
Biocatalytic Cascade of Sebacic Acid Production with In Situ Co-Factor Regeneration Enabled by Engineering of an Alcohol Dehydrogenase
 ,
,
Abstract
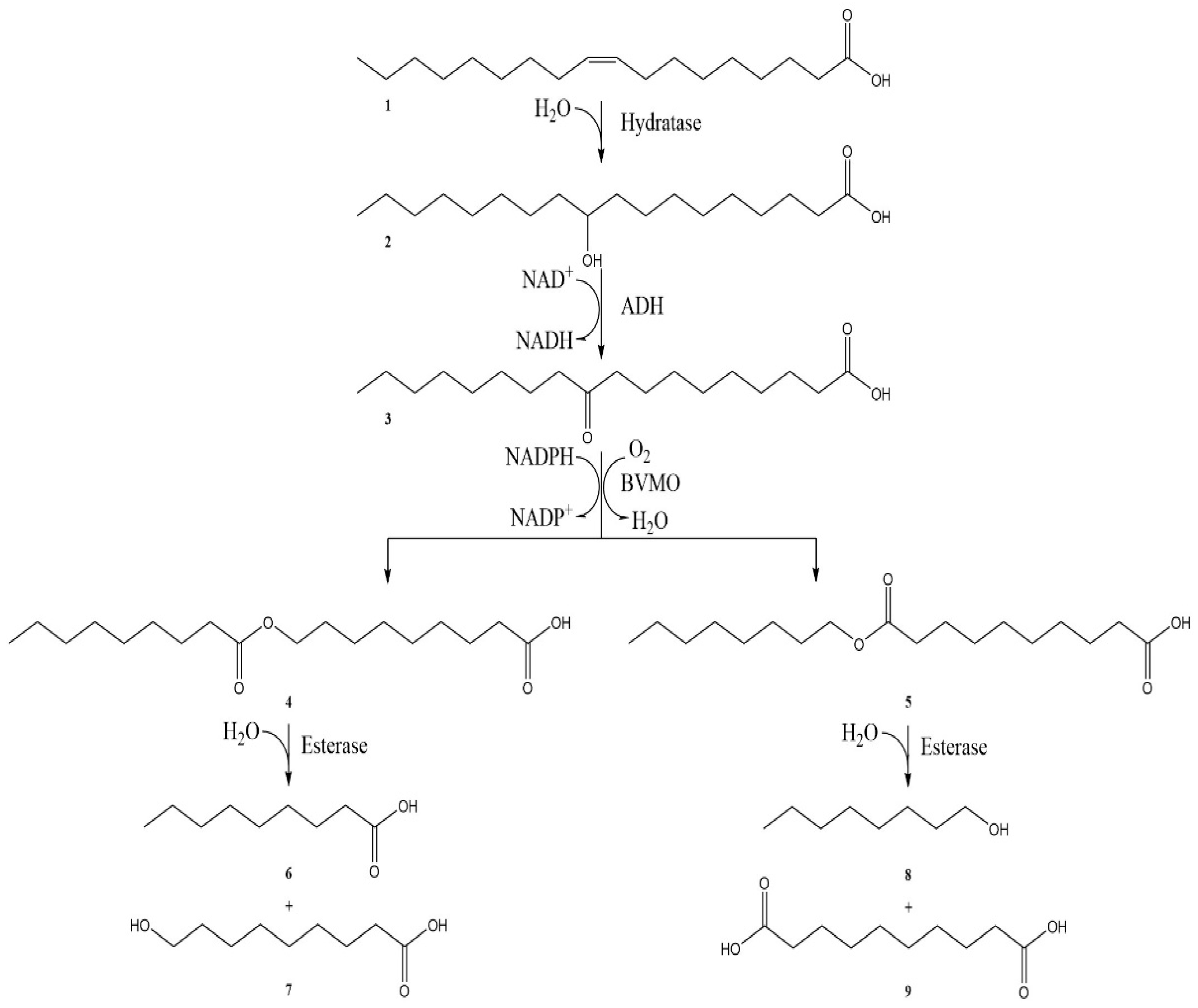
:1. Introduction
2. Results and Discussion
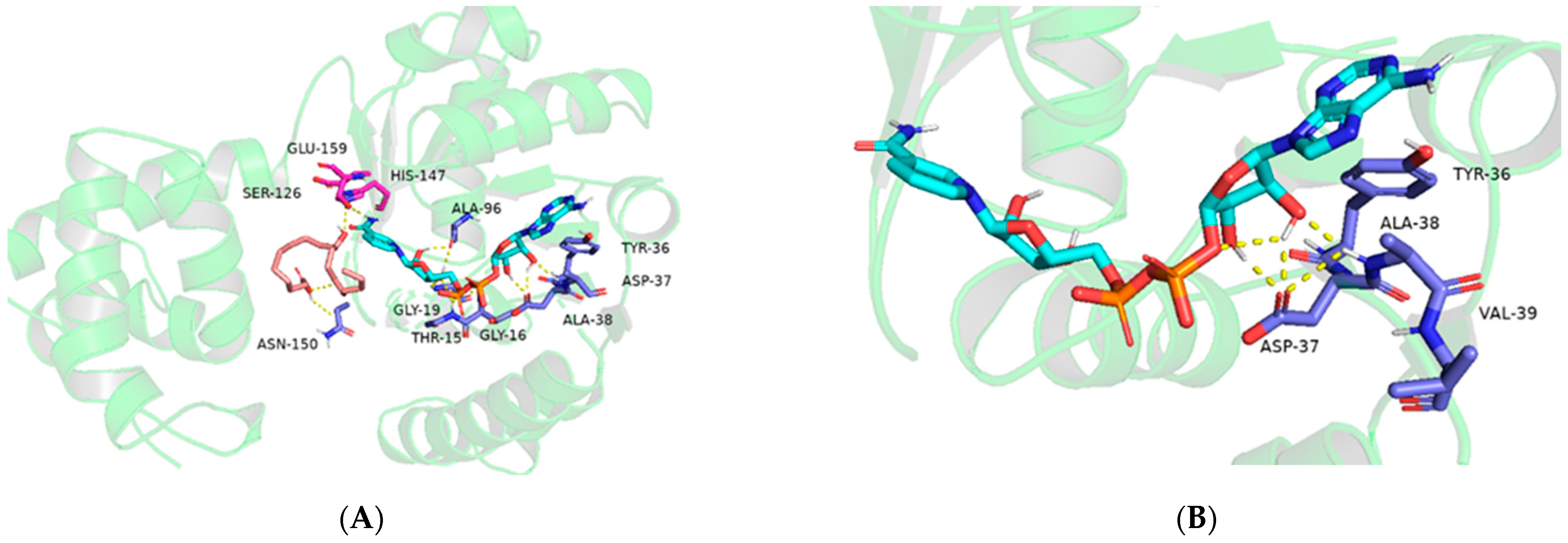
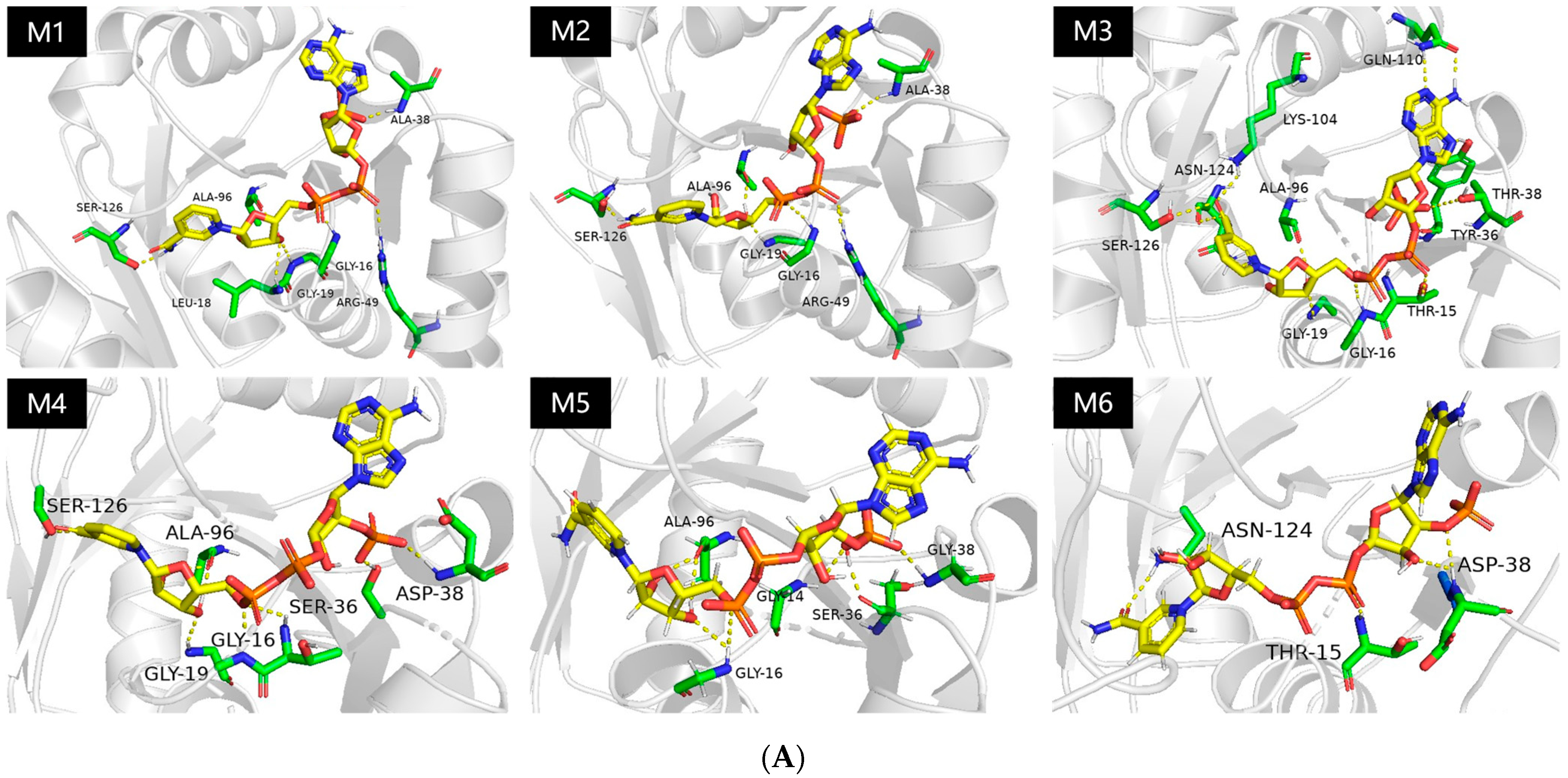
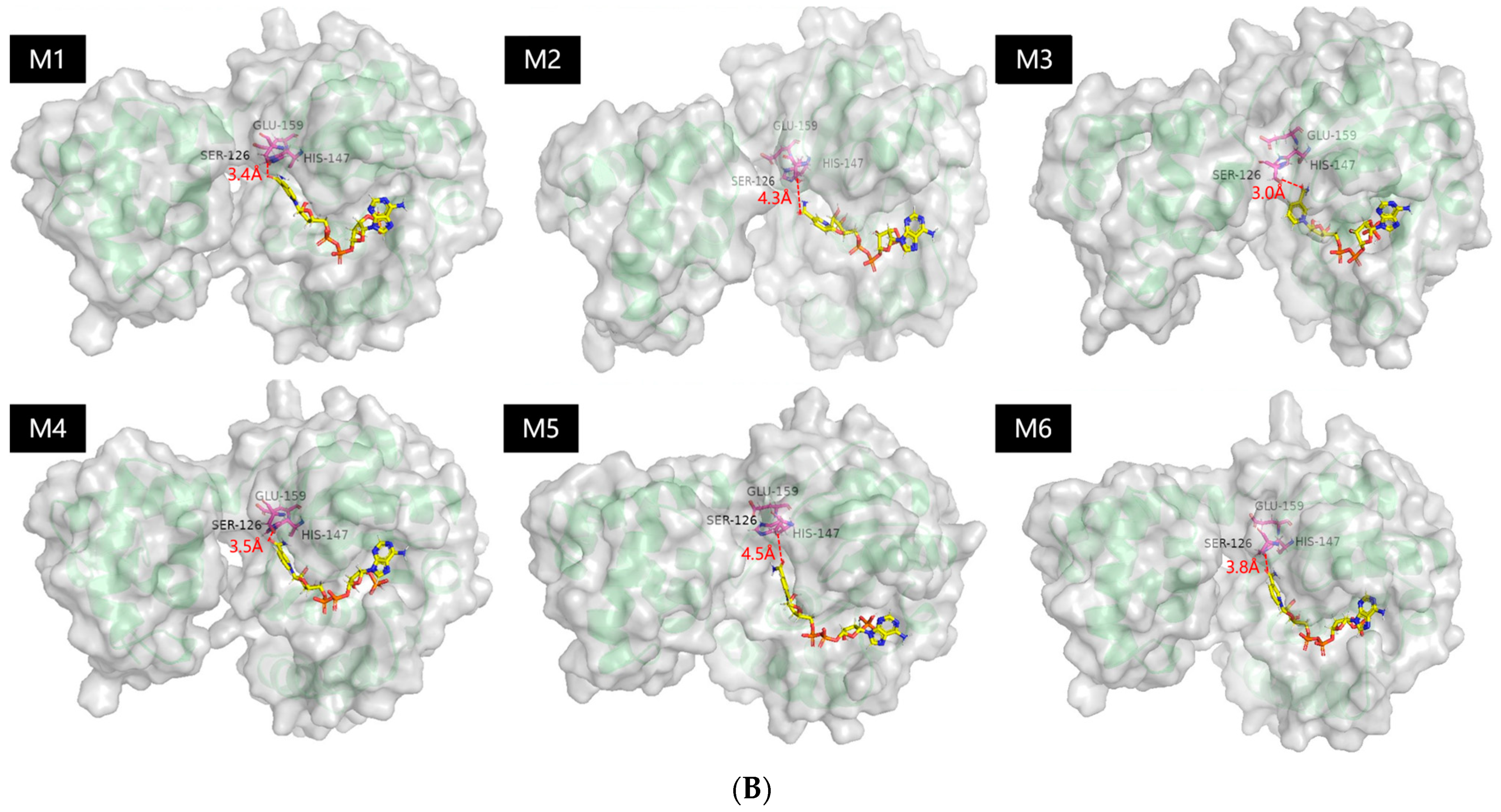
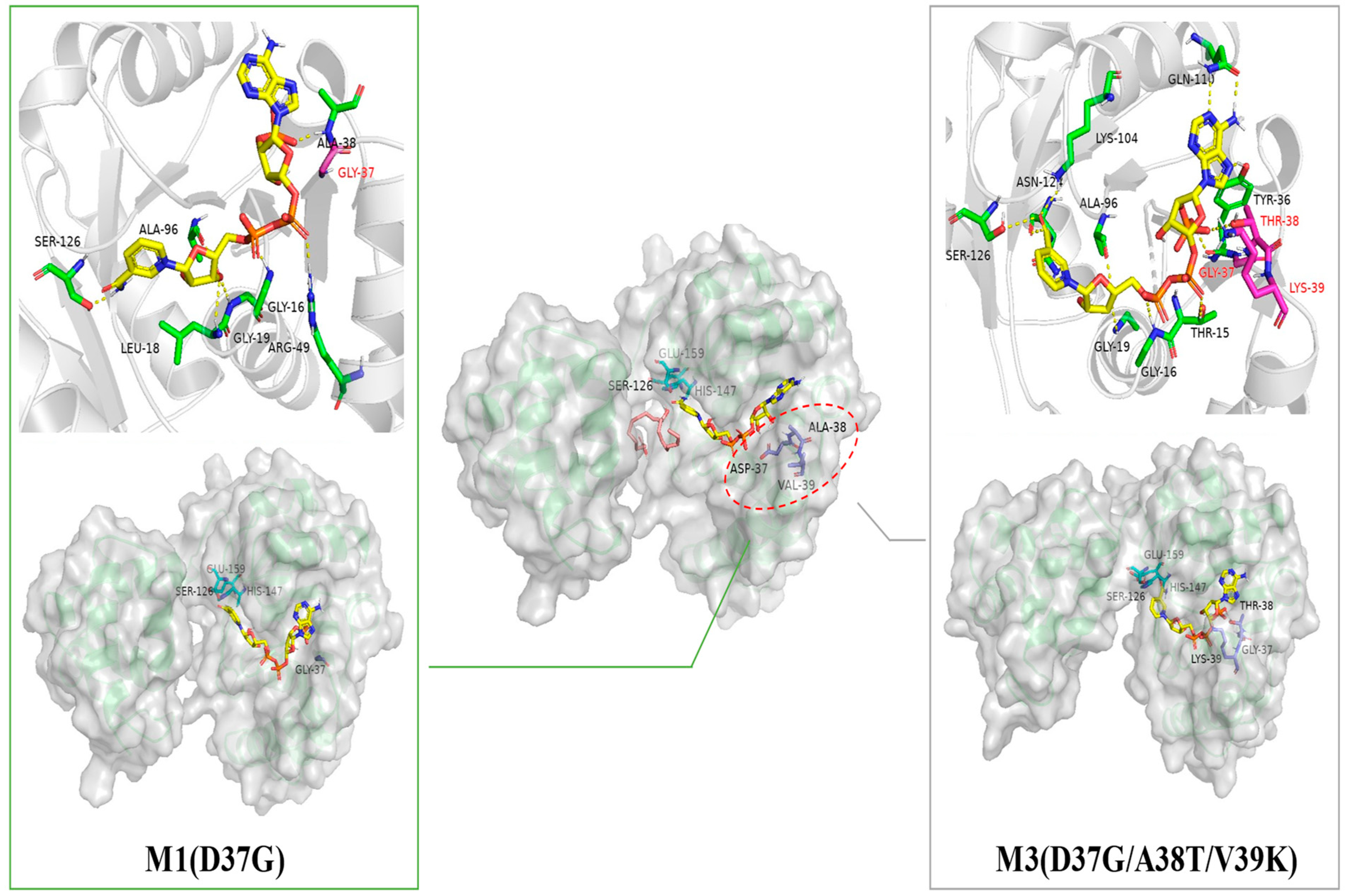
2.1. Probing the Targeted Residues that Determining the Co-Factor Specificity of MlADH
2.2. Enzyme kinetics
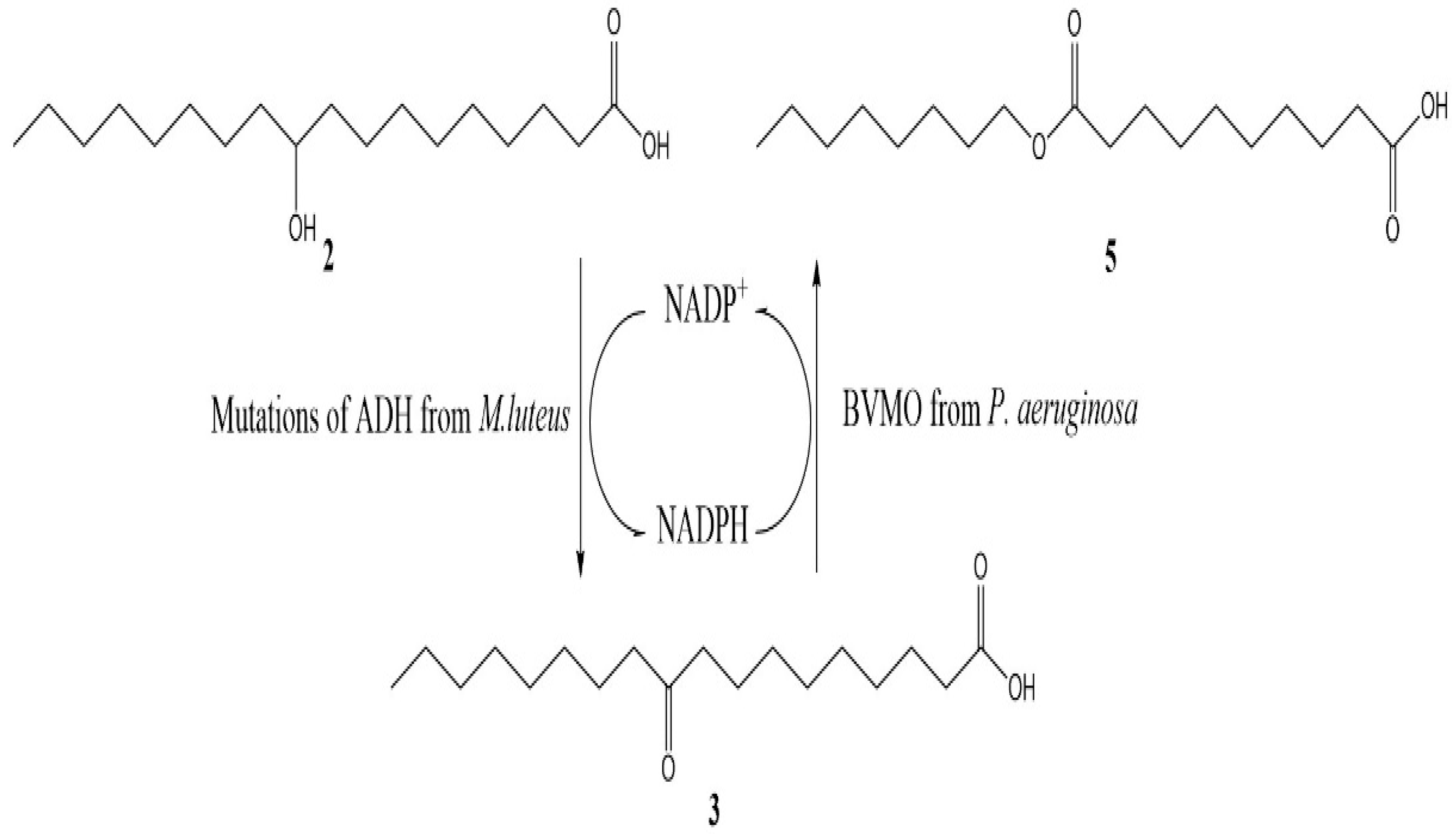
2.3. The Efficiency of In Situ NADP+ Regeneration System for Sebacic Acid Production
3. Materials and Methods
3.1. Materials
3.2. Molecular Modeling
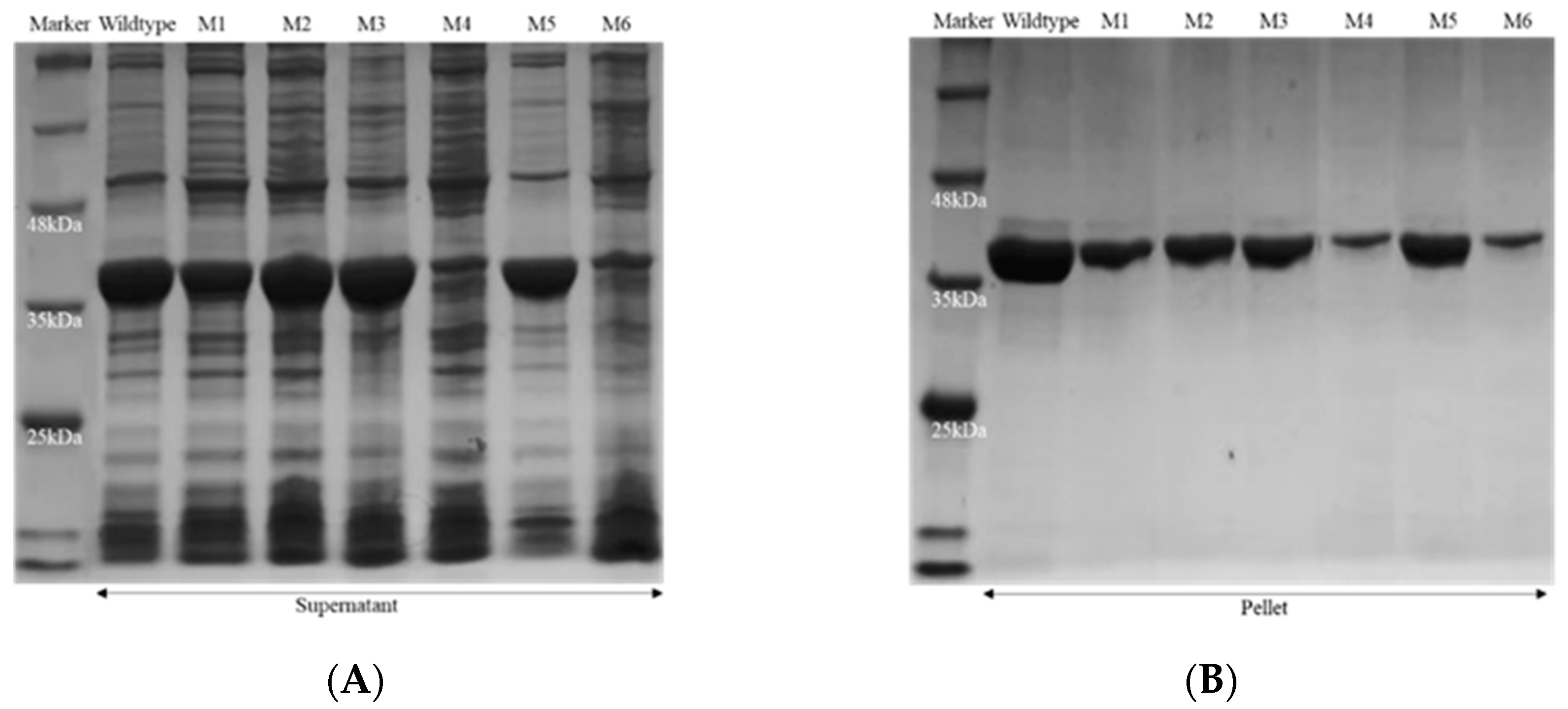
3.3. Gene Cloning and Expression of Recombinant Enzymes
3.4. Enzyme Kinetics Assay
3.5. Sebacic acid Production with In Situ NADP+ Regeneration System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, F.; Huang, C.; Xu, T. Production of sebacic acid using two-phase bipolar membrane electrodialysis. Ind. Eng. Chem. Res. 2009, 48, 7482–7488. [Google Scholar] [CrossRef]
- Köckritz, A.; Martin, A. Synthesis of azelaic acid from vegetable oil-based feedstocks. Eur. J. Lipid Sci. Technol. 2011, 113, 83–91. [Google Scholar] [CrossRef]
- Kroha, K. Industrial biotechnology provides opportunities for commercial production of new long-chain dibasic acids. Inform 2004, 15, 568–571. [Google Scholar]
