
Photocatalytic and Sonocatalytic Degradation of EDTA and Rhodamine B over Ti0 and Ti@TiO2 Nanoparticles

Abstract
:1. Introduction
2. Results and Discussion
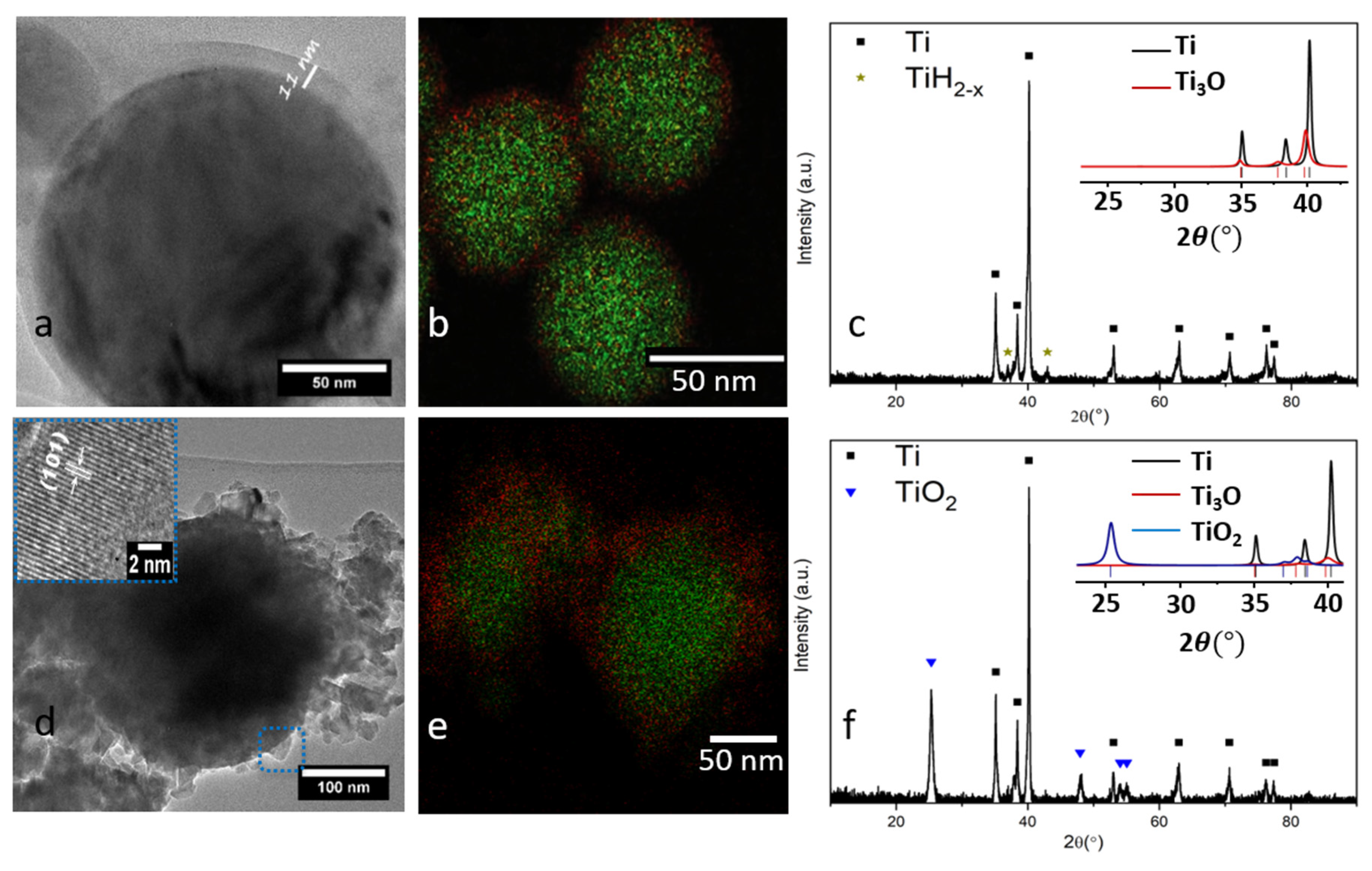
2.1. Structural, Chemical and Optical Properties of Ti0 and Ti@TiO2 NPs
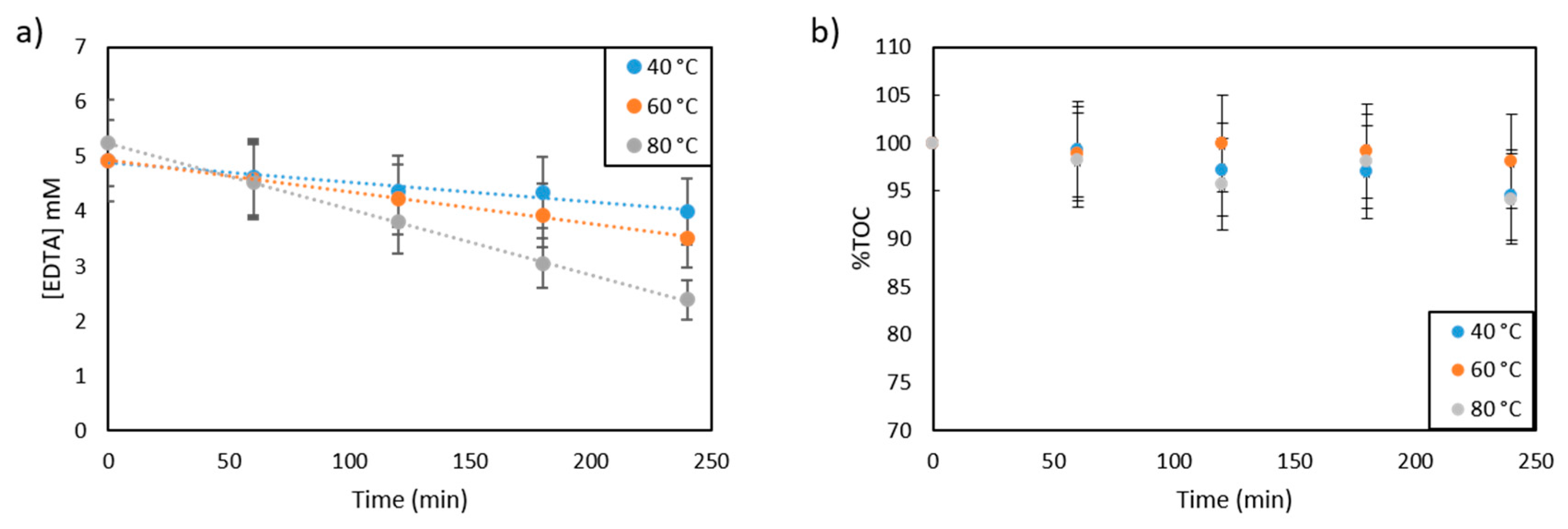
2.2. EDTA Study
2.2.1. Kinetics of Photocatalytic EDTA Degradation
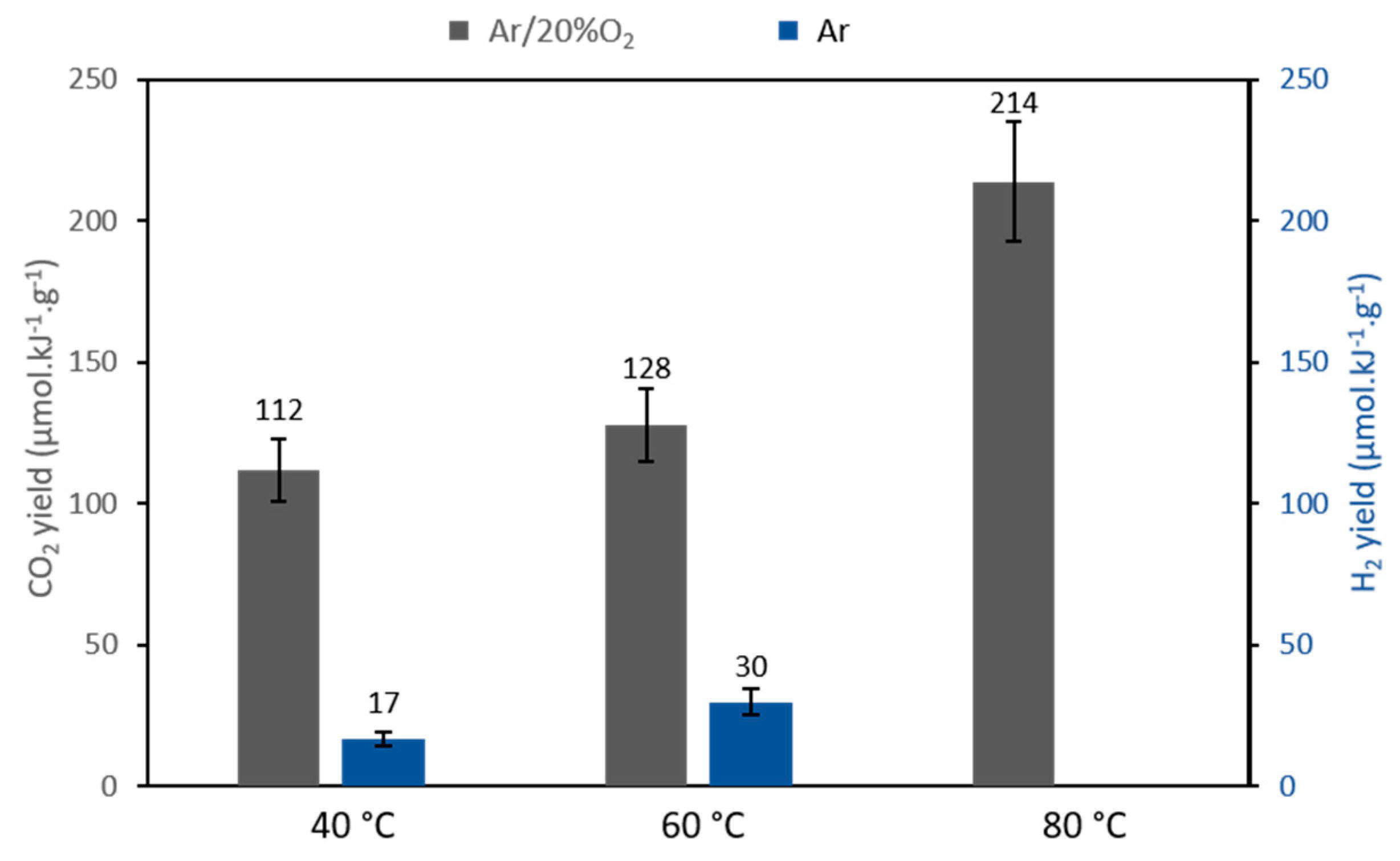
2.2.2. Gaseous Products Formation upon Photocatalytic EDTA Degradation
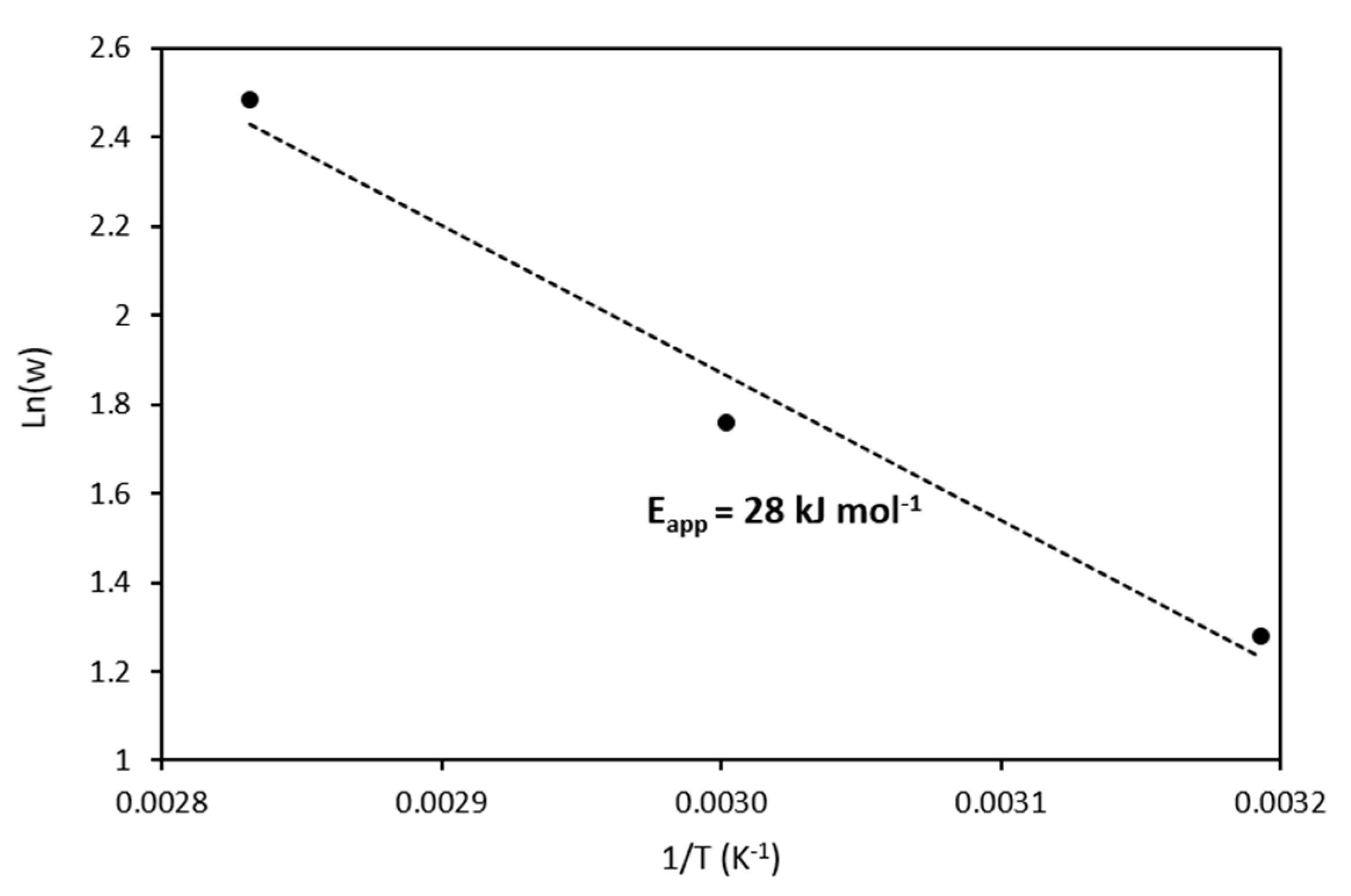
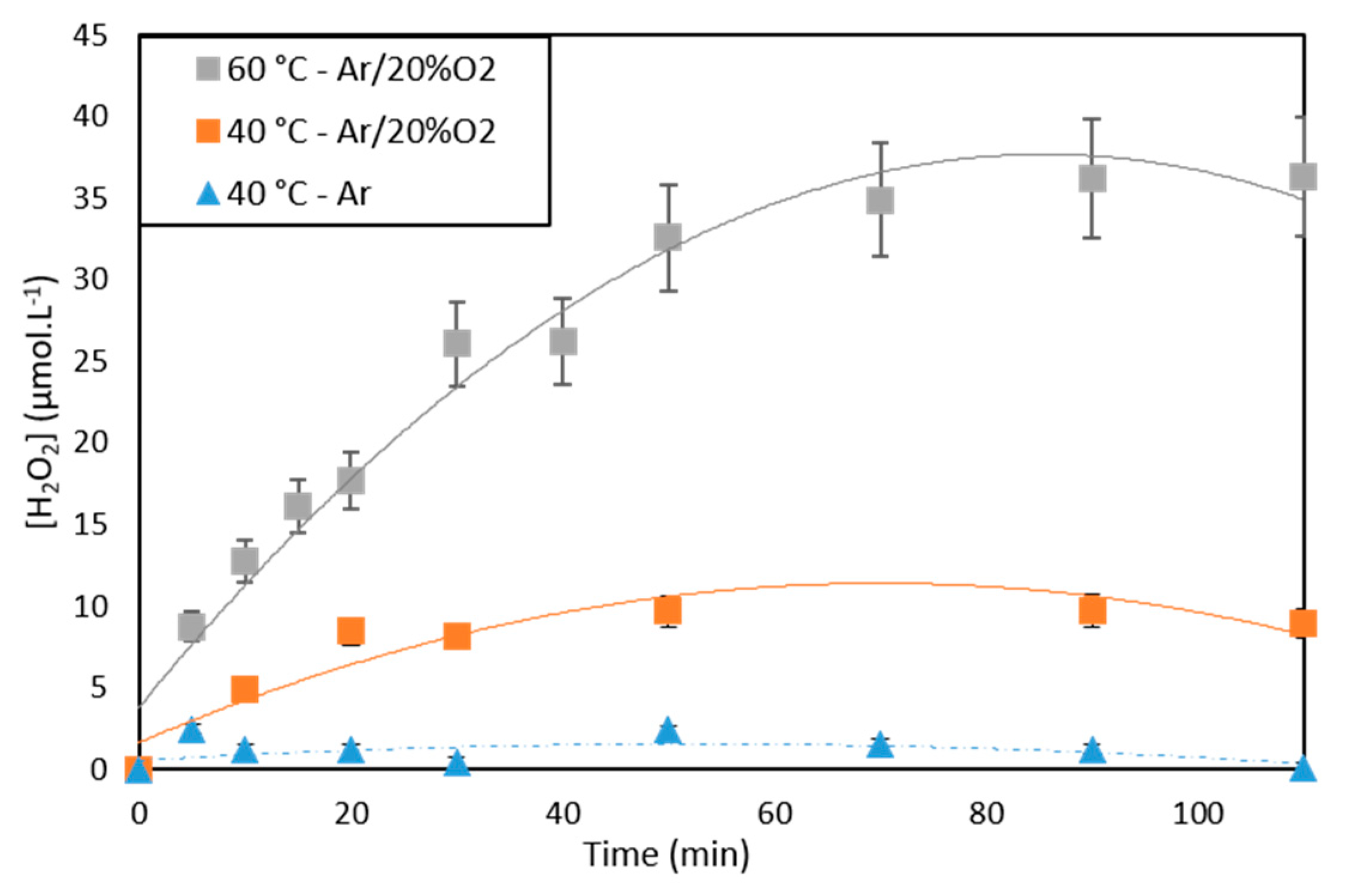
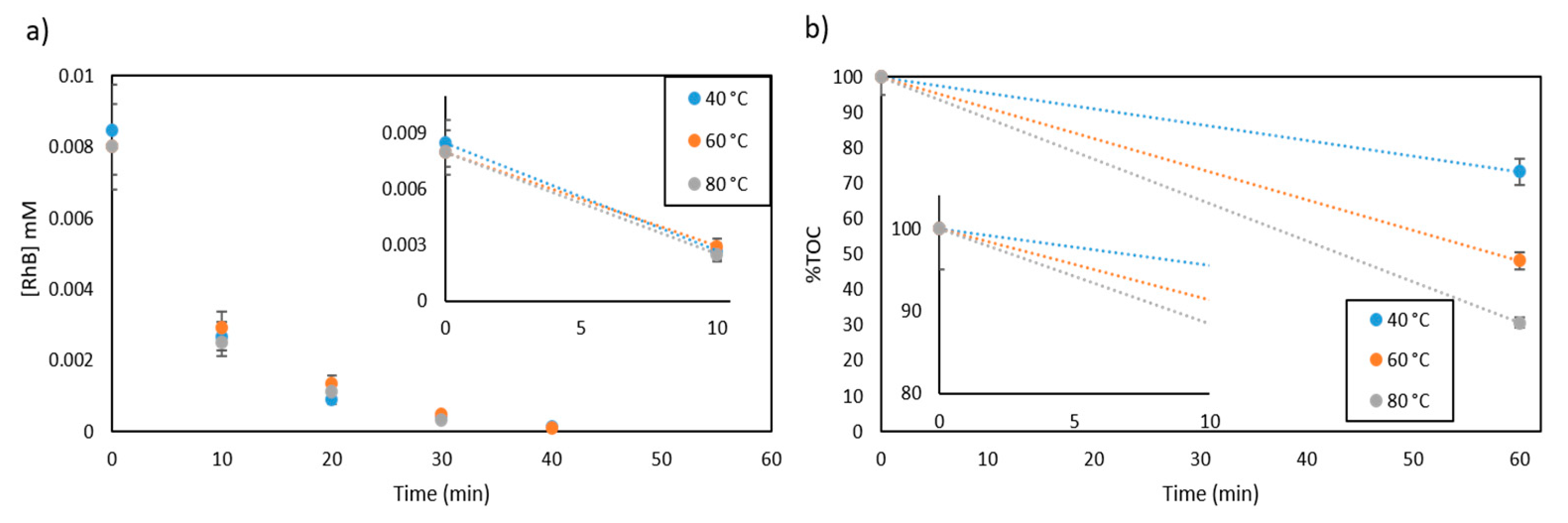
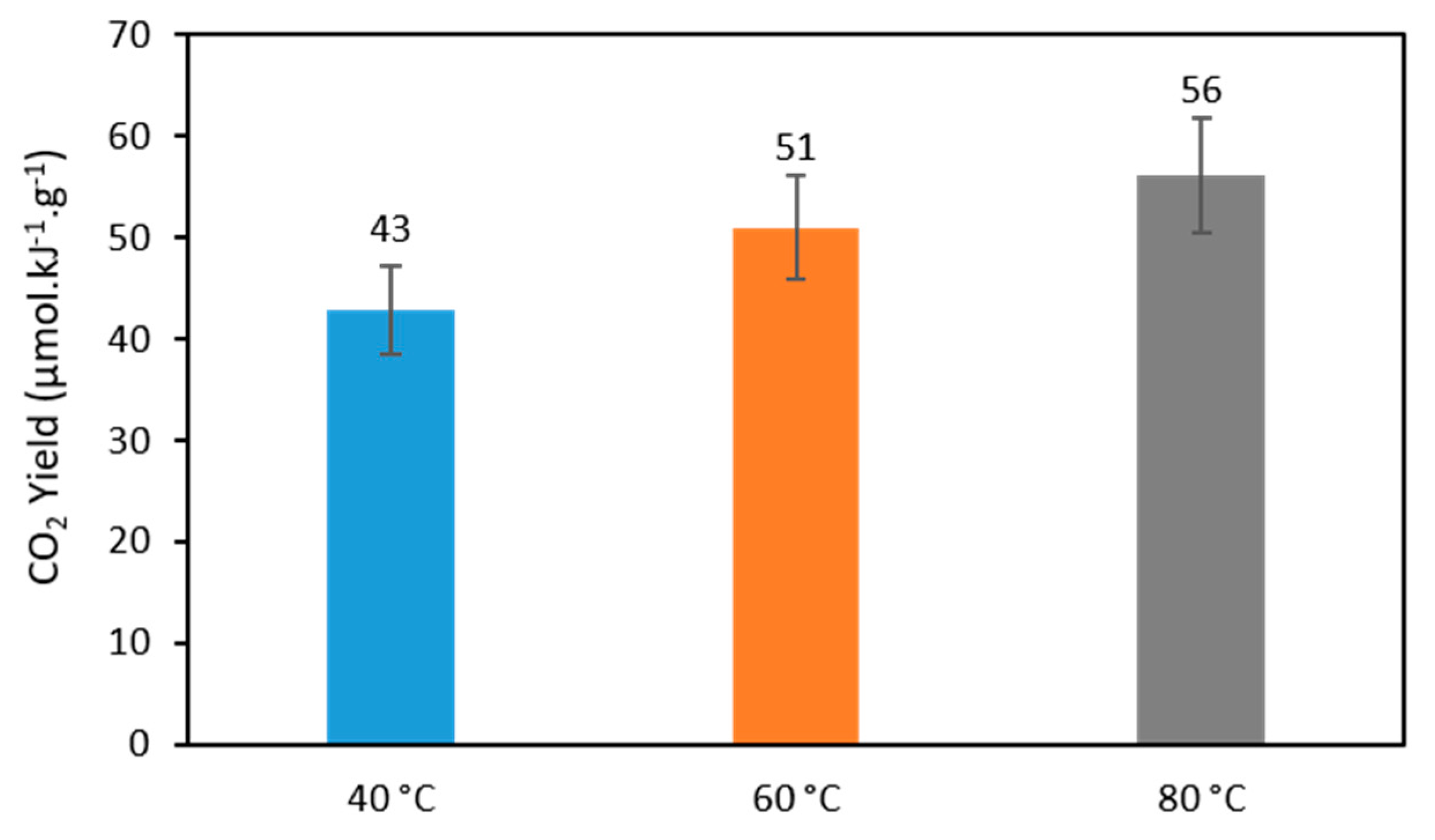
2.2.3. Effect of Temperature
2.2.4. Comparison of Sonocatalytic and Photocatalytic EDTA Degradation
2.2.5. Rhodamine B Degradation
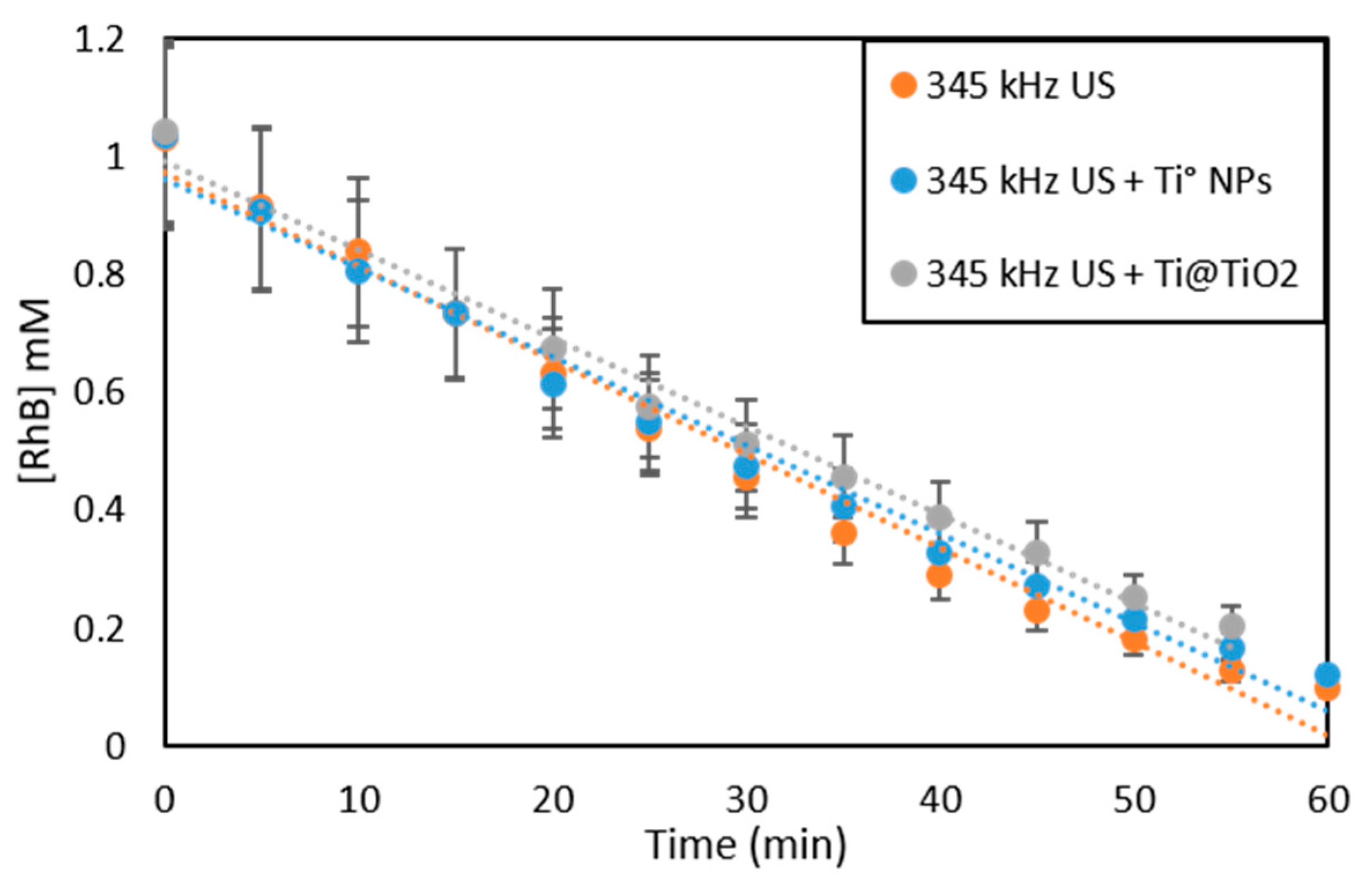
RhB Sonochemical Degradation
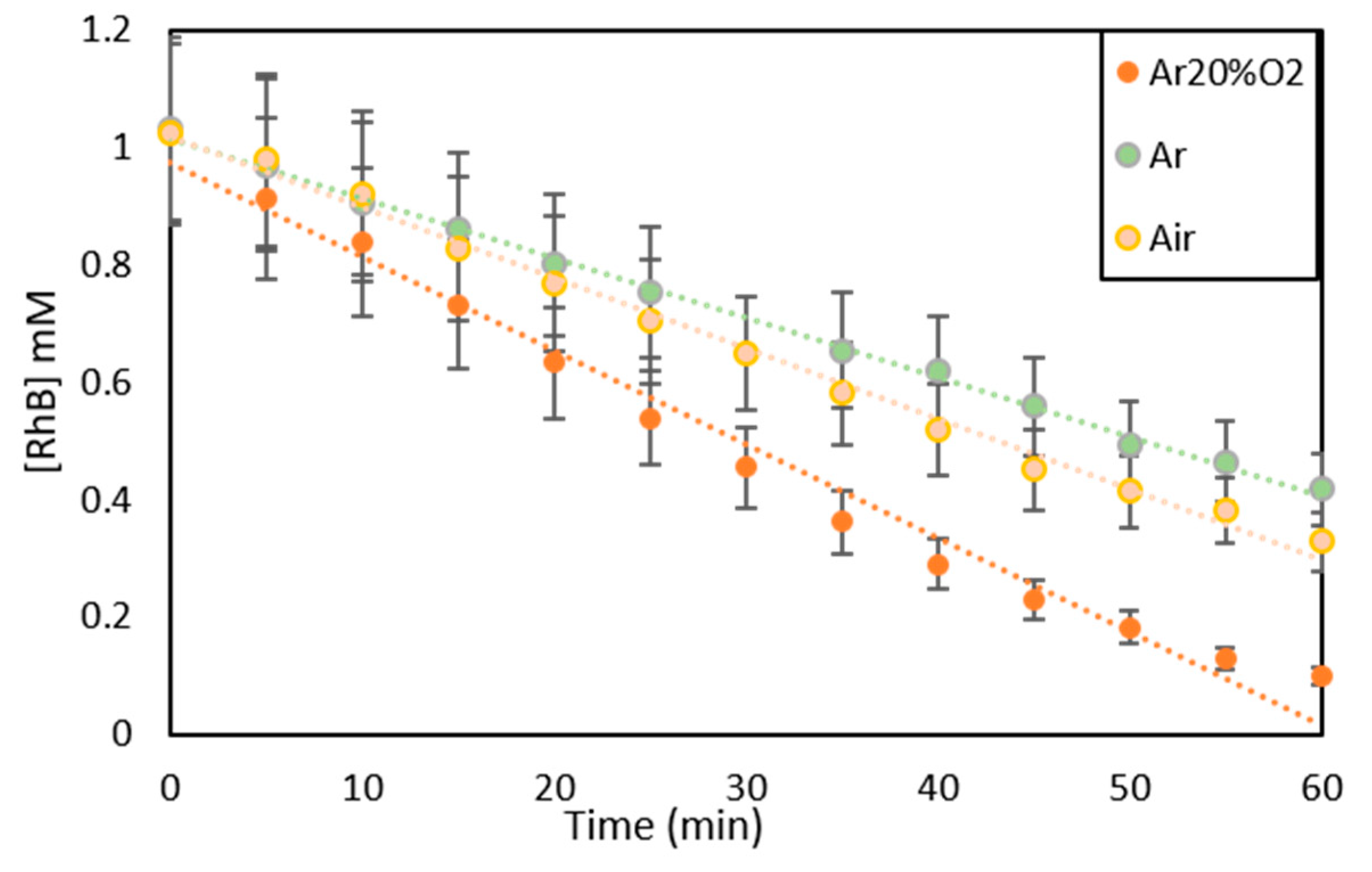
2.2.6. RhB Photocatalytic Degradation
3. Materials and Methods
3.1. Chemical Reagents
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.4. Experimental Setups
3.4.1. Photocatalytic Experiments
