
Ex Situ Upgrading of Extra Heavy Oil: The Effect of Pore Shape of Co-Mo/γ-Al2O3 Catalysts
 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
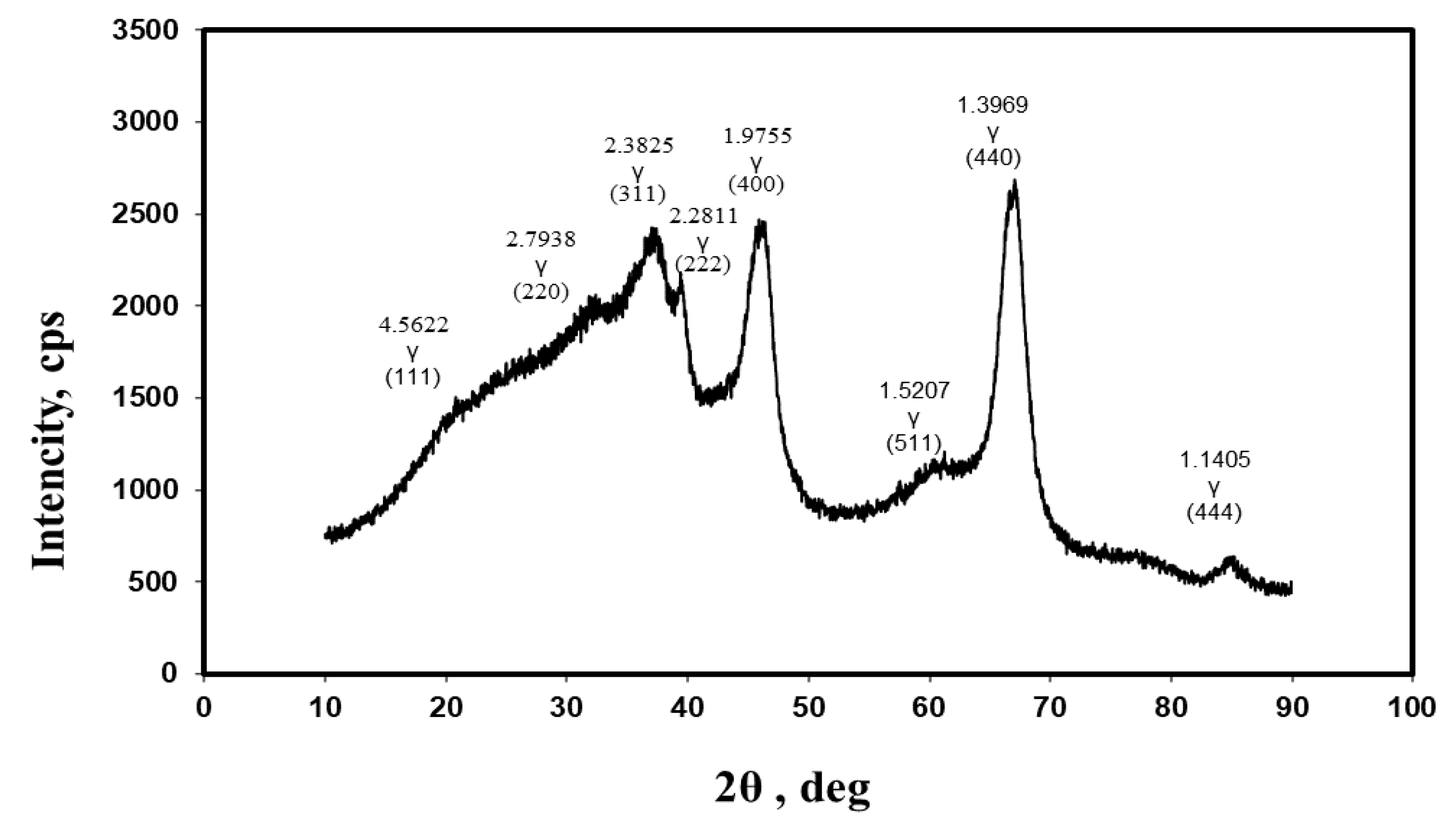
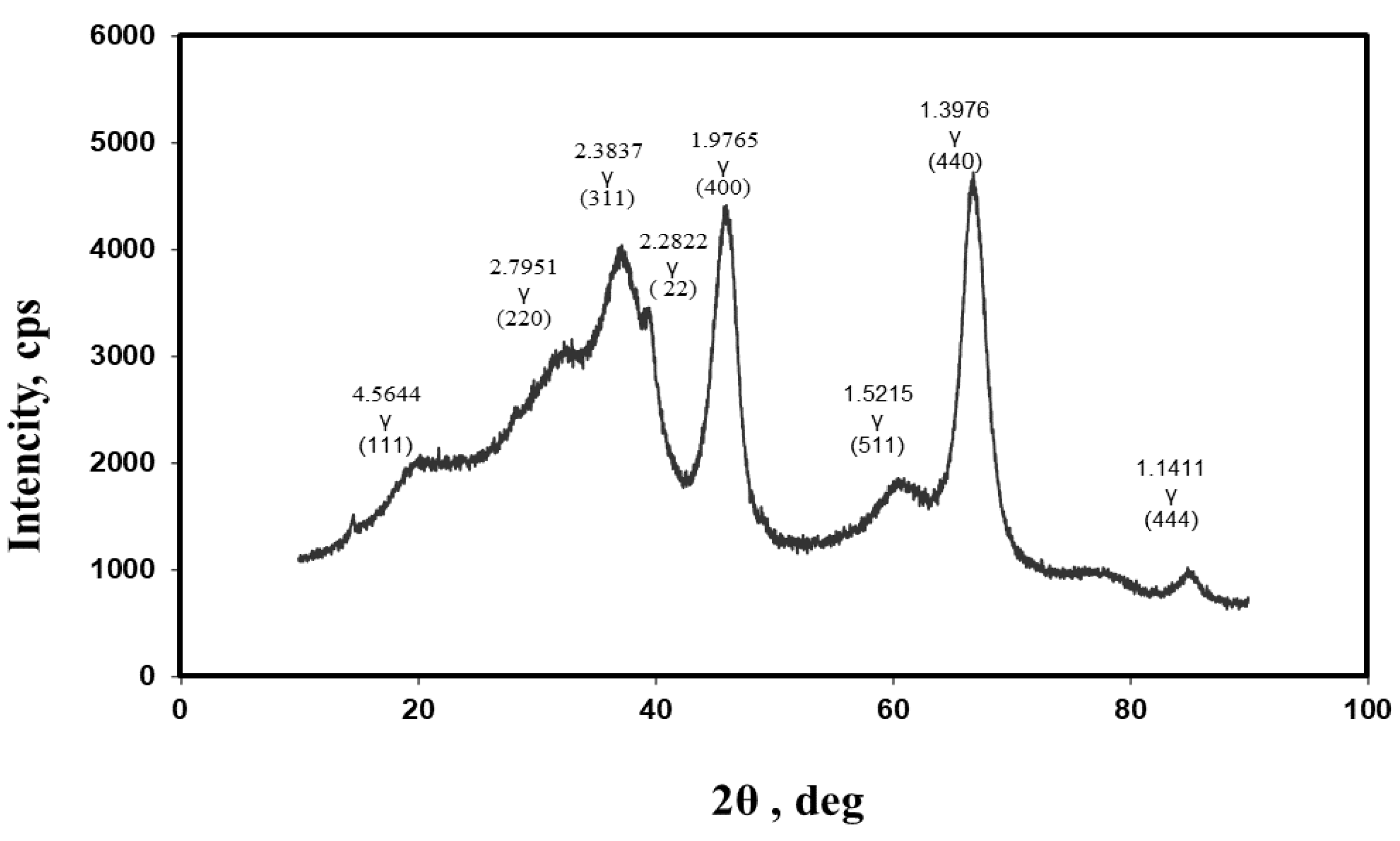
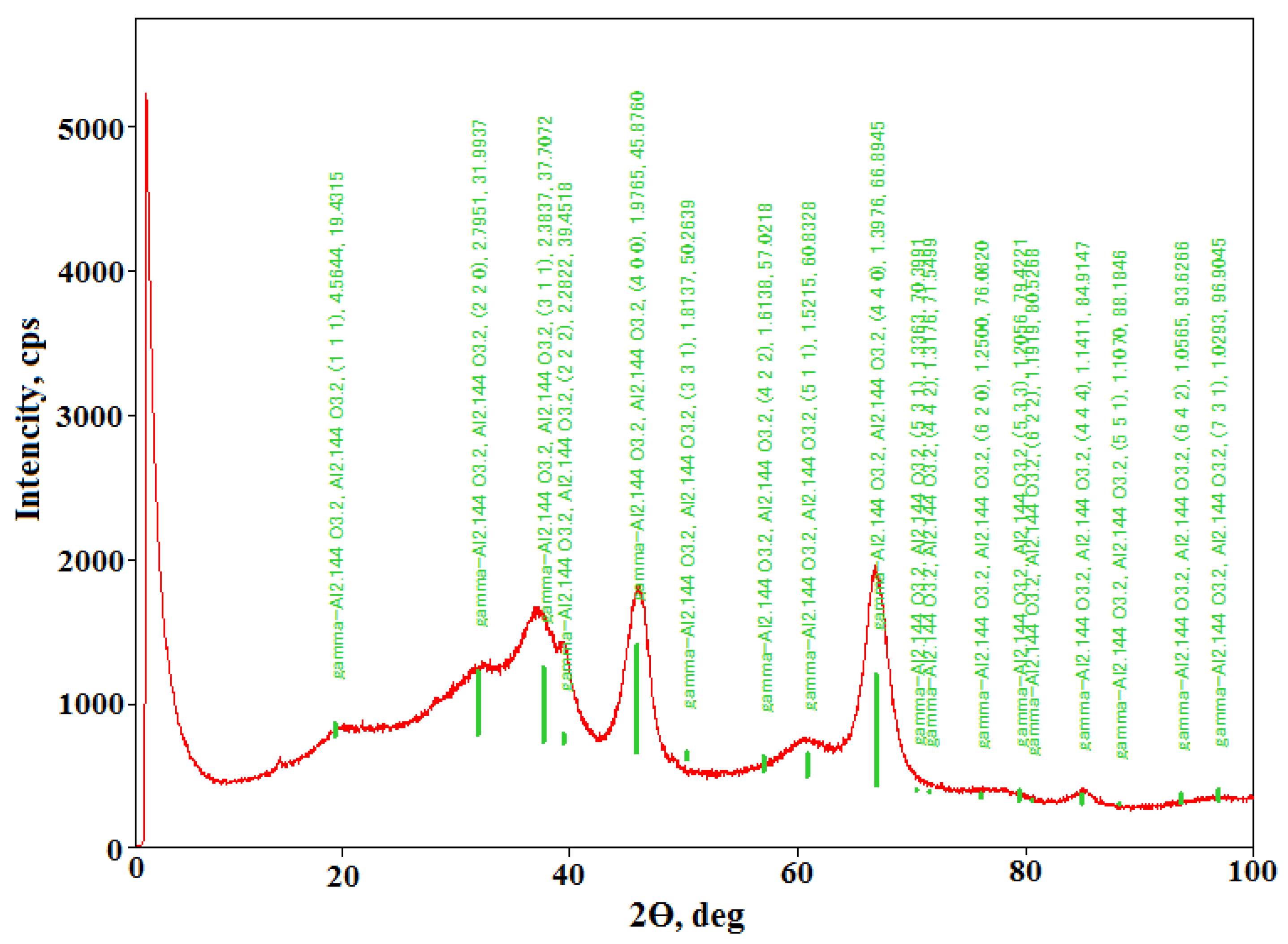
2.1. Phase Analysis of Aluminum Oxides
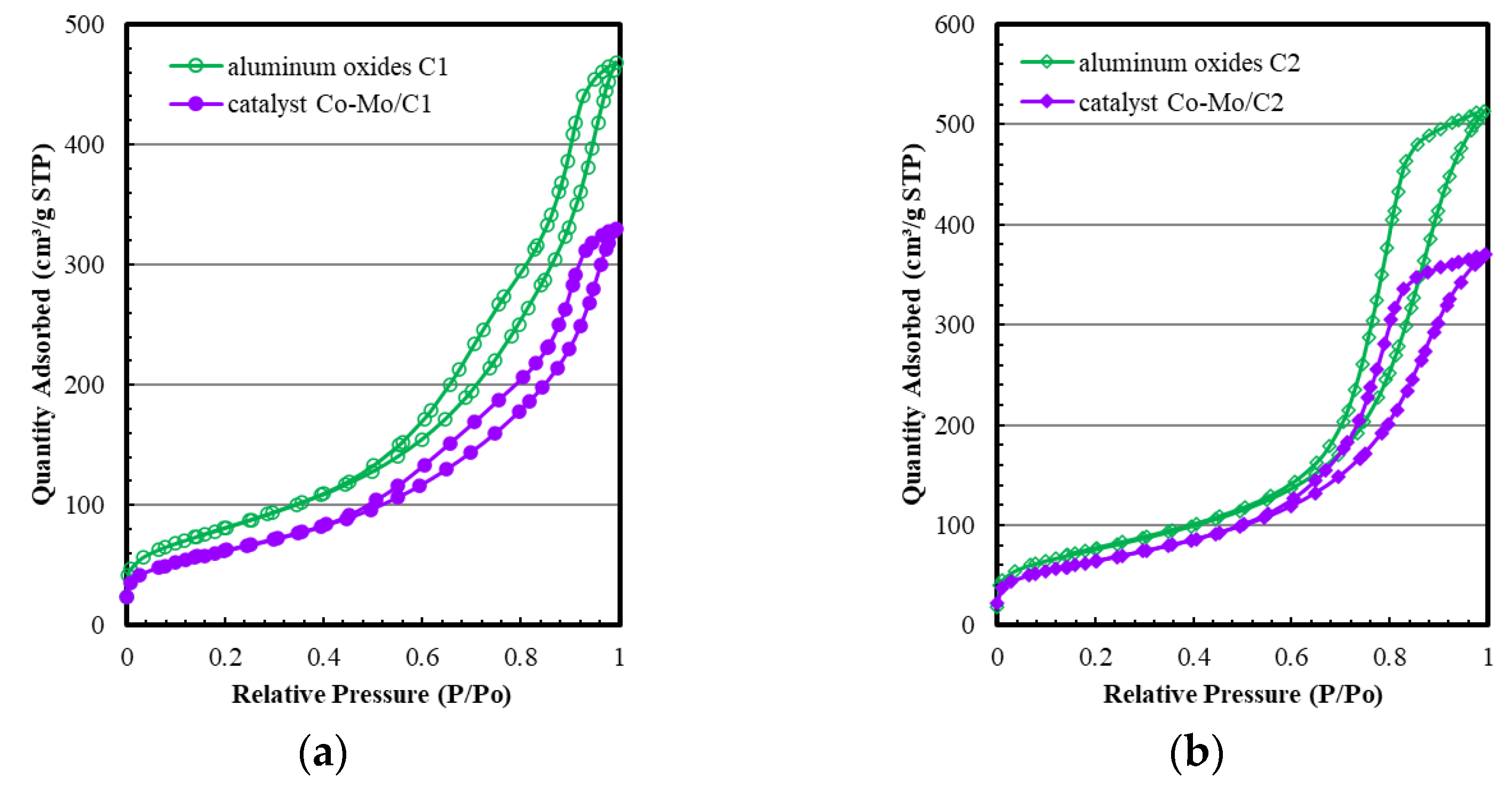
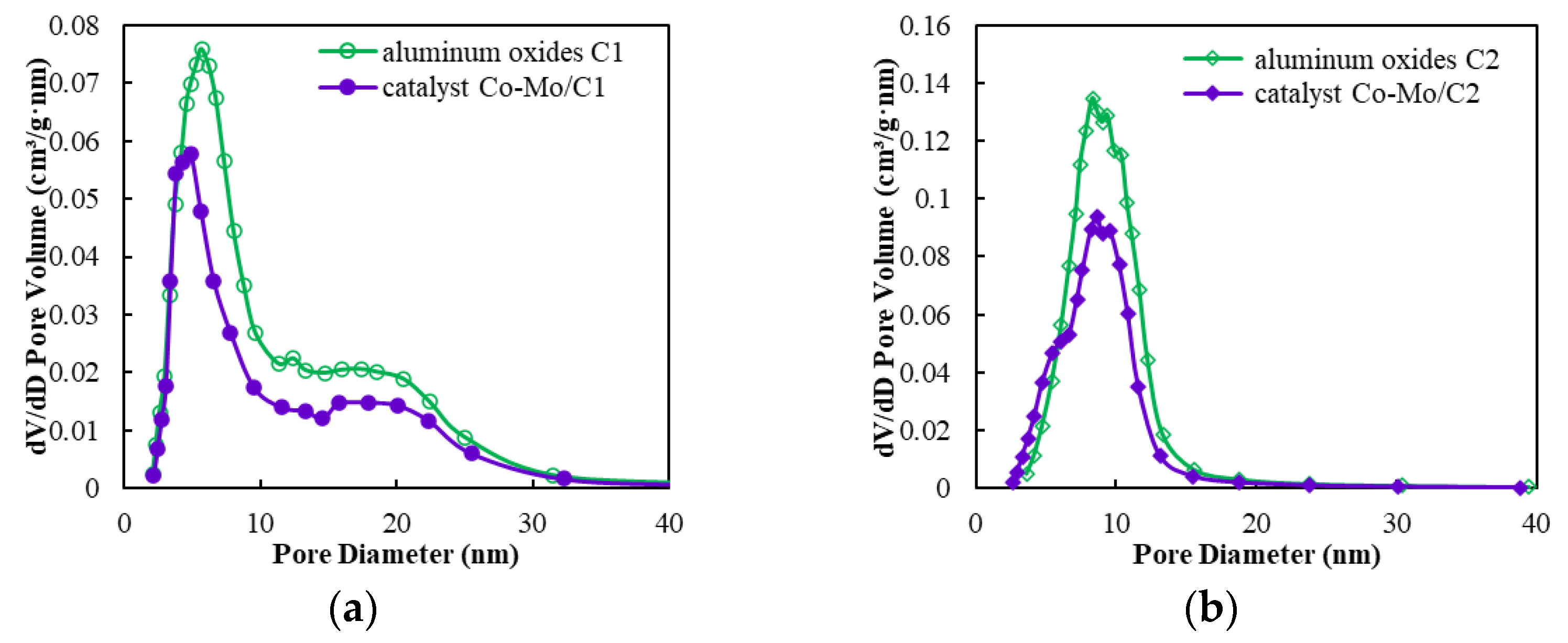
2.2. The Pore Structure of Aluminum Oxides
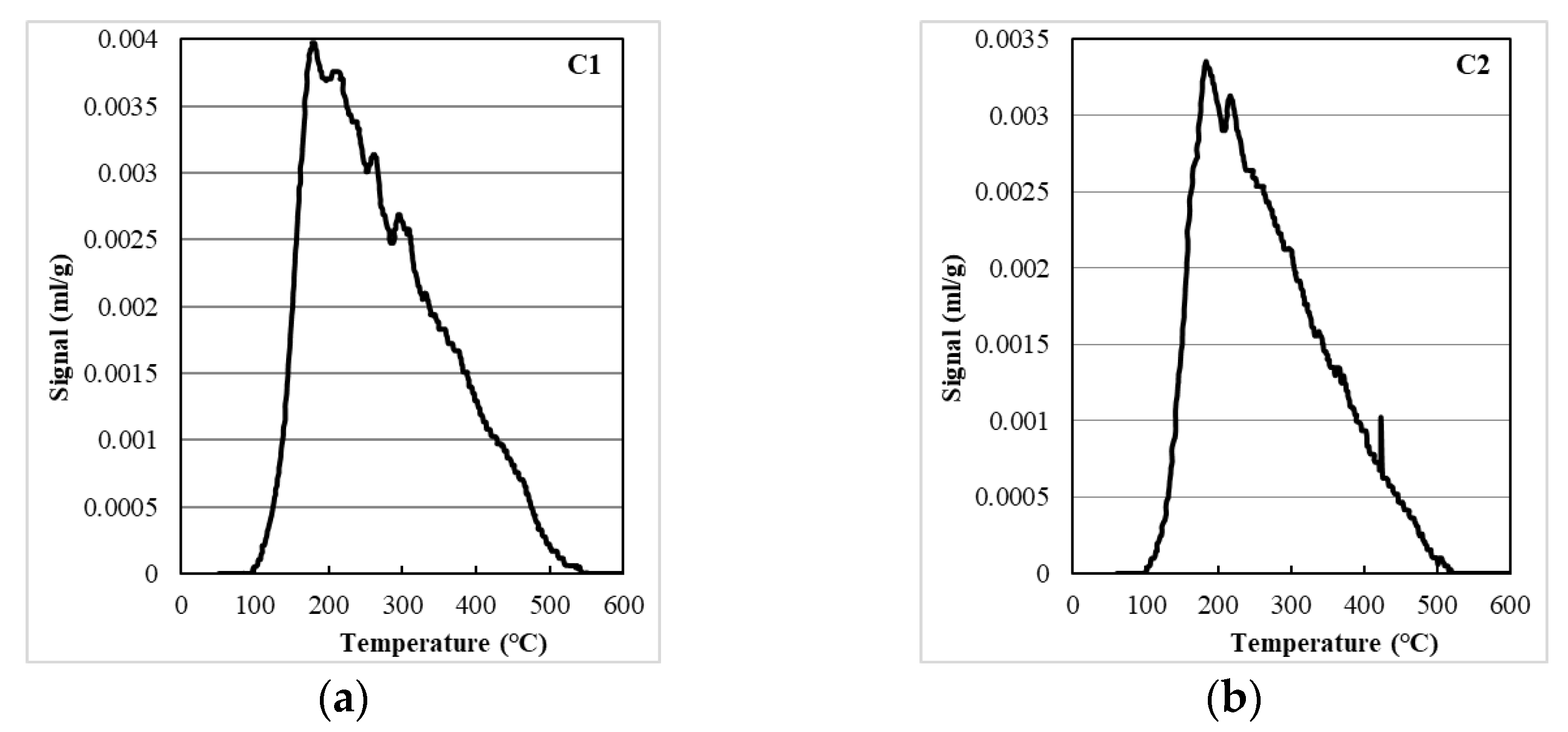
2.3. The Total Acidity of the Bronsted and Lewis Centers of Aluminum Oxides and Catalysts
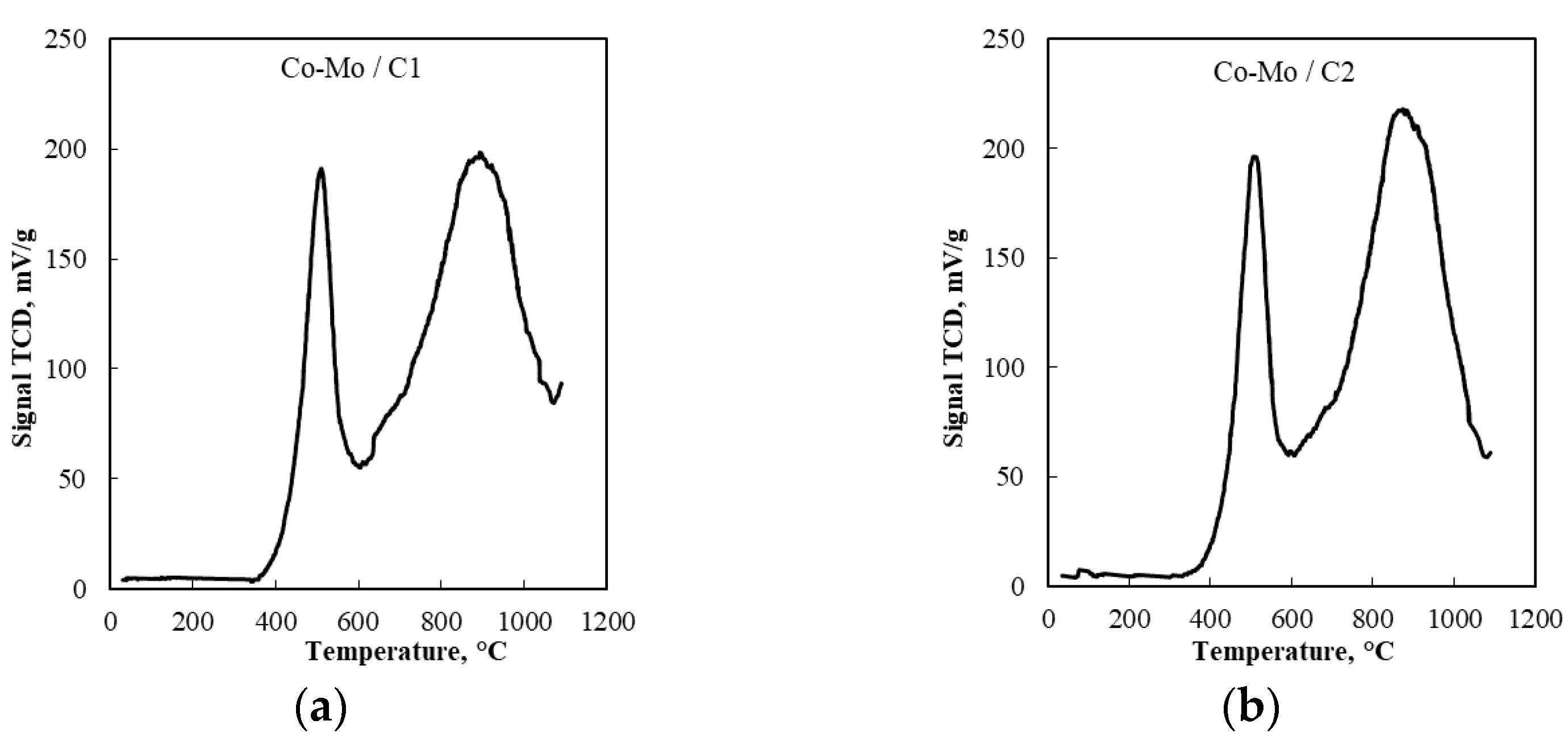
2.4. Temperature-Programmable Catalyst Reduction
2.5. Catalytic Effect of Catalysts
3. Materials and Methods
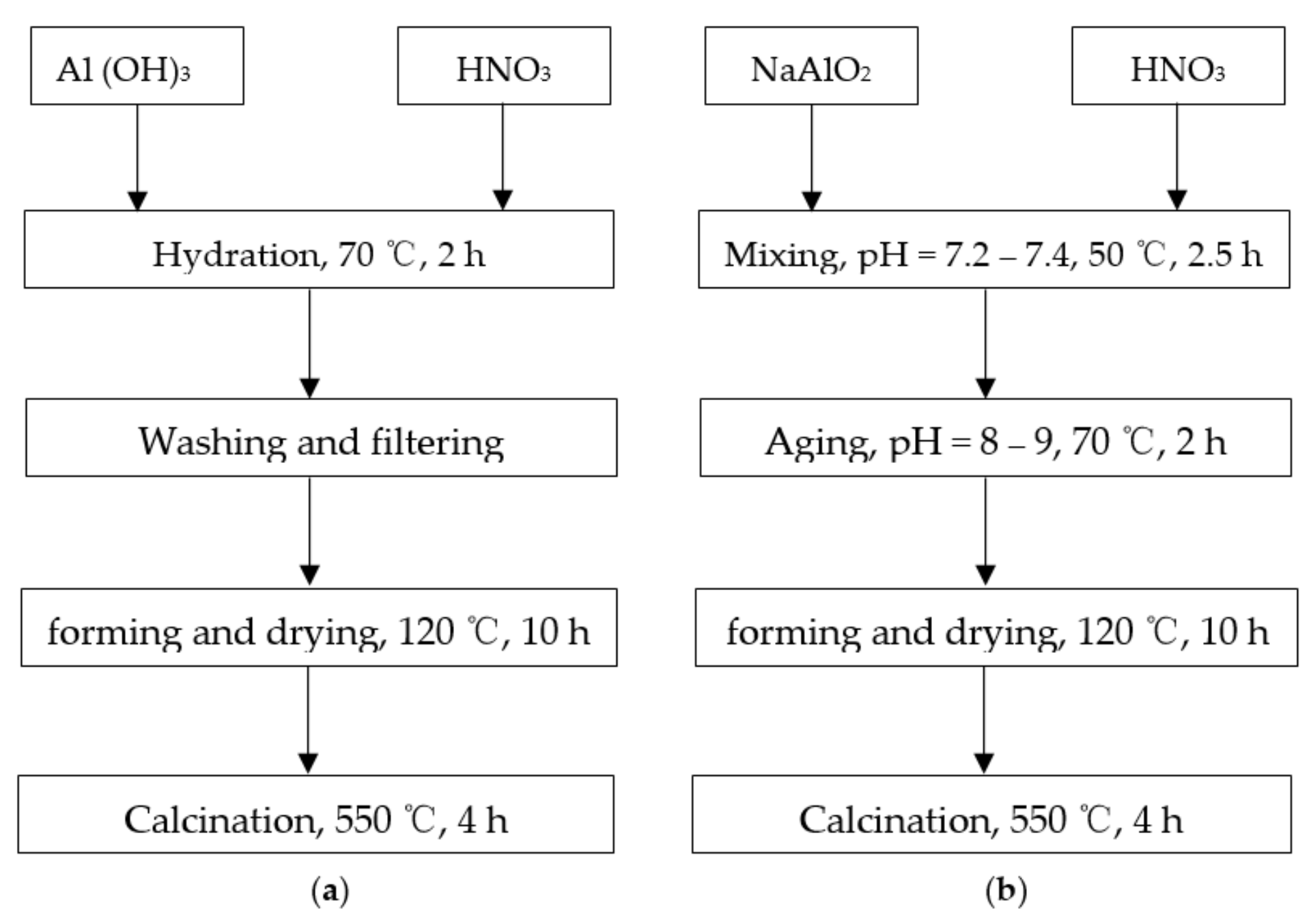
3.1. Synthesis of Aluminum Oxides and Catalysts
3.2. Characterization of Aluminum Oxides and Catalysts
3.3. Upgrading Experiments for Reducing Oil Viscosity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghanavati, M.; Shojaei, M.J.; Ahmad Ramazani, S.A. Effects of Asphaltene Content and Temperature on Viscosity of Iranian Heavy Crude Oil: Experimental and Modeling Study. Energy Fuels 2013, 27, 7217–7232. [Google Scholar] [CrossRef]
- Nassar, N.N.; Hassan, A.; Pereira-Almao, P. Effect of the Particle Size on Asphaltene Adsorption and Catalytic Oxidation onto Alumina Particles. Energy Fuels 2011, 25, 3961–3965. [Google Scholar] [CrossRef]
