
The Amino Acids Motif -32GSSYN36- in the Catalytic Domain of E. coli Flavorubredoxin NO Reductase Is Essential for Its Activity

 , , and
, , and
Abstract
:1. Introduction
2. Results
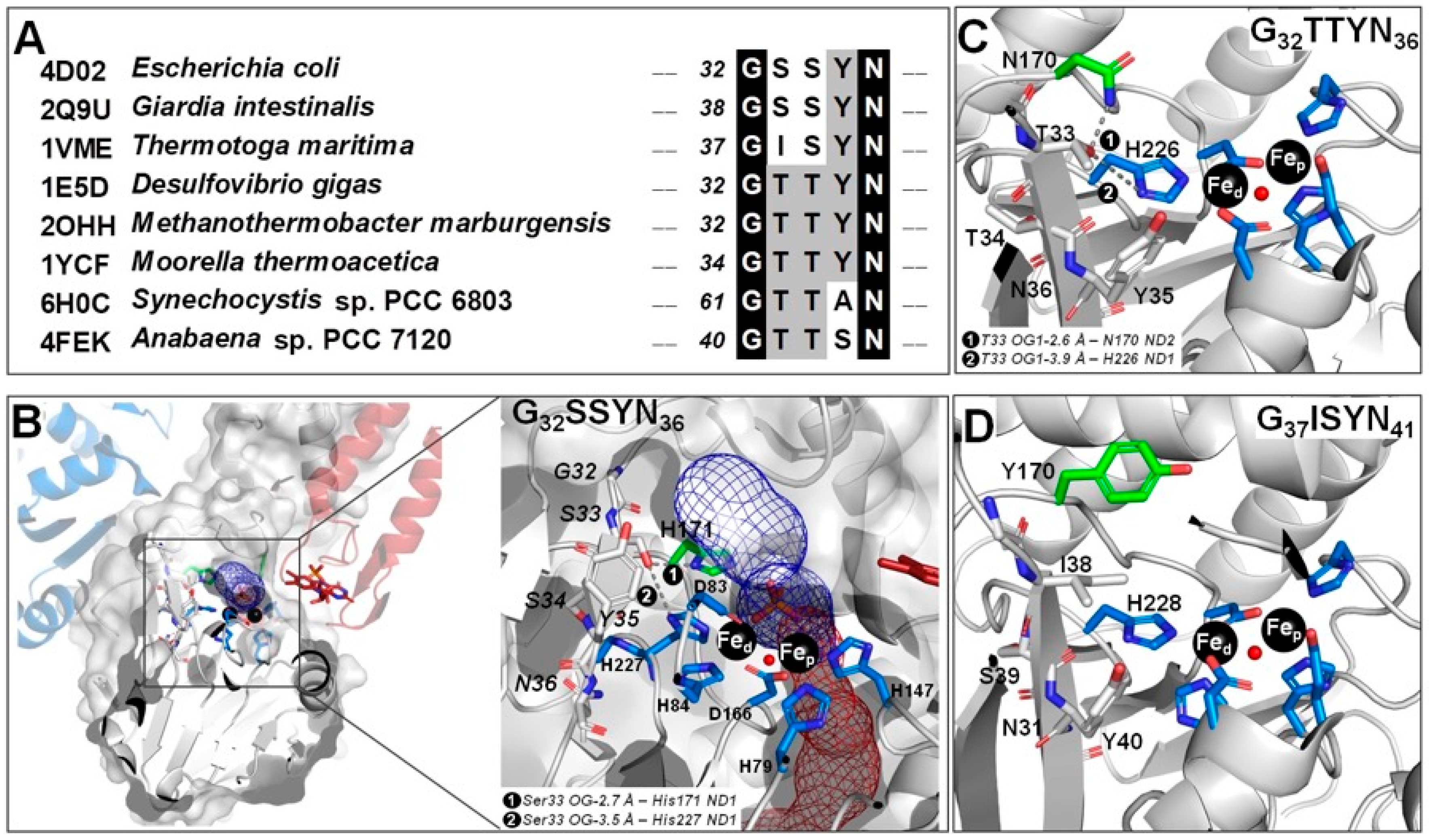
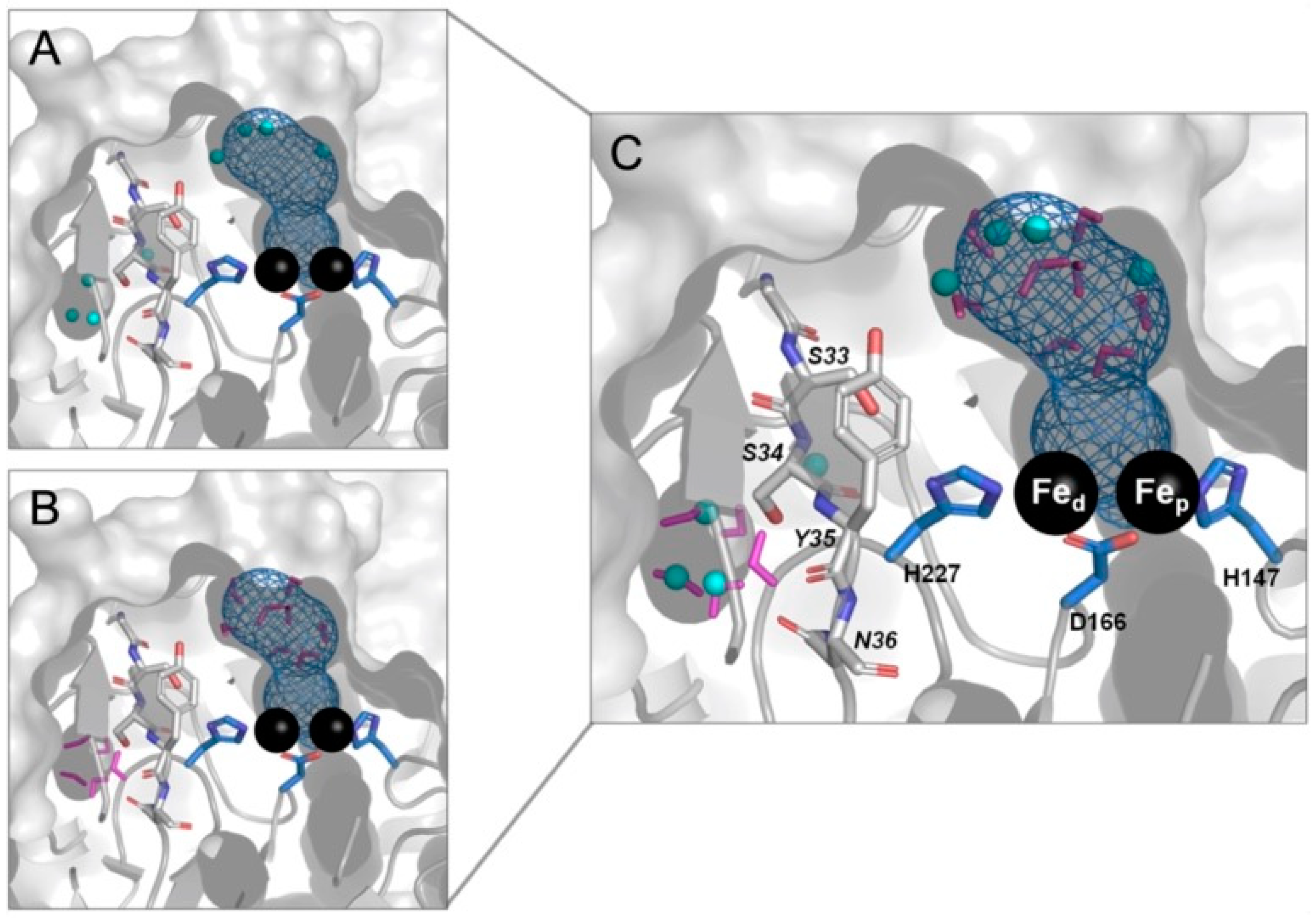
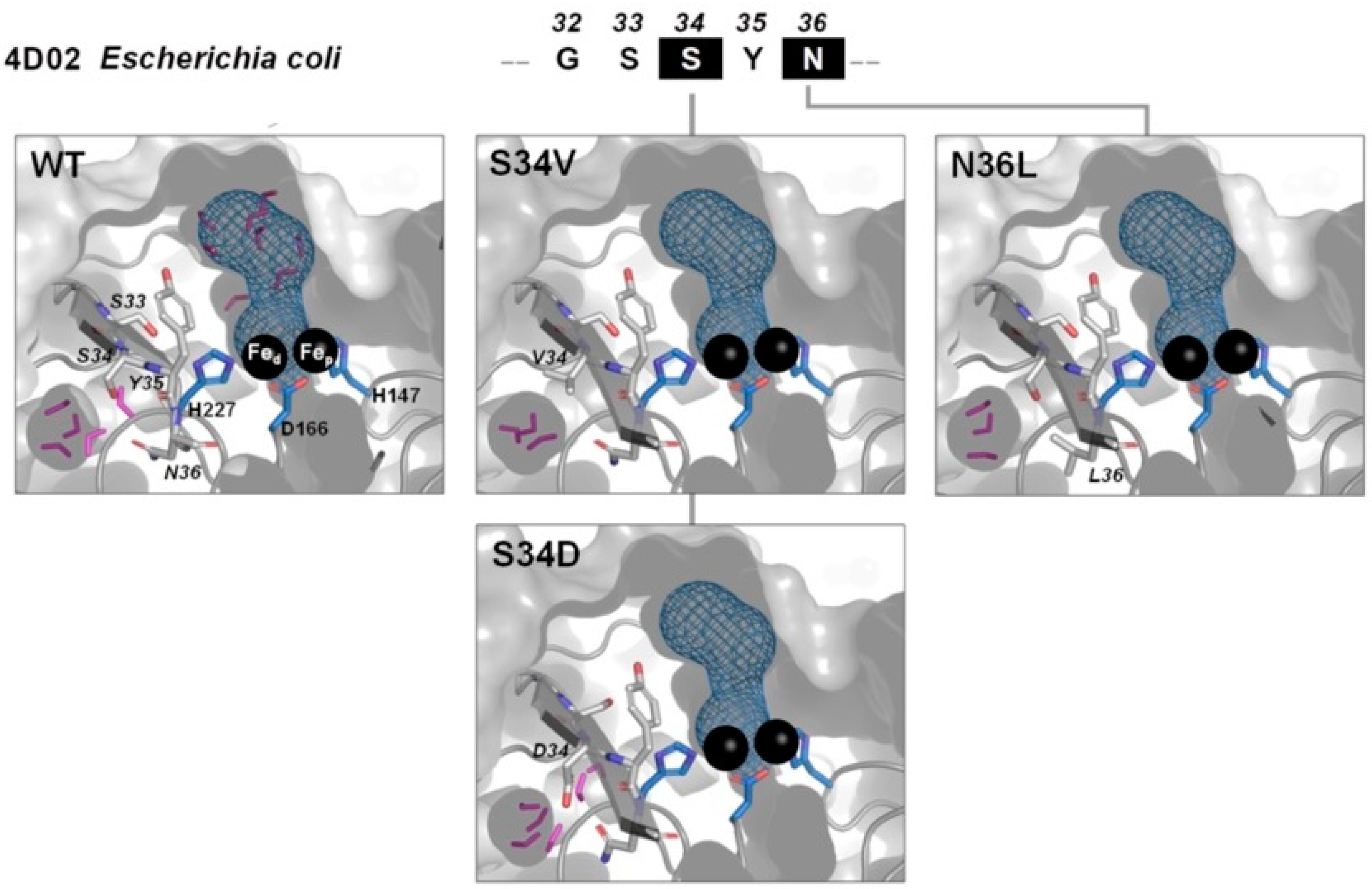
2.1. The -G[S,T]SYN- Motif
2.2. Proteins Characterization
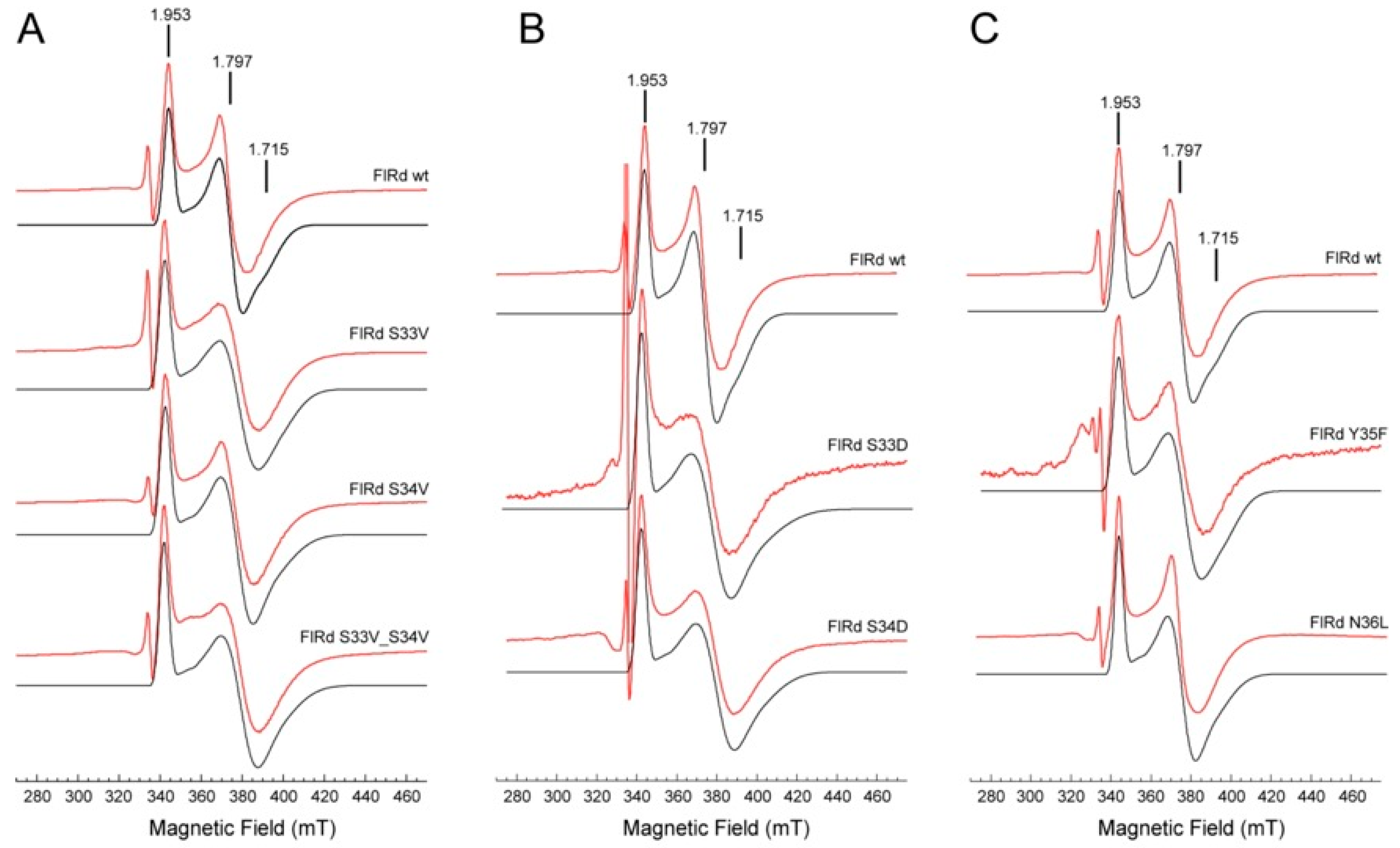
2.3. Spectroscopic Characterization
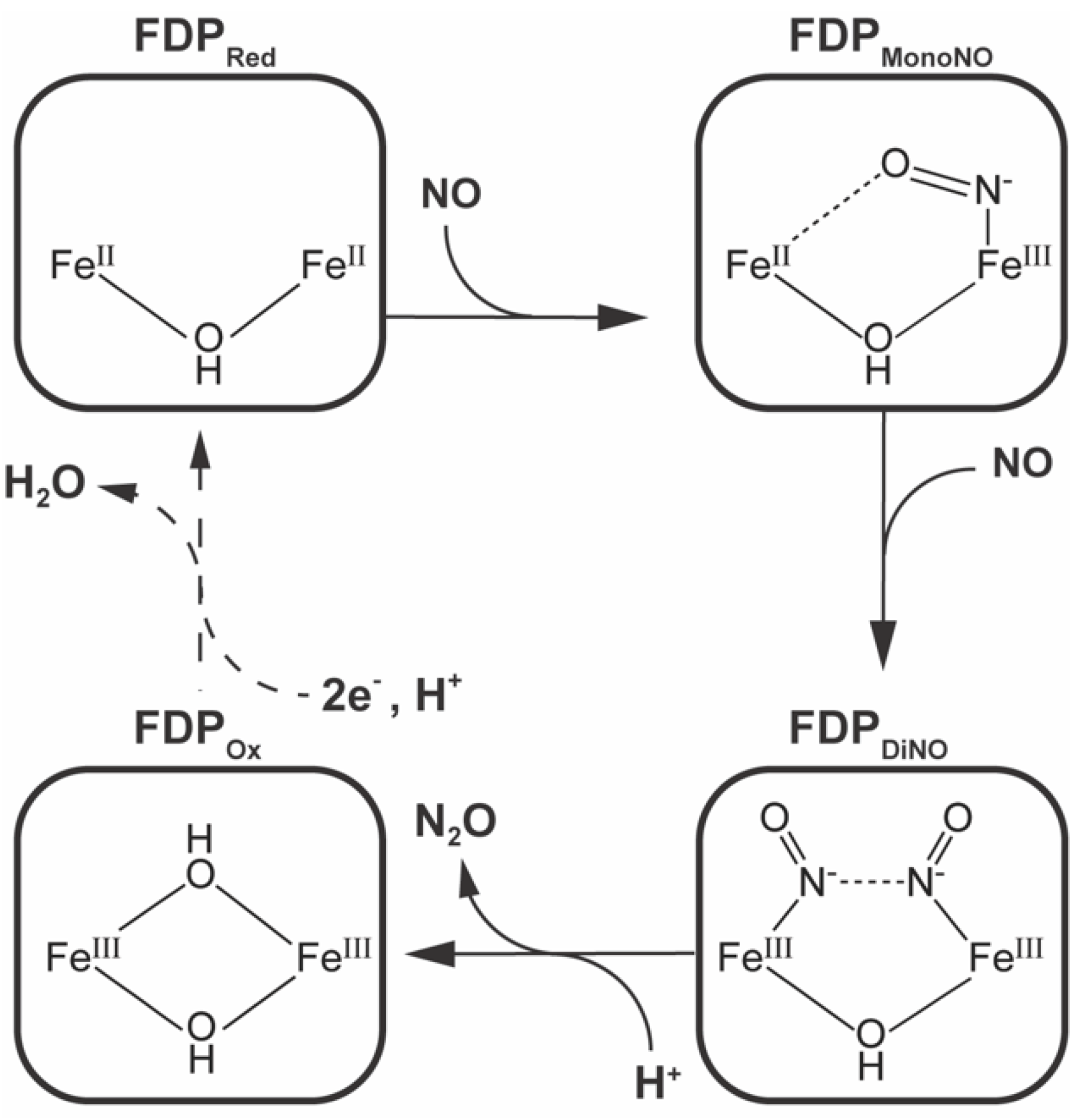
2.4. Kinetics
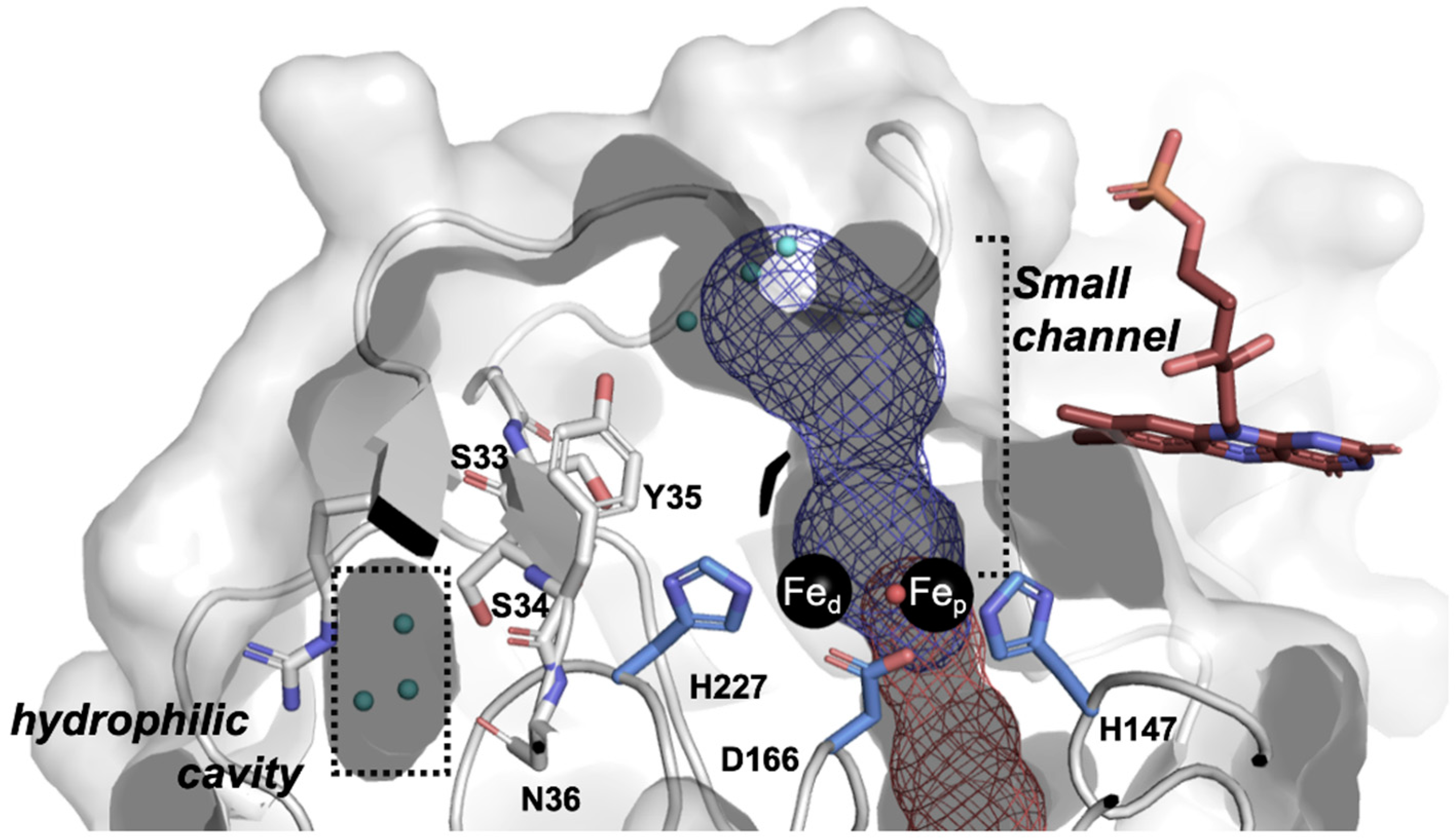
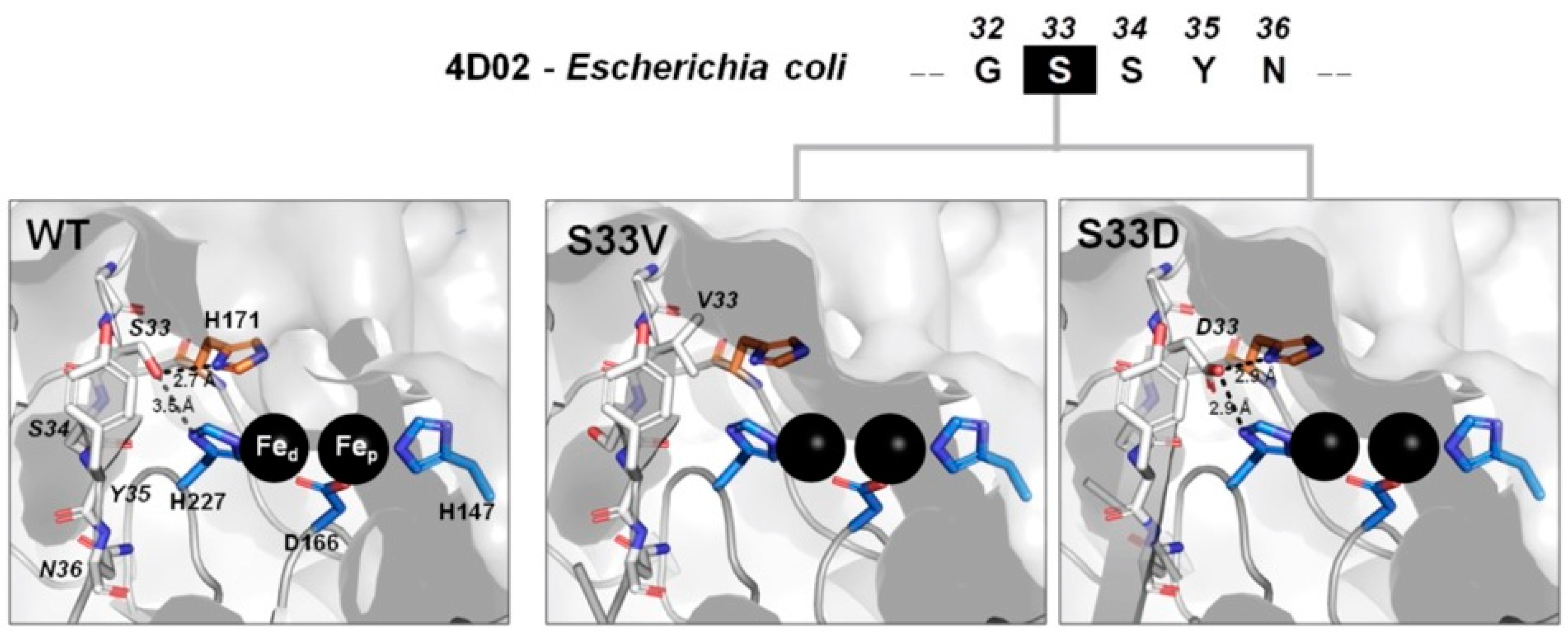
2.5. Molecular Modeling
3. Materials and Methods
3.1. Protein Expression and Purification
3.2. Protein, Metal and Flavin Quantification
3.3. Spectroscopic Methods
3.4. Protein Quaternary Structure Determination
3.5. Amperometric Measurements of O2 and NO Reductase Activities
3.6. Thermal Stability
3.7. Homology-Based Modeling and Prediction of Water Molecules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ignarro, L.; Freeman, B. (Eds.) Nitric Oxide, 3rd ed.; Academic Press: London, UK, 2017; ISBN 9780128042731. [Google Scholar]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.S.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Imlay, J.A. When anaerobes encounter oxygen: Mechanisms of oxygen toxicity, tolerance and defence. Nat. Rev. Microbiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Morvan, C.; Folgosa, F.; Kint, N.; Teixeira, M.; Martin-Verstraete, I. Responses of Clostridia to oxygen: From detoxification to adaptive strategies. Environ. Microbiol. 2021. [Google Scholar] [CrossRef]
