
Electronic and Structural Properties of the Double Cubane Iron-Sulfur Cluster

Abstract
:1. Introduction
2. Discussion
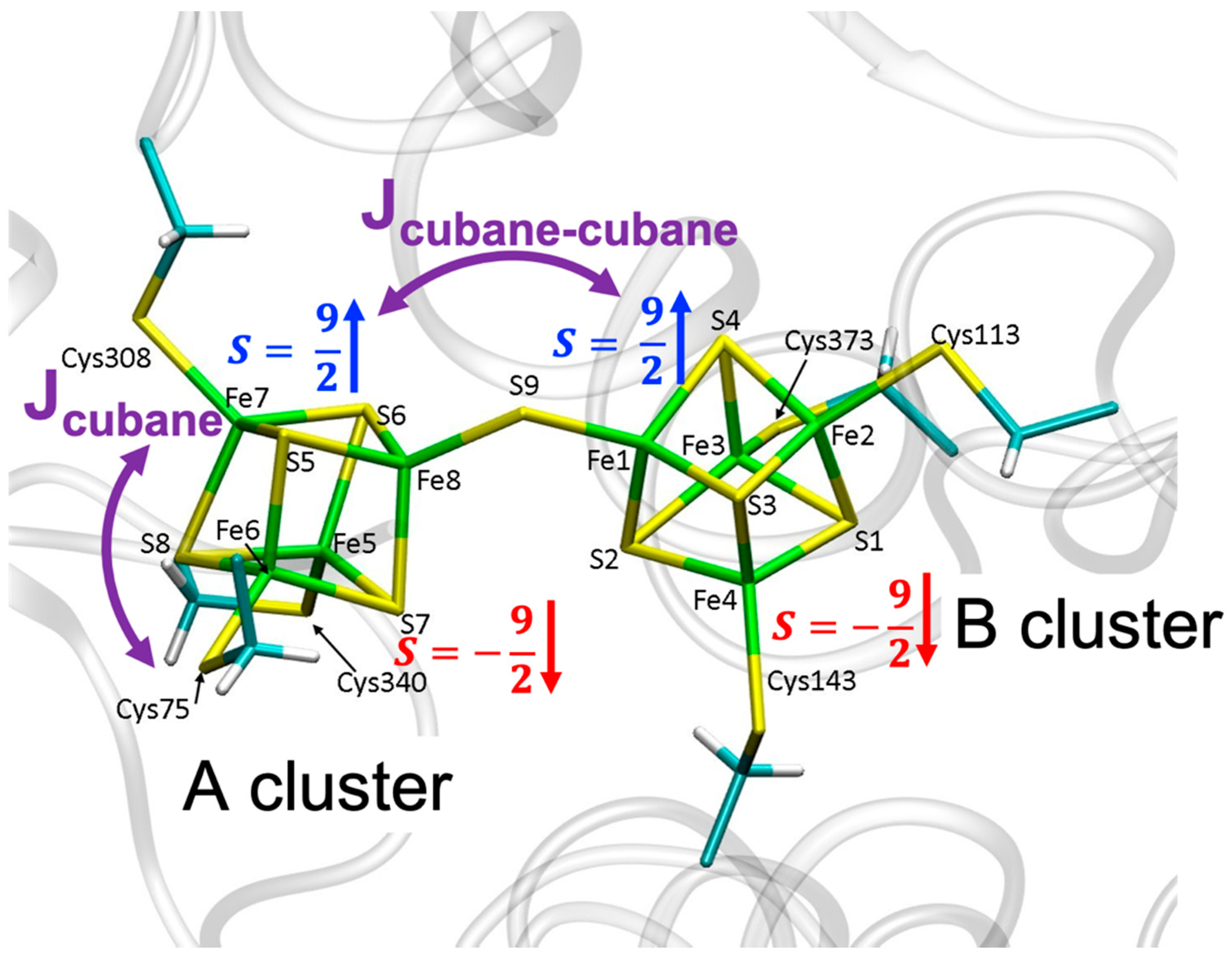
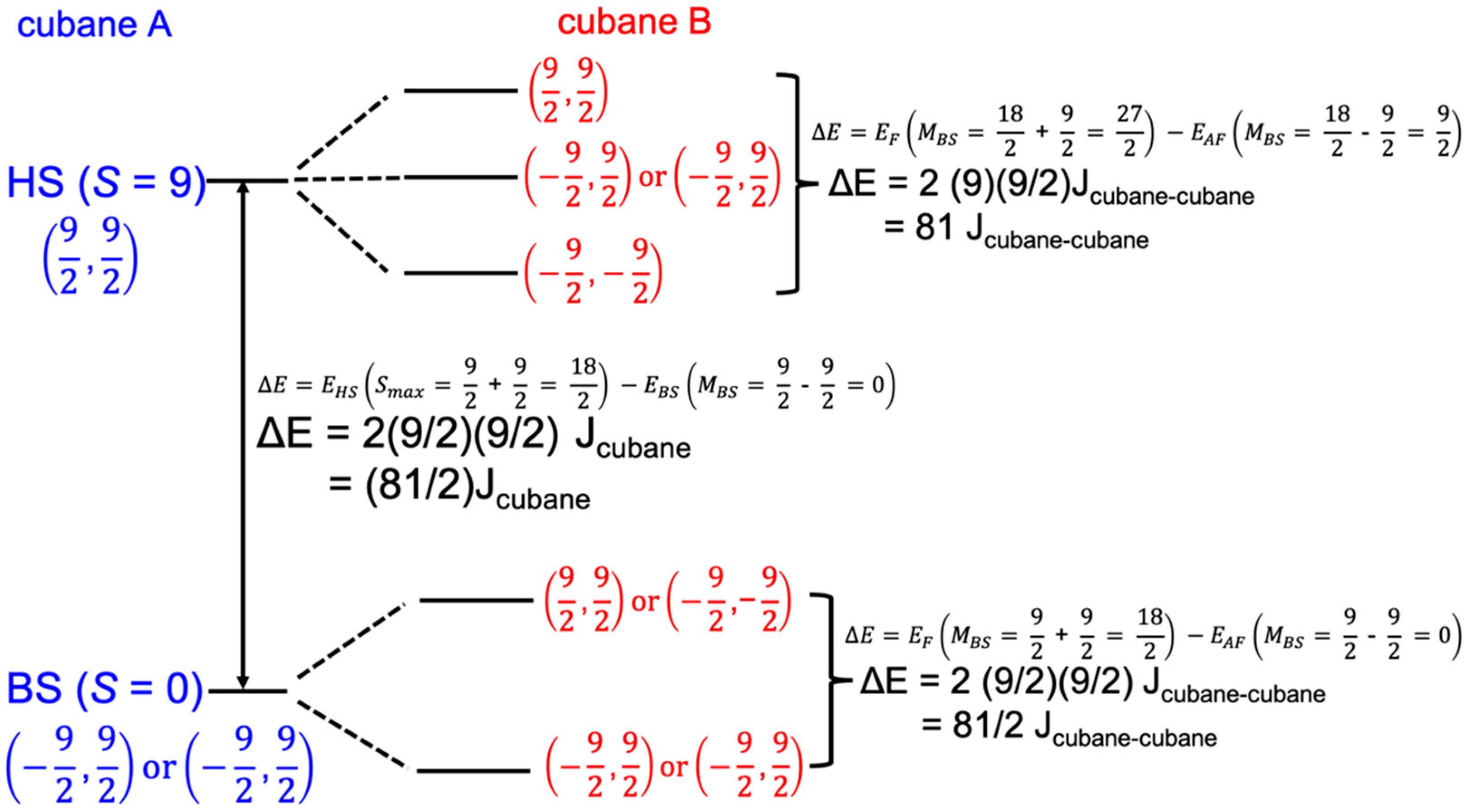
2.1. Exchange Parameters Jcubane and Jcubane−cubane
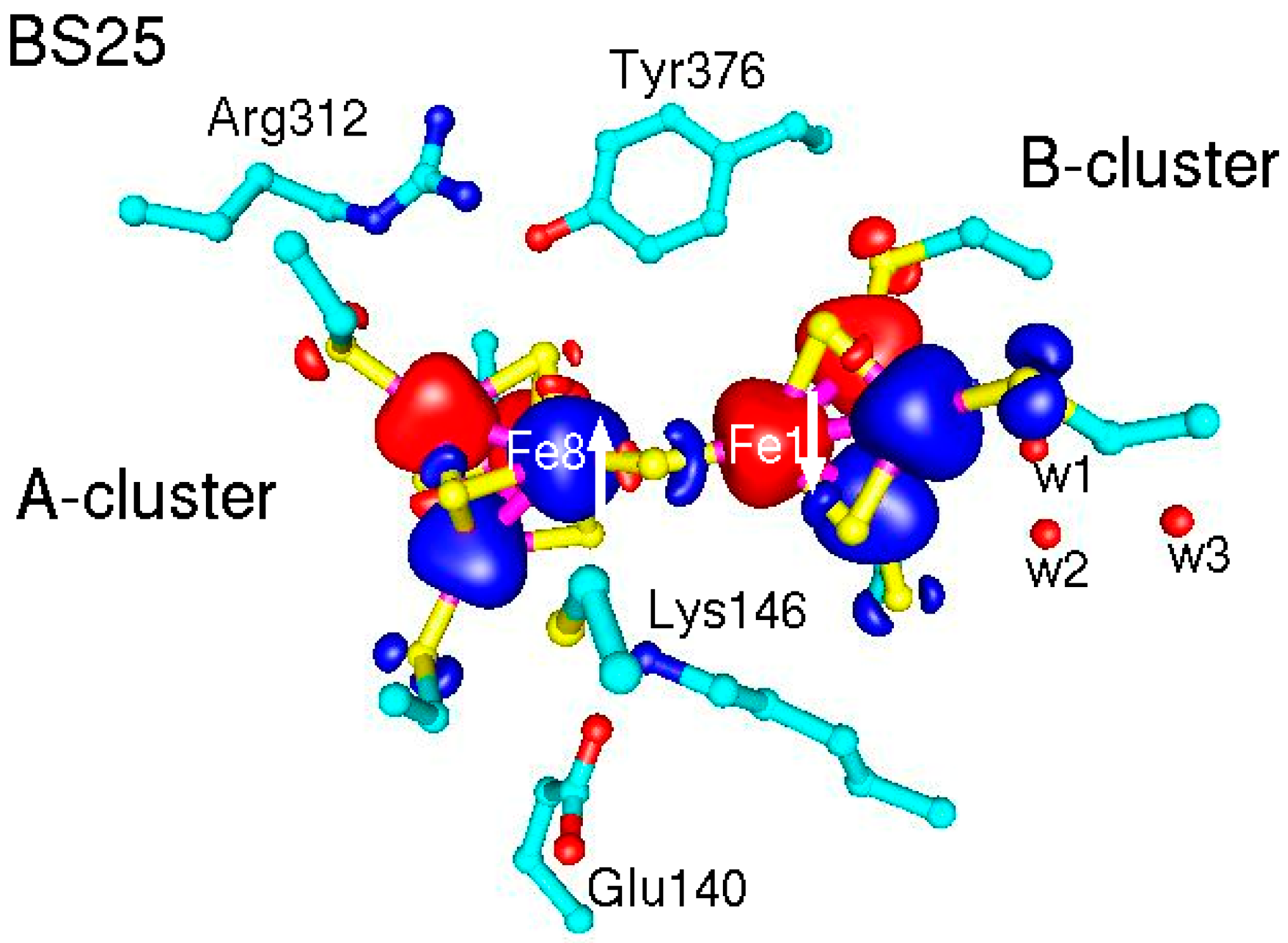
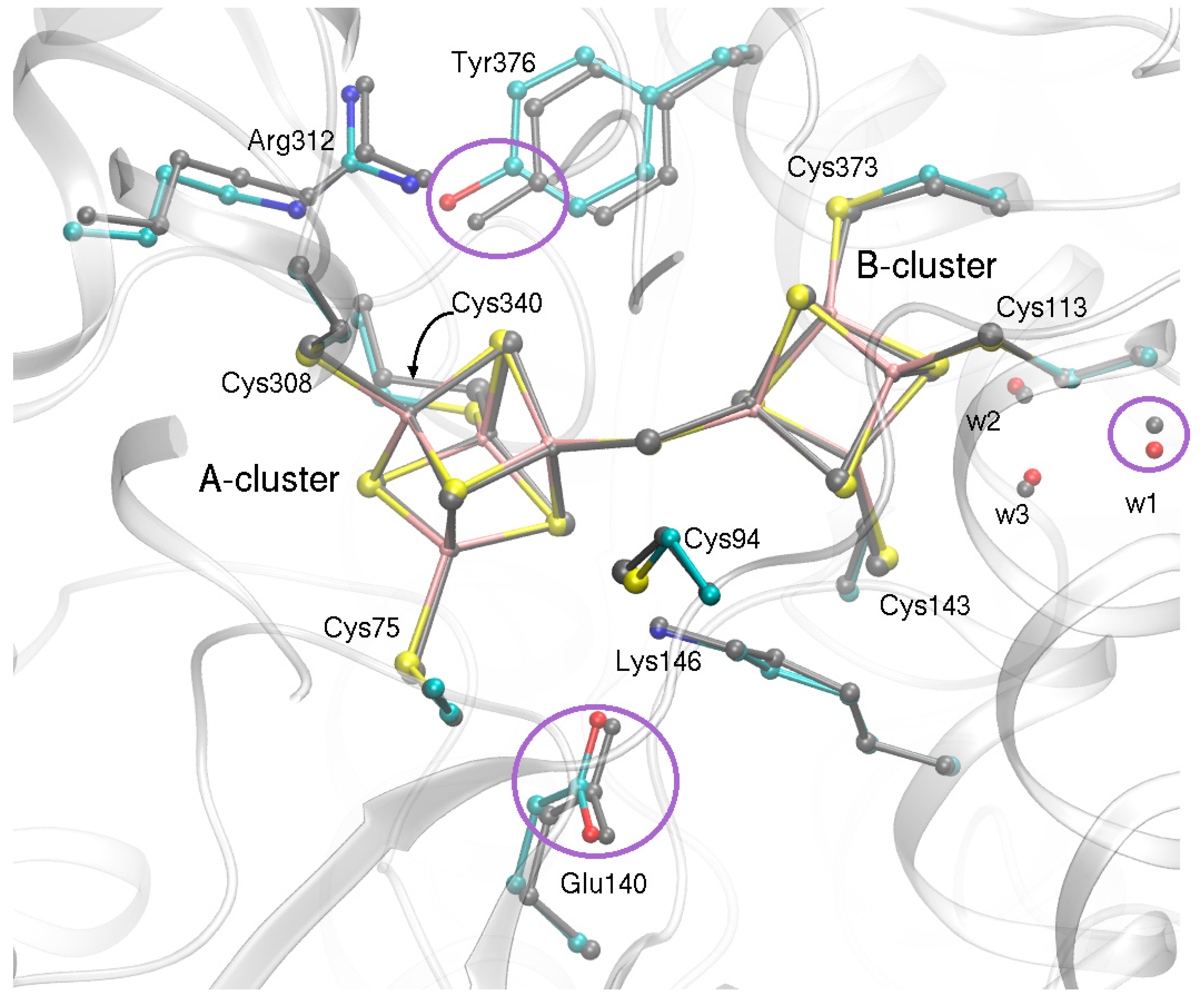
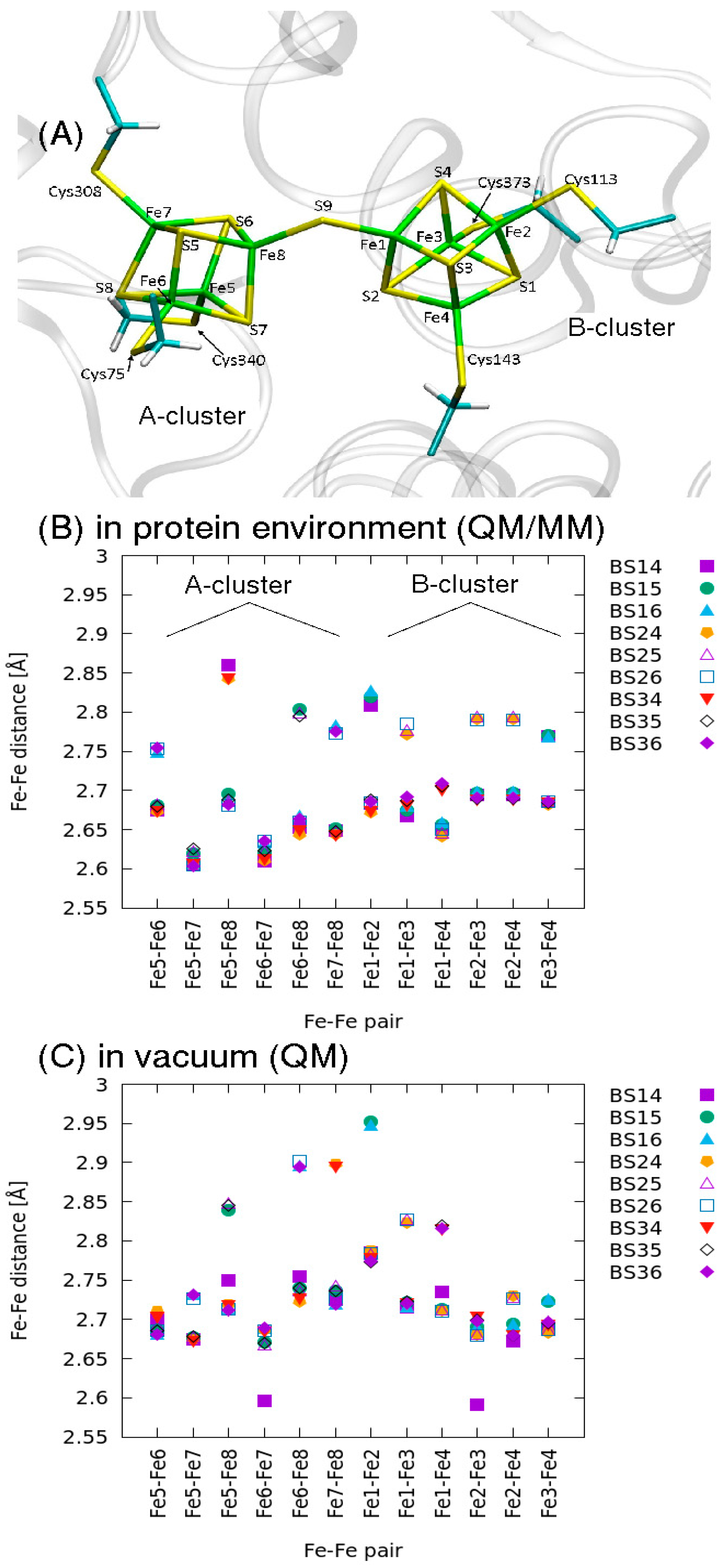
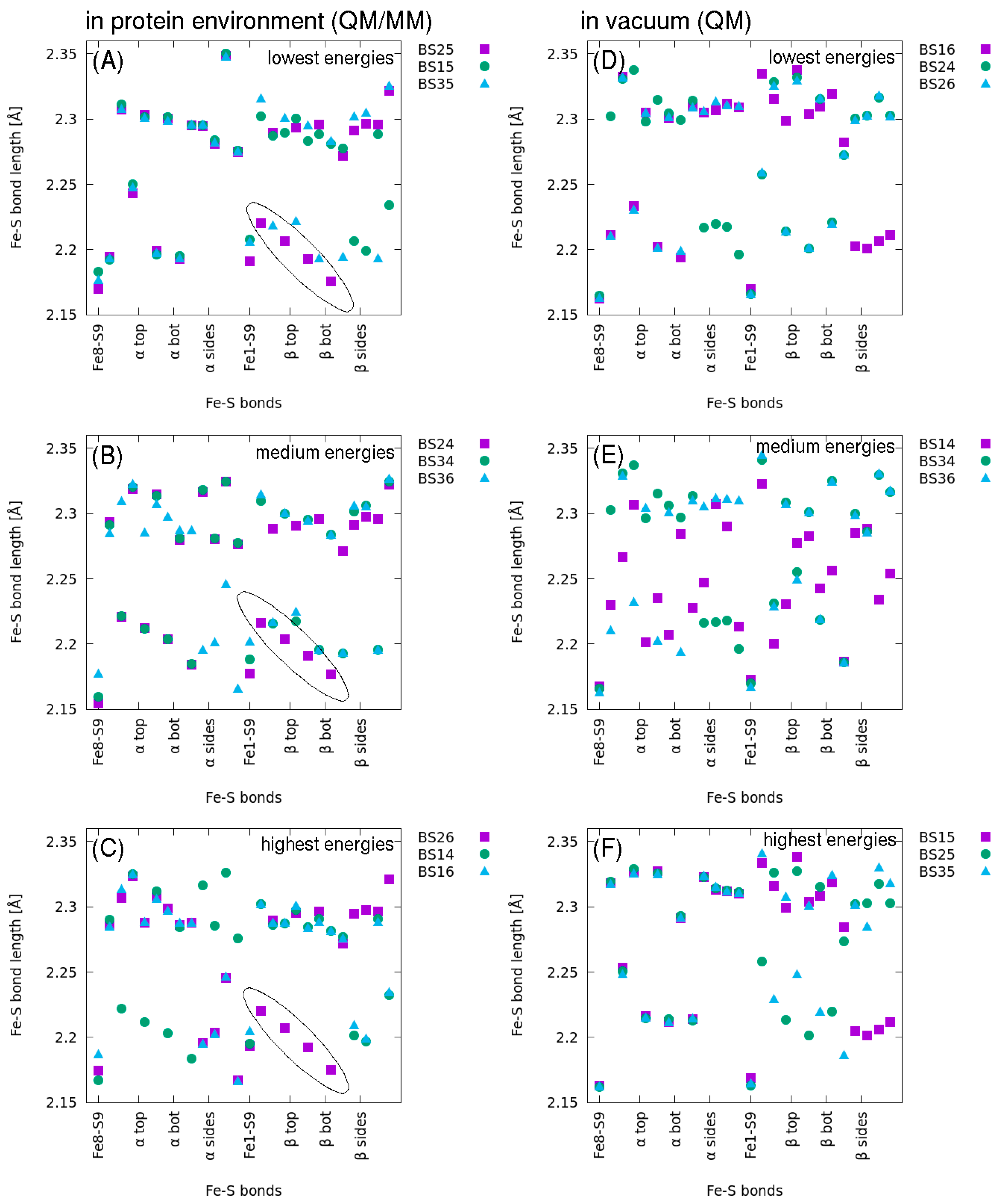
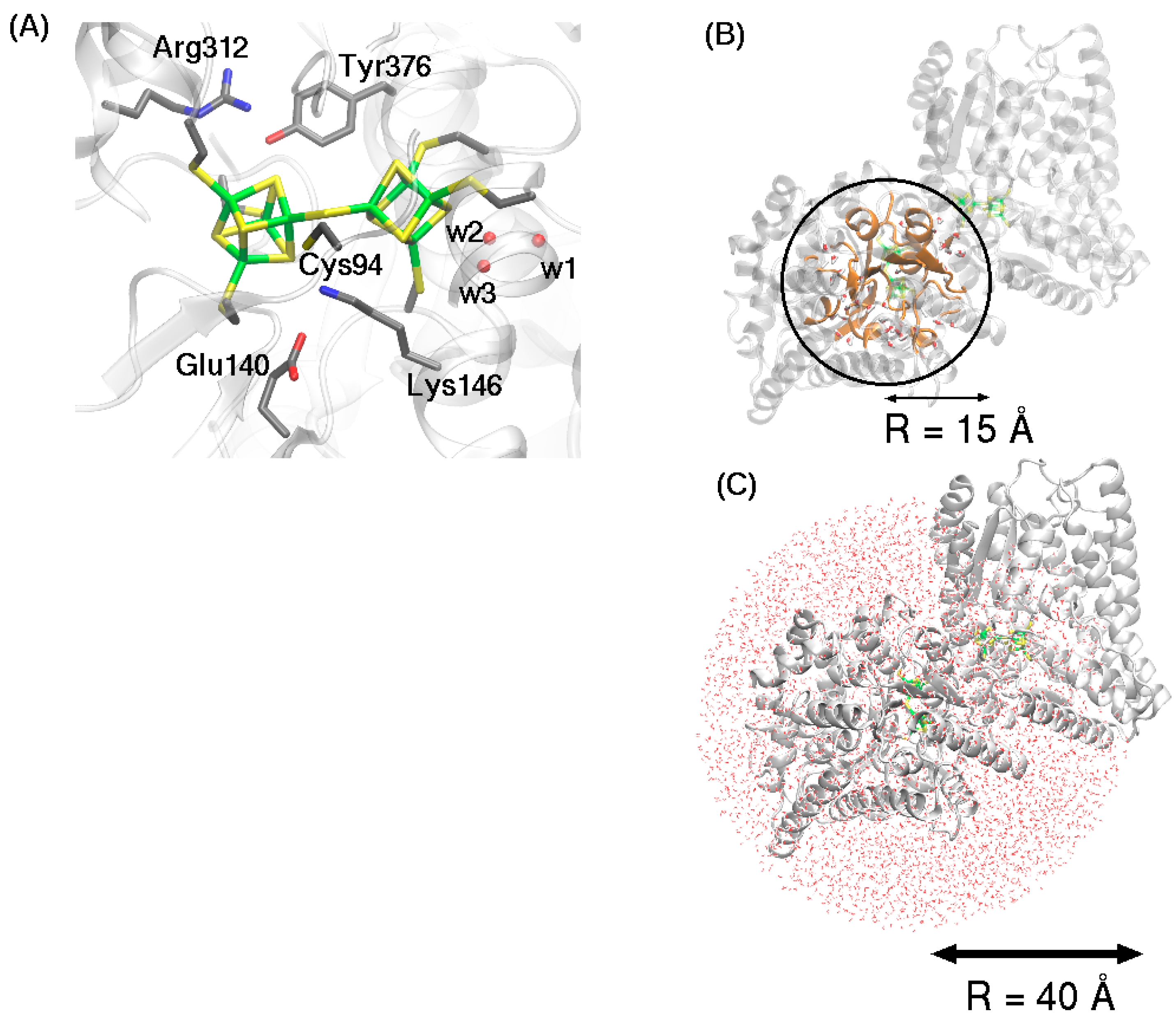
2.2. QM/MM Optimized Geometries
3. Methods
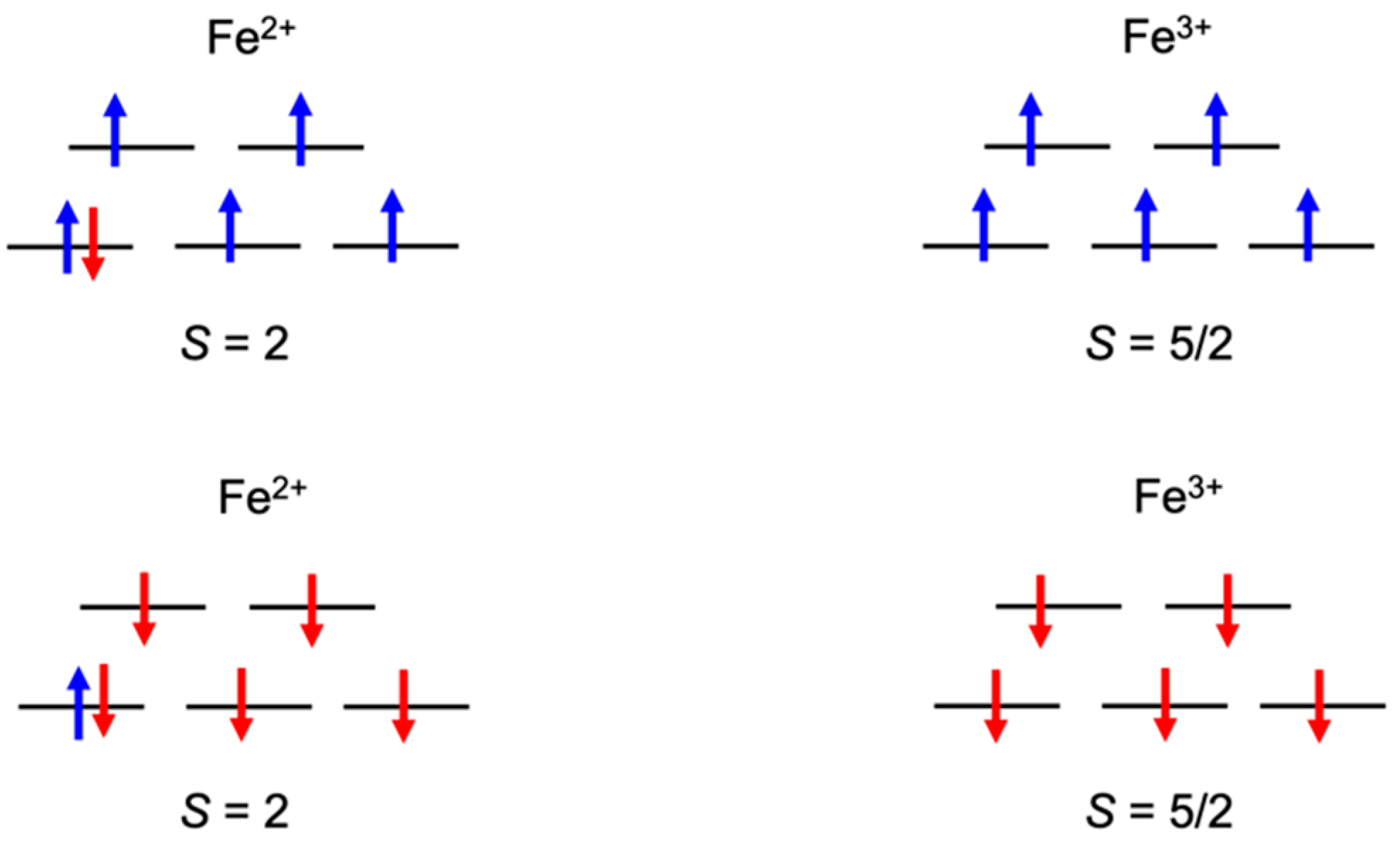
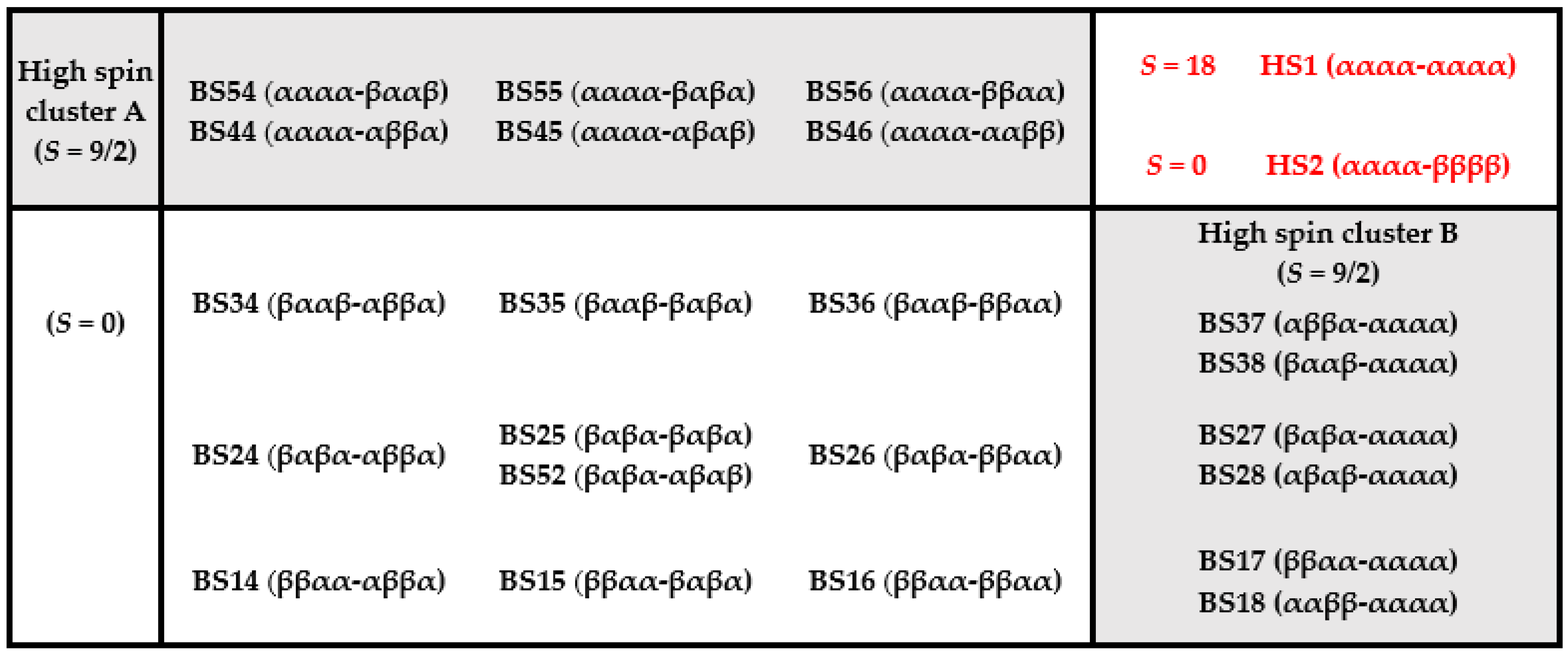
3.1. Broken Symmetry States
3.2. Model for QM Geometry Optimization
3.3. Model for QM/MM Geometry Optimization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeoung, J.-H.; Dobbek, H. ATP-dependent substrate reduction at an [Fe8S9] double-cubane cluster. Proc. Natl. Acad. Sci. USA 2018, 115, 2994–2999. [Google Scholar] [CrossRef] [Green Version]
- Jeoung, J.; Martins, B.M.; Dobbek, H. Double-Cubane [8Fe9S] Clusters: A Novel Nitrogenase-Related Cofactor in Biology. ChemBioChem 2020, 21, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Stack, T.D.; Carney, M.J.; Holm, R.H. Formation of Bridged [4Fe-4S]2+ Double Cubanes by Site-Specific Reactions: Elec-tron-Transfer Coupling Across Sulfur Containing Bridges of Variable Length. J. Am. Chem. Soc. 1989, 111, 1670–1676. [Google Scholar] [CrossRef]
