
Organocatalysis for the Asymmetric Michael Addition of Cycloketones and α, β-Unsaturated Nitroalkenes


Abstract
:1. Introduction
2. Results and Discussion
2.1. Asymmetric Michael Reaction of Various Ketones and α, β-Unsaturated Nitroalkenes Using a Thiourea Catalyst
2.2. Reaction According to the Type of Acetone and α, β-Unsaturated Nitroalkynes
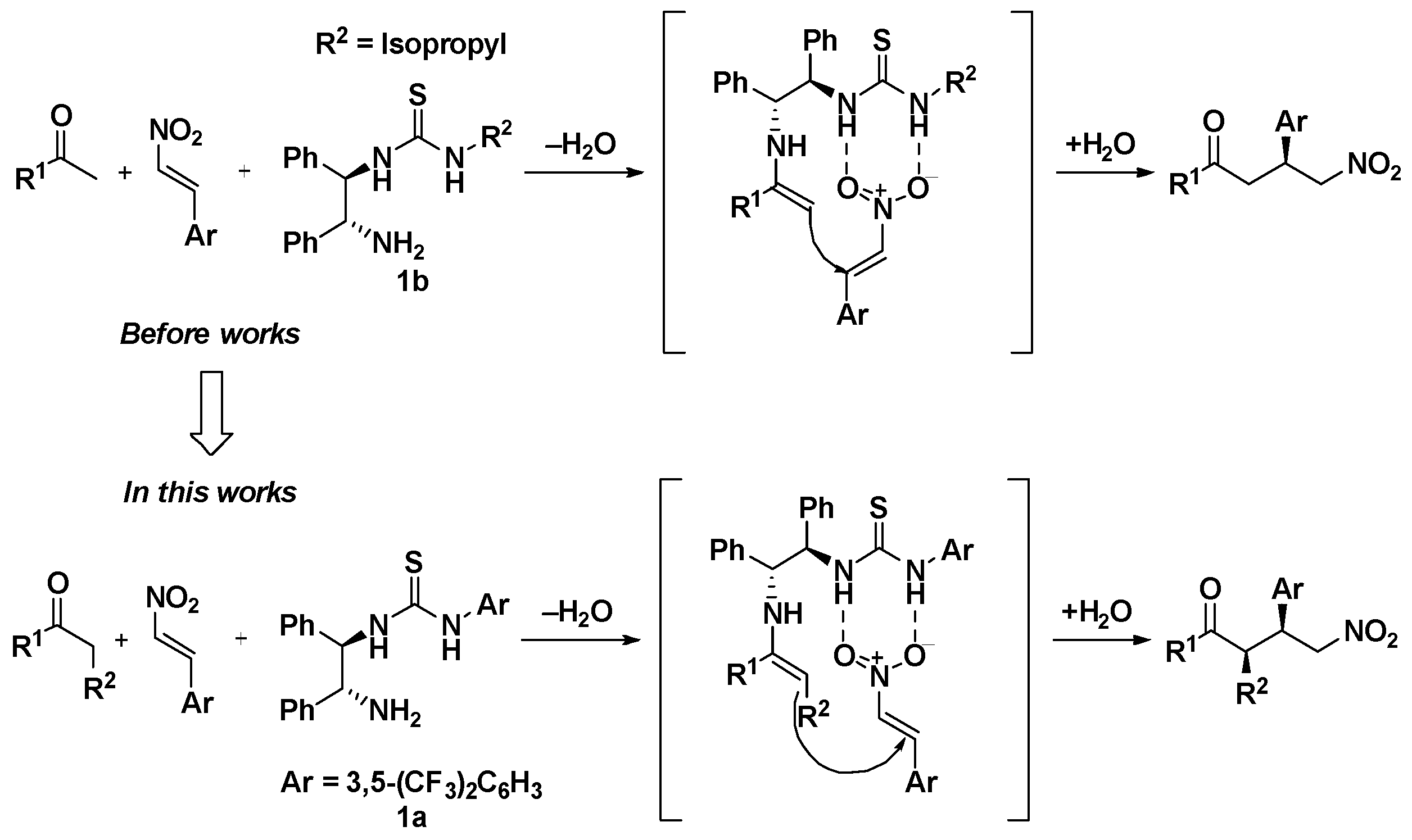
2.3. Asymmetric Michael Reaction Using a Thiourea Catalyst
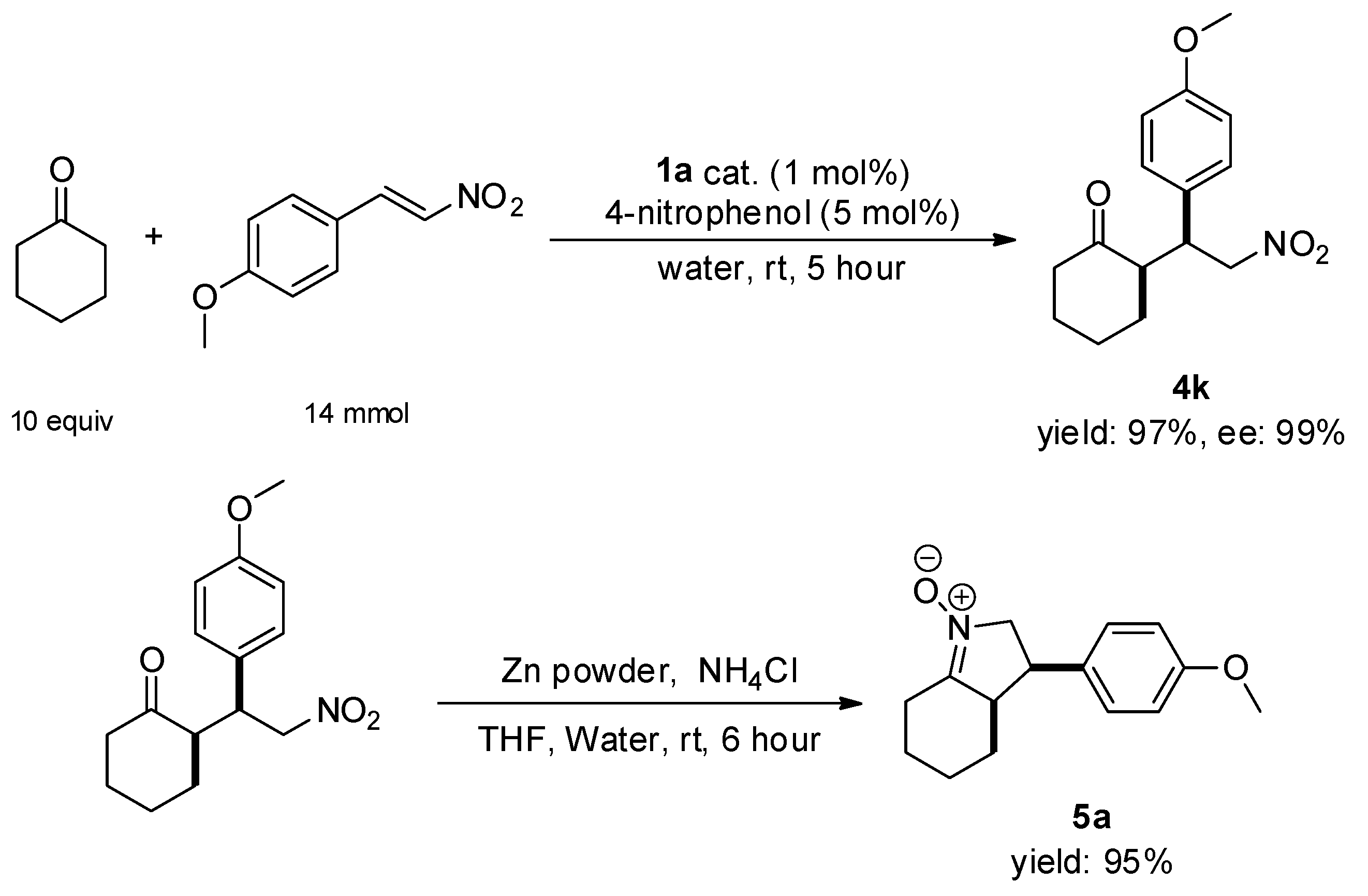
2.4. Application of Pancracin Intermediate Products to the Asymmetric Michael Reaction of Ketones with α, β-Unsaturated Nitroalkenes
3. Materials and Methods
3.1. Instruments and Reagents
3.2. Experimental Method
3.2.1. Synthesis of N-Mono-Thiourea Catalyst
3.2.2. Asymmetric Michael Reaction of Chitons and α,β-Unsaturated Nitroalkenes Using a Chiral Thiourea Catalyst
3.2.3. Synthesis of 3-Arylhexahydroindole 1-Oxides
3.2.4. General Procedure of the Racemic Michael Addition
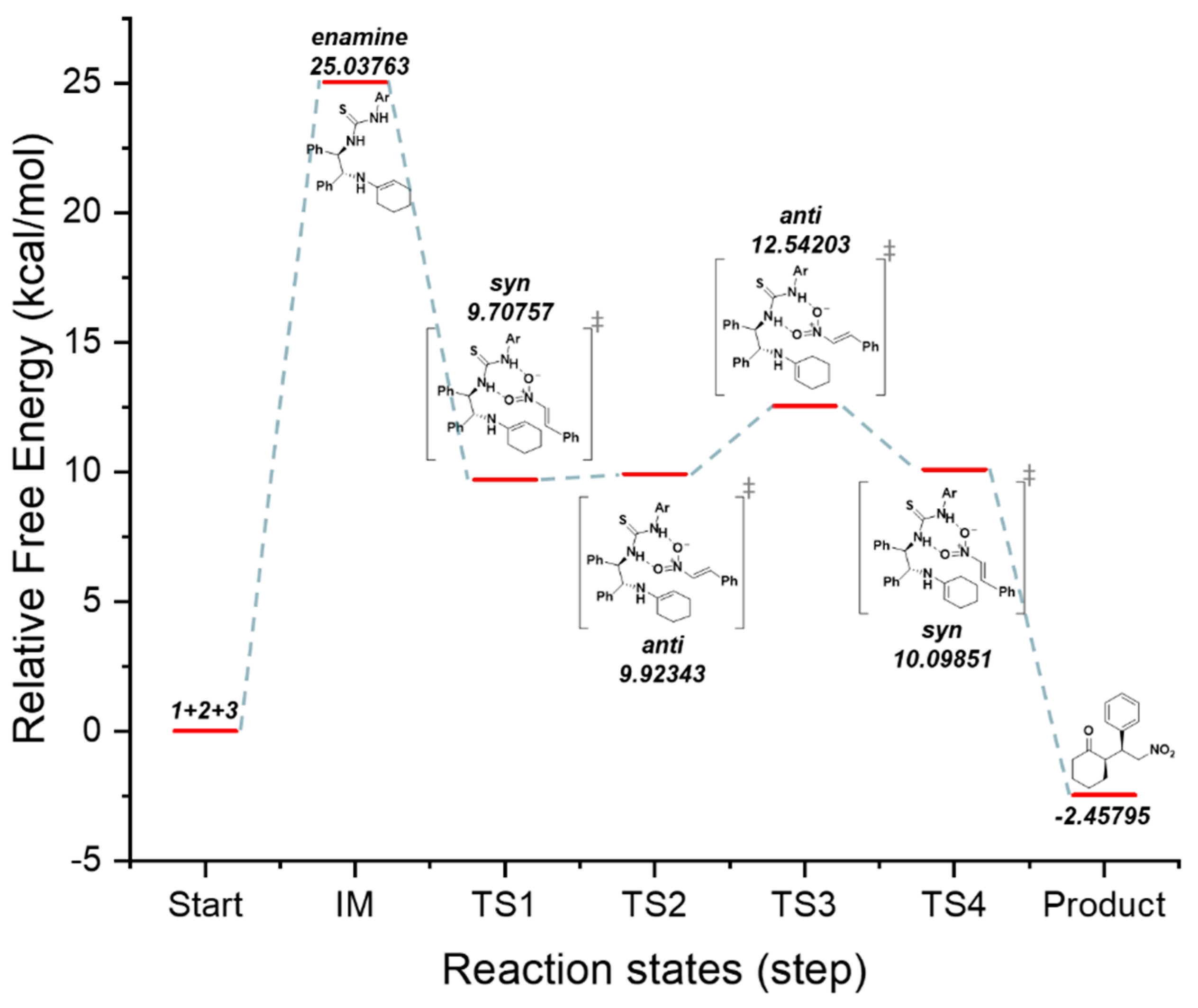
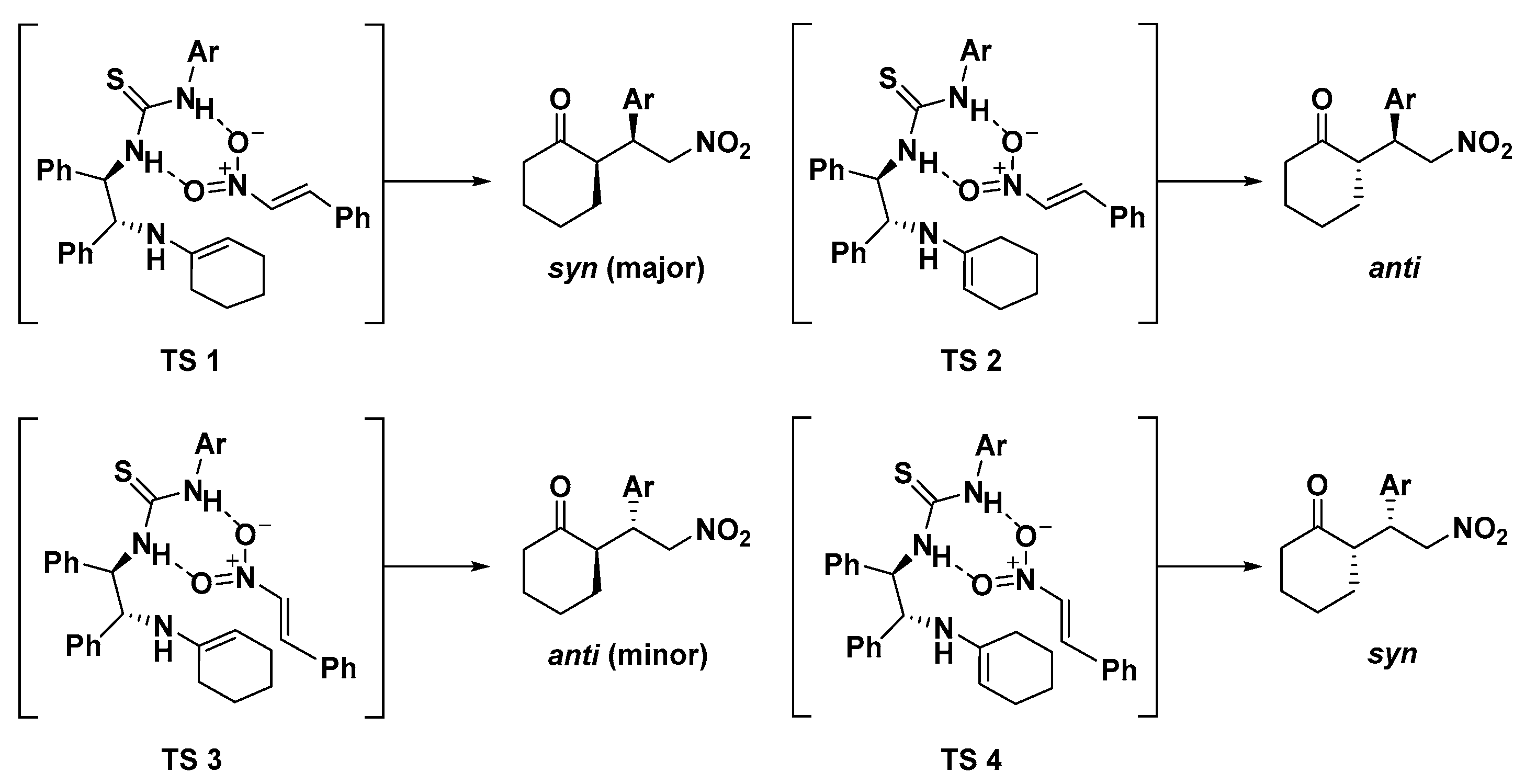
3.3. Results of DFT Calculations and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fubini, B.; Otero Arean, C. Chemical Aspects of the Toxicity of Inhaled Mineral Dusts. Chem. Soc. Rev. 1999, 28, 373–381. [Google Scholar] [CrossRef]
