

Ultrasound/Chlorine: A Novel Synergistic Sono-Hybrid Process for Allura Red AC Degradation
 , and
, and 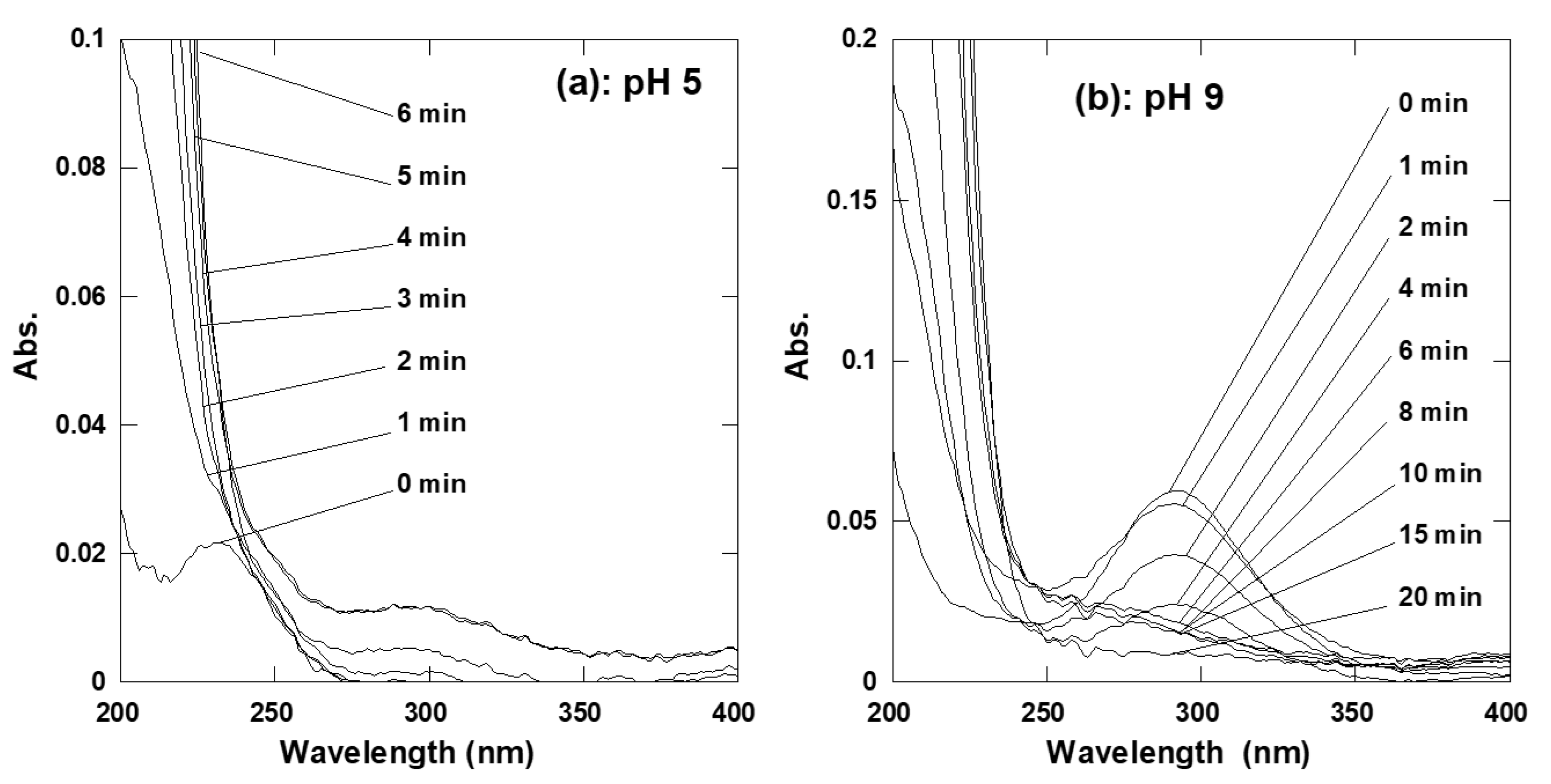{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Chlorine is easy to handle, with more safety, as it is less harmful than other oxidants. Furthermore, the liquid phase of chlorine simplifies its use.
- Chlorine is more available and less expensive than other oxidants that require in situ production via a sophisticated expensive device. Consequently, the cost of the US/chlorine technique could be lower than other processes for similar experimental conditions.
- The US/chlorine treatment does not necessitate elimination of the residual chlorine as it is originally employed as a disinfectant.
