

Na Promotion of Pt/m-ZrO2 Catalysts for the Steam Reforming of Formaldehyde


 ,
, 
Abstract
:1. Introduction
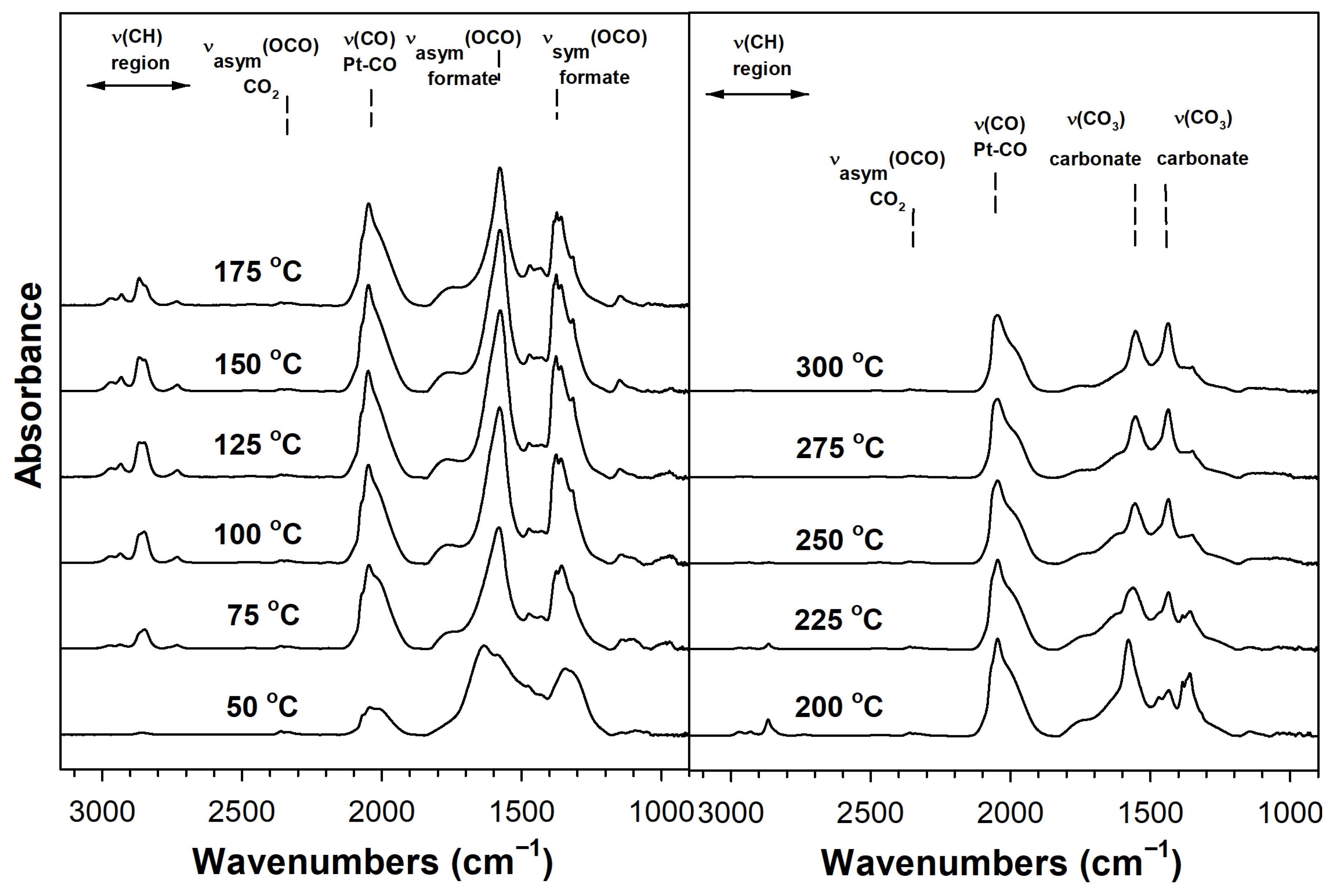
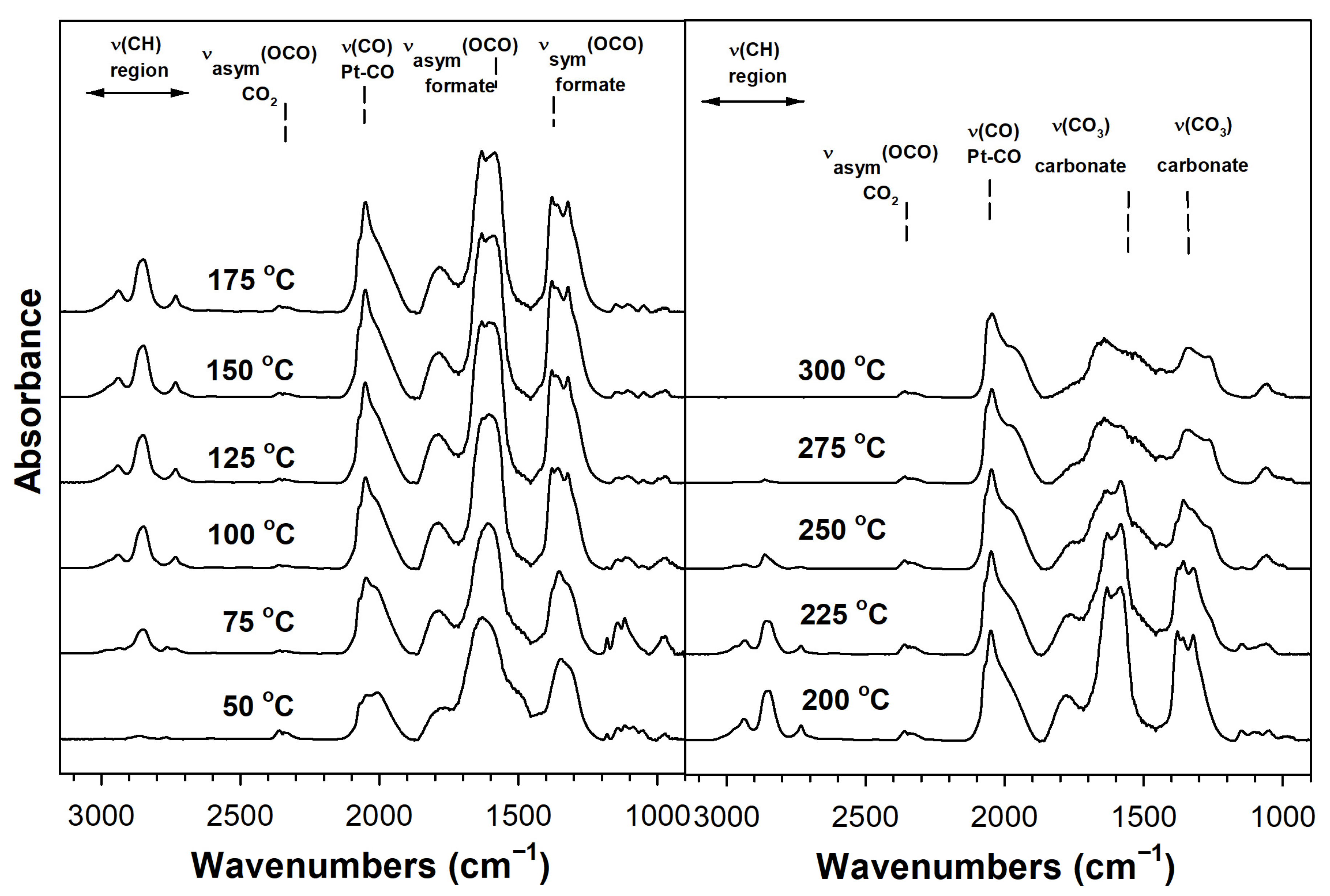
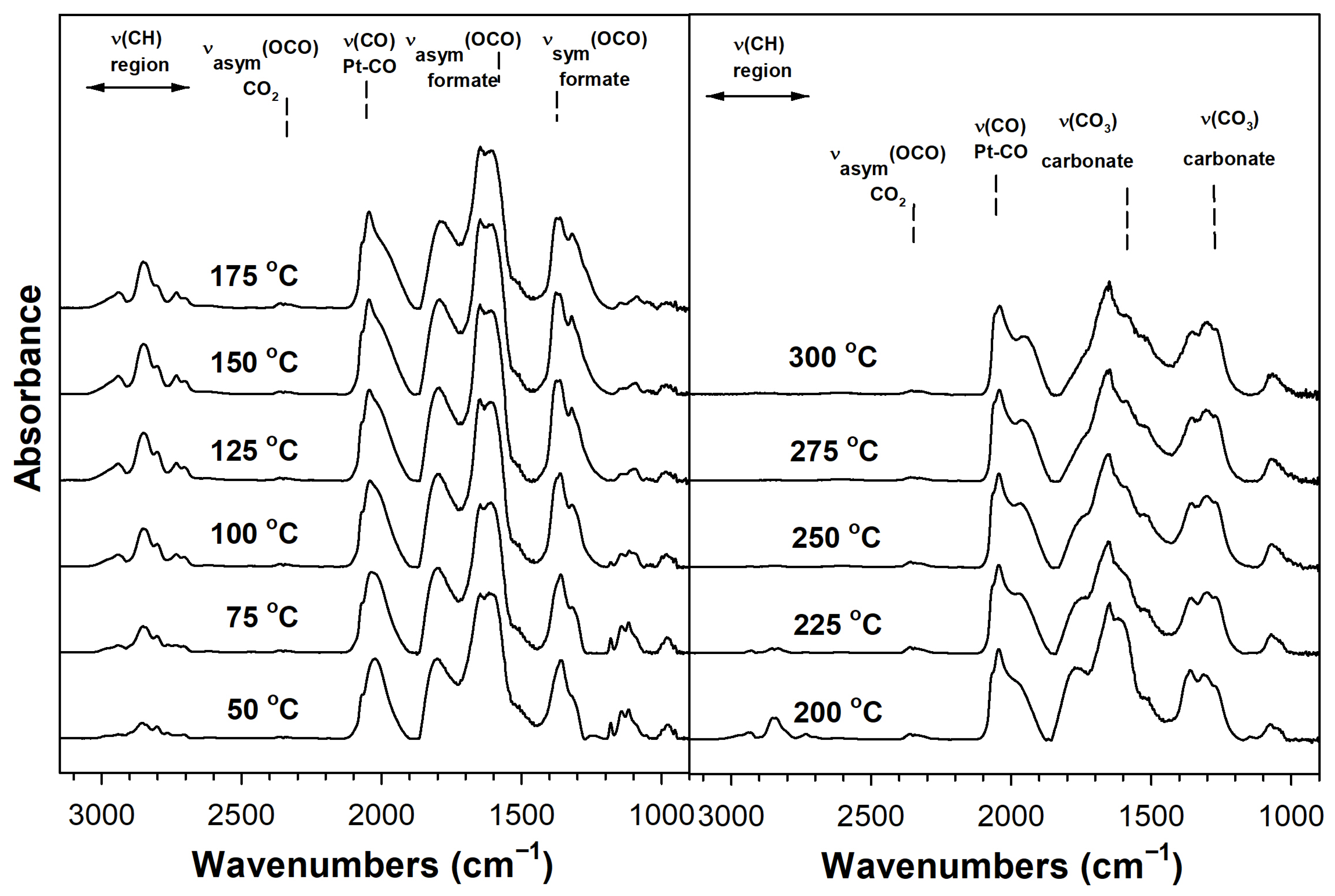
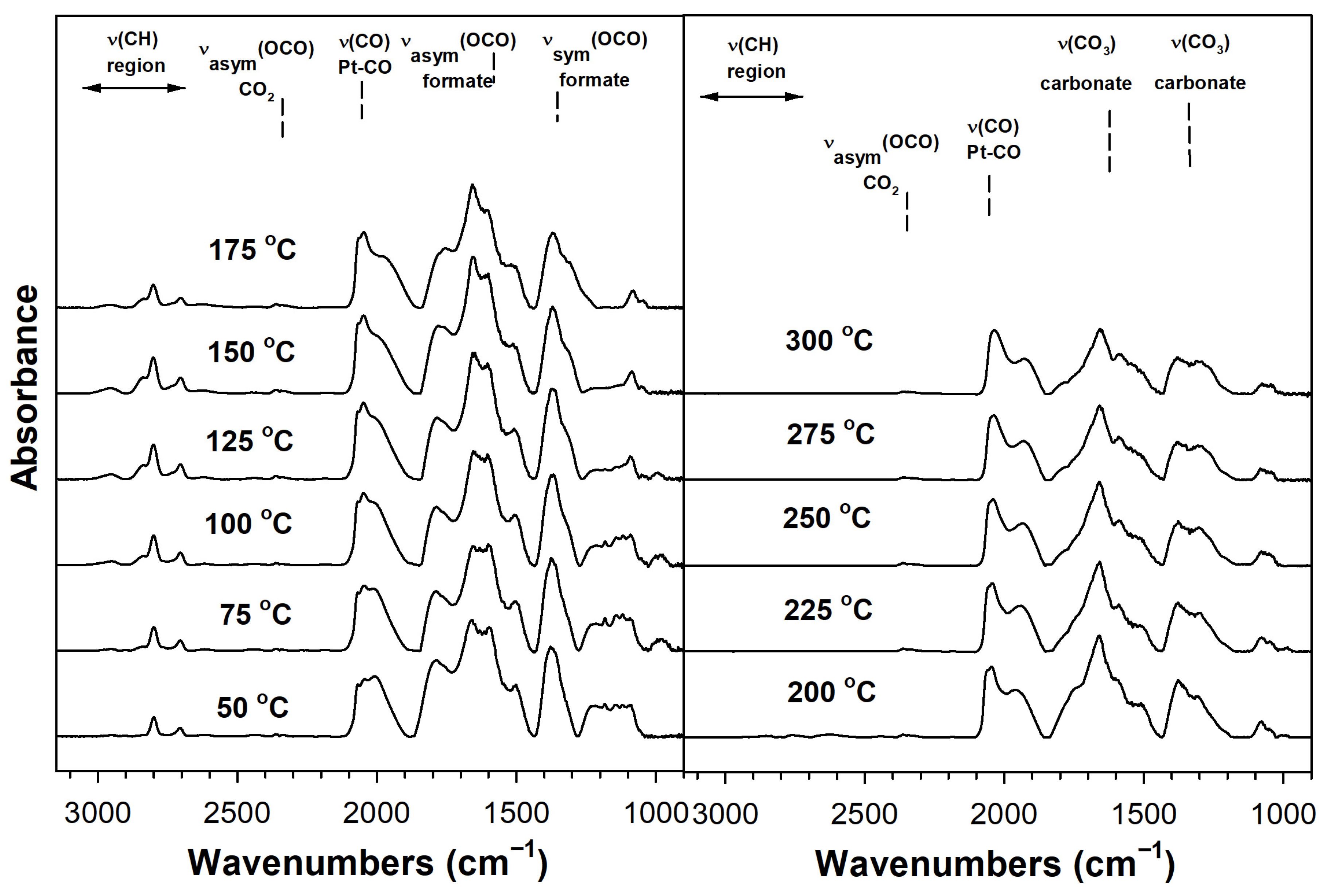
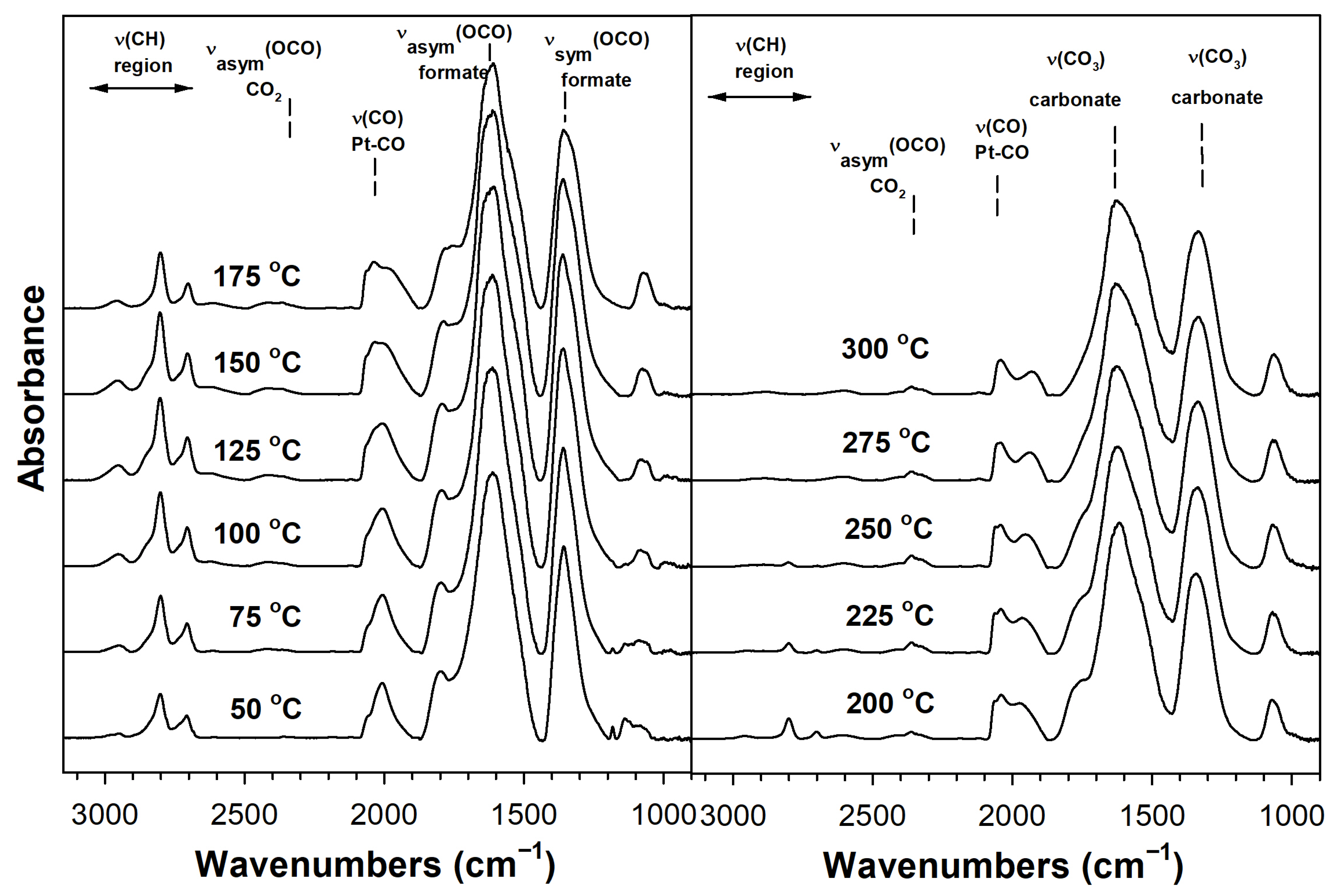
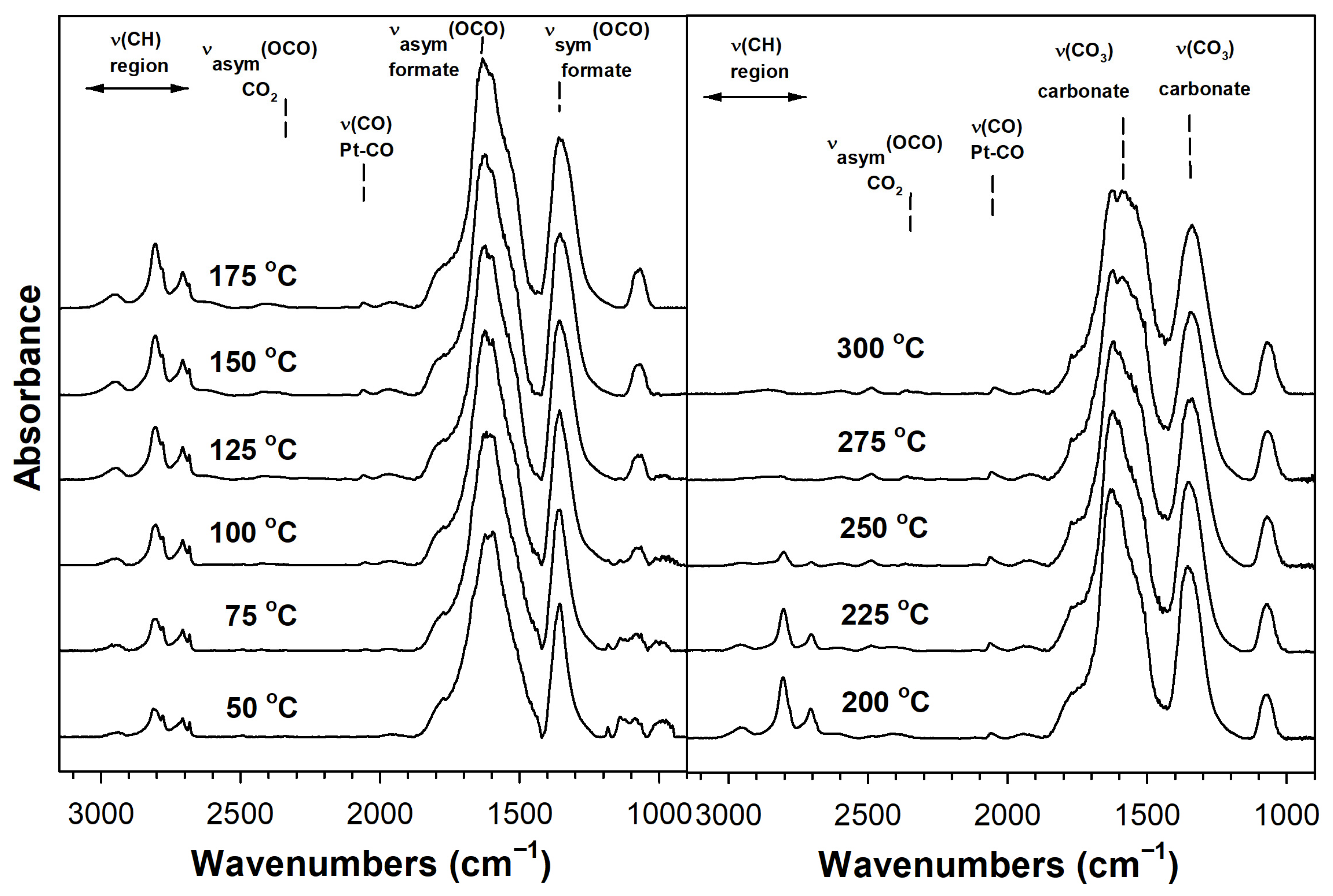
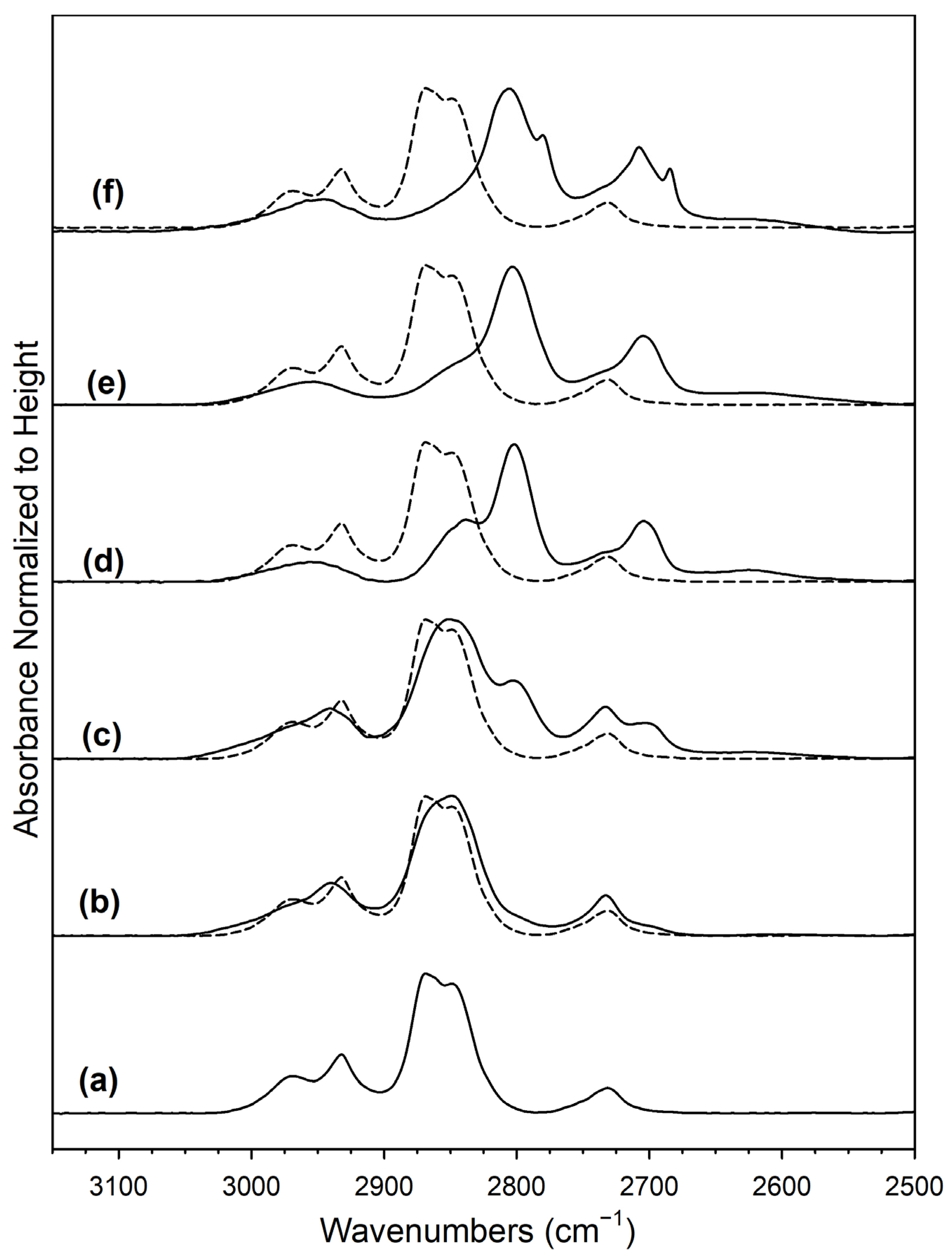
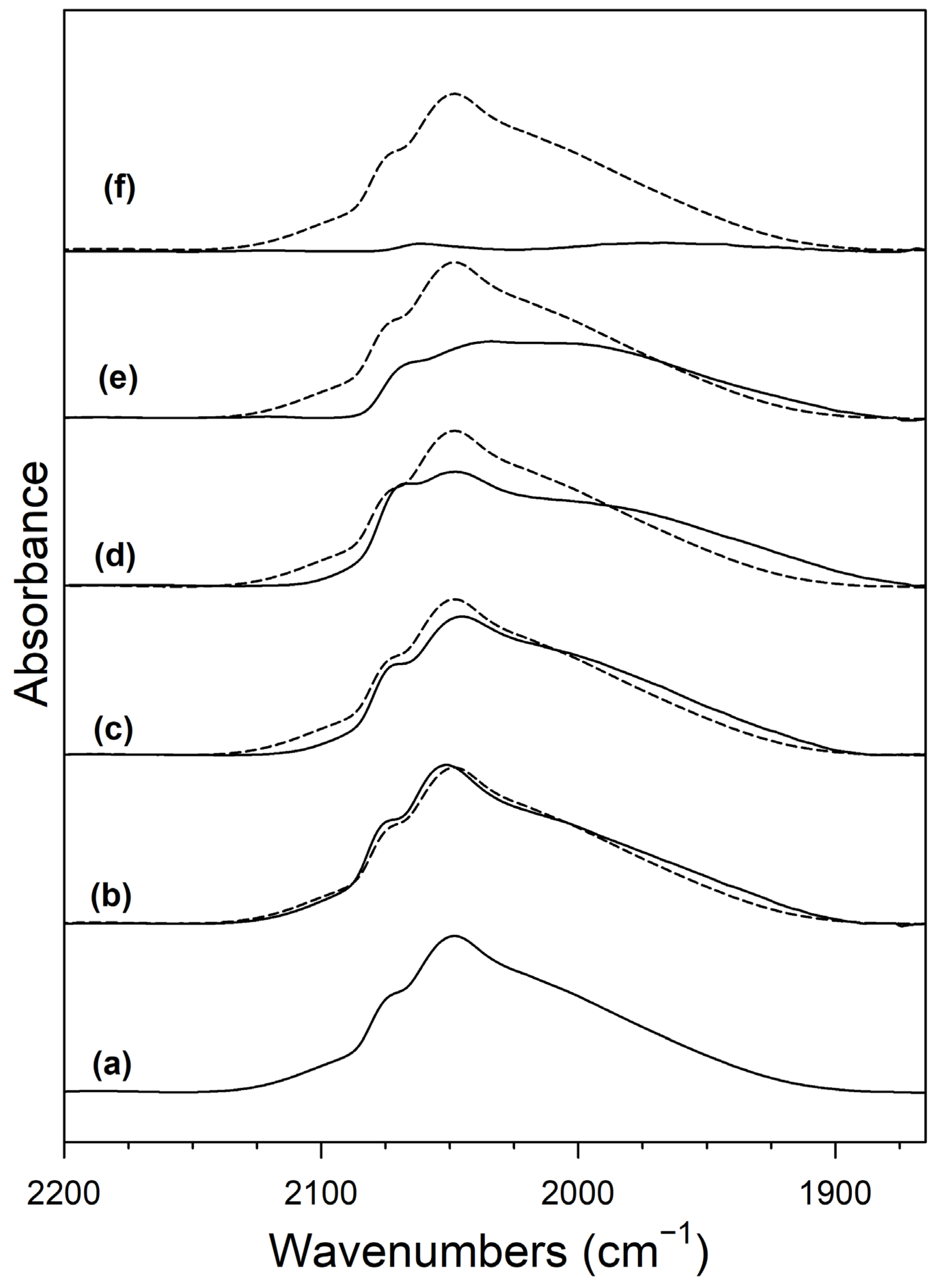
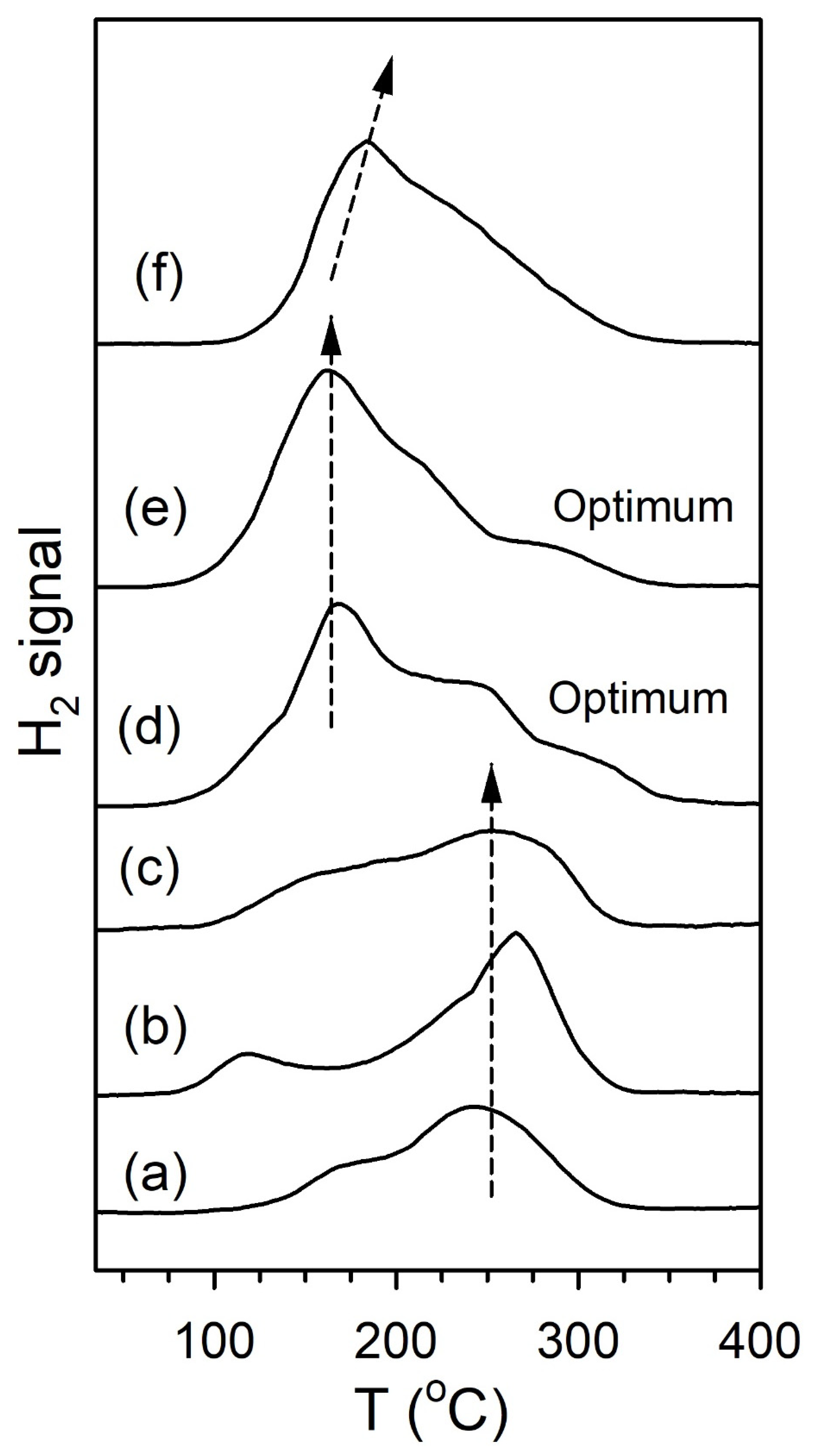
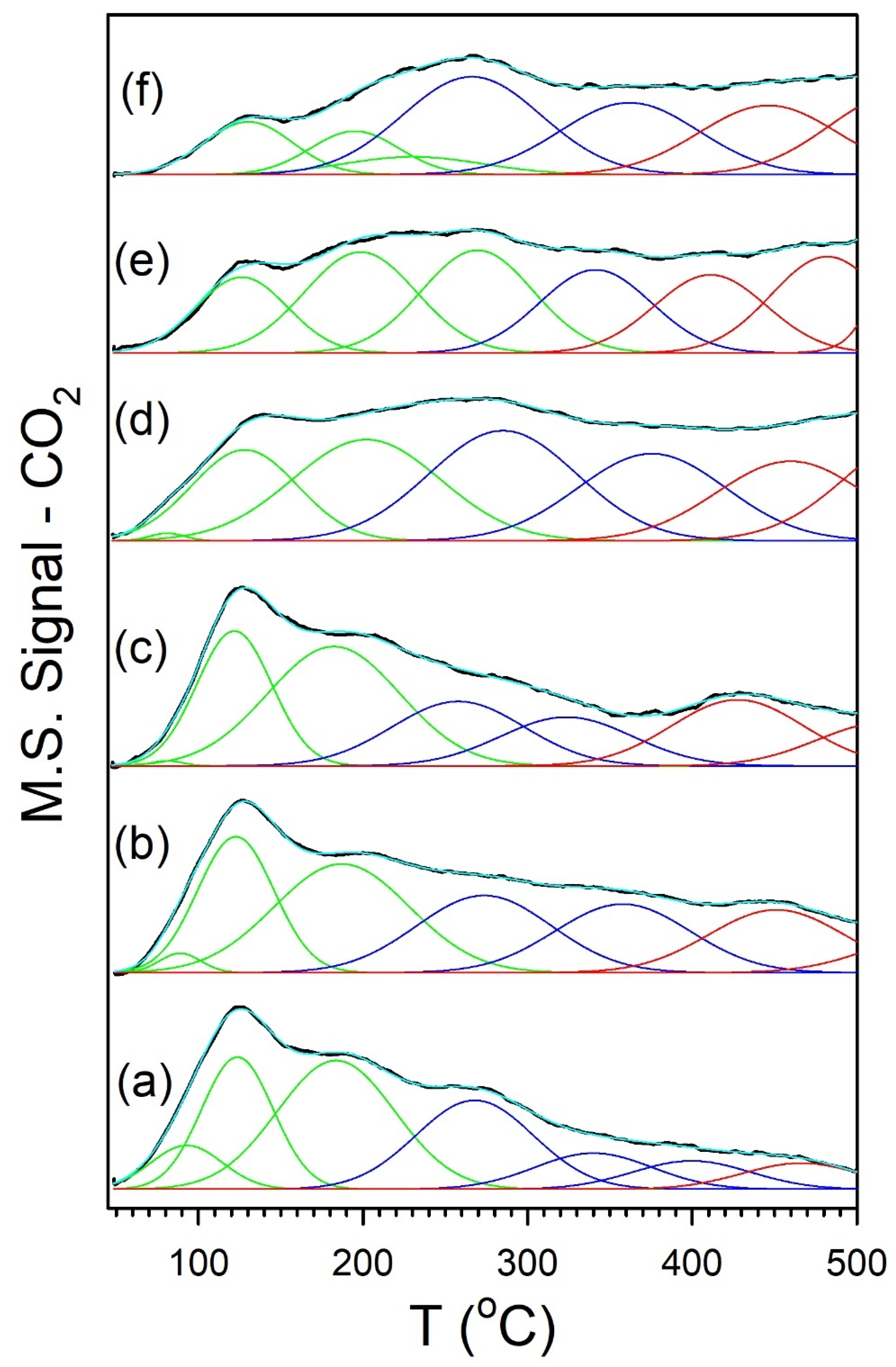
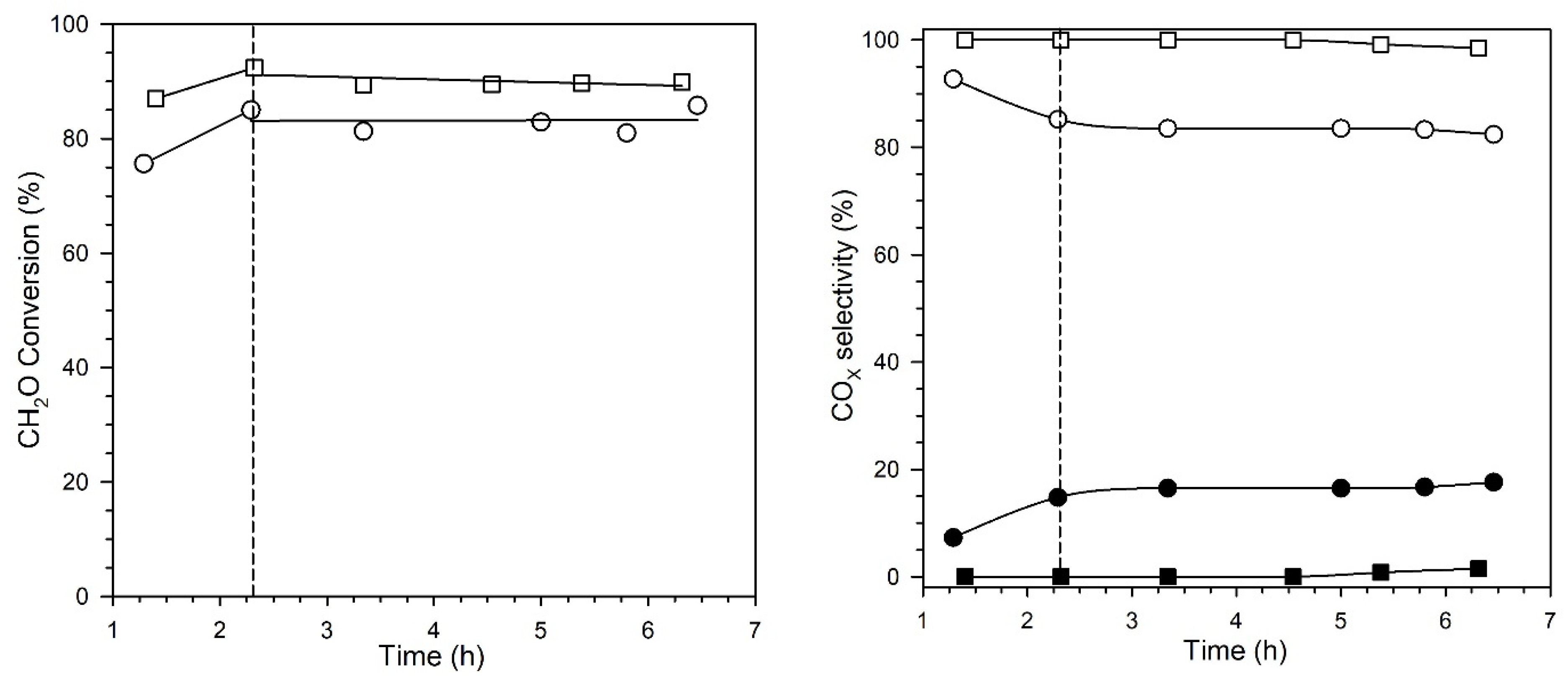
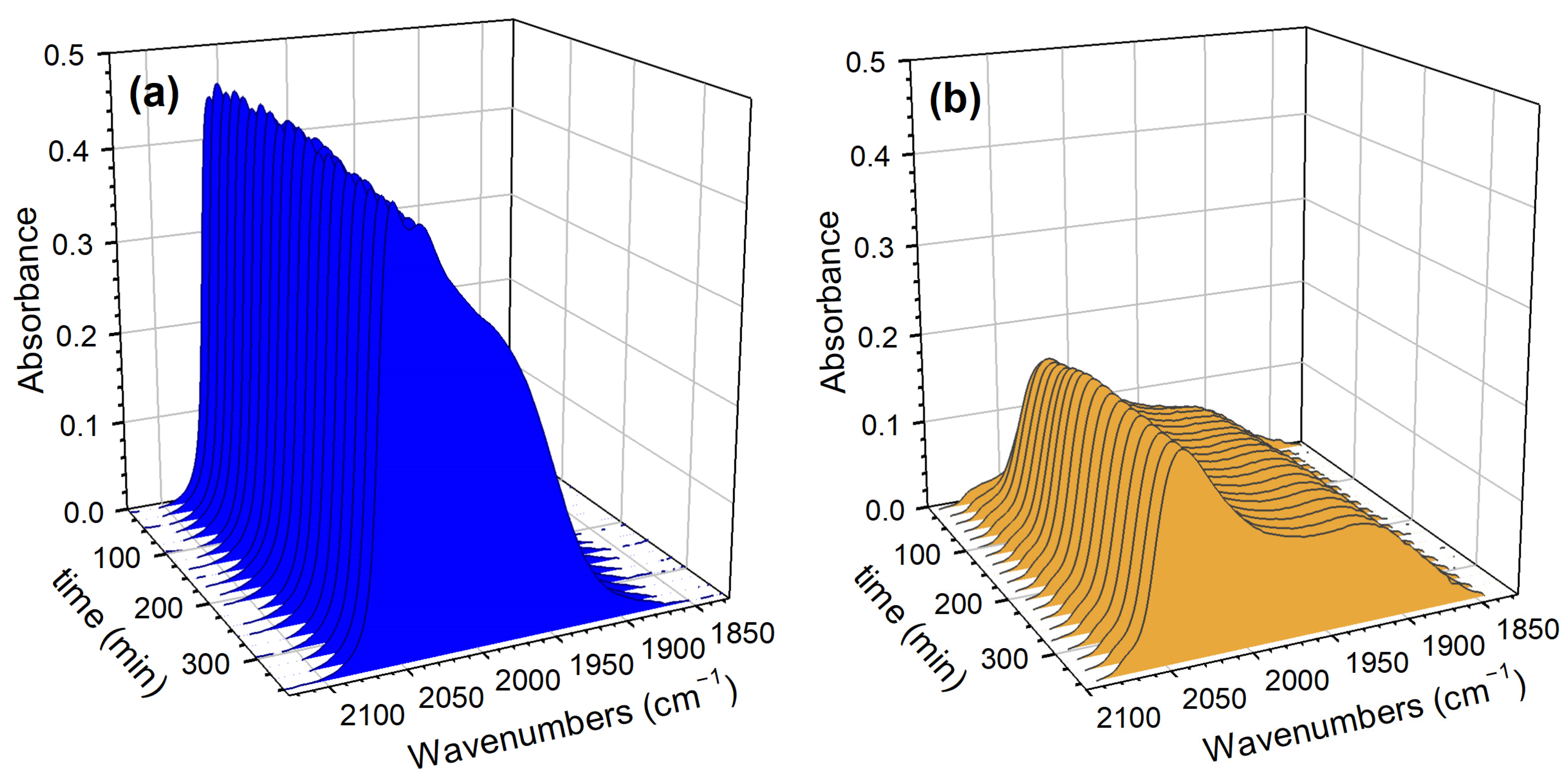
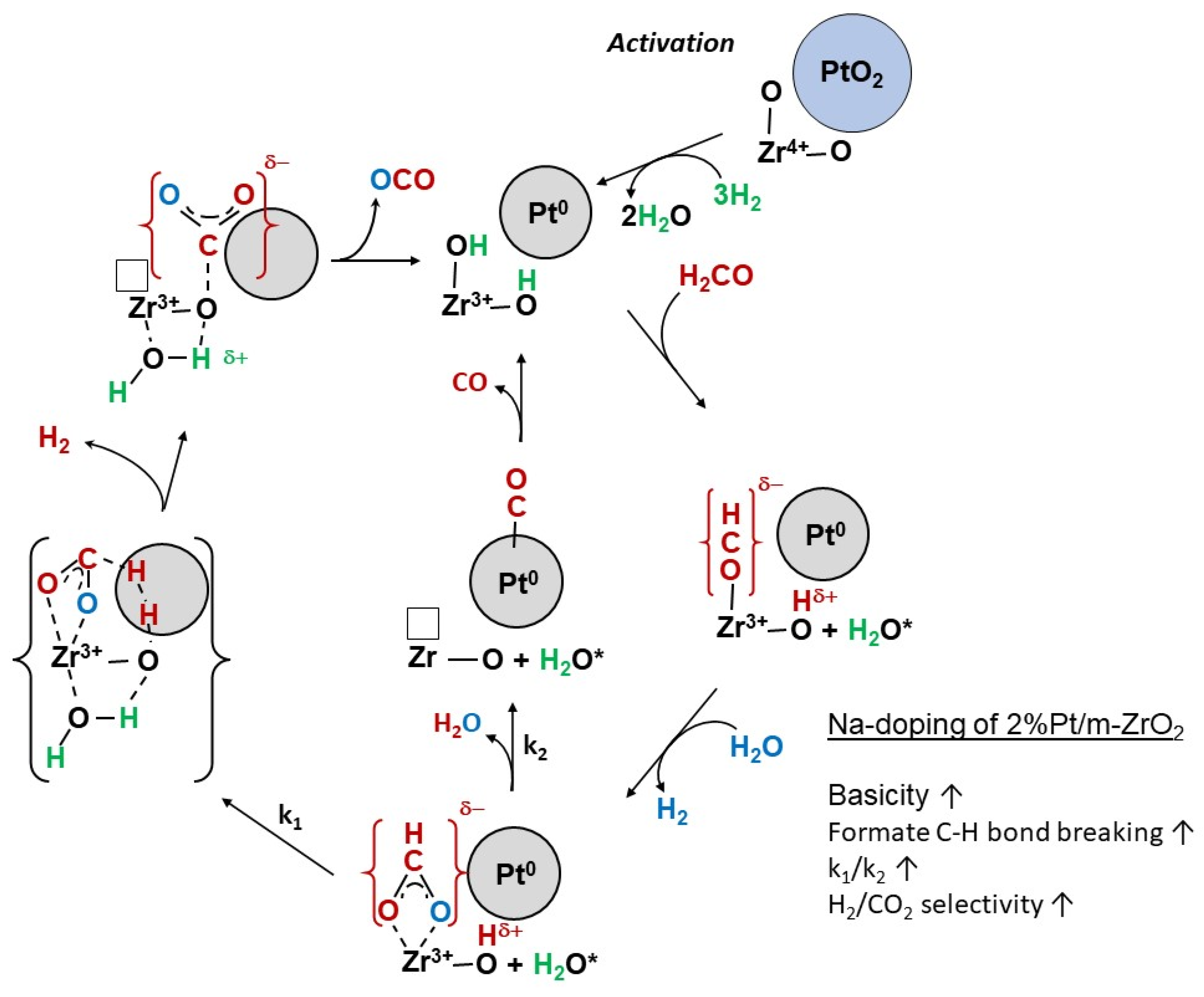
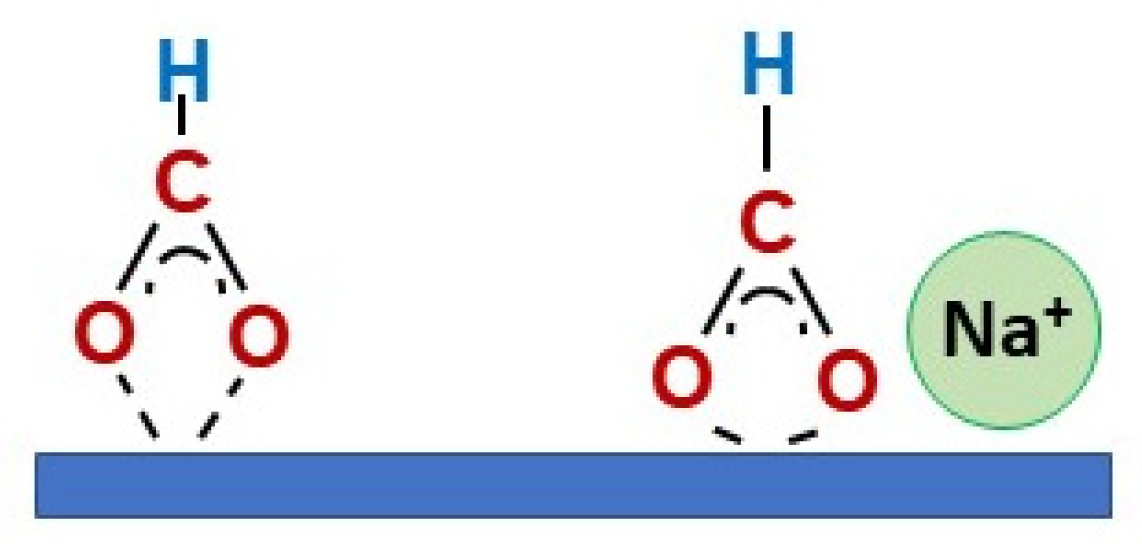
2. Results and Discussion
Catalyst Characterization
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Characterization
3.2.1. BET Surface Area
3.2.2. TEM
3.2.3. Temperature Programmed Reduction/Mass Spectrometry
3.2.4. Temperature Programmed Desorption, Reaction, and Oxidation
3.2.5. DRIFTS
3.2.6. Reaction Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anil, S.; Indraja, S.; Singh, R.; Appari, S.; Roy, B. A review on ethanol steam reforming for hydrogen production over Ni/Al2O3 and Ni/CeO2 based catalyst powders. Int. J. Hydrog. Energy 2022, 47, 8177–8213. [Google Scholar] [CrossRef]
- Ogo, S.; Sekine, Y. Recent progress in ethanol steam reforming using non-noble transition metal catalysts: A review. Fuel Proc. Tech. 2020, 199, 106238. [Google Scholar] [CrossRef]