- Muraza, O.; Galadima, A. Aquathermolysis of Heavy Oil: A Review and Perspective on Catalyst Development. Fuel 2015, 157, 219–231. [Google Scholar] [CrossRef]
- Lesueur, D. The Colloidal Structure of Bitumen: Consequences on the Rheology and on the Mechanisms of Bitumen Modification. Adv. Colloid Interface Sci. 2009, 145, 42–82. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Gu, Y. Effects of Asphaltene Content on the Heavy Oil Viscosity at Different Temperatures. Fuel 2007, 86, 1069–1078. [Google Scholar] [CrossRef]
- Sharma, A.; Mullins, O.C. Insights into Molecular and Aggregate Structures of Asphaltenes Using HRTEM. In Asphaltenes, Heavy Oils, and Petroleomics; Springer: Berlin/Heidelberg, Germany, 2007; pp. 205–229. [Google Scholar] [CrossRef]
- Ancheyta, J.; Speight, J.G. Heavy Oils and Residua. In Hydroprocessing of Heavy Oils and Residua; CRC Press: Boca Raton, FL, USA, 2007; pp. 1–14. [Google Scholar] [CrossRef]
- Al-Muntaser, A.A.; Varfolomeev, M.A.; Suwaid, M.A.; Feoktistov, D.A.; Yuan, C.; Klimovitskii, A.E.; Gareev, B.I.; Djimasbe, R.; Nurgaliev, D.K.; Kudryashov, S.I.; et al. Hydrogen Donating Capacity of Water in Catalytic and Non-Catalytic Aquathermolysis of Extra-Heavy Oil: Deuterium Tracing Study. Fuel 2021, 283, 118957. [Google Scholar] [CrossRef]
- Suwaid, M.A.; Varfolomeev, M.A.; Al-muntaser, A.A.; Yuan, C.; Starshinova, V.L.; Zinnatullin, A.; Vagizov, F.G.; Rakhmatullin, I.Z.; Emelianov, D.A.; Chemodanov, A.E. In-Situ Catalytic Upgrading of Heavy Oil Using Oil-Soluble Transition Metal-Based Catalysts. Fuel 2020, 281, 118753. [Google Scholar] [CrossRef]