3.4.2. Sonocatalytic Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Roy, M.; Saha, R. Dyes and their removal technologies from wastewater: A critical review. In Intelligent Environmental Data Monitoring for Pollution Management; Elsevier BV: Amsterdam, The Netherlands, 2021; pp. 127–160. [Google Scholar]
- Ortiz, I.; Rivero, M.J.; Margallo, M. Advanced oxidative and catalytic processes. In Sustainable Water and Wastewater Processing; Elsevier BV: Amsterdam, The Netherlands, 2019; pp. 161–201. [Google Scholar]
- Ullah, H.; Viglašová, E.; Galamboš, M. Visible Light-Driven Photocatalytic Rhodamine B Degradation Using CdS Nanorods. Processes 2021, 9, 263. [Google Scholar] [CrossRef]
- Severa, J.; Bàr, J. Handbook of Radioactive Contamination and Decontamination; Elsevier Science: Amsterdam, The Netherlands, 1991; Volume 47. [Google Scholar]
- Hinck, M.L.; Ferguson, J.; Puhaakka, J. Resistance of EDTA and DTPA to aerobic biodegradation. Water Sci. Technol. 1997, 35, 25–31. [Google Scholar] [CrossRef]
- Motekaitis, R.J.; Cox, X.B., III; Taylor, P.; Martell, A.E.; Miles, B.; Tvedt, T.J., Jr. Thermal degradation of EDTA chelates in aqueous solution. Can. J. Chem. 1982, 60, 1207–1213. [Google Scholar] [CrossRef]
- Oturan, M.A.; Aaron, J.-J. Advanced oxidation processes in water/wastewater treatment: Principles and applications. A review. Crit. Rev. Environ. Sci. Technol. 2014, 44, 2577–2641. [Google Scholar] [CrossRef]