- Romão, C.V.; Vicente, J.B.; Borges, P.T.; Frazão, C.; Teixeira, M. The dual function of flavodiiron proteins: Oxygen and/or nitric oxide reductases. J. Biol. Inorg. Chem. 2016, 21, 39–52. [Google Scholar] [CrossRef]
- Martins, M.C.; Romão, C.V.; Folgosa, F.; Borges, P.T.; Frazão, C.; Teixeira, M. How superoxide reductases and flavodiiron proteins combat oxidative stress in anaerobes. Free Radic. Biol. Med. 2019, 140, 36–60. [Google Scholar] [CrossRef]
- Chen, L.; Liu, M.Y.; LeGall, J.; Fareleira, P.; Santos, H.; Xavier, A.V. Rubredoxin Oxidase, a New Flavo-Hemo-Protein, Is the Site of Oxygen Reduction to Water by the “Strict Anaerobe” Desulfovibrio gigas. Biochem. Biophys. Res. Commun. 1993, 193, 100–105. [Google Scholar] [CrossRef]
- Gardner, A.M.; Helmick, R.A.; Gardner, P.R. Flavorubredoxin, an inducible catalyst for nitric oxide reduction and detoxification in Escherichia coli. J. Biol. Chem. 2002, 277, 8172–8177. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.M.; Gardner, P.R. Flavohemoglobin Detoxifies Nitric Oxide in Aerobic, but Not Anaerobic, Escherichia coli. J. Biol. Chem. 2002, 277, 8166–8171. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.M.; Giuffrè, A.; Forte, E.; Vicente, J.B.; Saraiva, L.M.; Brunori, M.; Teixeira, M. A Novel Type of Nitric-oxide Reductase. J. Biol. Chem. 2002, 277, 25273–25276. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, R.; Vicente, J.B.; Félix, R.; Oliveira, S.; Teixeira, M.; Rodrigues-Pousada, C. Desulfovibrio gigas flavodiiron protein affords protection against nitrosative stress in vivo. J. Bacteriol. 2006, 8, 2745–2751. [Google Scholar] [CrossRef] [Green Version]