2. Results and Discussion
2.1. Aqueous Chlorine Chemistry and ARAC Chlorination Tests
2.2. Chlorine Sonolysis
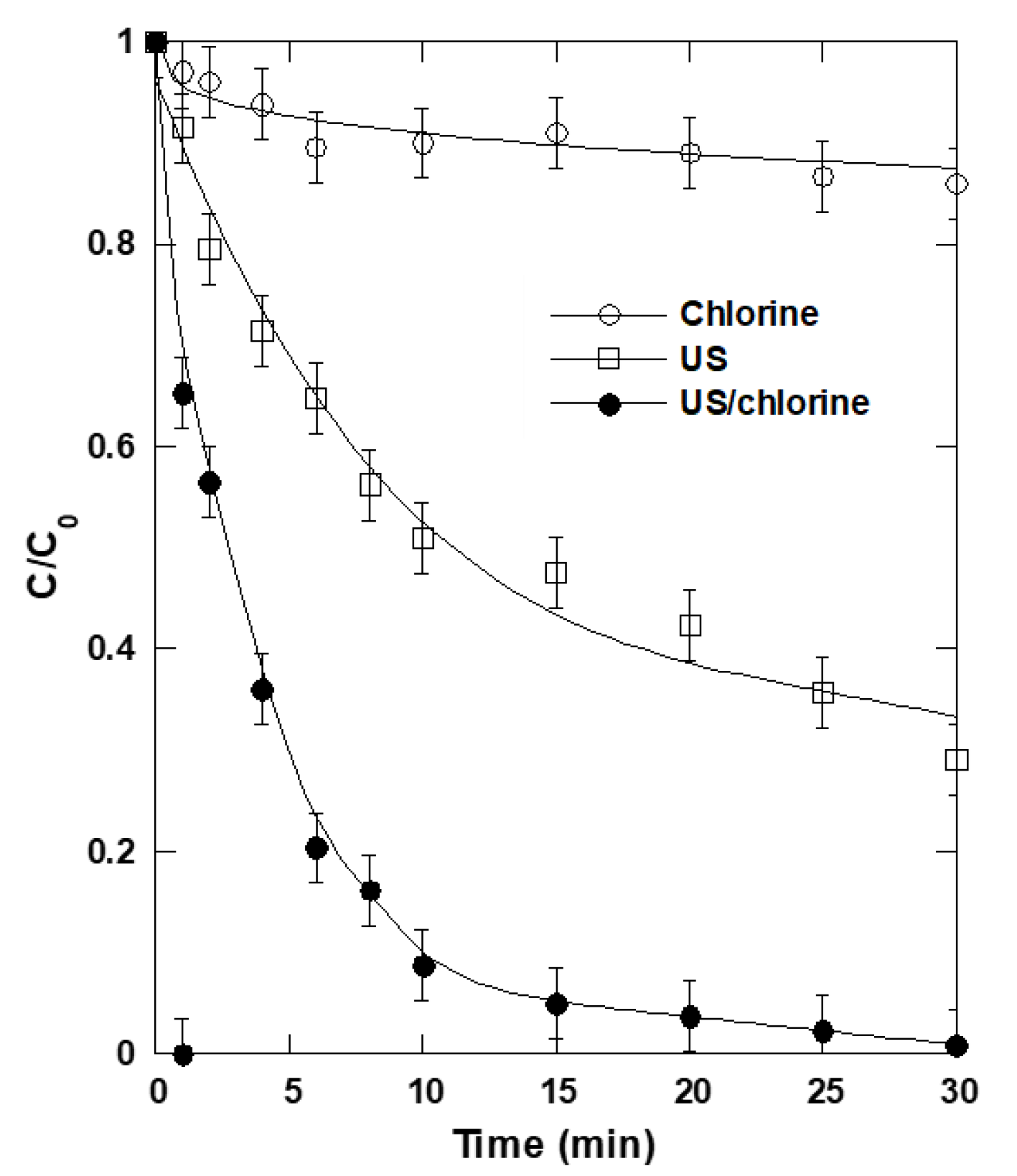
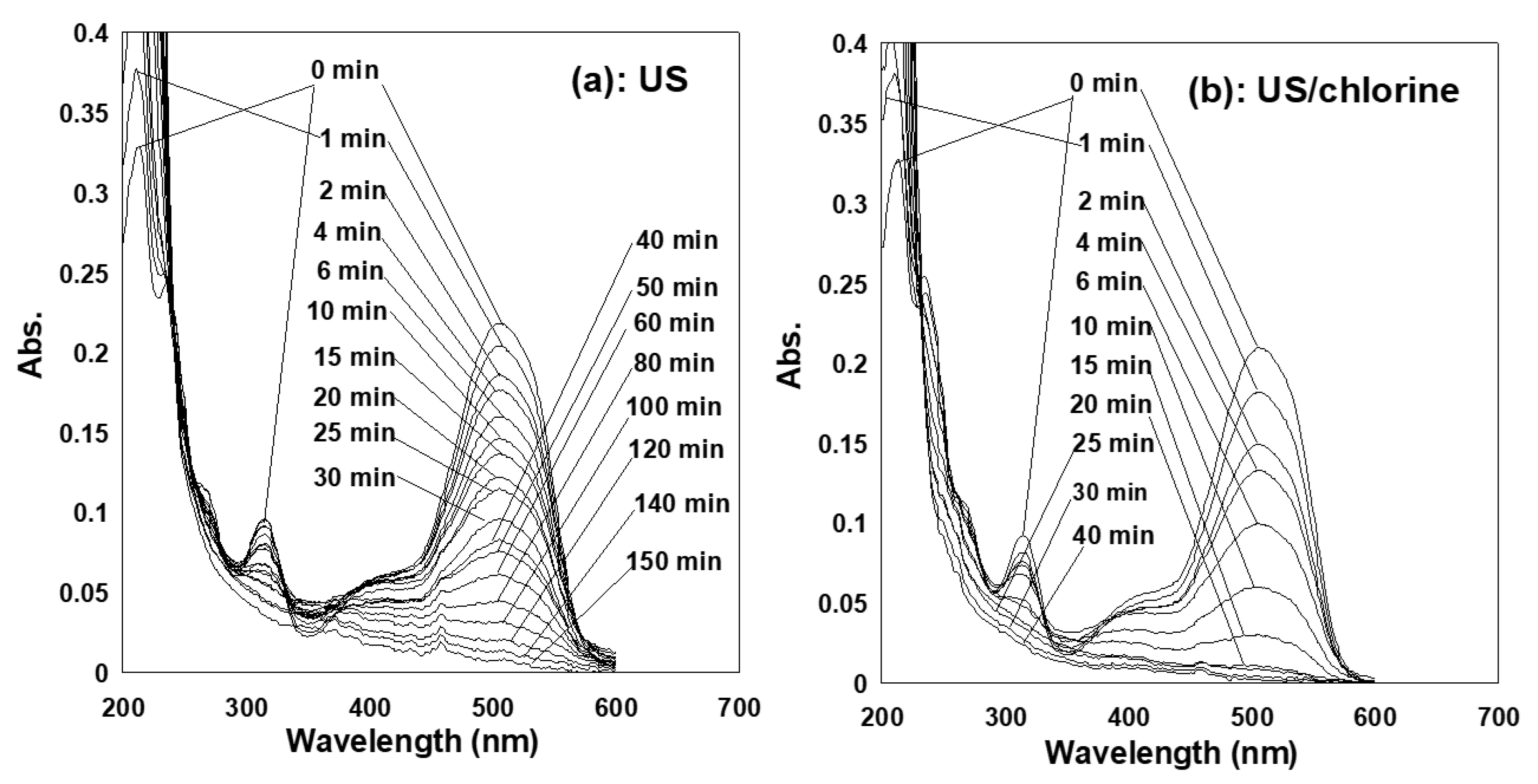
2.3. Synergism of Coupling Ultrasound and Chlorine Treatments
2.4. Source of the Synergistic Effect
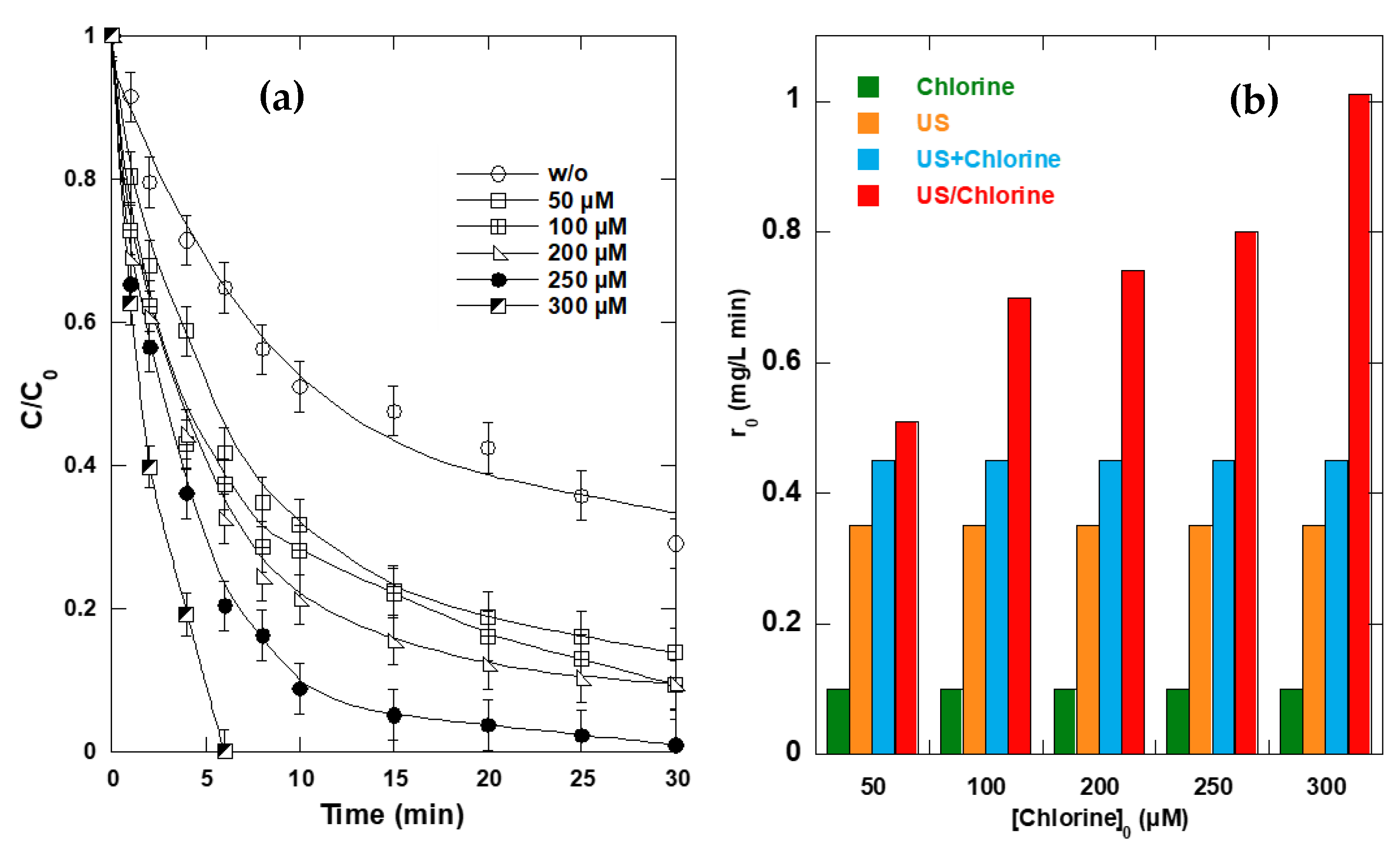
2.5. Synergism Dependence of Chlorine Dosage
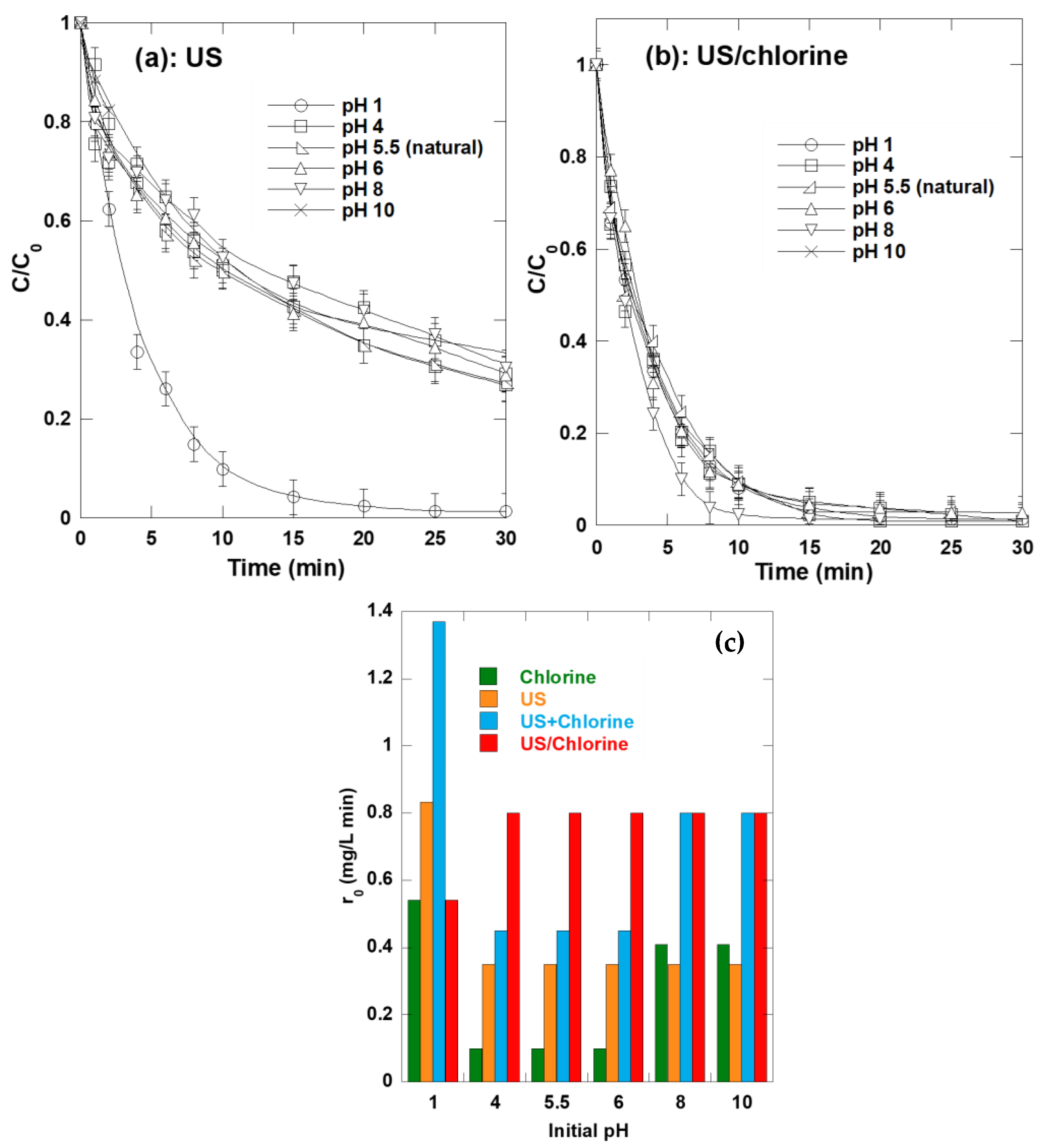
2.6. Synergism Dependence of pH
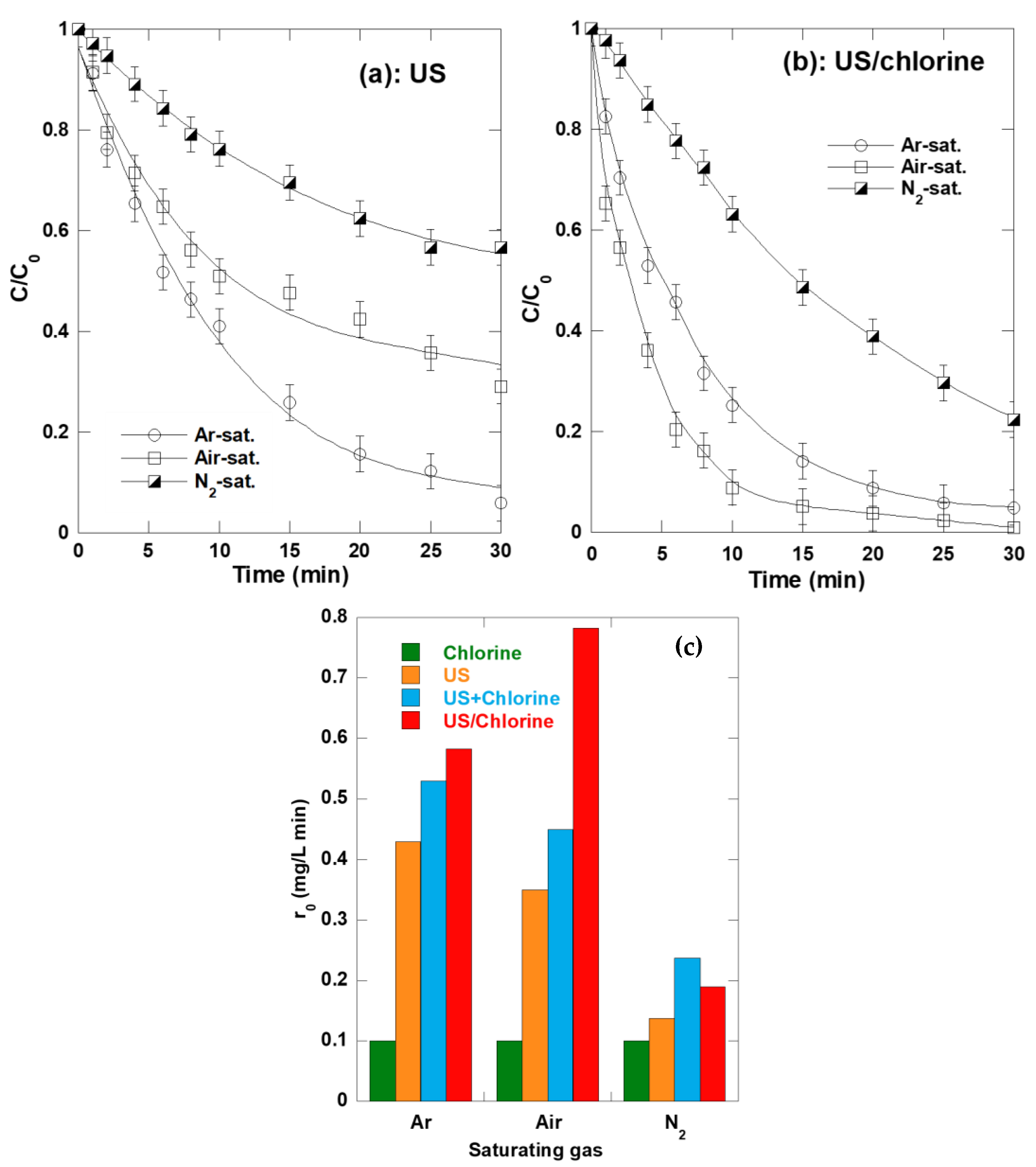
2.7. Synergism Dependence of Saturating Gases
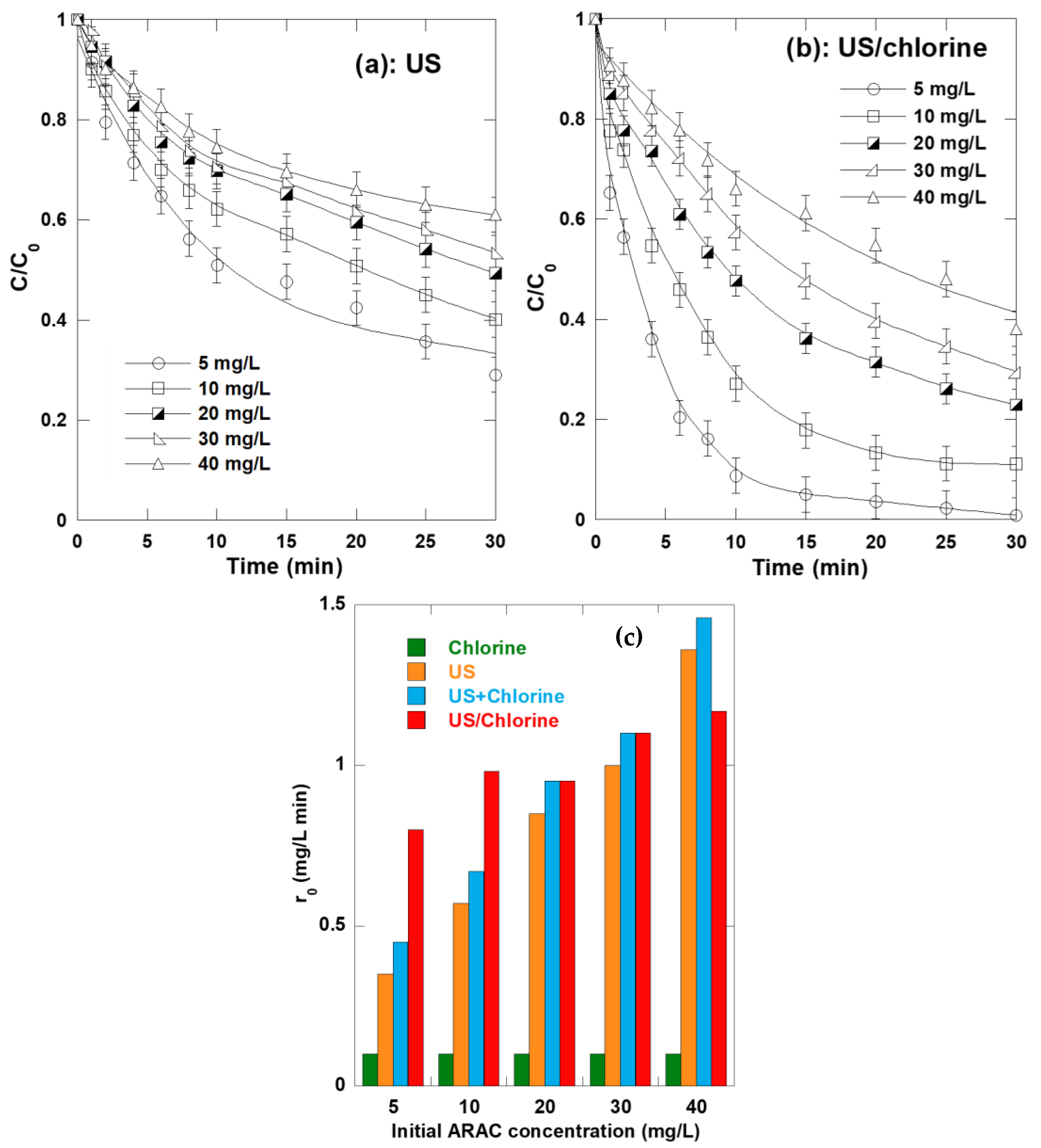
2.8. Synergism Dependence of Initial ARAC Concentration
3. Materials and Methods
4. Conclusions
- TOC, BOD5 and toxicity evolutions;
- Radicals’ identification and contribution to the overall degradation rate;
- The effect of processing conditions (e.g., pH, temperature, chlorine and pollutant concentrations) on radicals’ distribution;
- The identification of degradation by-products because of the possible formation of toxic trihalomethanes;
- The reaction mechanism and scheme for ARAC degradation.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Boczkaj, G.; Fernandes, A. Wastewater treatment by means of advanced oxidation processes at basic pH conditions: A review. Chem. Eng. J. 2017, 320, 608–633. [Google Scholar] [CrossRef]
- Kanakaraju, D.; Glass, B.D.; Oelgemöller, M. Advanced oxidation process-mediated removal of pharmaceuticals from water: A review. J. Environ. Manag. 2018, 219, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Ameta, S.; Ameta, R. Advanced Oxidation Processes for Wastewater Treatment: Emerging Green Chemical Technology; Elsevier Science: London, UK, 2018; ISBN 9780128105252. [Google Scholar]
- Stefan, M.I. Advanced Oxidation Processes for Water Treatment: Fundamentals and Applications; IWA Publishing: London, UK, 2017. [Google Scholar]
- Pétrier, C. The use of power ultrasound for water treatment. In Power Ultrasonics: Applications of High-Intensity Ultrasound; Gallego-Juarez, J.A., Graff, K., Eds.; Elsevier: Cambridge, MA, USA, 2015; pp. 939–963. [Google Scholar]
