
Integral Characteristic of Complex Catalytic Reaction Accompanied by Deactivation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Phenomenological Models
1.2. Detailed Kinetic Models
1.3. Semi-Phenomenological Models
- the number of parameters is much smaller;
- this model allows the derivation of interesting analytical results, as will be demonstrated in this paper;
- potentially, this simpler model can be useful for the design of catalytic reactors with deactivation and optimization of industrial regimes.
- a large difference in kinetic parameters
- a large difference between the total amounts of main reactants and the total amount of intermediates. For the ‘gas-solid’ catalytic reaction, the latter corresponds to the case when the total number of active catalytic centers is much smaller than the total number of reactant and product molecules, see [22] (Chapter 3).
2. Goal of the Paper: General Problems and Specific Problems of This Paper
- the small parameter which is caused by the difference between the number of catalyst active sites and the number of gaseous molecules (“the first small parameter”)
- the small parameter caused by the difference between the deactivation parameters and kinetic coefficients of the main catalytic cycle (“the second small parameter”)
- Kinetic models of the batch reactor (BR) and continuously stirred tank reactor (CSTR).
- Kinetic models of typical heterogeneous catalytic mechanisms:
- (a)
- The n-step single-route complex catalytic reaction with a linear mechanism.
- (b)
- Models with reversible and irreversible steps in the catalytic cycle.