- Terada, T.; Wakimoto, T.; Nakamura, T.; Hirabayashi, K.; Tanaka, K.; Li, J.; Matsumoto, T.; Tatsumi, K.; Matsumoto, T.; Tatsumi, K. Tridentate Thiolate Ligands: Application to the Synthesis of the Site-Differentiated [4Fe-4S] Cluster having a Hydrosulfide Ligand at the Unique Iron Center. Chem. Asian J. 2012, 7, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, D.L.; Coucouvanis, D.; Kampf, J.; Lehnert, N. Isolation and Characterization of Single and Sulfide-Bridged Double [4Fe-4S] Cubane Clusters with 4-Pyridinethiolato Ligands. Eur. J. Inorg. Chem. 2013, 2013, 5253–5264. [Google Scholar] [CrossRef] [Green Version]
- Challen, P.R.; Koo, S.-M.; Dunham, R.; Coucouvanis, D. New µ2-S2-–Coupled, Singly Bridged Double Cubane with the [(Fe4S4Cl3)2S]4- Core. The Stepwise Synthesis and Structural Characterization of (n -Bu4N)2 (Ph4P)2[(Fe4S4C13)2S]. J. Am. Chem. Soc. 1990, 112, 2455–2456. [Google Scholar] [CrossRef]
- Lee, S.C.; Lo, W.; Holm, R.H. Developments in the Biomimetic Chemistry of Cubane-Type and Higher Nuclearity Iron–Sulfur Clusters. Chem. Rev. 2014, 114, 3579–3600. [Google Scholar] [CrossRef] [Green Version]
- Rao, P.V.; Holm, R.H. Synthetic Analogues of the Active Sites of Iron−Sulfur Proteins. Chem. Rev. 2004, 104, 527–560. [Google Scholar] [CrossRef]
- Somasegaran, P.; Hoben, H. Handbook for Rhizobia: Methods in Legume-rhizobium Technology; Springer Lab Manuals Series; Springer: Berlin/Heidelberg, Germany, 1994. [Google Scholar]
- Lee, H.-I.; Sørlie, M.; Christiansen, J.; Song, R.; Dean, D.R.; Hales, B.J.; Hoffman, B.M. Characterization of an Intermediate in the Reduction of Acetylene by the Nitrogenase α-Gln195MoFe Protein by Q-band EPR and13C,1H ENDOR. J. Am. Chem. Soc. 2000, 122, 5582–5587. [Google Scholar] [CrossRef]