- Suslick, K.S.; Flannigan, D.J. Inside a Collapsing Bubble: Sonoluminescence and the Conditions During Cavitation. Annu. Rev. Phys. Chem. 2008, 59, 659–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasui, K.; Tuziuti, T.; Lee, J.; Kozuka, T.; Towata, A.; Iida, Y. The range of ambient radius for an active bubble in sonoluminescence and sonochemical reactions. J. Chem. Phys. 2008, 128, 184705. [Google Scholar] [CrossRef]
- Yasui, K.; Tuziuti, T.; Kozuka, T.; Towata, A.; Iida, Y. Relationship between the bubble temperature and main oxidant created inside an air bubble under ultrasound. J. Chem. Phys. 2007, 127, 154502. [Google Scholar] [CrossRef]
- Merouani, S.; Hamdaoui, O.; Rezgui, Y.; Guemini, M. Sensitivity of free radicals production in acoustically driven bubble to the ultrasonic frequency and nature of dissolved gases. Ultrason. Sonochem. 2015, 22, 41–50. [Google Scholar] [CrossRef]
- Makino, K.; Mossoba, M.M.; Riesz, P. Chemical effects of ultrasound on aqueous solutions. Evidence for •OH an •H by spin trapping. J. Am. Chem. Soc. 1982, 104, 3537–3539. [Google Scholar] [CrossRef]
- Hart, E.J.; Henglein, A. Sonochemistry of aqueous solutions: H2-O2 combustion in cavitation bubbles. J. Phys. Chem. 1987, 91, 3654–3656. [Google Scholar] [CrossRef]
- Henglein, A. Chemical effects of continuous and pulsed ultrasound in aqueous solutions. Ultrason. Sonochem. 1995, 2, 115–121. [Google Scholar] [CrossRef]
- Thompson, L.H.; Doraiswamy, L.K. Sonochemistry: Science and Engineering. Ind. Eng. Chem. Res. 1999, 38, 1215–1249. [Google Scholar] [CrossRef]
- Laat, J.D.E.; Stefan, M. UV/chlorine process. In Advanced Oxidation Processes for Water Treatment; Stefan, M.I., Ed.; IWA Publishing: London, UK, 2017; pp. 383–428. [Google Scholar]
- Remucal, C.K.; Manley, D. Emerging investigators series: The efficacy of chlorine photolysis as an advanced oxidation process for drinking water treatment. Environ. Sci. Water Res. Technol. 2016, 2, 565–579. [Google Scholar] [CrossRef]