- Yi, S.; Babadagli, T.; Li, H.A. Use of Nickel Nanoparticles for Promoting Aquathermolysis Reaction during Cyclic Steam Stimulation. SPE J. 2018, 23, 145–156. [Google Scholar] [CrossRef]
- Rodríguez, E.; Félix, G.; Ancheyta, J.; Trejo, F. Modeling of Hydrotreating Catalyst Deactivation for Heavy Oil Hydrocarbons. Fuel 2018, 225, 118–133. [Google Scholar] [CrossRef]
- Trimm, D.L. Deactivation, Regeneration and Disposal of Hydroprocessing Catalysts. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1989; Volume 53, pp. 41–60. [Google Scholar] [CrossRef]
- Sheu, E.Y. Petroleum Asphaltene—Properties, Characterization, and Issues. Energy Fuels 2002, 16, 74–82. [Google Scholar] [CrossRef]
- Ohtsuica, T. Catalyst for Hydrodesulfurization of Petroleum Residua. Catal. Rev. 1977, 16, 291–325. [Google Scholar] [CrossRef]
- Beaton, W.I.; Bertolacini, R.J. Resid Hydroprocessing at Amoco. Catal. Rev. 1991, 33, 281–317. [Google Scholar] [CrossRef]
- Tropsoe, H.; Clausen, B.S.; Massoth, F.E. Hydrotreating Catalysis: Science and Technology; Springer: Berlin/Heidelberg, Germany, 1996. [Google Scholar]
- Kohli, K.; Prajapati, R.; Maity, S.K.; Sau, M.; Garg, M.O. Deactivation of Hydrotreating Catalyst by Metals in Resin and Asphaltene Parts of Heavy Oil and Residues. Fuel 2016, 175, 264–273. [Google Scholar] [CrossRef]
