
Indium-Catalyzed Cycloisomerization of 1,6-Cyclohexenylalkynes

 ,
,
Abstract
:
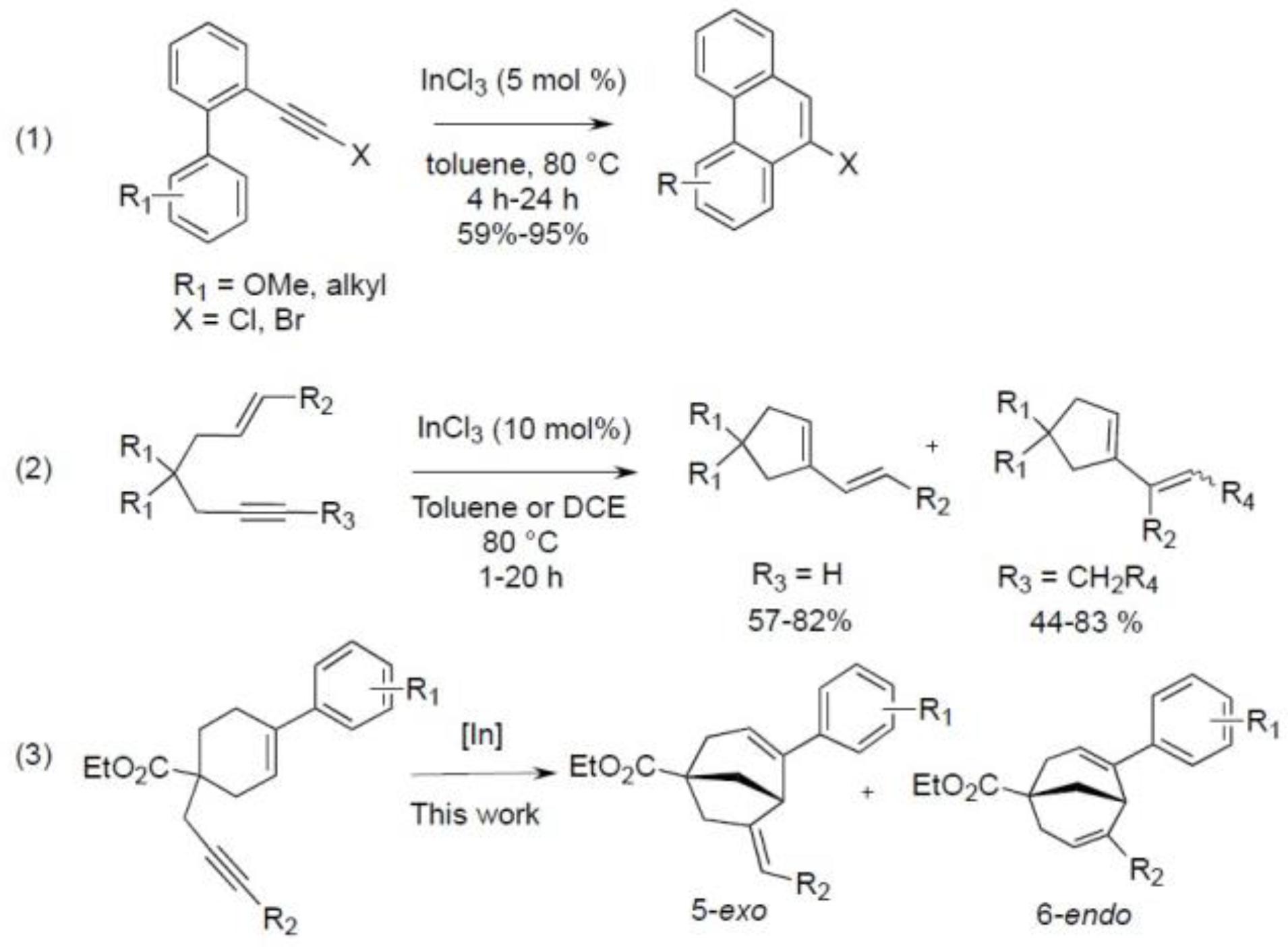
1. Introduction
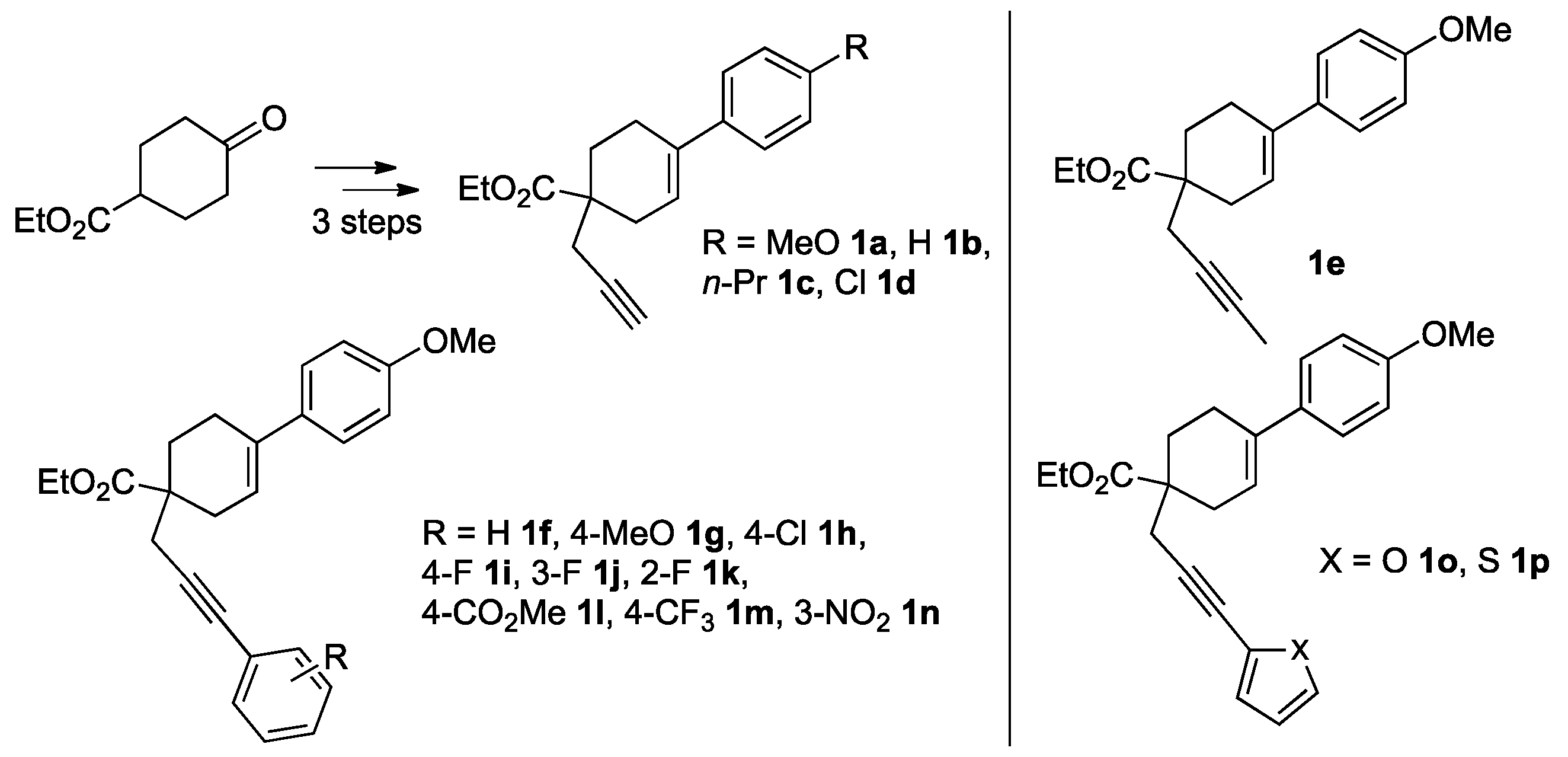
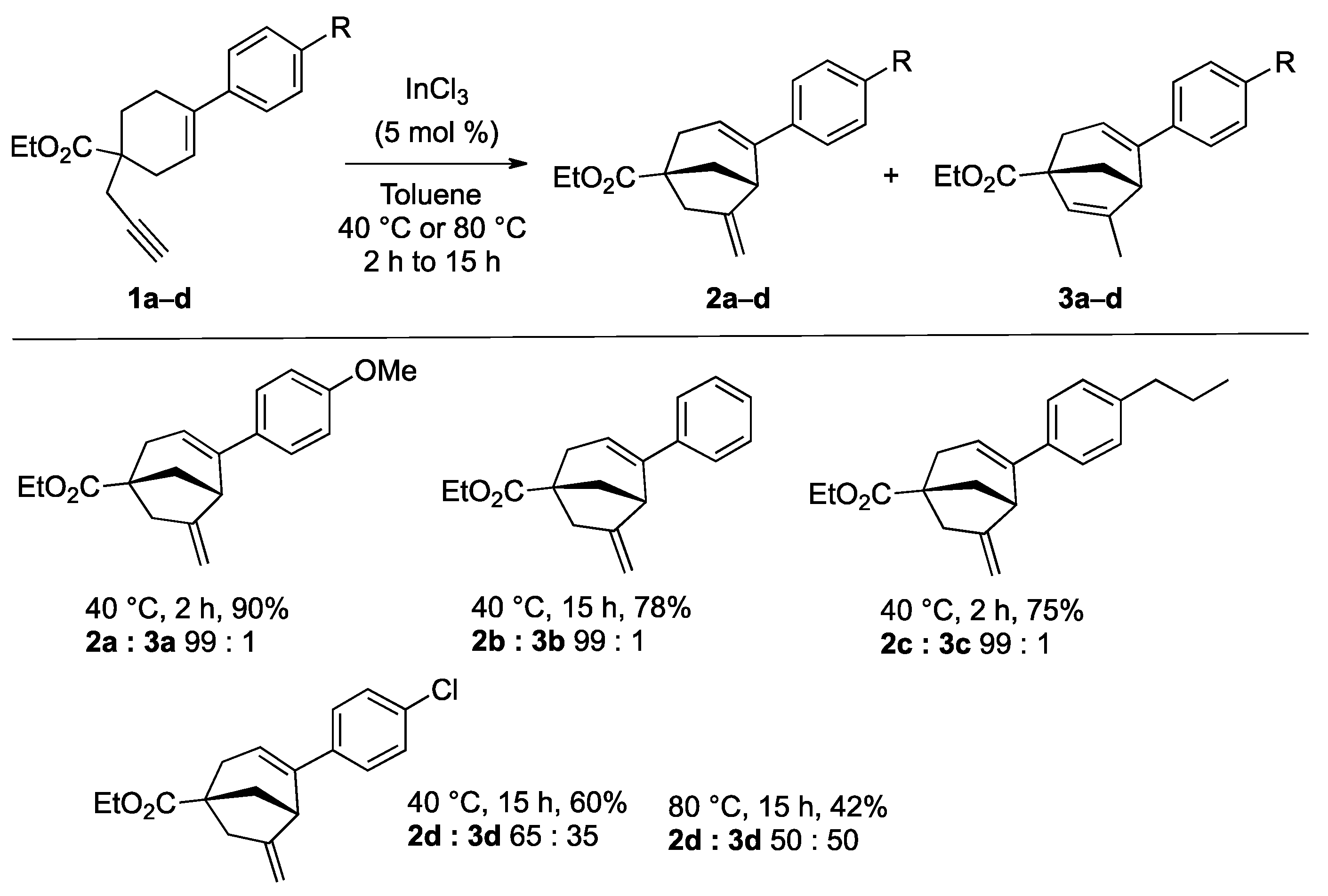
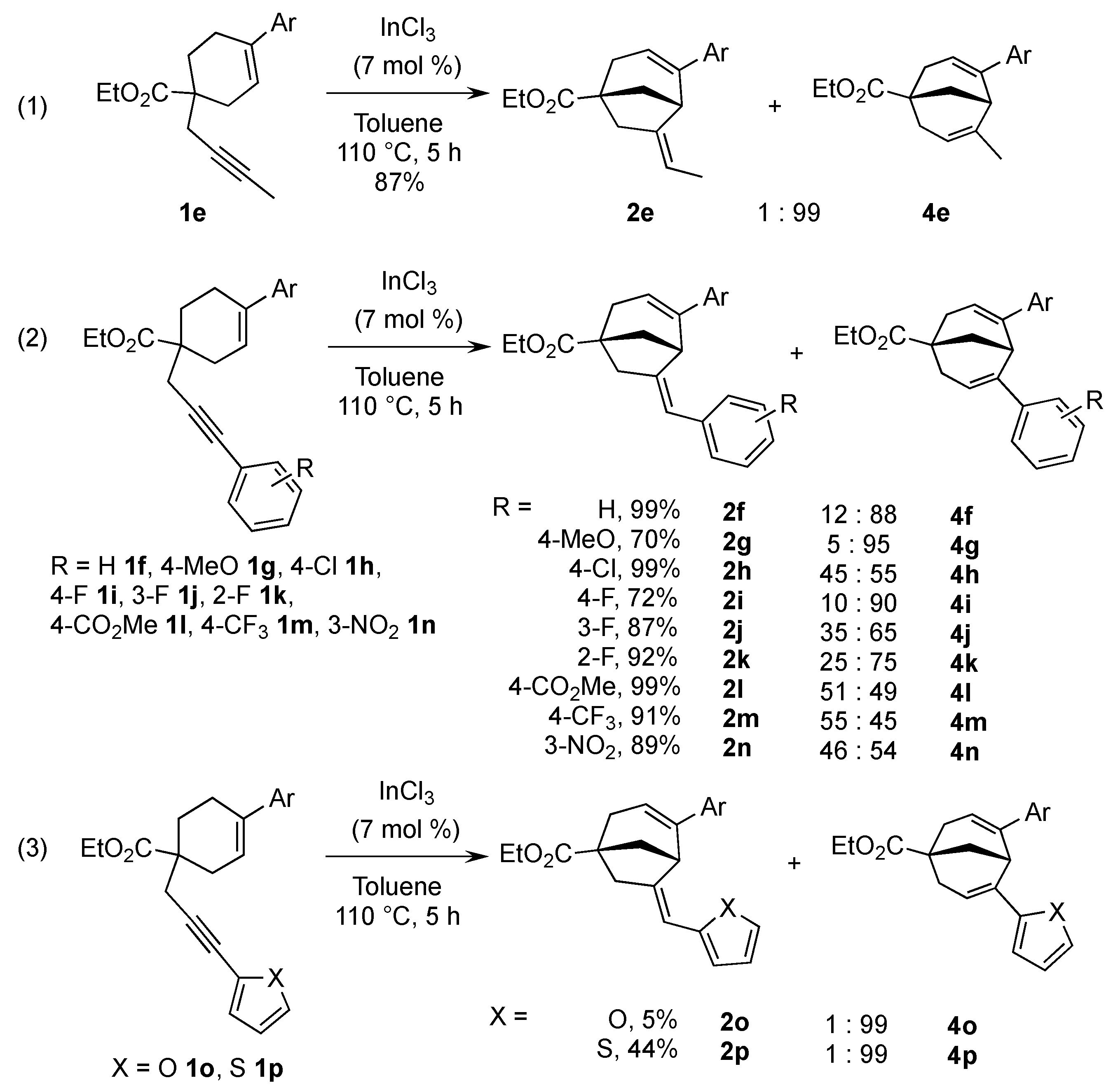
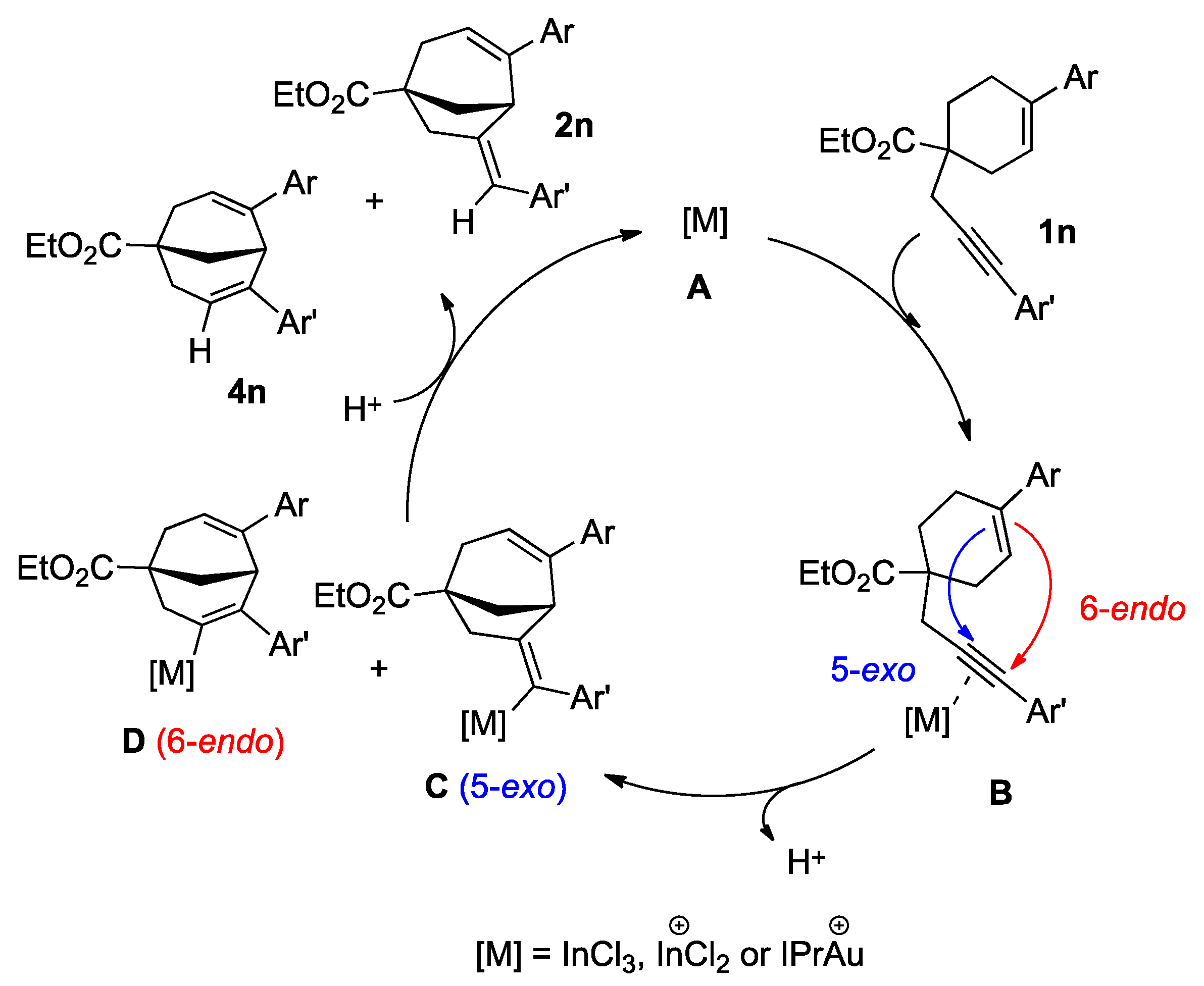
2. Results
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Reich, F.; Richter, T. Ueber das Indium. J. Prakt. Chem. 1863, 89, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Cintas, P. Synthetic Organoindium Chemistry: What Makes Indium so Appealing? Synlett 1995, 1995, 1087–1096. [Google Scholar] [CrossRef]
- Li, C.J.; Chan, T.H. Organic syntheses using indium-mediated and catalyzed reactions in aqueous media. Tetrahedron 1999, 55, 11149–11176. [Google Scholar] [CrossRef]
- Chauhan, K.K.; Frost, C.G. Advances in indium-catalysed organic synthesis. J. Chem. Soc. Perkin Trans. 2000, 1, 3015–3019. [Google Scholar] [CrossRef]