- Brook, M.A.; Seebach, D. Cyclic Nitronates from the Diastereoselective Addition of 1-Trimethylsilyloxycyclohexene to Nitroolefins. Starting Materials for Stereoselective Henry Reactions and 1,3-Dipolar Cycloadditions. Can. J. Chem. 1987, 65, 836–850. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Nishii, S. The Anti-Selective Michael Addition of Allylic Organometals to Ethylidenemalonates and Related Compound. J. Org. Chem. 1988, 53, 3597–3603. [Google Scholar] [CrossRef]
- Martens, J.; Lubben, S. Enantiomerenreine Bicyclische Pyrrolidin-Derivate:: Herstellung und Verwendung in Asymmetrischen Synthesen. Tetrahedron Lett. 1991, 47, 1205–1214. [Google Scholar] [CrossRef]
- Saraswathy, V.G.; Sankararaman, S.J. Chemoselectivity in the Michael Addition of Silyl Enol Ethers in Lithium Perchlorate-Diethyl Ether Medium. Evidence for Facile Silyl Group Transfer to Michael Acceptors. J. Org. Chem. 1995, 60, 5024–5028. [Google Scholar] [CrossRef]
- Alexakis, A.; Andrey, O. Diamine-Catalyzed Asymmetric Michael Additions of Aldehydes and Ketones to Nitrostyrene. Org. Lett. 2002, 4, 3611–3614. [Google Scholar] [CrossRef]
- Dalko, P.I.; Moisan, L. In the Golden Age of Organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef]
- Shim, J.H.; Nam, S.H.; Kim, B.S.; Ha, D.C. Organocatalytic Asymmetric Michael Addition of Ketones to α, β-Unsaturated Nitro Compounds. Catalysts 2020, 10, 618. [Google Scholar] [CrossRef]
- Shim, J.H.; Kim, M.J.; Lee, J.Y.; Kim, K.H.; Ha, D.C. Organocatalytic Asymmetric Aldol Reaction Using Protonated Chiral 1,2-Diamines. Tetrahedron Lett. 2020, 61, 152295. [Google Scholar] [CrossRef]