- Meghlaoui, F.Z.; Merouani, S.; Hamdaoui, O.; Bouhelassa, M.; Ashokkumar, M. Rapid catalytic degradation of refractory textile dyes in Fe (II)/chlorine system at near neutral pH: Radical mechanism involving chlorine radical anion (Cl2•−)-mediated transformation pathways and impact of environmental matrices. Sep. Purif. Technol. 2019, 227, 115685. [Google Scholar] [CrossRef]
- Bulman, D.M.; Mezyk, S.P.; Remucal, C.K. The Impact of pH and Irradiation Wavelength on the Production of Reactive Oxidants during Chlorine Photolysis. Environ. Sci. Technol. 2019, 53, 4450–4459. [Google Scholar] [CrossRef] [PubMed]
- Behin, J.; Akbari, A.; Mahmoudi, M.; Khajeh, M. Sodium hypochlorite as an alternative to hydrogen peroxide in Fenton process for industrial scale. Water Res. 2017, 121, 120–128. [Google Scholar] [CrossRef]
- Meghlaoui, F.Z.; Merouani, S.; Hamdaoui, O.; Alghyamah, A.; Bouhelassa, M.; Ashokkumar, M. Fe(III)-catalyzed degradation of persistent textile dyes by chlorine at slightly acidic conditions: The crucial role of Cl2•− radical in the degradation process and impacts of mineral and organic competitors. Asia-Pacific J. Chem. Eng. 2020, 16, e2553. [Google Scholar] [CrossRef]
- Belghit, A.; Merouani, S.; Hamdaoui, O.; Alghyamah, A.; Bouhelassa, M. Influence of processing conditions on the synergism between UV irradiation and chlorine toward the degradation of refractory organic pollutants in UV/chlorine advanced oxidation system. Sci. Total Environ. 2020, 736, 139623. [Google Scholar] [CrossRef]
- Wang, W.L.; Wu, Q.Y.; Huang, N.; Wang, T.; Hu, H.Y. Synergistic effect between UV and chlorine (UV/chlorine) on the degradation of carbamazepine: Influence factors and radical species. Water Res. 2016, 98, 190–198. [Google Scholar] [CrossRef]