- Song, J.W.; Lee, J.H.; Bornscheuer, U.T.; Park, J.B. Microbial synthesis of medium-chain α, ω-dicarboxylic acids and ω-aminocarboxylic acids from renewable long-chain fatty acids. Adv. Synth. Catal. 2014, 356, 1782–1788. [Google Scholar] [CrossRef]
- Lim, S.; Yoo, H.W.; Sarak, S.; Kim, B.G.; Yun, H. A multi-enzyme cascade reaction for the production of α, ω-dicarboxylic acids from free fatty acids. J. Ind. Eng. Chem. 2021, 98, 358–365. [Google Scholar] [CrossRef]
- Garcia-Junceda, E. Multi-Step Enzyme Catalysis: Biotransformations and Chemoenzymatic Synthesis; Wiley-VCH: Weinheim, Germany, 2008; pp. 83–107. [Google Scholar]
- Mutlu, H.; Meier, M.A.R. Castor oil as a renewable resource for the chemical industry. Eur. J. Lipid Sci. Technol. 2010, 112, 10–30. [Google Scholar] [CrossRef]
- Song, J.W.; Jeon, E.Y.; Song, D.H.; Jang, H.Y.; Bornscheuer, U.T.; Oh, D.K.; Park, J.B. Multistep enzymatic synthesis of long-chain α, ω-dicarboxylic and ω-hydroxycarboxylic acids from renewable fatty acids and plant oils. Angew. Chem. Int. Ed. 2013, 52, 2534–2537. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Huang, Y.; White, M.A.; Wu, X.; Liu, N.; Cheng, X.; Chen, Y. Dual catalysis mode for the dicarbonyl reduction catalyzed by diketoreductase. Chem. Commun. 2012, 48, 11352–11354. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-M.; Liu, Y.-Y.; Zheng, Y.-C.; Li, H.; Zhang, X.-Y.; Zheng, G.-W.; Li, C.-X.; Bai, Y.-P.; Xu, J.-H. Direct access to medium-chain α, ω-dicarboxylic acids by using a Baeyer–Villiger monooxygenase of abnormal regioselectivity. ChemBioChem 2018, 19, 2049–2054. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, A.; Altenbuchner, J.; Bornscheuer, U.T. Cloning, expression, and characterization of a Baeyer–Villiger monooxygenase from Pseudomonas fluorescens DSM 50106 in E. coli. Appl. Microbiol. Biotechnol. 2007, 73, 1065–1072. [Google Scholar] [CrossRef]
- Wichmann, R.; Vasic-Racki, D. Co-factor regeneration at the lab scale. Technol. Transf. Biotechnol. 2005, 92, 225–260. [Google Scholar]
- Liu, W.; Wang, P. Co-factor regeneration for sustainable enzymatic biosynthesis. Biotechnol. Adv. 2007, 25, 369–384. [Google Scholar] [CrossRef]
- Zhou, H.; Meng, L.; Yin, X.; Liu, Y.; Xu, G.; Wu, J.; Wu, M.; Yang, L. Artificial biocatalytic cascade with three enzymes in one pot for asymmetric synthesis of chiral unnatural amino acids. Eur. J. Org. Chem. 2019, 2019, 6470–6477. [Google Scholar] [CrossRef]
- Xu, Z.; Jing, K.; Liu, Y.; Cen, P. High-level expression of recombinant glucose dehydrogenase and its application in NADPH regeneration. J. Ind. Microbiol. Biotechnol. 2007, 34, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Bommarius, A.S.; Schwarm, M.; Stingl, K.; Kottenhahn, M.; Huthmacher, K.; Drauz, K. Synthesis and use of enantiomerically pure tert-leucine. Tetrahedron: Asymmetry 1995, 6, 2851–2888. [Google Scholar] [CrossRef]