- Shim, J.H.; Lee, M.J.; Lee, M.H.; Kim, B.S.; Ha, D.C. Enantioselective Organocatalytic Michael Reactions Using Chiral (R,R)-1,2-diphenylethylenediamine-derived Thioureas. RSC Adv. 2020, 10, 31808–31814. [Google Scholar] [CrossRef]
- Seebach, D.; Golinski, J. Synthesis of Open-Chain 2,3-Disubstituted 4-Nitroketones by Diastereoselective Michael-Addition of (E)-Enamines to (E)-Nitroolefins. A Topological Rule for C, C-Bond Forming Processes Between Prochiral Centers. Helv. Chim. Acta 1981, 64, 1413–1423. [Google Scholar] [CrossRef]
- Blarer, S.J.; Schweizer, W.B.; Seebach, D. Asymmetrische Micheal-Additionen. Praktisch Vollständigdiastereo- und Enantioselektive Alkylierungen des Enamins aus Cyclohexanon und Prolinylmethyläther. durch ω-Nitrostyrole zu u-2-(l′-Aryl-2′-nitroäthyl)cyclohexanonen. Helv. Chim. Acta 1982, 65, 1637–1654. [Google Scholar] [CrossRef]
- List, B.; Pojarlier, P.; Martin, H.J. Efficient Proline-Catalyzed Michael Additions of Unmodified Ketones to Nitro Olefins. Org. Lett. 2001, 3, 2423–2425. [Google Scholar] [CrossRef]
- Seayad, J.; List, B. Asymmetric Organocatalysis. Org. Biomol. Chem. 2005, 3, 719–724. [Google Scholar] [CrossRef]
- Mukherjee, S.; List, B.; Yang, J.W. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef]
- Jasinski, R.; Dresler, E. On the Question of Zwitterionic Intermediates in the [3+2] Cycloaddition Reactions: A Critical Review. Organics 2020, 1, 49–69. [Google Scholar] [CrossRef]