- Silaghi-Dumitrescu, R.; Coulter, E.D.; Das, A.; Ljungdahl, L.G.; Jameson, G.N.L.; Huynh, B.H.; Kurtz, D.M. A flavodiiron protein and high molecular weight rubredoxin from Moorella thermoacetica with nitric oxide reductase activity. Biochemistry 2003, 42, 2806–2815. [Google Scholar] [CrossRef]
- Justino, M.C.; Vicente, J.B.; Teixeira, M.; Saraiva, L.M. New Genes Implicated in the Protection of Anaerobically Grown Escherichia coli against Nitric Oxide. J. Biol. Chem. 2005, 280, 2636–2643. [Google Scholar] [CrossRef] [Green Version]
- Mehta, H.H.; Liu, Y.; Zhang, M.Q.; Spiro, S. Genome-wide analysis of the response to nitric oxide in uropathogenic Escherichia coli CFT073. Microb. Genomics 2015, 1, e000031. [Google Scholar] [CrossRef] [Green Version]
- Hutchings, M.I.; Mandhana, N.; Spiro, S. The NorR Protein of Escherichia coli Activates Expression of the Flavorubredoxin Gene norV in Response to Reactive Nitrogen Species. J. Bacteriol. 2002, 184, 4640–4643. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, P.; Zheng, M.; Bedzyk, L.A.; LaRossa, R.A.; Storz, G. Prominent roles of the NorR and Fur regulators in the Escherichia coli transcriptional response to reactive nitrogen species. Proc. Natl. Acad. Sci. USA 2004, 101, 745–750. [Google Scholar] [CrossRef] [Green Version]