- Zhuang, M.-Y.; Jiang, X.-P.; Ling, X.-M.; Xu, M.-Q.; Zhu, Y.-H.; Zhang, Y.-W. Immobilization of glycerol dehydrogenase and NADH oxidase for enzymatic synthesis of 1, 3-dihydroxyacetone with in situ co-factor regeneration. J. Chem. Technol. Biotechnol. 2018, 93, 2351–2358. [Google Scholar] [CrossRef]
- Xu, M.Q.; Li, F.L.; Yu, W.Q.; Li, R.F.; Zhang, Y.W. Combined cross-linked enzyme aggregates of glycerol dehydrogenase and NADH oxidase for high efficiency in situ NAD+ regeneration. Int. J. Biol. Macromol. 2020, 144, 1013–1021. [Google Scholar] [CrossRef]
- Chánique, A.M.; Parra, L.P. Protein engineering for nicotinamide coenzyme specificity in oxidoreductases: Attempts and challenges. Front. Microbiol. 2018, 9, 194. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, C.; Lavandera, I.; Gotor, V. Recent Advances in Cofactor Regeneration Systems Applied to Biocatalyzed Oxida-tive Processes. Curr. Org. Chem. 2012, 16, 2525–2541. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann-Chen, S.; Flock, T.; Cahn, J.K.B.; Snow, C.D.; Brustad, E.M.; McIntosh, J.A.; Meinhold, P.; Zhang, L.; Arnold, F.H. General approach to reversing ketol-acid reductoisomerase cofactor dependence from NADPH to NADH. Proc. Natl. Acad. Sci. USA 2013, 110, 10946–10951. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Liu, N.; Chen, Y.; Wang, S.; Liu, J.; Zhao, L.; Ma, X.; Cai, D.; Chen, S. Rational engineering of cofactor specificity of glutamate dehydrogenase for poly-γ-glutamic acid synthesis in Bacillus licheniformis. Enzym. Microb. Technol. 2022, 155, 109979. [Google Scholar] [CrossRef]