- Bajpai, P. Emerging technologies for waste water treatment. In Ind Finish; Pulp and Paper Industry Emerging Waste Water Treatment Technologies: Kanpur, India, 2017; Volume 51, pp. 93–179. [Google Scholar]
- Wang, J.; Wang, Z.; Vieira, C.L.; Wolfson, J.M.; Pingtian, G.; Huang, S. Review on the treatment of organic pollutants in water by ultrasonic technology. Ultrason. Sonochemistry 2019, 55, 273–278. [Google Scholar] [CrossRef]
- Chitra, S.; Paramasivan, K.; Sinha, P.; Lal, K. Ultrasonic treatment of liquid waste containing EDTA. J. Clean. Prod. 2004, 12, 429–435. [Google Scholar] [CrossRef]
- Jorfi, S.; Pourfadakari, S.; Kakavandi, B. A new approach in sono-photocatalytic degradation of recalcitrant textile wastewater using MgO@Zeolite nanostructure under UVA irradiation. Chem. Eng. J. 2018, 343, 95–107. [Google Scholar] [CrossRef]
- Mason, T.J.; Lorimer, J.P. Applied Sonochemistry: The Uses of Power Ultrasound in Chemistry and Processing; Wiley: Weinhim, Germany, 2002. [Google Scholar]