- Mitchell, P.C.H. Hydrodemetallisation of Crude Petroleum: Fundamental Studies. Catal. Today 1990, 7, 439–445. [Google Scholar] [CrossRef]
- Pham, H.H.; Kim, K.H.; Go, K.S.; Nho, N.S.; Kim, W.; Kwon, E.H.; Jung, R.H.; Lim, Y.-i.; Lim, S.H.; Pham, D.A. Hydrocracking and Hydrotreating Reaction Kinetics of Heavy Oil in CSTR Using a Dispersed Catalyst. J. Pet. Sci. Eng. 2021, 197, 107997. [Google Scholar] [CrossRef]
- Sámano, V.; Rana, M.S.; Ancheyta, J. An Easy Approach Based on Textural Properties to Evaluate Catalyst Deactivation during Heavy Oil Hydrotreating. Catal. Commun. 2020, 133, 105823. [Google Scholar] [CrossRef]
- Badoga, S.; Ganesan, A.; Dalai, A.K.; Chand, S. Effect of Synthesis Technique on the Activity of CoNiMo Tri-Metallic Catalyst for Hydrotreating of Heavy Gas Oil. Catal. Today 2017, 291, 160–171. [Google Scholar] [CrossRef]
- Rana, M.S.; Ancheyta, J.; Rayo, P.; Maity, S.K. Effect of Alumina Preparation on Hydrodemetallization and Hydrodesulfurization of Maya Crude. Catal. Today 2004, 98, 151–160. [Google Scholar] [CrossRef]
- Salimi, M.; Tavasoli, A.; Rosendahl, L. Optimization of γ-Alumina Porosity via Response Surface Methodology: The Influence of Engineering Support on the Performance of a Residual Oil Hydrotreating Catalyst. Microporous Mesoporous Mater. 2020, 299, 110124. [Google Scholar] [CrossRef]