- Models with reversible and irreversible deactivation process.
- The initial non-steady-state kinetic regime caused by the intrinsic catalytic cycle.
- The quasi-steady-state regime regarding the catalytic intermediates with insignificant deactivation (’no deactivation’ regime). This regime is caused by the difference between the number of catalyst active sites and the number of gaseous molecules (“the first small parameter”).
- The quasi-steady-state regime regarding the intermediates in which the deactivation process is significant. Within this domain, the total number of active sites is decreased, and the quasi-steady-state regime becomes more pronounced.
- In which domain will the catalyst composition be nearly constant, i.e., despite the change in the number of active sites the relative concentrations of catalytic intermediates are remaining approximately the same?
- How to analyze the long-term behavior of the catalytic system with deactivation based on its integral characteristic?
- What is the best strategy for the increase of catalytic efficiency based on the kinetic description?
3. Theoretical Analysis
- Only the main catalytic cycle model; the non-steady-state case.
- Only the main catalytic cycle model; the quasi-steady-state case.
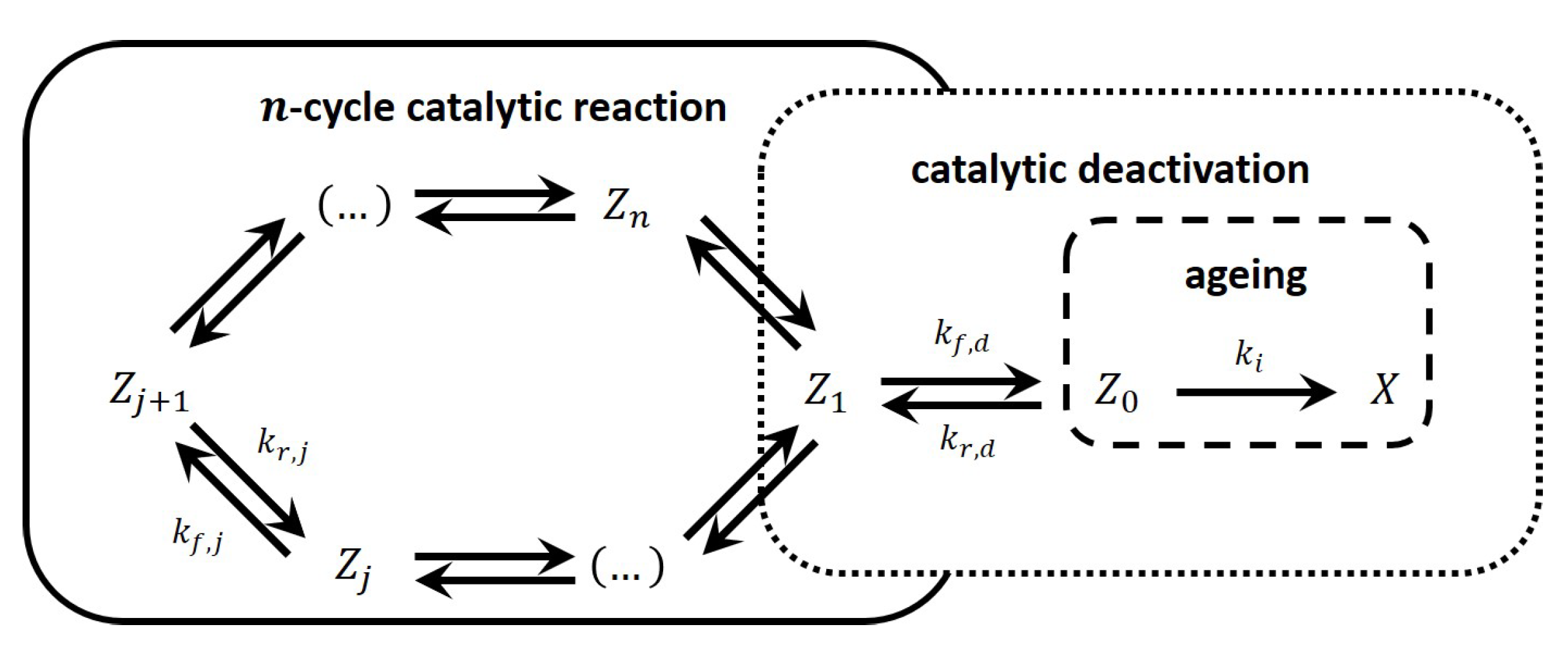
3.1. The Full Model of the Two-Step Irreversible Catalytic Cycle with Irreversible Deactivation
3.2. The Initial Non-Steady-State Domain; Gas Concentration Is Abundant, and Deactivation Is Negligible
3.3. The Quasi-Steady-State Domain; Deactivation Is Absent