- Qiu, P.; Park, B.; Choi, J.; Thokchom, B.; Pandit, A.B.; Khim, J. A review on heterogeneous sonocatalyst for treatment of organic pollutants in aqueous phase based on catalytic mechanism. Ultrason. Sonochem. 2018, 45, 29–49. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, D.; Hong, S.; Khan, S.; Darya, B.; Lee, J.-Y.; Chung, J.; Cho, S.-H. Investigation of the synergistic effect of sonolysis and photocatalysis of titanium dioxide for organic dye degradation. Catalysts 2020, 10, 500. [Google Scholar] [CrossRef]
- Babey, P.; Emilio, C.; Ferreyra, R.; Gautier, E.; Gettar, R.; Litter, M. Kinetics and mechanisms of EDTA photocatalytic degradation with TiO2. Water Sci. Technol. 2001, 44, 179–185. [Google Scholar] [CrossRef]
- Godt, J.; Scheidig, F.; Grosse-Siestrup, C.; Esche, V.; Brandenburg, P.; Reich, A.; Groneberg, D. The toxicity of cadmium and resulting hazards for human health. J. Occup. Med. Toxicol. 2006, 1, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hakim, S.; Chave, T.; Nikitenko, S.I. Sonocatalytic degradation of EDTA in the presence of Ti and Ti@TiO2 nanoparticles. Ultrason. Sonochem. 2021, 70, 105336. [Google Scholar] [CrossRef] [PubMed]
- El Hakim, S.; Chave, T.; Nada, A.A.; Roualdes, S.; Nikitenko, S.I. Tailoring noble metal-free Ti@TiO2 photocatalyst for boosting photothermal hydrogen production. Front. Catal. 2021, 1, 669260. [Google Scholar] [CrossRef]