- Frazão, C.; Silva, G.; Gomes, C.M.; Matias, P.; Coelho, R.; Sieker, L.; Macedo, S.; Liu, M.Y.; Oliveira, S.; Teixeira, M.; et al. Structure of a dioxygen reduction enzyme from Desulfovibrio gigas. Nat. Struct. Biol. 2000, 7, 1041–1045. [Google Scholar] [CrossRef]
- Silaghi-Dumitrescu, R.; Kurtz Donald, M.; Ljungdahl, L.G.; Lanzilotta, W.N. X-ray Crystal Structures of Moorella thermoacetica FprA. Novel Diiron Site Structure and Mechanistic Insights into a Scavenging Nitric Oxide Reductase. Biochemistry 2005, 44, 6492–6501. [Google Scholar] [CrossRef]
- Romão, C.V.; Vicente, J.B.; Borges, P.T.; Victor, B.L.; Lamosa, P.; Silva, E.; Pereira, L.; Bandeiras, T.M.; Soares, C.M.; Carrondo, M.A.; et al. Structure of Escherichia coli Flavodiiron Nitric Oxide Reductase. J. Mol. Biol. 2016, 428, 4686–4707. [Google Scholar] [CrossRef]
- Di Matteo, A.; Scandurra, F.M.; Testa, F.; Forte, E.; Sarti, P.; Brunori, M.; Giuffrè, A. The O2-scavenging Flavodiiron Protein in the Human Parasite Giardia intestinalis. J. Biol. Chem. 2008, 283, 4061–4068. [Google Scholar] [CrossRef] [Green Version]
- Folgosa, F.; Martins, M.C.; Teixeira, M. Diversity and complexity of flavodiiron NO/O2 reductases. FEMS Microbiol. Lett. 2018, 365, fnx267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, C.M.; Vicente, J.B.; Wasserfallen, A.; Teixeira, M. Spectroscopic studies and characterization of a novel electron-transfer chain from Escherichia coli involving a flavorubredoxin and its flavoprotein reductase partner. Biochemistry 2000, 39, 16230–16237. [Google Scholar] [CrossRef]