3.3.1. Introduction to the Lambert W function
3.4. The Quasi-Steady-State Domain of the Cyclic Reaction Accompanied by Deactivation
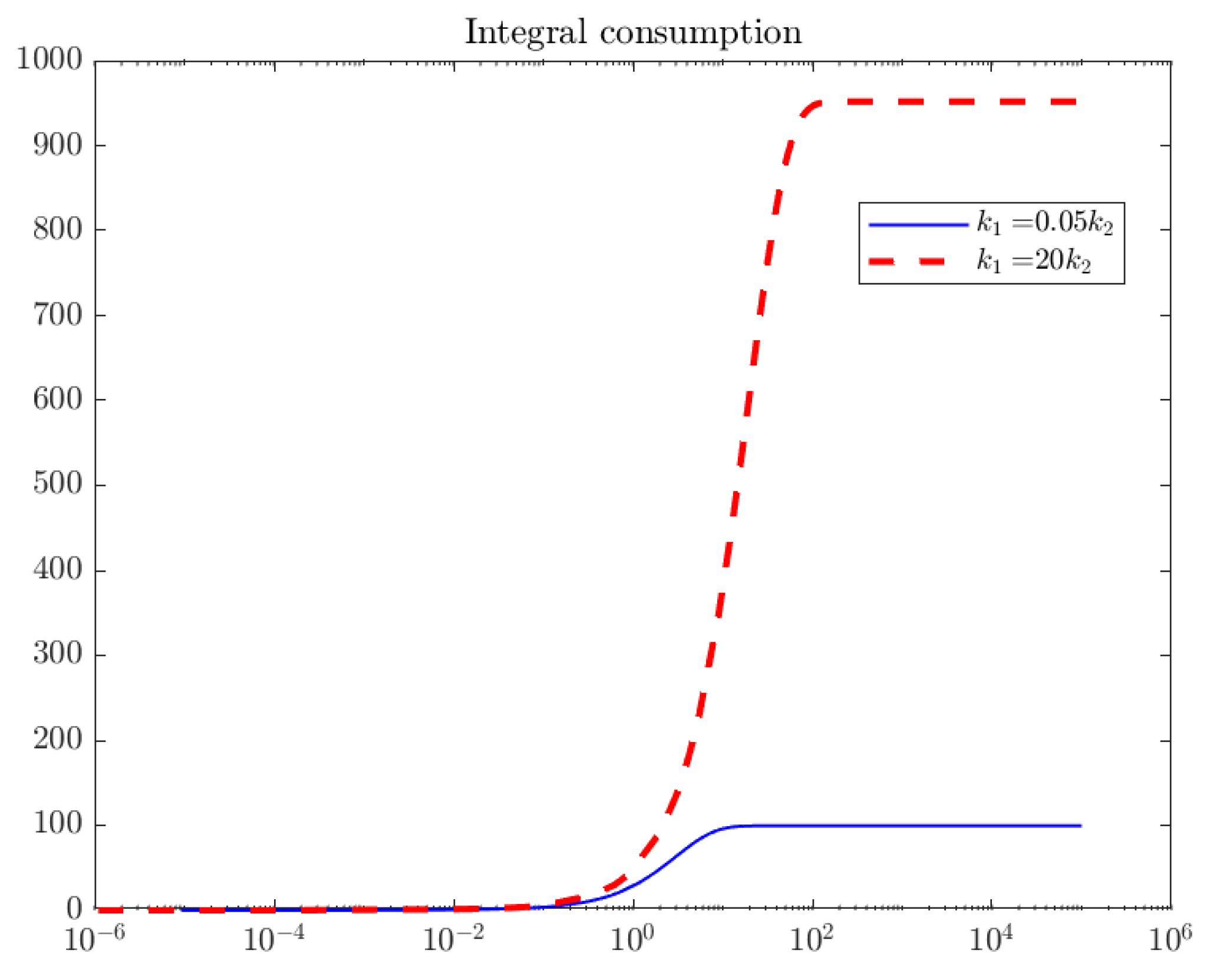
3.5. Integral Consumption
- The limit of the integral consumption as time goes to infinity, ,We find the limit of integral consumption is equal to the product of the number of active sites for the fresh catalyst multiplied by the ratio of the kinetic coefficients of the two reactions competing for the free active site Z. One reaction belongs to the catalytic cycle,The other reaction belongs to the irreversible deactivation step,
- The Taylor approximation for at small values of . If the term is very small, i.e., , the Taylor approximation of will be
3.6. Combining the Integral Consumption and the Quasi-Steady-State Equation Rate
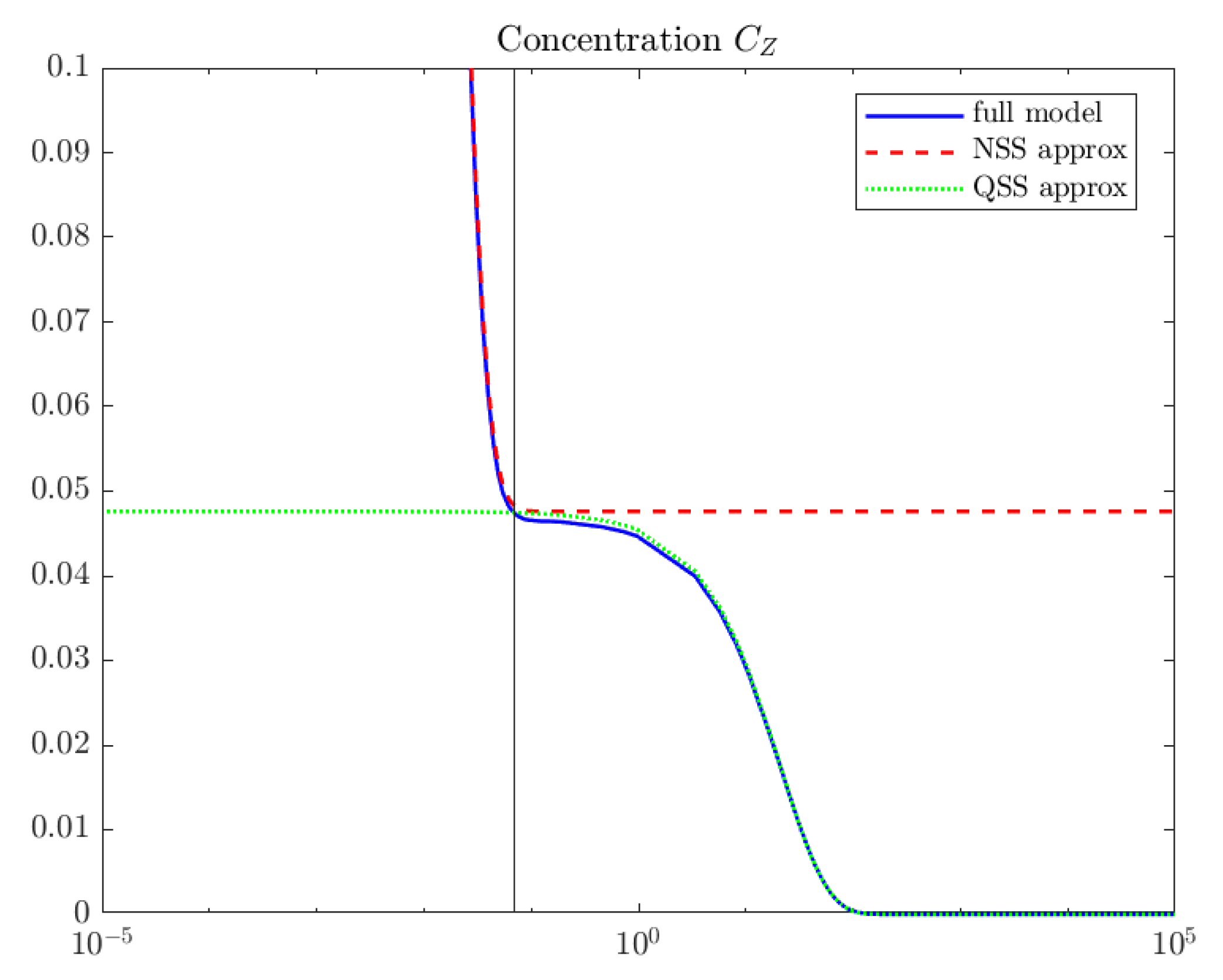
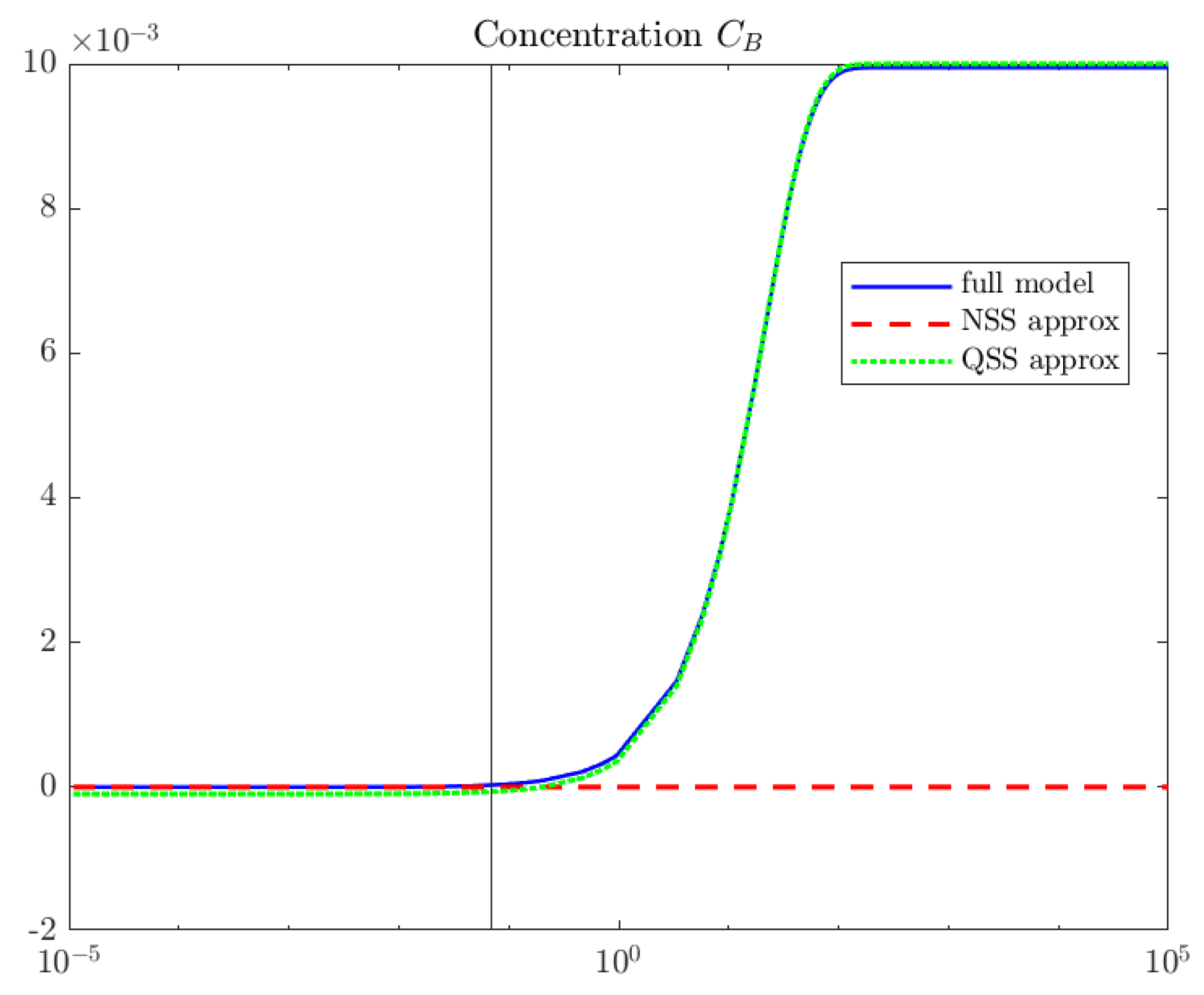
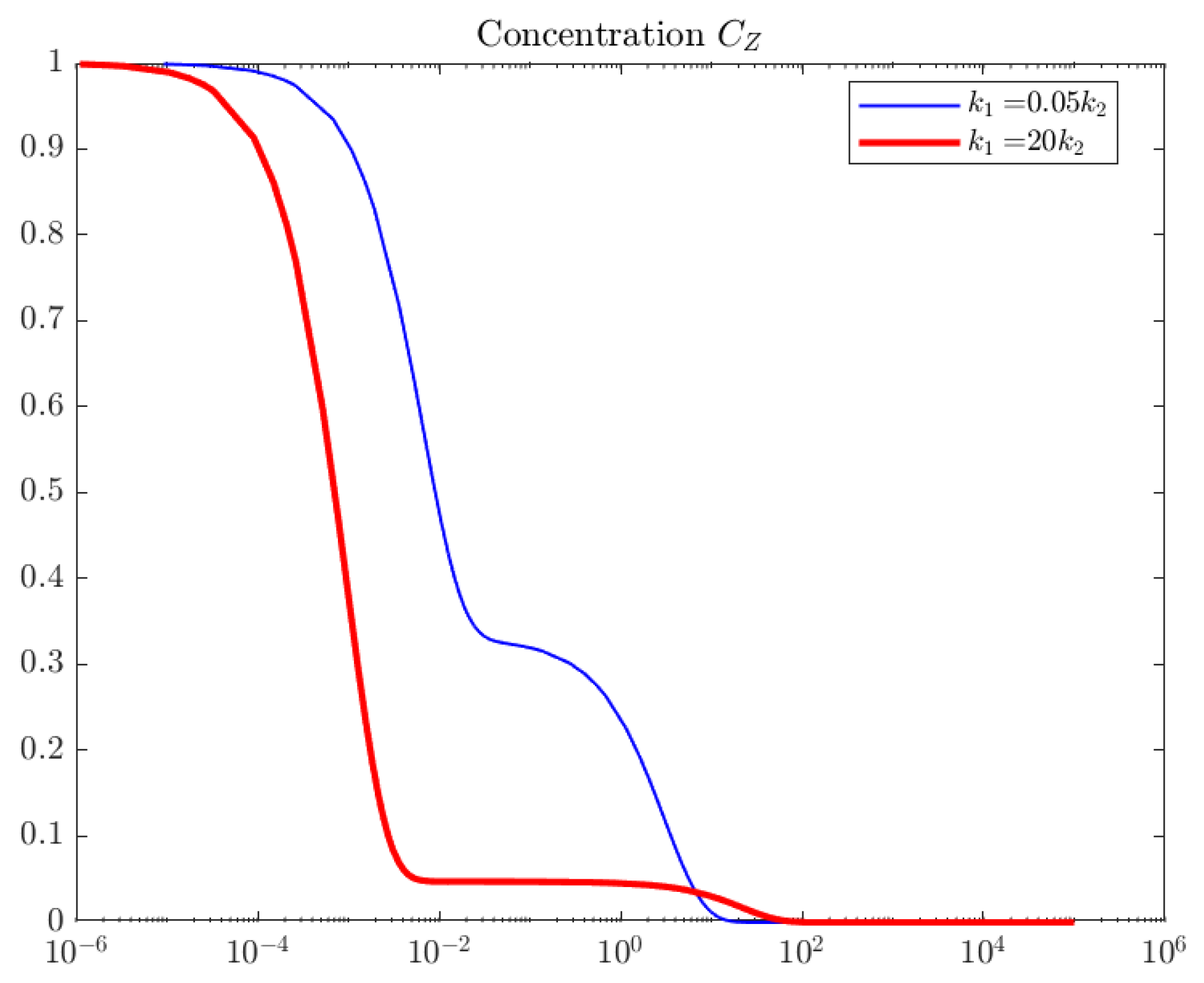
4. Computations
5. Interpretation and Discussion
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| QSS | quasi-steady state |
| NSS | non-steady state |
| SS | steady-state |
Appendix A. Epsilon Analysis
- Only one small parameter is present, i.e., and . As a result there is no deactivation and or equivalently .
- Two small parameters are present, i.e., .
- (a)
- (b)
- (c)
Appendix A.1. Case 2a: Two Small Parameters and ε1 ≫ ε2
Appendix A.2. Case 2b: There Are Two Small Parameters ε1 ≪ ε2
Appendix A.3. Case 2c: There Are Two Small Parameters ε1 ≈ ε2
References