- Bac, S.; Keskin, S.; Avci, A.K. Recent advances in materials for high purity H2 production by ethanol and glycerol steam reforming. Int. J. Hydrog. Energy 2020, 45, 34888–34917. [Google Scholar] [CrossRef]
- Bepari, S.; Kuila, D. Steam reforming of methanol, ethanol and glycerol over nickel-based catalyst: A review. Int. J. Hydrog. Energy 2020, 45, 18090–18113. [Google Scholar] [CrossRef]
- Mattos, L.V.; Jacobs, G.; Davis, B.H.; Noronha, F.B. Production of hydrogen from ethanol: Review of reaction mechanism and catalyst deactivation. Chem. Rev. 2012, 112, 4094–4123. [Google Scholar] [CrossRef] [PubMed]
- Sharma, Y.C.; Kumar, A.; Prasad, R.; Upadhyay, S.N. Ethanol steam reforming for hydrogen production: Latest and effective catalyst modification strategies to minimize carbonaceous deactivation. Renew. Sust. Energy Rev. 2017, 74, 89–103. [Google Scholar] [CrossRef]
- Iulianelli, A.; Ribeirinha, P.; Mendes, A.; Basile, A. Methanol steam reforming for hydrogen generation via conventional and membrane reactors: A review. Renew. Sust. Energy Rev. 2014, 29, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol steam reforming for hydrogen production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef]
- Jacobs, G.; Davis, B.H. In situ DRIFTS investigation of the steam reforming of methanol over Pt/ceria. Appl. Catal. A Gen. 2005, 285, 43–49. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Polychronopoulou, K.; Asif, A.; Goula, M.A. The potential of glycerol and phenol towards H2 production using steam reforming reaction: A review. Surf. Coat. Tech. 2018, 352, 92–111. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Dou, B.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. Ni/Y2O3–ZrO2 catalyst for hydrogen production through the glycerol steam reforming reaction. Int. J. Hydrogen Energy 2020, 45, 10442–10460. [Google Scholar] [CrossRef]
- Martinelli, M.; Jacobs, G.; Shafer, W.D.; Davis, B.H. Effect of alkali on C-H bond scission over Pt/YSZ catalyst during water-gas shift, steam-assisted formic acid decomposition and methanol steam reforming. Catal. Today 2017, 291, 29–35. [Google Scholar] [CrossRef]
- Chu, L.; Gu, S.; Jin, Q.; Zhu, P.; Shen, Y.; Li, P. Hydrogen production from formaldehyde steam reforming using recyclable NiO/NaF catalyst. Int. J. Hydrog. Energy 2020, 45, 28752–28763. [Google Scholar] [CrossRef]
- Chu, L.; Shen, Y. Hydrogen production from formaldehyde steam reforming using recyclable NiO/NaCl catalyst. Appl. Surf. Sci. 2020, 532, 147376. [Google Scholar] [CrossRef]
- Lorenz, H.; Friedrich, M.; Armbruester, M.; Kloetzer, B.; Penner, S. ZnO is a CO2-selective steam reforming catalyst. J. Catal. 2013, 297, 151–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielz, T.; Lorenz, H.; Amann, P.; Klotzer, B.; Penner, S. Water-gas shift and formaldehyde reforming activity determined by defect chemistry of polycrystalline In2O3. J. Phys. Chem. 2011, 115, 6622–6628. [Google Scholar] [CrossRef]
- Jin, Q.; Wang, A.; Lu, B.; Xu, X.; Shen, Y.; Zeng, Y. Steam reforming of formaldehyde for generating hydrogen and coproducing carbon nanotube for enhances photosynthesis. Catal. Sci. Tech. 2020, 10, 4436–4447. [Google Scholar] [CrossRef]