- Shlmura, M.; Shlroto, Y.; Takeuchi, C. Effect of Catalyst Pore Structure on Hydrotreating of Heavy Oil. Ind. Eng. Chem. Fundam. 1986, 25, 330–337. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kushiyama, S.; Aizawa, R.; Koinuma, Y.; Inoue, K.; Shimizu, Y.; Egi, K. Kinetic Study on the Hydrotreating of Heavy Oil. 2. Effect of Catalyst Pore Size. Ind. Eng. Chem. Res. 1987, 26, 2245–2250. [Google Scholar] [CrossRef]
- Quann, R.J.; Ware, R.A.; Hung, C.W.; Wei, J. Catalytic Hydrodemetallation of Petroleum. In Advances in Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 1988; Volume 14, pp. 95–259. [Google Scholar] [CrossRef]
- Ancheyta, J. Deactivation of Heavy Oil Hydroprocessing Catalysts; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar] [CrossRef]
- Rudolph, M.; Motylenko, M.; Rafaja, D. Structure Model of γ-Al2O3 Based on Planar Defects. IUCrJ 2019, 6, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregg, S.J.; Sing, K.S.W. Adsorption, Surface Area, and Porosity; Academic Press: London, UK, 1982. [Google Scholar]
- Al-Dalama, K.; Stanislaus, A. Temperature Programmed Reduction of SiO2-Al2O3 Supported Ni, Mo and NiMo Catalysts Prepared with EDTA. Thermochim. Acta 2011, 520, 67–74. [Google Scholar] [CrossRef]
- Hassan, A.; Ahmed, S.; Ali, M.A.; Hamid, H.; Inui, T. A Comparison between β- and USY-Zeolite-Based Hydrocracking Catalysts. Appl. Catal. A Gen. 2001, 220, 59–68. [Google Scholar] [CrossRef]