- Seefeldt, L.C.; Hoffman, B.; Dean, D.R. Mechanism of Mo-Dependent Nitrogenase. Annu. Rev. Biochem. 2009, 78, 701–722. [Google Scholar] [CrossRef]
- Gao, H.; Azam, T.; Randeniya, S.; Couturier, J.; Rouhier, N.; Johnson, M.K. Function and maturation of the Fe–S center in dihydroxyacid dehydratase fromArabidopsis. J. Biol. Chem. 2018, 293, 4422–4433. [Google Scholar] [CrossRef] [Green Version]
- Katz, S.; Noth, J.; Horch, M.; Shafaat, H.; Happe, T.; Hildebrandt, P.; Zebger, I. Vibrational spectroscopy reveals the initial steps of biological hydrogen evolution. Chem. Sci. 2016, 7, 6746–6752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubitz, W.; Reijerse, E.J.; Van Gastel, M. [NiFe] and [FeFe] Hydrogenases Studied by Advanced Magnetic Resonance Techniques. Chem. Rev. 2007, 107, 4331–4365. [Google Scholar] [CrossRef]
- DeLacey, A.L.; Fernández, V.M.; Rousset, M.; Cammack, R. Activation and inactivation of hydrogenase function and the catalytic cycle: Spectrochemical studies. Chem. Rev. 2007, 107, 4304–4330. [Google Scholar] [CrossRef]
- Spiro, T.G.; Czernuszewicz, R.S. Biochemical Spectroscopy; Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1995; Volume 246, pp. 416–460. [Google Scholar]