- Kącka-Zych, A.; Ríos-Gutiérrez, M.; Domingo, L.R. A molecular electron density theory study of the Lewis acid–catalyzed decomposition reaction of nitroethyl benzoate using aluminum derivatives. J. Phys. Org. Chem. 2019, 32, e3938. [Google Scholar] [CrossRef]
- Jasinski, R. β-Trifluoromethylated nitroethenes in Diels-Alder reaction with cyclopentadiene: A DFT computational study. J. Fluor. Chem. 2018, 206, 1–7. [Google Scholar] [CrossRef]
- Kacka, A.; Jasinski, R. A dramatic change of kinetic conditions and molecular mechanism of decomposition processes of nitroalkyl carboxylates catalyzed by ethylammonium cations. Comput. Theor. Chem. 2017, 1104, 37–42. [Google Scholar] [CrossRef]
- Nobuyuki, M.; Watanabe, K.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C.F. Organocatalytic Direct Michael Reaction of Ketones and Aldehydes with β-Nitrostyrene in Brine. J. Am. Chem. Soc. 2006, 128, 4966–4967. [Google Scholar] [CrossRef] [Green Version]
- Notz, W.; Tanaka, F.; Barbas, C.F. Enamine-Based Organocatalysis with Proline and Diamines: The Development of Direct Catalytic Asymmetric Aldol, Mannich, Michael, and Diels−Alder Reactions. Acc. Chem. Res. 2004, 34, 580–591. [Google Scholar] [CrossRef]
- Cao, C.-L.; Ye, M.-C.; Sun, X.-L.; Tang, Y. Pyrrolidine−Thiourea as a Bifunctional Organocatalyst: Highly Enantioselective Michael Addition of Cyclohexanone to Nitroolefins. Org. Lett. 2006, 8, 2901–2904. [Google Scholar] [CrossRef]