- Cahn, J.K.B.; Werlang, C.A.; Baumschlager, A.; Brinkmann-Chen, S.; Mayo, S.L.; Arnold, F.H. A general tool for engineering the NAD/NADP cofactor preference of oxidoreductases. ACS Synth. Biol. 2017, 6, 326–333. [Google Scholar] [CrossRef]
- Seo, E.-J.; Kim, H.-J.; Kim, M.-J.; Kim, J.-S.; Park, J.-B. Cofactor specificity engineering of a long-chain secondary alcohol dehydrogenase from Micrococcus luteus for redox-neutral biotransformation of fatty acids. Chem. Commun. 2019, 55, 14462–14465. [Google Scholar] [CrossRef]
- Bommareddy, R.R.; Chen, Z.; Rappert, S.; Zeng, A.P. A de novo NADPH generation pathway for improving lysine production of Corynebacterium glutamicum by rational design of the coenzyme specificity of glyceraldehyde 3-phosphate dehydrogenase. Metab. Eng. 2014, 25, 30–37. [Google Scholar] [CrossRef]
- Beier, A.; Bordewick, S.; Genz, M.; Schmidt, S.; Van Den Bergh, T.; Peters, C.; Joosten, H.J.; Bornscheuer, U.T. Switch in co-factor specificity of a baeyer–villiger monooxygenase. ChemBioChem 2016, 17, 2312–2315. [Google Scholar] [CrossRef]
- Borlinghaus, N.; Nestl, B.M. Switching the co-factor specificity of an imine reductase. ChemCatChem 2018, 10, 183–187. [Google Scholar] [CrossRef]
- Thompson, M.P.; Turner, N.J. Two-enzyme hydrogen-borrowing amination of alcohols enabled by a co-factor-switched alcohol dehydrogenase. ChemCatChem 2017, 9, 3833–3836. [Google Scholar] [CrossRef]