- Fang, J.; Fu, Y.; Shang, C. The roles of reactive species in micropollutant degradation in the UV/free chlorine system. Environ. Sci. Technol. 2014, 48, 1859–1868. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, X.; Wu, Q.; Du, Y.; Hu, H. Degradation of natural organic matter by UV/chlorine oxidation: Molecular decomposition, formation of oxidation byproducts and cytotoxicity. Water Res. 2017, 124, 251–258. [Google Scholar] [CrossRef]
- Wang, A.; Lin, Y.; Xu, B.; Hu, C.; Xia, S.; Zhang, T.; Chu, W. Kinetics and modeling of iodoform degradation during UV/chlorine advanced oxidation process. Chem. Eng. J. 2017, 323, 312–319. [Google Scholar] [CrossRef]
- Kong, X.; Wu, Z.; Ren, Z.; Guo, K.; Hou, S.; Hua, Z.; Li, X.; Fang, J. Degradation of lipid regulators by the UV/chlorine process: Radical mechanisms, chlorine oxide radical (ClO•)-mediated transformation pathways and toxicity changes. Water Res. 2018, 137, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Fang, J.; Shang, C. Kinetics and pathways of ibuprofen degradation by the UV/chlorine advanced oxidation process. Water Res. 2016, 90, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Wu, Z.; Fang, J. UV-based advanced oxidation process for the treatment of pharmaceuticals and personal care products. In Contaminants of Emerging Concern in Water and Wastewater; Hernández-Maldonado, A.J., Blaney, L., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; pp. 367–408. ISBN 9780128135617. [Google Scholar]
- Wang, W.L.; Wu, Q.Y.; Li, Z.M.; Lu, Y.; Du, Y.; Wang, T.; Huang, N.; Hu, H.Y. Light-emitting diodes as an emerging UV source for UV/chlorine oxidation: Carbamazepine degradation and toxicity changes. Chem. Eng. J. 2017, 310, 148–156. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, K.; Fang, J.; Yang, X.; Xiao, H.; Hou, S.; Kong, X.; Shang, C.; Yang, X.; Meng, F.; et al. Factors affecting the roles of reactive species in the degradation of micropollutants by the UV/chlorine process. Water Res. 2017, 126, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wu, G.; Yuan, S.; Zhan, X.; Wang, W.; Hu, Z.H. Ciprofloxacin degradation in UV/chlorine advanced oxidation process: Influencing factors, mechanisms and degradation pathways. J. Photochem. Photobiol. A Chem. 2019, 371, 151–158. [Google Scholar] [CrossRef]