- Henker, M.; Wendlandt, K.P.; Anisimov, A.V.; Karakhanov, E.A. Molybdenum-Containing Catalysts in the Reactions of Cumene Cracking and Thiophene Hydrocracking. Pet. Chem. USSR 1990, 30, 135–140. [Google Scholar] [CrossRef]
- Danilevich, V.V.; Klimov, O.V.; Nadeina, K.A.; Gerasimov, E.Y.; Cherepanova, S.V.; Vatutina, Y.V.; Noskov, A.S. Novel Eco-Friendly Method for Preparation of Mesoporous Alumina from the Product of Rapid Thermal Treatment of Gibbsite. Superlattices Microstruct. 2018, 120, 148–160. [Google Scholar] [CrossRef]
- Hart, A.; Leeke, G.; Greaves, M.; Wood, J. Downhole Heavy Crude Oil Upgrading UsingCAPRI: Effect of Steam upon Upgrading and Coke Formation. Energy Fuels 2014, 28, 1811–1819. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | S, m2/g | V, cm3/g | D, nm | Pore Volume | |||||
|---|---|---|---|---|---|---|---|---|---|
| 2–5 nm | 5–11 nm | ˃11 nm | |||||||
| N | % | N | % | N | % | ||||
| C1 | 294 | 0.74 | 8.3 | 0.130 | 17.6 | 0.270 | 36.5 | 0.340 | 45.9 |
| Co-Mo/C1 | 224 | 0.52 | 8.0 | 0.118 | 22.7 | 0.158 | 30.5 | 0.240 | 46.8 |
| C2 | 277 | 0.81 | 9.0 | 0.024 | 3.0 | 0.553 | 68.5 | 0.230 | 28.5 |
| Co-Mo/C2 | 232 | 0.58 | 8.0 | 0.052 | 9.0 | 0.417 | 72.5 | 0.106 | 18.5 |
| Sample | ∑N | Weak Td < 250 | Medium 250 ≤ Td < 350 | Strong 350 ≤ Td < 450 | Very Strong Td ≥ 450 | ||||
|---|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | N | % | ||
| C1 | 475 | 230 | 49 | 149 | 31 | 79 | 17 | 17 | 4 |