- Szépe, S.; Levenspiel, O. Optimal temperature policies for reactors subject to catalyst deactivation—I Batch reactor. Chem. Eng. Sci. 1968, 23, 881–894. [Google Scholar] [CrossRef]
- Corella, J.; Asua, J. Kinetic equations of mechanistic type with nonseparable variables for catalyst deactivation by coke. Models and data analysis methods. Ind. Eng. Chem. Process Des. Dev. 1982, 21, 55–61. [Google Scholar] [CrossRef]
- Corella, J.; Adanez, J.; Monzon, A. Some intrinsic kinetic equations and deactivation mechanisms leading to deactivation curves with a residual activity. Ind. Eng. Chem. Res. 1988, 27, 375–381. [Google Scholar] [CrossRef]
- Froment, G.; Bischoff, K. Non-steady state behaviour of fixed bed catalytic reactors due to catalyst fouling. Chem. Eng. Sci. 1961, 16, 189–201. [Google Scholar] [CrossRef]
- Froment, G.; Bischoff, K. Kinetic data and product distributions from fixed bed catalytic reactors subject to catalyst fouling. Chem. Eng. Sci. 1962, 17, 105–114. [Google Scholar] [CrossRef]
- Beeckman, J.; Froment, G. Catalyst deactivation by site coverage and pore blockage: Finite rate of growth of the carbonaceous deposit. Chem. Eng. Sci. 1980, 35, 805–815. [Google Scholar] [CrossRef]
- Marin, G.; Beeckman, J.; Froment, G. Rigorous kinetic models for catalyst deactivation by coke deposition: Application to butene dehydrogenation. J. Catal. 1986, 97, 416–426. [Google Scholar] [CrossRef]
- Tsheglove, G.; Gel’bshtein, A.; Yablonskii, G.; Kamenko, B. Dynamic kinetic model of vinyl chlorice synthesis. Ind. Eng. Chem. Res. 1972, 27, 709–718. [Google Scholar]