- Guo, K.; Wu, Z.; Shang, C.; Yao, B.; Hou, S.; Yang, X.; Song, W.; Fang, J. Radical Chemistry and Structural Relationships of PPCP Degradation by UV/Chlorine Treatment in Simulated Drinking Water. Environ. Sci. Technol. 2017, 51, 10431–10439. [Google Scholar] [CrossRef]
- Guo, Z.; Lin, Y.; Xu, B.; Huang, H.; Zhang, T.; Tian, F.; Gao, N. Degradation of chlortoluron during UV irradiation and UV/chlorine processes and formation of disinfection by-products in sequential chlorination. Chem. Eng. J. 2016, 283, 412–419. [Google Scholar] [CrossRef]
- Dong, H.; Qiang, Z.; Hu, J.; Qu, J. Degradation of chloramphenicol by UV/chlorine treatment: Kinetics, mechanism and enhanced formation of halonitromethanes. Water Res. 2017, 121, 178–185. [Google Scholar] [CrossRef]
- Alegre, M.L.; Geronees, M.; Rosso, J.A.; Bertolotti, S.G.; Braun, A.M.; Marrtire, D.O.; Gonzalez, M.C. Kinetic Study of the Reactions of Chlorine Atoms and Cl2•− Radical Anions in Aqueous Solutions. 1. Reaction with Benzene. J. Phys. Chem. A 2000, 104, 3117–3125. [Google Scholar] [CrossRef]
- Deborde, M.; Gunten, U. Von Reactions of chlorine with inorganic and organic compounds during water treatment—Kinetics and mechanisms: A critical review. Water Res. 2008, 42, 13–51. [Google Scholar] [CrossRef]
- Zou, H.; Tang, H. Comparison of different bacteria inactivation by a novel continuous-flow ultrasound/chlorination water treatment system in a pilot scale. Water 2019, 11, 258. [Google Scholar] [CrossRef] [Green Version]
- Blume, T.; Neis, U. Improving chlorine disinfection of wastewater by ultrasound application. Water Sci. Technol. 2005, 52, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Lambert, N.; Rediers, H.; Hulsmans, A.; Joris, K.; Declerck, P.; De Laedt, Y.; Liers, S. Evaluation of ultrasound technology for the disinfection of process water and the prevention of biofilm formation in a pilot plant. Water Sci. Technol. 