| Co-Mo/C1 | 324 | 157 | 48 | 101 | 31 | 53 | 16 | 13 | 5 |
| C2 | 371 | 186 | 50 | 122 | 33 | 54 | 15 | 9 | 2 |
| Co-Mo/C2 | 229 | 115 | 50 | 80 | 35 | 30 | 13 | 4 | 2 |
| Sample | Amount of Absorbed Hydrogen (μmol/g)/Temperature of Peak Maximum (°C) | Ratio of Peak Intensities III/(I + II) | Total Absorbed Hydrogen, μmol/g | ||
|---|---|---|---|---|---|
| I | II | III | |||
| Co-Mo/C1 | 345.3/512 | - | 1182.9/897 | 3.4 | 1528.2 |
| Co-Mo/C2 | 342.2/511 | - | 1142.1/875 | 3.3 | 1484.3 |
| Sample | Hydrocarbon Content, wt.% | |||
|---|---|---|---|---|
| Saturated | Aromatic | Resins | Asphaltenes | |
| Extra heavy oil + diesel fraction | 65.59 | 14.75 | 7.98 | 11.68 |
| Co-Mo/C1 | 83.14 ± 0.65 | 13.45 ± 0.23 | 2.09 ± 0.07 | 2.42 ± 0.09 |
| Co-Mo/C2 | 80.09 ± 0.53 | 15.31 ± 0.32 | 2.36 ± 0.09 | 2.24 ± 0.09 |
| Sample | Element Content, wt.% | ||
|---|---|---|---|
| Sulfur | Vanadium | Nickel | |
| Extra heavy oil + diesel fraction | 2.18 | 0.080 | 0.010 |
| Co-Mo/C1 | 0.67 ± 0.05 | 0.015 ± 0.0015 | 0.006 ± 0.0007 |
| Co-Mo/C2 | 0.43 ± 0.03 | 0.010 ± 0.0010 | 0.008 ± 0.0006 |
| Sample | Viscosity, mPa∙s at 50 °C |
|---|---|
| The initial extra heavy oil | 28,200 |
| The upgraded oil by Co-Mo/C1 | 485 ± 7.3 |
| The upgraded oil by Co-Mo/C2 | 449 ± 5.1 |
| Sample | Yield, wt.% | ||
|---|---|---|---|
| Liquid | Coke | Gas | |
| Co-Mo/C1 | 96.73 ± 0.28 | 0.54 ± 0.07 | 2.73 ± 0.12 |
| Co-Mo/C2 | 97.72 ± 0.37 | 0.33 ± 0.05 | 1.95 ± 0.10 |