- Todorovic, S.; Teixeira, M. Resonance Raman spectroscopy of Fe–S proteins and their redox properties. J. Biol. Inorg. Chem. 2018, 23, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Glaser, T. Mössbauer Spectroscopy and Transition Metal Chemistry. Fundamentals and Applications; Gütlich, P., Bill, E., Trautwein, A.X., Eds.; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Frielingsdorf, S.; Fritsch, J.; Schmidt, A.; Hammer, M.; Löwenstein, J.; Siebert, E.; Pelmenschikov, V.; Jaenicke, T.; Kalms, J.; Rippers, Y.; et al. Reversible [4Fe-3S] cluster morphing in an O2-tolerant [NiFe] hydrogenase. Nat. Chem. Biol. 2014, 10, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Roessler, M.M.; Salvadori, E. Principles and applications of EPR spectroscopy in the chemical sciences. Chem. Soc. Rev. 2018, 47, 2534–2553. [Google Scholar] [CrossRef]
- Noodleman, L. Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys. 1981, 74, 5737–5743. [Google Scholar] [CrossRef]
- Noodleman, L.; Baerends, E.J. Electronic structure, magnetic properties, ESR, and optical spectra for 2-iron ferredoxin models by LCAO-Xα valence bond theory. J. Am. Chem. Soc. 1984, 106, 2316–2327. [Google Scholar] [CrossRef]
- Noodleman, L.; Peng, C.; Case, D.; Mouesca, J.-M. Orbital interactions, electron delocalization and spin coupling in iron-sulfur clusters. Co-ord. Chem. Rev. 1995, 144, 199–244. [Google Scholar] [CrossRef]
- Roos, B.O.; Taylor, P.R.; Sigbahn, P.E.M. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Sharma, S.; Sivalingam, K.; Neese, F.; Chan, G.K.-L. Low-energy spectrum of iron–sulfur clusters directly from many-particle quantum mechanics. Nat. Chem. 2014, 6, 927–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, D.; Rouxel, J.R.; Mukamel, S.; Chan, G.K.-L.; Li, Z. Stimulated X-ray Raman and Absorption Spectroscopy of Iron–Sulfur Dimers. J. Phys. Chem. Lett. 2019, 10, 6664–6671. [Google Scholar] [CrossRef]
