCAGACCAACTGC |
| f_hheG_A217K | GATGGTGATCCGGAACGTCGTAAAATGATTGAAGCACAGGTTC |
| r_hheG_A217K | GAACCTGTGCTTCAATCATTTTACGACGTTCCGGATCACCATC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staar, M.; Staar, S.; Schallmey, A. Crystal Contact Engineering for Enhanced Cross-Linking Efficiency of HheG Crystals. Catalysts 2022, 12, 1553. https://doi.org/10.3390/catal12121553
Staar M, Staar S, Schallmey A. Crystal Contact Engineering for Enhanced Cross-Linking Efficiency of HheG Crystals. Catalysts. 2022; 12(12):1553. https://doi.org/10.3390/catal12121553
Chicago/Turabian StyleStaar, Marcel, Sophie Staar, and Anett Schallmey. 2022. "Crystal Contact Engineering for Enhanced Cross-Linking Efficiency of HheG Crystals" Catalysts 12, no. 12: 1553. https://doi.org/10.3390/catal12121553