| Sample | Molybdenum Concentration | Cobalt Concentration |
|---|---|---|
| Co-Mo/C1 | 16.4 | 4.2 |
| Co-Mo/C2 | 16.5 | 4.1 |
| SARA Fractions, wt.% | ||||||
|---|---|---|---|---|---|---|
| Saturated | Aromatic | Resins | Asphaltenes | |||
| 38.72 | 20.36 | 17.75 | 23.12 | |||
| Viscosity, mPa∙s | ||||||
| 40 °C | 50 °C | 60 °C | 70 °C | 80 °C | 90 °C | 95 °C |
| 244,800 | 28,200 | 13,300 | 3800 | 1800 | 800 | 580 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirgizov, A.Y.; Ding, B.; Spiridonov, A.A.; Liu, L.; Laskin, A.I.; Cao, C.; Il’yasov, I.R.; Al-Muntaser, A.A.; Zhou, X.; Zinnatov, R.A.; et al. Ex Situ Upgrading of Extra Heavy Oil: The Effect of Pore Shape of Co-Mo/γ-Al2O3 Catalysts. Catalysts 2022, 12, 1271. https://doi.org/10.3390/catal12101271
Kirgizov AY, Ding B, Spiridonov AA, Liu L, Laskin AI, Cao C, Il’yasov IR, Al-Muntaser AA, Zhou X, Zinnatov RA, et al. Ex Situ Upgrading of Extra Heavy Oil: The Effect of Pore Shape of Co-Mo/γ-Al2O3 Catalysts. Catalysts. 2022; 12(10):1271. https://doi.org/10.3390/catal12101271
Chicago/Turabian StyleKirgizov, Alexey Y., Baodong Ding, Artur A. Spiridonov, Lei Liu, Artem I. Laskin, Chang Cao, Il’dar R. Il’yasov, Ameen A. Al-Muntaser, Xiaodong Zhou, Radik A. Zinnatov, and et al. 2022. "Ex Situ Upgrading of Extra Heavy Oil: The Effect of Pore Shape of Co-Mo/γ-Al2O3 Catalysts" Catalysts 12, no. 10: 1271. https://doi.org/10.3390/catal12101271