- Höbel, B.; Von Sonntag, C. OH-Radical induced degradation of ethylenediaminetetraacetic acid (EDTA) in aqueous solution: A pulse radiolysis study. J. Chem. Soc. Perkin Trans. 1998, 2, 509–514. [Google Scholar] [CrossRef]
- Nikitenko, S.I.; Chave, T.; Cau, C.; Brau, H.-P.; Flaud, V. Photothermal Hydrogen production using noble-metal-free Ti@TiO2 core–shell nanoparticles under visible–NIR light irradiation. ACS Catal. 2015, 5, 4790–4795. [Google Scholar] [CrossRef]
- Parizot, L.; Chave, T.; Galvez, M.-E.; Dutilleul, H.; Da Costa, P.; Nikitenko, S.I. Sonocatalytic oxidation of EDTA in aqueous solutions over noble metal-free Co3O4/TiO2 catalyst. Appl. Catal. B Environ. 2019, 241, 570–577. [Google Scholar] [CrossRef]
- Babay, P.A.; Emilio, C.A.; Ferreyra, R.E.; Gautier, E.A.; Gettar, R.T.; Litter, M.I. Kinetics and mechanisms of EDTA photocatalytic degradation with TiO2 under different experimental conditions. Int. J. Photoenergy 2001, 3, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Chen, J.; Yin, Z.; Sheng, W.; Lin, F.; Xu, H.; Cao, S. Complete removal of phenolic contaminants from bismuth-modified TiO2 single-crystal photocatalysts. Chin. J. Catal. 2021, 42, 347–355. [Google Scholar] [CrossRef]