- Butt, J.; Petersen, E. Activation, Deactivation and Poisoning of Catalysts; Academic Press: San Diego, CA, USA, 1988; 495p. [Google Scholar]
- Bartholomew, C. Mechanisms of catalyst deactivation. Appl. Catal. Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Gromotka, Z.; Yablonsky, G.; Ostrovskii, N.; Constales, D. Three-Factor Kinetic Equation of Catalyst Deactivation. Entropy 2021, 23, 818. [Google Scholar] [CrossRef]
- Ostrovskii, N.; Yablonskii, G. Kinetic equation for catalyst deactivation. React. Kinet. Catal. Lett. 1989, 39, 287–292. [Google Scholar] [CrossRef]
- Ostrovskii, N. Catalyst Deactivation Kinetics: Mathematical Models and Their Applications; NAUKA: Moscow, Russia, 2001; 334p. (In Russian) [Google Scholar]
- Ostrovskii, N. General equation for linear mechanisms of catalyst deactivation. Chem. Eng. J. 2006, 120, 73–82. [Google Scholar] [CrossRef]
- Yablonsky, G.; Constales, D.; Marin, G. Single-Route Linear Catalytic Mechanism: A New, Kinetico-Thermodynamic Form of the Complex Reaction Rate. Symmetry 2020, 12, 1748. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Aguayo, A.; Gayubo, A.; Castaño, P.; Bilbao, J. Simultaneous modeling of the kinetics for n-pentane cracking and the deactivation of a HZSM-5 based catalyst. Chem. Eng. J. 2018, 331, 818–830. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Hita, I.; Garcia-Mateos, F.; Castaño, P.; Rodriguez-Mirasol, J.; Cordero, T.; Bilbao, J. Adaptable kinetic model for the transient and pseudo-steady states in the hydrodeoxygenation of raw bio-oil. Chem. Eng. J. 2020, 400, 124679. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Aguayo, A.; Gayubo, A.; Castaño, P.; Bilbao, J. A comprehensive approach for designing different configurations of isothermal reactors with fast catalyst deactivation. Chem. Eng. J. 2020, 379, 122260. [Google Scholar] [CrossRef]