- Ranu, B.C. Indium Metal and Its Halides in Organic Synthesis. Eur. J. Org. Chem. 2000, 2347–2356. [Google Scholar] [CrossRef]
- Ghosh, R. InCl3 in organic syntheses. Indian J. Chem. 2001, 40B, 550–557. [Google Scholar]
- Mamane, V.; Hannen, P.; Fürstner, A. Synthesis of Phenanthrenes and Polycyclic Heteroarenes by Transition-Metal Catalyzed Cycloisomerization Reactions. Chem. Eur. J. 2004, 10, 4556–4575. [Google Scholar] [CrossRef]
- Miyanohana, Y.; Chatani, N. Skeletal Reorganization of Enynes Catalyzed by InCl3. Org. Lett. 2006, 8, 2155–2158. [Google Scholar] [CrossRef]
- Michelet, B.; Colard-Itté, J.R.; Thiery, G.; Guillot, R.; Bour, C.; Gandon, V. Dibromoindium(iii) cations as a π-Lewis acid: Characterization of [IPr·InBr2][SbF6] and its catalytic activity towards alkynes and alkenes. Chem. Commun. 2015, 51, 7401–7404. [Google Scholar] [CrossRef] [Green Version]
- Surendra, K.; Qiu, W.; Corey, E.J. A Powerful New Construction of Complex Chiral Polycycles by an Indium(III)-Catalyzed Cationic Cascade. J. Am. Chem. Soc. 2011, 133, 9724–9726. [Google Scholar] [CrossRef]
- Nakamura, M.; Endo, K.; Nakamura, E. Indium-Catalyzed Addition of Active Methylene Compounds to 1-Alkynes. J. Am. Chem. Soc. 2003, 125, 13002–13003. [Google Scholar] [CrossRef]
- Itoh, Y.; Tsuji, H.; Yamagata, K.I.; Endo, K.; Tanaka, I.; Nakamura, M.; Nakamura, E. Efficient Formation of Ring Structures Utilizing Multisite Activation by Indium Catalysis. J. Am. Chem. Soc. 2008, 130, 17161–17167. [Google Scholar] [CrossRef]
- Urabe, F.; Nagashima, S.; Takahashi, K.; Ishihara, J.; Hatakeyama, S. Total Synthesis of (−)-Cinatrin C1 Based on an In(OTf)3-Catalyzed Conia-Ene Reaction. J. Org. Chem. 2013, 78, 3847–3857. [Google Scholar] [CrossRef]