- Li, Z.; Guo, S.; Sun, Q.; Chan, G.K.-L. Electronic landscape of the P-cluster of nitrogenase as revealed through many-electron quantum wavefunction simulations. Nat. Chem. 2019, 11, 1026–1033. [Google Scholar] [CrossRef]
- Kubas, A. Characterization of charge transfer excited states in [2Fe–2S] iron–sulfur clusters using conventional configuration interaction techniques. Theor. Chem. Accounts 2020, 139, 1–7. [Google Scholar] [CrossRef]
- Papaefthymiou, V.; Millar, M.M.; Münck, E. Mössbauer and EPR studies of a synthetic analogue for the Fe4S4 core of oxidized and reduced high-potential iron proteins. Inorg. Chem. 1986, 25, 3010–3014. [Google Scholar] [CrossRef]
- Cammack, R. Iron—Sulfur Clusters in Enzymes: Themes and Variations. Adv. Inorg. Chem. 1992, 38, 281–322. [Google Scholar] [CrossRef]
- Fiedler, A.T.; Brunold, T.C. Computational Studies of the H-Cluster of Fe-Only Hydrogenases: Geometric, Electronic, and Magnetic Properties and Their Dependence on the [Fe4S4] Cubane. Inorg. Chem. 2005, 44, 9322–9334. [Google Scholar] [CrossRef]
- Volbeda, A.; Amara, P.; Darnault, C.; Mouesca, J.-M.; Parkin, A.; Roessler, M.M.; Armstrong, F.A.; Fontecilla-Camps, J.C. X-ray crystallographic and computational studies of the O2-tolerant [NiFe]-hydrogenase 1 from Escherichia coli. Proc. Nat. Aca. Sci. USA 2012, 109, 5305–5310. [Google Scholar] [CrossRef] [Green Version]