- Sigman, M.S.; Jacobsen, E.N. Schiff Base Catalysts for the Asymmetric Strecker Reaction Identified and Optimized from Parallel Synthetic Libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly Enantioselective Conjugate Addition of Nitromethane to Chalcones Using Bifunctional Cinchona Organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef]
- McCooey, S.H.; Connon, S.J. Urea- and Thiourea-Substituted Cinchona Alkaloid Derivatives as Highly Efficient Bifunctional Organocatalysts for the Asymmetric Addition of Malonate to Nitroalkenes: Inversion of Configuration at C9 Dramatically Improves Catalyst Performance. Angew. Chem. Int. Ed. 2005, 44, 6367–6370. [Google Scholar] [CrossRef]
- Singh, V.; Singh, V.K. Highly Enantioselective Water-Compatible Organocatalyst for Michael Reaction of Ketones to Nitroolefins. Org. Lett. 2007, 9, 1117–1119. [Google Scholar] [CrossRef]
- Ni, B.; Zhang, Q.; Headley, A.D. Highly Enantioselective Michael addition of Ketones to Nitroolefins Catalyzed by (S)-pyrrolidine Arenesulfonamide. Tetrahedron Asymmetry 2007, 18, 1443–1447. [Google Scholar] [CrossRef]
- Xiong, Y.; Wen, Y.; Wang, F.; Gao, B.; Liu, X.; Huang, X.; Feng, X. A Chiral Functionalized Salt-Catalyzed Asymmetric Michael Addition of Ketones to Nitroolefins. Adv. Synth. Catal. 2007, 349, 2156–2166. [Google Scholar] [CrossRef]
- Yacob, Z.; Shah, J.; Leistner, J.; Liebscher, J. (S)-Pyrrolidin-2-ylmethyl-1, 2, 3-triazolium Salts-Ionic Liquid Supported Organocatalysts for Enantioselective Michael Additions to β-Nitrostyrenes. Synlett 2008, 15, 2342–2344. [Google Scholar] [CrossRef]