2010, 61, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Hamdaoui, O.; Merouani, S.; Ait Idir, M.; Benmahmoud, H.C.; Dehane, A.; Alghyamah, A. Ultrasound/chlorine sono-hybrid-advanced oxidation process: Impact of dissolved organic matter and mineral constituents. Ultrason. Sonochem. 2022, 83, 105918. [Google Scholar] [CrossRef] [PubMed]
- Borzelleca, J.F.; Olson, J.W.; Reno, F.E. Lifetime toxicity/carcinogenicity study of FD & C Red No. 40 (Allura Red) in Sprague-Dawley rats. Food Chem. Toxicol. 1989, 27, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Garole, V.J.; Choudhary, B.C.; Tetgure, S.R.; Garole, D.J.; Borse, A.U. Detoxification of toxic dyes using biosynthesized iron nanoparticles by photo-Fenton processes. Int. J. Environ. Sci. Technol. 2018, 15, 1649–1656. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Butcher, R.E.; Brunner, R.L.; Wootten, V.; Sobotka, T.J. Development toxicity and psychotoxicity of FD and C red dye no. 40 (Allura red AC) in rates. Toxicology 1983, 28, 207–217. [Google Scholar] [CrossRef]
- Sun, Q.; Yang, L.; Yang, J.; Liu, S.; Hu, X. Study on the interaction between Rhodamine dyes and Allura Red based on fluorescence spectra and its analytical application in soft Drinks. Anal. Sci. 2017, 33, 1181–1187. [Google Scholar] [CrossRef] [Green Version]
- Vogt, R.; Schindler, R.N. Product channels in the photolysis of HOCl. J. Photochem. Photobiol. A Chem. 1992, 66, 133–140. [Google Scholar] [CrossRef]
- Buxton, G.V.; Bydder, M.; Arthur Salmon, G. Reactivity of chlorine atoms in aqueous solution Part 1. The equilibrium ClMNsbd+Cl−Cl2−. J. Chem. Soc. Faraday Trans. 1998, 94, 653–657. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated Electrons, hydrogen atoms and hydroxyl radicals (•OH/O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 515–886. [Google Scholar] [CrossRef] [Green Version]
- Pubchem. Allura Red AC. 2020. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Allura-Red-AC (accessed on 9 September 2022).