- Jin, Q.; Shen, Y.; Cai, Y.; Chu, L.; Zeng, Y. Resource utilization of waste V2O5-based deNOX catalysts for hydrogen production from formaldehyde and water via steam reforming. J. Hazard. Mat. 2020, 381, 120934. [Google Scholar] [CrossRef]
- Li, X.; Lim, K.H. DFT studies of steam reforming of formaldehyde on Cu, PdZn and Ir. ChemCatChem 2012, 4, 1311–1320. [Google Scholar] [CrossRef]
- Bo, J.-Y.; Zhang, S.; Lim, K.H. Steam reforming of formaldehyde on Cu(100) surface: A density functional study. Catal. Lett. 2009, 129, 444–448. [Google Scholar] [CrossRef]
- Lim, K.H.; Chen, Z.-X.; Neyman, K.M.; Roesch, N. Comparative theoretical study of formaldehyde decomposition on PdZn, Cu, and Pd Surfaces. J. Phys. Chem. B 2006, 110, 14890–14897. [Google Scholar] [CrossRef] [PubMed]
- Chenu, E.; Jacobs, G.; Crawford, A.C.; Keogh, R.A.; Patterson, P.M.; Sparks, D.E.; Davis, B.H. Water-gas Shift: An examination of unpromoted and Pt promoted MgO and tetragonal and monoclinic ZrO2 by in-situ DRIFTS. Appl. Catal. B Environ. 2005, 59, 45–56. [Google Scholar] [CrossRef]
- Azzam, K.G.; Babich, I.V.; Seshan, K.; Lefferts, L. Bifunctional catalysts for single-stage water-gas shift reaction in fuel cell applications. J. Catal. 2007, 251, 153–162. [Google Scholar] [CrossRef]
- Petallidou, K.C.; Kalamaras, C.M.; Efstathiou, A.M. The effect of La3+, Ti4+ and Zr4+ dopants on the mechanism of WGS on ceria-doped supported Pt catalysts. Catal. Today 2014, 228, 183–193. [Google Scholar] [CrossRef]