- Andrs, J.M.; Manzano, R.; Pedrosa, R. Novel Bifunctional Chiral Urea and Thiourea Derivatives as Organocatalysts: Enantioselective Nitro-Michael Reaction of Malonates and Diketones. Chem. Eur. J. 2008, 14, 5116–5119. [Google Scholar] [CrossRef]
- Puleo, G.L.; Iuliano, A. Substrate Control by Means of the Chiral Cavity of Prolinamide Derivatives of Cholic Acid in the Organocatalyzed Michael Addition of Cyclohexanone to Nitroolefins. Tetrahedron Asymmetry 2008, 19, 2045–2050. [Google Scholar] [CrossRef]
- Diez, D.; Anton, A.B.; Garcia, P.; Marcos, I.S.; Babase, P.; Urones, J.G. Synthesis of a New Organocatalyst for Michael Addition. Tetrahedron Asymmetry 2008, 19, 2088–2091. [Google Scholar] [CrossRef]
- Mandal, T.; Zhao, C.G. Modularly Designed Organocatalyst Assemblies for Direct Nitro-Michael Addition Reactions. Angew. Chem. Int. Ed. 2008, 47, 7714–7717. [Google Scholar] [CrossRef] [PubMed]
- Rasappan, R.; Reiser, O. Cyclohexane-1,2-diamines: Efficient Catalysts for the Enantioselective Conjugate Addition of Ketones to Nitro Olefins. Eur. J. Org. Chem. 2009, 1305–1308. [Google Scholar] [CrossRef]
- Tan, B.; Zeng, X.; Lu, Y.; Chua, P.J.; Zhong, G. Rational Design of Organocatalyst: Highly Stereoselective Michael Addition of Cyclic Ketones to Nitroolefins. Org. Lett. 2009, 11, 1927–1930. [Google Scholar] [CrossRef]