- Papaefthymiou, G.C.; Laskowski, E.J.; Frota-Pessôa, S.; Frankel, R.B.; Holm, R.H. Antiferromagnetic exchange interactions in [Fe4S4(SR)4]2-,3- clusters. Inorg. Chem. 1982, 21, 1723–1728. [Google Scholar] [CrossRef]
- Laskowski, E.J.; Frankel, R.B.; Gillum, W.O.; Papaefthymiou, G.C.; Renaud, J.; Ibers, J.A.; Holm, R.H. Synthetic analogs of the 4-Fe active sites of reduced ferredoxins. Electronic properties of the tetranuclear trianions [Fe4S4(SR)4]3- and the structure of [(C2H5)3(CH3)N]3[Fe4S4(SC6H5)4]. J. Am. Chem. Soc. 1978, 100, 5322–5337. [Google Scholar] [CrossRef]
- Pereira, A.S.; Tavares, P.; Moura, I.; Moura, J.J.; Huynh, B.H. Mössbauer characterization of the iron-sulfur clusters in Desulfovibrio vulgaris hydrogenase. J. Am. Chem. Soc. 2001, 123, 2771–2782. [Google Scholar] [CrossRef] [PubMed]
- Popescu, C.V.; Münck, E. Electronic Structure of the H Cluster in [Fe]-Hydrogenases. J. Am. Chem. Soc. 1999, 121, 7877–7884. [Google Scholar] [CrossRef]
- Greco, C.; Bruschi, M.; De Gioia, L.; Ryde, U. A QM/MM Investigation of the Activation and Catalytic Mechanism of Fe-Only Hydrogenases. Inorg. Chem. 2007, 46, 5911–5921. [Google Scholar] [CrossRef]
- Mouesca, J.-M.; Chen, J.L.; Noodleman, L.; Bashford, N.; Case, D.A. Density Functional/Poisson-Boltzmann Calculations of Redox Potentials for Iron-Sulfur Clusters. J. Am. Chem. Soc. 1994, 116, 11898–11914. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, C.K.H. Electronic structure calculations on work-station computers: The computer system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- TURBOMOLE V6.2 2010, A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. 2010. Available online: http://www.turbomole.com (accessed on 9 February 2021).