- Martinelli, M.; Jacobs, G.; Graham, U.M.; Shafer, W.D.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L.; Khalid, S.; Visconti, C.G.; Lietti, L.; et al. Water-gas shift: Characterization and testing of nanoscale YSZ supported Pt catalysts. Appl. Catal. A Gen. 2015, 497, 184–197. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, M.; Jacobs, G.; Graham, U.M.; Davis, B.H. Methanol steam reforming: Na doping of Pt/YSZ provides fine tuning of selectivity. Catalysts 2017, 7, 148. [Google Scholar] [CrossRef] [Green Version]
- Pigos, J.M.; Brooks, C.J.; Jacobs, G.; Davis, B.H. Low temperature water-gas shift: Assessing formates as potential intermediates over Pt/ZrO2 and Na doped Pt/ZrO2 catalysts employing the SSITKA-DRIFTS technique. In Advances in Fischer-Tropsch Synthesis, Catalysts and Catalysis; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2010; Chapter 19; pp. 365–394. [Google Scholar] [CrossRef]
- Pigos, J.M.; Brooks, C.J.; Jacobs, G.; Davis, B.H. Low temperature water-gas shift: Characterization of Pt-based ZrO2 catalyst promoted with Na discovered by combinatorial methods. Appl. Catal. A General 2007, 319, 47–57. [Google Scholar] [CrossRef]
- Martinelli, M.; Alhraki, N.; Castro, J.D.; Matamoros, M.E.; Jacobs, G. Effect of Na loading on Pt/ZrO2 catalysts for low temperature water-gas shift for the production and purification of hydrogen. In New Dimensions in Production and Utilization of Hydrogen; Nanda, S., Vo, D.-V., Tri, P.N., Eds.; Elsevier Books: Amsterdam, The Netherlands, 2020; Chapter 6; pp. 143–160. ISBN 9780128195536. [Google Scholar]