- Ni, B.; Zhang, Q.; Dhungana, K.; Headley, A.D. Ionic Liquid-Supported (ILS) (S)-Pyrrolidine Sulfonamide, a Recyclable Organocatalyst for the Highly Enantioselective Michael Addition to Nitroolefins. Org. Lett. 2009, 11, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Freund, M.; Schenker, S.; Tsogoeva, S.B. Enantioselective nitro-Michael Reactions Catalyzed by Short Peptides on Water. Org. Biomol. Chem. 2009, 7, 4279–4284. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, S.; Lee, D.W.; Ko, D.H.; Ha, D.C. Organocatalysis Using Protonated 1,2-Diamino-1,2-diphenylethane for Asymmetric Diels–Alder reaction. Tetrahedron Lett. 2005, 46, 5991–5994. [Google Scholar] [CrossRef]
- Wang, L.; Xu, X.; Huang, J.; Peng, L.; Huang, Q.; Wang, L. Asymmetric Michael Addition of Aromatic Ketones to Nitroolefins Catalyzed by Simple Chiral Bifunctional Primary Amine-Thioureas. Lett. Org. Chem. 2010, 7, 367–372. [Google Scholar]
- Pansare, S.V.; Ligampally, R.; Kirby, R.L. Stereoselective Synthesis of 3-Aryloctahydroindoles and Application in a Formal Synthesis of (−)-Pancracine. Org. Lett. 2010, 12, 556–559. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
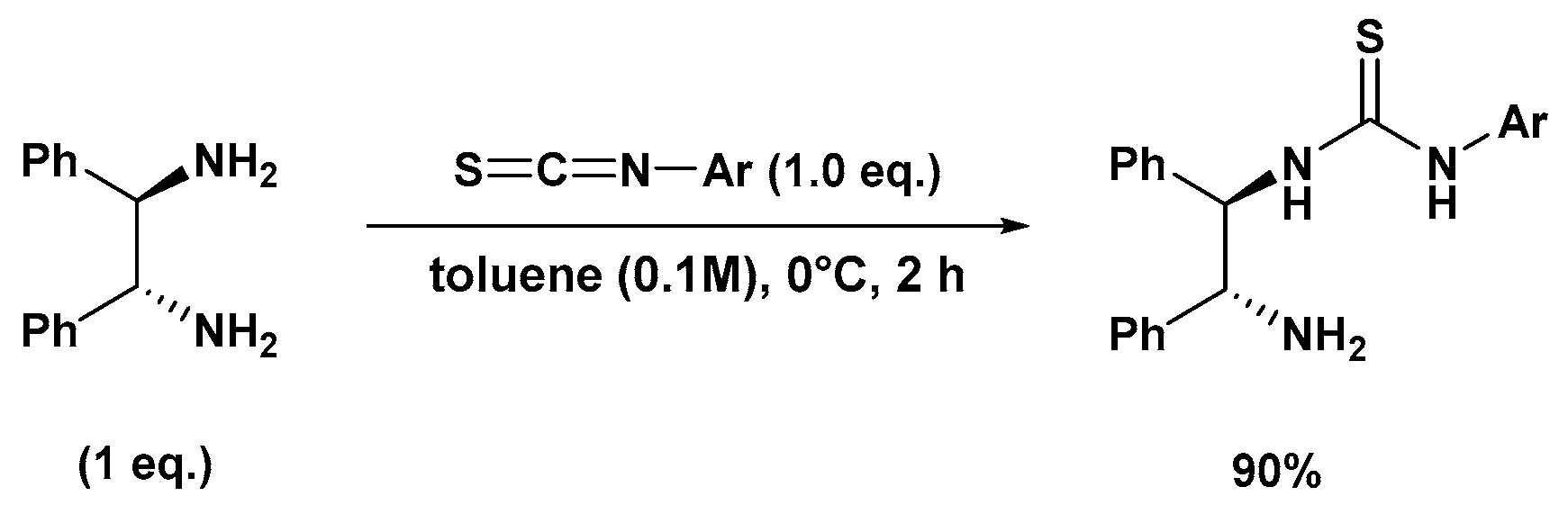{kind=link}

| Entry | R1 | R2 | Yield a (%) | syn/anti b | ee (syn) c (%) |
|---|---|---|---|---|---|
| 1 d | CH3 | H | 72 | - | 72 |
| 2 e | CH3 | H | 81 | - | 98 |
| 3 f | CH3 | H | 90 | - | 99 |
| 4 g | CH3 | H | 98 | - | 99 |
| 5 | C6H5 | H | 95 | - | 98 |
| 6 | –(CH2)4– | 99 | 90/10 | 99 | |
| 7 | –(CH2)3– | 98 | 80/20 | 88 | |
| 8 | –CH2CH2OCH2– | 87 | 82/18 | 66 | |
| 9 | –CH2CH2SCH2– | 85 | 66/34 | 94 | |

| Entry | Ar | Time (h) | Yield (%) a | ee (%) b |