- Wang, F.; Feng, Y.; Chen, P.; Wang, Y.; Su, Y.; Zhang, Q.; Zeng, Y.; Xie, Z.; Liu, H.; Liu, Y.; et al. Photocatalytic degradation of fluoroquinolone antibiotics using ordered mesoporous g-C3N4 under simulated sunlight irradiation: Kinetics, mechanism, and antibacterial activity elimination. Appl. Catal. B Environ. 2018, 227, 114–122. [Google Scholar] [CrossRef]
- Liu, S.-H.; Tang, W.-T.; Chou, P.-H. Microwave-assisted synthesis of triple 2D g-C3N4/Bi2WO6/rGO composites for ibuprofen photodegradation: Kinetics, mechanism and toxicity evaluation of degradation products. Chem. Eng. J. 2020, 387, 124098. [Google Scholar] [CrossRef]
- O’Sullivan, D.W.; Tyree, M. The kinetics of complex formation between Ti(IV) and hydrogen peroxide. Int. J. Chem. Kinet. 2007, 39, 457–461. [Google Scholar] [CrossRef]
- Setiawan, D.; Kazaryan, A.; Martoprawiro, M.; Filatov, M. A first principles study of fluorescence quenching in rhodamine B dimers: How can quenching occur in dimeric species? Phys. Chem. Chem. Phys. 2010, 12, 11238–11244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiha, M.; Merouani, S.; Hamdaoui, O.; Baup, S.; Gondrexon, N.; Pétrier, C. Modeling of ultrasonic degradation of non-volatile organic compounds by Langmuir-type kinetics. Ultrason. Sonochem. 