- Bodenstein, M. Eine theorie der photochemischen reaktionsgeschwindigkeiten. Zeitschrift für Physikalische Chemie 1913, 85, 329–397. [Google Scholar] [CrossRef] [Green Version]
- Chapman, D.; Underhill, L. The interaction of chlorine and hydrogen. The influence of mass. J. Chem. Soc. Trans. 1913, 103, 496–508. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, J. The elucidation of reaction mechanisms by the method of intermediates in quasi-stationary concentrations. Adv. Catal. 1953, 5, 311–353. [Google Scholar]
- Yablonskii, G.; Bykov, V.; Elokhin, V.; Gorban, A. Kinetic Models of Catalytic Reactions; Elsevier: Amsterdam, The Netherlands, 1991; Volume 32, 396p. [Google Scholar]
- Marin, G.; Yablonsky, G.; Constales, D. Kinetics of Chemical Reactions: Decoding Complexity; Wiley-VCH: Weinheim, Germany, 2019; 442p. [Google Scholar]
- Michaelis, L.; Menten, M. Die kinetik der Invertinwirkung. Biochem. Z 1913, 49, 352. [Google Scholar]
- Gorban, A.; Shahzad, M. The Michaelis–Menten–Stueckelberg theorem. Entropy 2011, 13, 966–1019. [Google Scholar] [CrossRef] [Green Version]
- Briggs, G.; Haldane, J. A note on the kinetics of enzyme action. Biochem. J. 1925, 19, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikhonov, A. Systems of differential equations containing small parameters in the derivatives. Matematicheskii Sbornik 1952, 73, 575–586. [Google Scholar]