- Pétrier, C.; Francony, A. Ultrasonic waste-water treatment: Incidence of ultrasonic frequency on the rate of phenol and carbon tetrachloride degradation. Ultrason. Sonochem. 1997, 4, 295–300. [Google Scholar] [CrossRef]
- Merouani, S.; Hamdaoui, O.; Saoudi, F.; Chiha, M. Influence of experimental parameters on sonochemistry dosimetries: KI oxidation, Fricke reaction and H2O2 production. J. Hazard. Mater. 2010, 178, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Tauber, A.; Mark, G.; Schuchmann, H.-P.; von Sonntag, C. Sonolysis of tert-butyl alcohol in aqueous solution. J. Chem. Soc. Perkin Trans. 2 1999, 2, 1129–1136. [Google Scholar] [CrossRef]
- Kong, X.; Jiang, J.; Ma, J.; Yang, Y.; Liu, W.; Liu, Y. Degradation of atrazine by UV/chlorine: Efficiency, influencing factors, and products. Water Res. 2016, 90, 15–23. [Google Scholar] [CrossRef]
- Huang, N.; Wang, T.; Wang, W.; Wu, Q.; Li, A.; Hu, H. UV/chlorine as an advanced oxidation process for the degradation of benzalkonium chloride: Synergistic effect, transformation products and toxicity evaluation. Water Res. 2017, 114, 246–253. [Google Scholar] [CrossRef]
- Chadi, N.E.; Merouani, S.; Hamdaoui, O.; Bouhelassa, M. New aspect of the effect of liquid temperature on sonochemical degradation of nonvolatile organic pollutants in aqueous media. Sep. Purif. Technol. 2018, 200, 68–74. [Google Scholar] [CrossRef]
- Henglein, A. Sonolysis of carbon dioxide, nitrous oxide and methane in aqueous solution. Z. Naturforsch. B 1985, 40, 100–107. [Google Scholar] [CrossRef]
- Harada, H.; Ono, Y. Improvement of the rate of sono-oxidation in the presence of CO2. Jpn. J. Appl. Phys. 2015, 54, 52–55. [Google Scholar] [CrossRef]
- Rooze, J. Cavitation in Gas-Saturated Liquids; Technische Universiteit Eindhoven: Eindhoven, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Merouani, S.; Hamdaoui, O.; Al-Zahrani, S.M. Toward understanding the mechanism of pure CO2-quenching sonochemical processes. J. Chem. Technol. Biotechnol. 2020, 95, 553–566. [Google Scholar] [CrossRef]
- Gao, Y.Q.; Gao, N.Y.; Deng, Y.; Gu, J.S.; Gu, Y.L.; Zhang, D. Factors affecting sonolytic degradation of sulfamethazine in water. Ultrason. Sonochem. 2013, 20, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Ferkous, H.; Hamdaoui, O.; Merouani, S. Sonochemical degradation of naphthol blue black in water: Effect of operating parameters. Ultrason. Sonochem. 2015, 26, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Boutamine, Z.; Merouani, S.; Hamdaoui, O. Sonochemical degradation of Basic Red 29 in aqueous media. Turk. J. Chem. 2017, 41, 99–115. [Google Scholar] [CrossRef]
- Hamdaoui, O.; Merouani, S. Improvement of Sonochemical Degradation of Brilliant Blue R in Water Using Periodate Ions: Implication of Iodine Radicals in the Oxidation Process. Ultrason. Sonochem. 2017, 37, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Merouani, S.; Ferkous, H.; Hamdaoui, O.; Rezgui, Y.; Guemini, M. New interpretation of the effects of argon-saturating gas toward sonochemical reactions. Ultrason. Sonochem. 2015, 23, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Rooze, J.; Rebrov, E.V.; Schouten, J.C.; Keurentjes, J.T.F. Dissolved gas and ultrasonic cavitation—A review. Ultrason. Sonochem. 2013, 20, 1–11. [Google Scholar] [CrossRef]
- Rooze, J.; Rebrov, E.V.; Schouten, J.C.; Keurentjes, J.T.F. Effect of resonance frequency, power input, and saturation gas type on the oxidation efficiency of an ultrasound horn. Ultrason. Sonochem. 2011, 18, 209–215. [Google Scholar] [CrossRef]
- Chadi, N.E.; Merouani, S.; Hamdaoui, O.; Bouhelassa, M.; Ashokkumar, M. H2O2/Periodate (IO4−): A novel advanced oxidation technology for the degradation of refractory organic pollutants. Environ. Sci. Water Res. Technol. 2019, 5, 1113–1123. [Google Scholar] [CrossRef]
- Ferkous, H.; Merouani, S.; Hamdaoui, O.; Pétrier, C. Ultrasonics Sonochemistry Persulfate-enhanced sonochemical degradation of naphthol blue black in water: Evidence of sulfate radical formation. Ultrason. Sonochem. 2017, 34, 580–587. [Google Scholar] [CrossRef]
- Bekkouche, S.; Merouani, S.; Hamdaoui, O.; Bouhelassa, M. Efficient photocatalytic degradation of Safranin O by integrating solar-UV/TiO2/persulfate treatment: Implication of sulfate radical in the oxidation process and effect of various water matrix components. J. Photochem. Photobiol. A Chem. 2017, 345, 80–91. [Google Scholar] [CrossRef]
- Villaroel, E.; Silva-Agredo, J.; Petrier, C.; Taborda, G.; Torres-Palma, R.A. Ultrasonic degradation of acetaminophen in water: Effect of sonochemical parameters and water matrix. Ultrason. Sonochem. 2014, 21, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Merouani, S.; Hamdaoui, O.; Saoudi, F.; Chiha, M. Sonochemical degradation of Rhodamine B in aqueous phase: Effects of additives. Chem. Eng. J. 2010, 158, 550–557. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdaoui, O.; Merouani, S.; Benmahmoud, H.C.; Ait Idir, M.; Ferkous, H.; Alghyamah, A. Ultrasound/Chlorine: A Novel Synergistic Sono-Hybrid Process for Allura Red AC Degradation. Catalysts 2022, 12, 1171. https://doi.org/10.3390/catal12101171
Hamdaoui O, Merouani S, Benmahmoud HC, Ait Idir M, Ferkous H, Alghyamah A. Ultrasound/Chlorine: A Novel Synergistic Sono-Hybrid Process for Allura Red AC Degradation. Catalysts. 2022; 12(10):1171. https://doi.org/10.3390/catal12101171
Chicago/Turabian StyleHamdaoui, Oualid, Slimane Merouani, Hadjer C. Benmahmoud, Meriem Ait Idir, Hamza Ferkous, and Abdulaziz Alghyamah. 2022. "Ultrasound/Chlorine: A Novel Synergistic Sono-Hybrid Process for Allura Red AC Degradation" Catalysts 12, no. 10: 1171. https://doi.org/10.3390/catal12101171