- Montaignac, B.; Vitale, M.R.; Michelet, V.; Ratovelomanana-Vidal, V. Combined InCl3- and Amine-Catalyzed Intramolecular Addition of α-Disubstituted Aldehydes onto Unactivated Alkynes. Org. Lett. 2010, 12, 2582–2585. [Google Scholar] [CrossRef]
- Montaignac, B.; Vitale, M.R.; Ratovelomanana-Vidal, V.; Michelet, V. InCl3/CyNH2 Co-catalyzed Carbocyclization Reaction: An Entry to α-Disubstituted exo-Methylene Cyclopentanes. J. Org. Chem. 2010, 75, 8322–8325. [Google Scholar] [CrossRef]
- Praveen, C.; Montaignac, B.; Vitale, M.R.; Ratovelomanana-Vidal, V.; Michelet, V. Enantioselective Merger of Aminocatalysis with π-Lewis Acid Metal Catalysis: Asymmetric Preparation of Carbo-and Heterocycles. ChemCatChem 2013, 5, 2395–2404. [Google Scholar] [CrossRef]
- Tang, Y.; Benaissa, I.; Huynh, M.; Vendier, L.; Lugan, N.; Bastin, S.; Belmont, P.; César, V.; Michelet, V. An Original L-shape, Tunable N-Heterocyclic Carbene Platform for Efficient Gold(I) Catalysis. Angew. Chem. Int. Ed. 2019, 58, 7977–7981. [Google Scholar] [CrossRef]