- Xu, J.-Z.; Yang, H.-K.; Liu, L.-M.; Wang, Y.-Y.; Zhang, W.-G. Rational modification of Corynebacterium glutamicum dihydrodipicolinate reductase to switch the nucleotide-co-factor specificity for increasing l-lysine production. Biotechnol. Bioeng. 2018, 115, 1764–1777. [Google Scholar] [CrossRef]
- Bellamacina, C.R. The nicotinamide dinucleotide binding motif: A comparison of nucleotide binding proteins. FASEB J. 1996, 10, 1257–1269. [Google Scholar]
- Corbier, C.; Clermont, S.; Billard, P.; Skarzynski, T.; Branlant, C.; Wonacott, A.; Branlant, G. Probing the coenzyme specificity of glyceraldehyde-3-phosphate dehydrogenases by site-directed mutagenesis. Biochemistry 1990, 29, 7101–7106. [Google Scholar] [CrossRef]
- Ge, Y.; Song, P.; Cao, Z.; Wang, P.; Zhu, G. Alteration of coenzyme specificity of malate dehydrogenase from Streptomyces coelicolor A3 (2) by site-directed mutagenesis. Genet. Mol. Res. 2014, 13, 5758–5766. [Google Scholar] [CrossRef]
- De Ruyck, J.; Famerée, M.; Wouters, J.; Perpète, E.A.; Preat, J.; Jacquemin, D. Towards the understanding of the absorption spectra of NAD (P) H/NAD (P)+ as a common indicator of dehydrogenase enzymatic activity. Chem. Phys. Lett. 2007, 450, 119–122. [Google Scholar] [CrossRef]
- Qin, Y.Z.; Xia, J.; Wang, B.W. Biological Reaction Engineering, 2nd ed.; Chemical Industry Press: Beijing, China, 2009; pp. 9–14. [Google Scholar]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Co-Factors | IU (U·mg−1) | Vmax (mg·mL−1min−1) | Km (mM) | kcat (min−1) | kcat/Km (mM−1·min−1) |
|---|---|---|---|---|---|---|
| Wildtype | NAD+ | 13.56 | 4.13 | 2.08 | 8.05 | 3.86 |