- Shido, T.; Iwasawa, Y. Reactant-promoted reaction mechanism for water-gas shift reaction on Rh-doped CeO2. J. Catal. 1993, 141, 71–81. [Google Scholar] [CrossRef]
- Martinelli, M.; Castro, J.D.; Alhraki, N.; Matamoros, M.E.; Kropf, A.J.; Cronauer, D.C.; Jacobs, G. Effect of sodium loading on Pt/ZrO2 during ethanol steam reforming. Appl. Catal. A General 2021, 610, 117947. [Google Scholar] [CrossRef]
- Martinelli, M.; Watson, C.D.; Jacobs, G. Sodium doping of Pt/m-ZrO2 promotes C-C scission and decarboxylation during ethanol steam reforming. Internat. J. Hydrogen Energy 2020, 45, 18490–18501. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.-C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Zhu, X.; Xie, Y.; Liu, C.-J.; Zhang, Y.-P. Stability of Pt particles on ZrO2 support during partial oxidation of methane: DRIFT studies of adsorbed CO. J. Molec. Catal. A 2008, 282, 67–73. [Google Scholar] [CrossRef]
- Jongpatiwuta, S.; Trakarnroek, S.; Rirksomboon, T.; Osuwan, S.; Resasco, D.E. n-Octane aromatization on Pt-containing non-acidic large pore zeolite catalysts. Catal. Lett. 2005, 100, 7–15. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Expected As (m2/g) | Actual As (BET) (m2/g) | Vp (BJH Des) (cm3/g) | Dp (BJH Des) (Å) | Pt0 Size from EXAFS (nm) | Expected %Pt Disp. | Formate ν(CH) CO Ads. (cm−1) | Formate Decomp. T (°C) TP-rxn ** | XANES % of Pt L3 Minus L2 Area Rel. to No Na |