- Chen, X.; Martini, S.; Michelet, V. A mild and regioselective synthesis of α-fluoroketones via gold and Selectfluor partnership. Adv. Synth. Catal. 2019, 361, 3612–3618. [Google Scholar] [CrossRef]
- Rode, N.D.; Arcadi, A.; Di Nicola, A.; Marinelli, F.; Michelet, V. Gold-Catalyzed Cascade Reaction of β-2-Aminophenyl)-α,β-ynones with Ynamides: A Sequential Route to Polysubstituted 2‑Aminoquinolines. Org. Lett. 2018, 20, 5103–5106. [Google Scholar] [CrossRef]
- Tomas-Mendivil, E.; Heinrich, C.; Ortuno, J.-C.; Starck, J.; Michelet, V. Gold-catalyzed access to 1H-isochromenes: Reaction development and mechanistic insight. ACS Catal. 2017, 7, 380–387. [Google Scholar] [CrossRef]
- Laher, R.; Marin, C.; Michelet, V. When Gold Meets Perfumes: Synthesis of Olfactive Compounds via Gold-Catalyzed Cycloisomerization Reactions. Org. Lett. 2020, 22, 4058–4062. [Google Scholar] [CrossRef]
- Davenel, V.; Nisole, C.; Fontaine-Vive, F.; Fourquez, J.-M.; Chollet, A.-M.; Michelet, V. Gold-Catalyzed Cycloisomerization of 1,6-Cyclohexenylalkyne: An Efficient Entry to Bicyclo[3.2.1]oct-2-ene and Bicyclo[3.3.1]nonadiene. J. Org. Chem. 2020, 85, 12657–12669. [Google Scholar] [CrossRef]
- Presset, M.; Coquerel, Y.; Rodriguez, J. Syntheses and Applications of Functionalized Bicyclo[3.2.1]octanes: Thirteen Years of Progress. Chem. Rev. 2013, 113, 525–595. [Google Scholar] [CrossRef]
- Ciochina, R.; Grossman, R.B. Polycyclic Polyprenylated Acylphloroglucinols. Chem. Rev. 2006, 106, 3963–3986. [Google Scholar] [CrossRef]
- Butkus, E. Stereocontrolled Synthesis and Reactions of Bicyclo[3.3.1]nonanes. Synlett 2001, 1827–1835. [Google Scholar] [CrossRef]
- Spiegel, D.A.; Njardason, J.T.; McDonald, I.M.; Wood, J.L. The Art of Innovation in Organic Chemistry: Synthetic Efforts toward the Phomoidrides. Chem. Rev. 2003, 103, 2691–2728. [Google Scholar] [CrossRef]
- Boa, A.N.; Jenkins, P.R.; Lawrence, N.J. Recent progress in the synthesis of taxanes. Contemp. Org. Synth. 1994, 1, 47–75. [Google Scholar] [CrossRef]
- Filippini, M.H.; Rodriguez, J. Synthesis of Functionalized Bicyclo[3.2.1]octanes and Their Multiple Uses in Organic Chemistry. Chem. Rev. 1999, 99, 27–76. [Google Scholar] [CrossRef]
- Ondet, P.; Lemière, G.; Dunach, E. Cyclisations Catalysed by Bismuth(III) Triflate. Eur. J. Org. Chem. 2017, 2017, 761–780. [Google Scholar] [CrossRef]
- Noteworthy that this result contrasts with the behavior of the exo derivative 2a in the presence of the gold catalyst, stable in toluene and in dichloromethane after 2 h. In the presence of triflic acid, the isomerization was also very quick but sled to high degradation.