- Wasserfallen, A.; Ragettli, S.; Jouanneau, Y.; Leisinger, T. A family of flavoproteins in the domains Archaea and Bacteria. Eur. J. Biochem. 1998, 254, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.B.; Carrondo, M.A.; Teixeira, M.; Frazão, C. Flavodiiron Proteins: Nitric Oxide and/or Oxygen Reductases. In Handbook of Metalloproteins; Major Reference Works; John Wiley & Sons, Ltd.: Chichester, UK, 2007; ISBN 9780470028636. [Google Scholar]
- Solomon, E.I.; Brunold, T.C.; Davis, M.I.; Kemsley, J.N.; Lee, S.K.; Lehnert, N.; Neese, F.; Skulan, A.J.; Yang, Y.S.; Zhou, J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 2000, 100, 235–350. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.M.; Teixeira, M. The Family of A-type Flavoproteins: New Members and Definition of Unique Sequence Fingerprints. In Flavins and Flavoproteins; Ghisla, S., Kroneck, P.M.H., Macheroux, P., Sund, H., Eds.; Weber: Berlin, Germany, 1999; pp. 219–222. ISBN 3000051287. [Google Scholar]
- Pravda, L.; Sehnal, D.; Toušek, D.; Navrátilová, V.; Bazgier, V.; Berka, K.; Svobodová Vařeková, R.; Koča, J.; Otyepka, M. MOLEonline: A web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res. 2018, 46, W368–W373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petasis, D.T.; Hendrich, M.P. Quantitative Interpretation of Multifrequency Multimode EPR Spectra of Metal Containing Proteins, Enzymes, and Biomimetic Complexes. Methods Enzymol. 2015, 563, 171–208. [Google Scholar] [PubMed] [Green Version]