2010, 17, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Akbal, F. Photocatalytic degradation of organic dyes in the presence of titanium dioxide under UV and solar light: Effect of operational parameters. Environ. Prog. 2005, 24, 317–322. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, L.; Li, C.; Yu, J.; Jiao, F. Synthesis, characterization and enhanced visible light photocatalytic activity of Bi2WO6/Ni–Al layered double hydroxide composites. J. Mater. Sci. Mater. Electron. 2018, 29, 14008–14021. [Google Scholar] [CrossRef]
- Kratochvil, B.; White, M.C. Spectrophotometric determination of microgram quantities of (ethylenedinitrilo)tetraacetic acid with bis(2/4,6-tripyridyl-s-triazine)iron(ll). UTC, 1962. Anal. Chem. 1965, 37, 111–113. Available online: https://pubs.acs.org/sharingguidelines (accessed on 29 July 2021). [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | mol.% | ||
|---|---|---|---|
| Ti | Ti3O | TiO2 | |
| Air passivated Ti0 | 86 | 14 | - |
| Ti@TiO2 | 62 | 3 | 35 |
| Temperature (°C) | [Ti] (mg·L−1) | %Ti (± 5%) |
|---|---|---|
| 40 | 0.26 | 0.27 |
| 60 | 0.32 | 0.33 |
| 80 | 0.4 | 0.41 |
| Process | Catalyst | M (μmol) | P (kJ) | m (g) | Y (μmol·kJ−1·g−1) |
|---|---|---|---|---|---|
| Photocatalysis | Ti@TiO2 | 60 | 118 | 7.8 × 10−3 | 65 |
| Ti0 | <1 | 118 | 7.8 × 10−3 | <1 | |
| Sonocatalysis [17] | Ti@TiO2 | 354 | 720 | 0.4 | 1.5 |
| Ti0 | 780 | 720 | 0.4 | 2.7 |
| Ar 20%O2 | Ar | Air | |
|---|---|---|---|
| RRhB (µmol−1 L−1 min−1) | 16 ± 1.3 | 10 ± 0.3 | 12 ± 0.6 |
| REDTA (µmol−1 L−1 min−1) | 6.42 ± 0.43 | 4 ± 0.03 | Not calculated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Hakim, S.; Chave, T.; Nikitenko, S.I. Photocatalytic and Sonocatalytic Degradation of EDTA and Rhodamine B over Ti0 and Ti@TiO2 Nanoparticles. Catalysts 2021, 11, 928. https://doi.org/10.3390/catal11080928
El Hakim S, Chave T, Nikitenko SI. Photocatalytic and Sonocatalytic Degradation of EDTA and Rhodamine B over Ti0 and Ti@TiO2 Nanoparticles. Catalysts. 2021; 11(8):928. https://doi.org/10.3390/catal11080928
Chicago/Turabian StyleEl Hakim, Sara, Tony Chave, and Sergey I. Nikitenko. 2021. "Photocatalytic and Sonocatalytic Degradation of EDTA and Rhodamine B over Ti0 and Ti@TiO2 Nanoparticles" Catalysts 11, no. 8: 928. https://doi.org/10.3390/catal11080928