| NADP+ | *ND | *ND | *ND | *ND | *ND | |
| M1 (D37G) | NAD+ | 1.21 | 0.02 | 0.60 | 0.04 | 0.07 |
| NADP+ | 1.82 | 0.03 | 0.21 | 0.05 | 0.24 | |
| M2 (D37G-V39S) | NAD+ | 0.46 | 0.14 | 1.56 | 0.15 | 0.10 |
| NADP+ | 0.49 | 0.15 | 1.35 | 0.16 | 0.12 | |
| M3 (D37G-A38T-V39K) | NAD+ | 2.31 | 0.29 | 16.48 | 0.77 | 0.05 |
| NADP+ | 0.56 | 0.07 | 1.10 | 0.19 | 0.17 | |
| M4 (Y36S-D37G-A38D) | NAD+ | 0.19 | 0.06 | 0.19 | 0.06 | 0.30 |
| NADP+ | 0.19 | 0.06 | 0.64 | 0.06 | 0.09 | |
| M5 (Y36S-D37G-A38G-V39K) | NAD+ | 0.24 | 0.01 | 0.04 | 0.04 | 0.99 |
| NADP+ | *ND | *ND | *ND | *ND | *ND | |
| M6 (Y36S-D37G-A38D-V39K) | NAD+ | 0.30 | 0.01 | 0.01 | 0.05 | 4.22 |
| NADP+ | *ND | *ND | *ND | *ND | *ND |
| Enzymes | Co-Factors | Yields of Sebacic Acid (%) |
|---|---|---|
| Wildtype MlADH & PaBVMO & NOX & GDH | NAD+ & NADP+ | 41.2 ± 2.7 |
| Wildtype MlADH & PaBVMO | NAD+ | 6.7 ± 1.2 |
| NADP+ | 20.1 ± 3.9 | |
| M1(D37G) & PaBVMO | NAD+ | 2.1 ± 1.1 |
| NADP+ | 42.4 ± 2.2 | |
| M2(D37G-V39S) & PaBVMO | NAD+ | 5.3 ± 1.4 |
| NADP+ | 25.8 ± 3.2 | |
| M3(D37G-A38T-V39K) & PaBVMO | NAD+ | 2.4 ± 0.8 |
| NADP+ | 49.3 ± 3.6 |
| MlADH or Mutants | Co-Factors | Binding Energy (kJ·mol−1) |
|---|---|---|
| Wildtype | NAD+ | −34.60 |
| NADP+ | −14.31 | |
| M1(D37G) | NAD+ | −29.79 |
| NADP+ | −27.69 | |
| M2(D37G-V39S) | NAD+ | −30.87 |
| NADP+ | −32.56 | |
| M3(D37G-A38T-V39K) | NAD+ | −31.73 |
| NADP+ | −34.96 |
| Mutants | Primer | |
|---|---|---|
| M1(D37G) | F | CGGCACTGCACCATAGGCCATCAC |