- Marritt, S.J.; Lowe, T.G.; Bye, J.; McMillan, D.G.G.; Shi, L.; Fredrickson, J.; Zachara, J.; Richardson, D.J.; Cheesman, M.R.; Jeuken, L.J.C.; et al. A functional description of CymA, an electron-transfer hub supporting anaerobic respiratory flexibility in Shewanella. Biochem. J. 2012, 444, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Vicente, J.B.; Teixeira, M. Redox and Spectroscopic Properties of the Escherichia coli Nitric Oxide-detoxifying System Involving Flavorubredoxin and Its NADH-oxidizing Redox Partner. J. Biol. Chem. 2005, 280, 34599–34608. [Google Scholar] [CrossRef] [Green Version]
- Morozenko, A.; Stuchebrukhov, A.A. Dowser++, a new method of hydrating protein structures. Proteins Struct. Funct. Bioinform. 2016, 84, 1347–1357. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, O.; Kalimeri, M.; Melchionna, S.; Hénin, J.; Sterpone, F. Role of Internal Water on Protein Thermal Stability: The Case of Homologous G Domains. J. Phys. Chem. B 2015, 119, 8939–8949. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Taly, A.; Sterpone, F. Stay Wet, Stay Stable? How Internal Water Helps the Stability of Thermophilic Proteins. J. Phys. Chem. B 2015, 119, 12760–12770. [Google Scholar] [CrossRef] [Green Version]
- Folgosa, F.; Martins, M.C.; Teixeira, M. The multidomain flavodiiron protein from Clostridium difficile 630 is an NADH:oxygen oxidoreductase. Sci. Rep. 2018, 8, 10164. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.; Han, S.; Park, C.-J. Recognition and Unfolding of c-MYC and Telomeric G-Quadruplex DNAs by the RecQ C-Terminal Domain of Human Bloom Syndrome Helicase. ACS Omega 2020, 5, 14513–14522. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Weitz, A.C.; Giri, N.; Frederick, R.E.; Kurtz, D.M.; Bominaar, E.L.; Hendrich, M.P. Spectroscopy and DFT Calculations of Flavo–Diiron Nitric Oxide Reductase Identify Bridging Structures of NO-Coordinated Diiron Intermediates. ACS Catal. 2018, 8, 11704–11715. [Google Scholar] [CrossRef]
- Weitz, A.C.; Giri, N.; Caranto, J.D.; Kurtz, D.M.; Bominaar, E.L.; Hendrich, M.P. Spectroscopy and DFT Calculations of a Flavo-diiron Enzyme Implicate New Diiron Site Structures. J. Am. Chem. Soc. 2017, 139, 12009–12019. [Google Scholar] [CrossRef]
- Hayashi, T.; Caranto, J.D.; Matsumura, H.; Kurtz, D.M.; Moënne-Loccoz, P. Vibrational Analysis of Mononitrosyl Complexes in Hemerythrin and Flavodiiron Proteins: Relevance to Detoxifying NO Reductase. J. Am. Chem. Soc. 2012, 134, 6878–6884. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Caranto, J.D.; Wampler, D.A.; Kurtz, D.M.; Moënne-Loccoz, P. Insights into the Nitric Oxide Reductase Mechanism of Flavodiiron Proteins from a Flavin-Free Enzyme. Biochemistry 2010, 49, 7040–7049. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Iron/Protein | Flavin/Protein | Tm (°C) | g1 | g2 | g3 |
|---|---|---|---|---|---|---|
| WT | 2.3 ± 0.3 | 0.6 ± 0.1 | 53.3 ± 1.5 | 1.953 | 1.797 | 1.715 |
| S33V | 2.3 ± 0.3 | 0.5 ± 0.1 | 53.2 ± 0.8 | 1.961 | 1.780 | 1.685 |
| S33D | 2.3 ± 0.4 | 0.8 ± 0.1 | 50.1 ± 2.4 | 1.960 | 1.787 | 1.686 |
| S34V | 2.0 ± 0.3 | 0.4 ± 0.1 | 42.7 ± 1.2 | 1.961 | 1.779 | 1.692 |
| S34D | 2.1 ± 0.2 | 0.7 ± 0.1 | 57.9 ± 2.8 | 1.965 | 1.774 | 1.678 |
| S33V_S34V | 1.7 ± 0.2 | 0.5 ± 0.1 | 51.7 ± 0.8 | 1.963 | 1.773 | 1.686 |