|---|---|---|---|---|---|---|---|---|---|
| ZrO2 | 106.6 | 106.6 | 0.324 | 96 | - | - | - | - | - |
| 2%Pt/m-ZrO2 | 104.1 | 95.1 | 0.279 | 93 | 0.84 | 93 | 2868, 2863, 2848 | 304 | 100 |
| Pt/ZrO2 with | |||||||||
| 0.5%Na | 100.8–102.9 | 94.2 | 0.269 | 93 | 0.87 | 92 | (2868, 2856), 2845 | 275 | 108 |
| 1.0%Na | 97.5–101.7 | 94.1 | 0.272 | 94 | 0.92 | 90 | 2869, 2862, 2847 | (wk 190), 287 | 115 |
| 1.8%Na | 92.1–99.7 | 71.8 | 0.232 | 94 | 0.88 * | 91 * | (2850), 2833, 2800 | - | 156 * |
| 2.5%Na | 87.5–98.0 | 66.4 | 0.216 | 97 | 0.91 | 91 | 2802 | 190, 270 | 135 |
| 5.0%Na | 70.9–91.8 | 46.1 | 0.158 | 102 | 0.86 | 86 | 2803 | (wk 212), 280 | 180 |
| Catalyst | Band Position (cm−1) | Δ(OCO) Formate (cm−1) | |||||
|---|---|---|---|---|---|---|---|
| ν(CH) | δ(CH) + νsym(OCO) | 2δ(CH) | ν(OCO) Formate | ν(OCO) Carbonate | % of Formate Band Area at 200 °C Relative to Maximum Area (3050–2500 cm−1) | ||
| 2%Pt/ZrO2 (reference) | 2850, 2868 | 2968, 2934 | (2758), 2731 | sy 1377, 1359, 1317 asy 1576 | 1745, 1553, 1436, 1350, 1138–1025 | 199 | 27 |
| 0.5%Na-2%Pt/ZrO2 | 2849 (2862) | (2971), 2939 | 2733 (2702) | sy, 1379, 1359, 1322 asy 1632, 1586 | (1745), 1644, (1595–1460), 1441, 1341, (1309), 1268, 1060 | 253 | 94 |
| 1%Na-2%Pt/ZrO2 | 2851, 2803 | (2969), 2941 | 2733 (2625) | sy 1377, 1367, 1321 asy 1648, 1605, 1508 | (1745), 1650, (1591, 1531), 1354, 1299, 1272, 1069 | 271 | 33 |
| 1.8%Na-2%Pt/ZrO2 | 2838, 2802 | 2956 | (2735), 2704, 2625 | sy (1430–1382), 1371, (1340–1268) asy 1655, (1634–1550) | (1780, 1745), 1659, 1591, (1555–1460), 1382, 1315, 1294, 1079 | 284 | 15 |