|---|---|---|---|---|
| 1 | 4-MeC6H4 | 10 | 91 | 99 |
| 2 | 4-MeOC6H4 | 12 | 95 | 99 |
| 3 | 4-ClC6H4 | 9 | 96 | 99 |
| 4 | 4-BrC6H4 | 9 | 99 | 98 |
| 5 | 2-MeOC6H4 | 17 | 88 | 99 |
| 6 | 2-furyl | 10 | 95 | 98 |

| Entry | R | Yield a (%) | syn/anti b | ee(syn) c (%) |
|---|---|---|---|---|
| 1 | 4-Me-C6H4 | 98 | 86/14 | 99 |
| 2 | 4-i-Pr-C6H4 | 88 | 93/7 | 82 |
| 3 | 4-F-C6H4 | 92 | 88/12 | 99 |
| 4 | 4-Cl-C6H4 | 99 | 81/19 | 99 |
| 5 | 4-Br-C6H4 | 97 | 85/15 | 96 |
| 6 | 3-Cl-C6H4 | 93 | 91/9 | 96 |
| 7 | 2-furyl | 92 | 92/8 | 96 |
| 8 | 2-thienyl | 99 | 84/16 | 98 |
| 9 | 2-naphthyl | 92 | 89/11 | 92 |
| 10 | 4-MeO-C6H4 | 99 | 91/9 | 99 |
| 11 | 2-MeO-C6H4 | 94 | 91/9 | 99 |
| 12 | 2-NO2-C6H4 | 95 | 87/13 | 94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, J.H.; Ahn, B.K.; Lee, J.Y.; Kim, H.S.; Ha, D.-C. Organocatalysis for the Asymmetric Michael Addition of Cycloketones and α, β-Unsaturated Nitroalkenes. Catalysts 2021, 11, 1004. https://doi.org/10.3390/catal11081004
Shim JH, Ahn BK, Lee JY, Kim HS, Ha D-C. Organocatalysis for the Asymmetric Michael Addition of Cycloketones and α, β-Unsaturated Nitroalkenes. Catalysts. 2021; 11(8):1004. https://doi.org/10.3390/catal11081004
Chicago/Turabian StyleShim, Jae Ho, Byung Kook Ahn, Ji Yeon Lee, Hyeon Soo Kim, and Deok-Chan Ha. 2021. "Organocatalysis for the Asymmetric Michael Addition of Cycloketones and α, β-Unsaturated Nitroalkenes" Catalysts 11, no. 8: 1004. https://doi.org/10.3390/catal11081004