| Y35F | 1.8 ± 0.2 | 0.5 ± 0.04 | 40.4 ± 1.9 | 1.957 | 1.790 | 1.696 |
| N36L | 2.7 ± 0.1 | 0.7 ± 0.03 | 54.0 ± 1.4 | 1.953 | 1.795 | 1.715 |
| Protein | NO Reductase Activity (s−1) | O2 Reductase Activity (s−1) |
|---|---|---|
| WT | 15.3 ± 5 | 2.2 ± 0.7 |
| S33V | 2.0 ± 0.7 | 2.1 ± 0.7 |
| S33D | 6.5 ± 1 | 2.3 ± 0.4 |
| S34V | 0.3 ± 0.1 | 1.1 ± 0.5 |
| S34D | 7.6 ± 1 | 2.2 ± 0.4 |
| S33V_S34V | 0.2 ± 0.1 | 2.0 ± 0.7 |
| Y35F | 5.1 ± 2 | 1.2 ± 0.4 |
| N36L | 0.14 ± 0.02 | 0.63 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, M.C.; Fernandes, S.F.; Salgueiro, B.A.; Soares, J.C.; Romão, C.V.; Soares, C.M.; Lousa, D.; Folgosa, F.; Teixeira, M. The Amino Acids Motif -32GSSYN36- in the Catalytic Domain of E. coli Flavorubredoxin NO Reductase Is Essential for Its Activity. Catalysts 2021, 11, 926. https://doi.org/10.3390/catal11080926
Martins MC, Fernandes SF, Salgueiro BA, Soares JC, Romão CV, Soares CM, Lousa D, Folgosa F, Teixeira M. The Amino Acids Motif -32GSSYN36- in the Catalytic Domain of E. coli Flavorubredoxin NO Reductase Is Essential for Its Activity. Catalysts. 2021; 11(8):926. https://doi.org/10.3390/catal11080926
Chicago/Turabian StyleMartins, Maria C., Susana F. Fernandes, Bruno A. Salgueiro, Jéssica C. Soares, Célia V. Romão, Cláudio M. Soares, Diana Lousa, Filipe Folgosa, and Miguel Teixeira. 2021. "The Amino Acids Motif -32GSSYN36- in the Catalytic Domain of E. coli Flavorubredoxin NO Reductase Is Essential for Its Activity" Catalysts 11, no. 8: 926. https://doi.org/10.3390/catal11080926