- MacKerell, A.D., Jr.; Feig, M.; Brooks, C.L., III. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; MacKerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular simulation Program. J. Comp. Chem. 2009, 30, 1545–1615. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 Revision; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, P.; DeVries, A.H.; Guest, M.F.; Schreckenbach, G.; Catlow, C.R.A.; French, S.A.; Sokol, A.A.; Bromley, S.T.; Thiel, W.; Turner, A.J.; et al. QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis. J. Mol. Struct. 2003, 632, 1–28. [Google Scholar] [CrossRef]
- Metz, S.; Kästner, A.; Sokol, A.; Keal, T.W.; Sherwood, P. ChemShell—A modular software package for QM/MM simulations, Wiley Interdisc. Rev. Comp. Mol. Sci. 2014, 4, 101–110. [Google Scholar] [CrossRef]
- Ribbe, M.W.; Hu, Y.; Hodgson, K.O.; Hedman, B. Biosynthesis of Nitrogenase Metalloclusters. Chem. Rev. 2014, 114, 4063–4080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.C.L.; Hu, Y.; Ribbe, M.W. ATP-independent substrate reduction by nitrogenase P-cluster variant. Proc. Nat. Aca. Sci. USA 2012, 109, 6922–6926. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BS25 | Fe1 | Fe2 | Fe3 | Fe4 | Fe5 | Fe6 | Fe7 | Fe8 | S9 |
|---|---|---|---|---|---|---|---|---|---|
| −2.81 | 2.93 | −2.91 | 2.99 | −3.01 | 2.87 | −2.97 | 2.90 | 0.08 | |