- Birman, V.B.; Danishefsky, S.J. The total synthesis of (+/-)-merrilactone A. J. Am. Chem. Soc. 2002, 124, 2080–2081. [Google Scholar] [CrossRef]
- Bell, R.A.; Ireland, R.E.; Mander, L.N. Experiments Directed toward the Total Synthesis of Terpenes. IX. The Total Synthesis of Hibaene and the Oxygenation of Some Tetracyclic Diterpenes J. Org. Chem. 1966, 31, 2536–2542. [Google Scholar] [PubMed]
- Rojas, J.; Aparicio, R.; Villasmil, T.; Usubillaga, P.A. On the Isomerization of ent-Kaurenic Acid. Nat. Prod. Commun. 2011, 6, 935–938. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Peters-son, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Toste, F.D.; Michelet, V. (Eds.) Gold Catalysis: An Homogeneous, Approach; Imperial College Press: London, UK, 2014. [Google Scholar]
- Rappoport, Z.; Liebman, J.F.; Marek, I. (Eds.) The Chemistry of Organogold Compounds. In Patai’s Chemistry of Functional Groups; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014. [Google Scholar]
- Slaughter, L.M. (Ed.) Homogeneous Gold Catalysis. In Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Zhuo, L.-G.; Zhang, J.-J.; Yu, Z.-X. DFT and Experimental Exploration of the Mechanism of InCl3-Catalyzed Type II Cycloisomerization of 1,6-Enynes: Identifying InCl2+ as the Catalytic Species and Answering Why Nonconjugated Dienes Are Generated. J. Org. Chem. 2012, 77, 8527–8540. [Google Scholar] [CrossRef]
- Zhuo, L.-G.; Zhang, J.-J.; Yu, Z.-X. Mechanisms of the InCl3-Catalyzed Type-I, II, and III Cycloisomerizations of 1,6-Enynes. J. Org. Chem. 2014, 79, 3809–3820. [Google Scholar] [CrossRef]
- Zhuo, L.-G.; Shi, Y.-C.; Yu, Z.-X. Using the Type II Cycloisomerization Reaction of 1,6-Enynes as a Mechanistic Probe to Identify the Real Catalytic Species of GaX3 and InX3. Asian J. Org. Chem. 2014, 3, 842–846. [Google Scholar]
- Becke, A.D.J. Density-functional thermochemistry. III. The role of exact exchange. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154105. [Google Scholar] [CrossRef] [Green Version]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | [M] (x mol %) | Solvent | T (°C) | t (h) | 2a:3a Ratio (%) 1 | Yield 2 (Conv) (%) |
| 1 | (PPh3)AuNTf2 (2) | DCM | rt | 1 | 45:55 | 60 (81) |
| 2 | (PPh3)AuNTf2 (2) | toluene | rt | 0.5 | 53:47 | 50 (100) |
| 3 | IPrAuNTf2 (1) | toluene | rt | 0.75 | 99:1 | 90 (100) |
| 4 | InCl3 (1) | toluene | rt | 2 | / | / (0) |
| 5 | InCl3 (5) | toluene | 40 | 1 | 99:1 | 90 (96) |
| 6 | InCl3 (5) | DCE | 40 | 0.33 | 85:15 | 60 (100) |
| 7 | Bi(OTf)3 (5) | toluene | 40 | 1 | 80:20 | 35 (40) |
| Entry | Experimental endo:exo Ratio (%) | [M] | endo | exo | Theoretical endo:exo Ratio (%) |
|---|---|---|---|---|---|
| 1 | 54:46 | InCl3 |  |  | 43:57 |
| 2 | 54:46 | InCl2 |  |  | 96:4 |
| 3 | 70:30 | IPrAu |  |  | 99:1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davenel, V.; Puteaux, C.; Nisole, C.; Fontaine-Vive, F.; Fourquez, J.-M.; Michelet, V. Indium-Catalyzed Cycloisomerization of 1,6-Cyclohexenylalkynes. Catalysts 2021, 11, 546. https://doi.org/10.3390/catal11050546
Davenel V, Puteaux C, Nisole C, Fontaine-Vive F, Fourquez J-M, Michelet V. Indium-Catalyzed Cycloisomerization of 1,6-Cyclohexenylalkynes. Catalysts. 2021; 11(5):546. https://doi.org/10.3390/catal11050546
Chicago/Turabian StyleDavenel, Vincent, Chloé Puteaux, Christian Nisole, Fabien Fontaine-Vive, Jean-Marie Fourquez, and Véronique Michelet. 2021. "Indium-Catalyzed Cycloisomerization of 1,6-Cyclohexenylalkynes" Catalysts 11, no. 5: 546. https://doi.org/10.3390/catal11050546