- Sayasov, Y.; Vasil’eva, A. Semenov-Bodenstein’s method of quasi-steady-state concentrations: Justification and applicability to the gas chain reaction. Zh. Fiz. Khim. 1955, 29, 802–810. [Google Scholar]
- Butuzov, V.; Vasil’eva, A. Asymptotic Expansions for Singularly Perturbed Equations; NAUKA: Moscow, Russia, 1973. [Google Scholar]
- Tikhonov, N.; Vasil’eva, A.; Sveshnikov, A. Differential Equations; Springer: New York, NY, USA, 1985. [Google Scholar]
- Bowen, J.; Acrivos, A.; Oppenheim, A. Singular perturbation refinement to quasi-steady state approximation in chemical kinetics. Chem. Eng. Sci. 1963, 18, 177–188. [Google Scholar] [CrossRef]
- Heineken, F.; Tsuchiya, H.; Aris, R. On the mathematical status of the pseudo-steady state hypothesis of biochemical kinetics. Math. Biosci. 1967, 1, 95–113. [Google Scholar] [CrossRef]
- Yablonskii, G.; Bykov, V.; Gorban, A. Mathematical Models of Catalytic Reactions; Nauka: Novsibirsk, Russia, 1983. [Google Scholar]
- Gorban, A.; Karlin, I. Method of invariant manifold for chemical kinetics. Chem. Eng. Sci. 2003, 58, 4751–4768. [Google Scholar] [CrossRef] [Green Version]
- Gorban, A.; Radulescu, O.; Zinovyev, A. Asymptotology of chemical reaction networks. Chem. Eng. Sci. 2010, 65, 2310–2324. [Google Scholar] [CrossRef] [Green Version]
- Gorban, A. Model reduction in chemical dynamics: Slow invariant manifolds, singular perturbations, thermodynamic estimates, and analysis of reaction graph. Curr. Opin. Chem. Eng. 2018, 21, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Temkin, M. Relaxation rate of two-stage catalytic reaction. Kinetica Catal. 1976, 17, 945–949. [Google Scholar]