| β | α | β | α | β | α | β | α | - |
| BS State | Configuation Fe 1234-5678 | ΔEnergy (kcal/mol) QM/MM | ΔEnergy (kcal/mol) QM | Average Cluster A Bond Length (Å) | Average Cluster B Bond Length (Å) | Average Cluster Bond Length (Å) |
|---|---|---|---|---|---|---|
| BS25 | βαβα-βαβα | 0.00 | 2.037 | 2.269 | 2.263 | 2.266 |
| BS15 | ββαα-βαβα | 0.45 | 2.517 | 2.271 | 2.269 | 2.270 |
| BS35 | βααβ-βαβα | 1.34 | 2.443 | 2.267 | 2.270 | 2.268 |
| BS24 | βαβα-αββα | 2.30 | 0.517 | 2.269 | 2.262 | 2.265 |
| BS34 | βααβ-αββα | 2.37 | 1.347 | 2.269 | 2.262 | 2.265 |
| BS36 | βααβ-ββαα | 2.38 | 0.979 | 2.266 | 2.263 | 2.265 |
| BS26 | βαβα-ββαα | 2.59 | 0.556 | 2.266 | 2.263 | 2.265 |
| BS14 | ββαα-αββα | 2.95 | 0.812 | 2.270 | 2.269 | 2.269 |
| BS16 | ββαα-ββαα | 4.27 | 0.00 | 2.267 | 2.270 | 2.268 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elghobashi-Meinhardt, N.; Tombolelli, D.; Mroginski, M.A. Electronic and Structural Properties of the Double Cubane Iron-Sulfur Cluster. Catalysts 2021, 11, 245. https://doi.org/10.3390/catal11020245
Elghobashi-Meinhardt N, Tombolelli D, Mroginski MA. Electronic and Structural Properties of the Double Cubane Iron-Sulfur Cluster. Catalysts. 2021; 11(2):245. https://doi.org/10.3390/catal11020245
Chicago/Turabian StyleElghobashi-Meinhardt, Nadia, Daria Tombolelli, and Maria Andrea Mroginski. 2021. "Electronic and Structural Properties of the Double Cubane Iron-Sulfur Cluster" Catalysts 11, no. 2: 245. https://doi.org/10.3390/catal11020245