| 2.5%Na-2%Pt/ZrO2 | (2851) 2803 | 2954 | (2735), 2705, (2661–2538) | sy 1361 asy 1609 | (1745) 1625, 1335, 1064 | 248 | 17 |
| 5%Na-2%Pt/ZrO2 | 2805 (2784) | 2955 | (2737) 2708, 2685 | sy (1366–1423), 1360, 1348 asy (1651, 1642), 1632, (1605, 1557–1475) | (1772, 1745), 1625, 1590, 1565, (1540), 1447, 1339, 1070 | 272 | 78 |
| Catalyst | % T < 250 °C | % 250 °C < T < 400 °C | % T > 400 °C |
|---|---|---|---|
| 2%Pt/m-ZrO2 | 54 | 34 | 12 |
| 0.5%Na-2%Pt/m-ZrO2 | 48 | 37 | 16 |
| 1%Na-2%Pt/m-ZrO2 | 51 | 29 | 20 |
| 1.8%Na-2%Pt/m-ZrO2 | 37 | 44 | 19 |
| 2.5%Na-2%Pt/m-ZrO2 | 33 | 37 | 30 |
| 5%Na-2%Pt/m-ZrO2 | 23 | 50 | 26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinelli, M.; Garcia, E.S.; Rajabi, Z.; Watson, C.D.; Kropf, A.J.; Cronauer, D.C.; Jacobs, G. Na Promotion of Pt/m-ZrO2 Catalysts for the Steam Reforming of Formaldehyde. Catalysts 2022, 12, 1294. https://doi.org/10.3390/catal12111294
Martinelli M, Garcia ES, Rajabi Z, Watson CD, Kropf AJ, Cronauer DC, Jacobs G. Na Promotion of Pt/m-ZrO2 Catalysts for the Steam Reforming of Formaldehyde. Catalysts. 2022; 12(11):1294. https://doi.org/10.3390/catal12111294
Chicago/Turabian StyleMartinelli, Michela, Elijah S. Garcia, Zahra Rajabi, Caleb D. Watson, A. Jeremy Kropf, Donald C. Cronauer, and Gary Jacobs. 2022. "Na Promotion of Pt/m-ZrO2 Catalysts for the Steam Reforming of Formaldehyde" Catalysts 12, no. 11: 1294. https://doi.org/10.3390/catal12111294