- Boudart, M. Two-step catalytic reactions. AIChE J. 1972, 18, 465–478. [Google Scholar] [CrossRef]
- Marin, G.; Galvita, V.; Yablonsky, G. Kinetics of chemical processes: From molecular to industrial scale. J. Catal. 2021, 404, 745–759. [Google Scholar] [CrossRef]
- Mezo, I. The Lambert W Function: Its Generalizations and Applications; Chapman and Hall/CRC: New York, NY, USA, 2022. [Google Scholar] [CrossRef]
- Temkin, O.; Pozdeev, P. Homogeneous Catalysis with Metal Complexes: Kinetic Aspects and Mechanisms; John Wiley & Sons: Hoboken, NJ, USA, 2012; 860p. [Google Scholar]
- Kagan, Y.; Rozovskij, A.; Slivinskij, E.; Korneeva, G. Autoregulatory effect in the hydroformylation reaction of olefins. Kinetika I Kataliz 1987, 28, 1508–1511. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gromotka, Z.; Yablonsky, G.; Ostrovskii, N.; Constales, D. Integral Characteristic of Complex Catalytic Reaction Accompanied by Deactivation. Catalysts 2022, 12, 1283. https://doi.org/10.3390/catal12101283
Gromotka Z, Yablonsky G, Ostrovskii N, Constales D. Integral Characteristic of Complex Catalytic Reaction Accompanied by Deactivation. Catalysts. 2022; 12(10):1283. https://doi.org/10.3390/catal12101283
Chicago/Turabian StyleGromotka, Zoë, Gregory Yablonsky, Nickolay Ostrovskii, and Denis Constales. 2022. "Integral Characteristic of Complex Catalytic Reaction Accompanied by Deactivation" Catalysts 12, no. 10: 1283. https://doi.org/10.3390/catal12101283