| R | GGCCTATGGTGCAGTGCCGGC | |
| M2(D37G-V39S) | F | GCTGCCGGGCTTGCACCATAGGCCATCAC |
| R | GATGGCCTATGGTGCAAGCCCGGCAGCAC | |
| M3(D37G-A38T-V39K) | F | GTGCTGCCGGCTTTGTACCATAGGCCATCAC |
| R | GATGGCCTATGGTACAAAGCCGGCAGCAC | |
| M4(Y36S-D37G-A38D) | F | CTGCCGGCACGTCACCACTGGCCATCACTTTTTTG |
| R | GTGATGGCCAGTGGTGACGTGCCGGCAGCAC | |
| M5(Y36S-D37G-A38G-V39K) | F | GTGCTGCCGGGCTTCCACCACTGGCCATCACTTTTTTG |
| R | GTGATGGCCAGTGGTGGAAGCCCGGCAGCACTGG | |
| M6(Y36S-D37G-A38D-V39K) | F | GTGCTGCCGGGCTGTCACCACTGGCCATCACTTTTTTG |
| R | GTGATGGCCAGTGGTGACAGCCCGGCAGCACTGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, J.; Lu, D.; Wu, Q.; Jin, S.; Liu, J.; Qin, M.; Deng, L.; Wang, F.; Nie, K. Biocatalytic Cascade of Sebacic Acid Production with In Situ Co-Factor Regeneration Enabled by Engineering of an Alcohol Dehydrogenase. Catalysts 2022, 12, 1318. https://doi.org/10.3390/catal12111318
Lu J, Lu D, Wu Q, Jin S, Liu J, Qin M, Deng L, Wang F, Nie K. Biocatalytic Cascade of Sebacic Acid Production with In Situ Co-Factor Regeneration Enabled by Engineering of an Alcohol Dehydrogenase. Catalysts. 2022; 12(11):1318. https://doi.org/10.3390/catal12111318
Chicago/Turabian StyleLu, Jie, Dong Lu, Qiuyang Wu, Shuming Jin, Junfeng Liu, Meng Qin, Li Deng, Fang Wang, and Kaili Nie. 2022. "Biocatalytic Cascade of Sebacic Acid Production with In Situ Co-Factor Regeneration Enabled by Engineering of an Alcohol Dehydrogenase" Catalysts 12, no. 11: 1318. https://doi.org/10.3390/catal12111318