
Hydrogenation of α,β-Unsaturated Carbonyl Compounds over Covalently Heterogenized Ru(II) Diphosphine Complexes on AlPO4-Sepiolite Supports

 , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
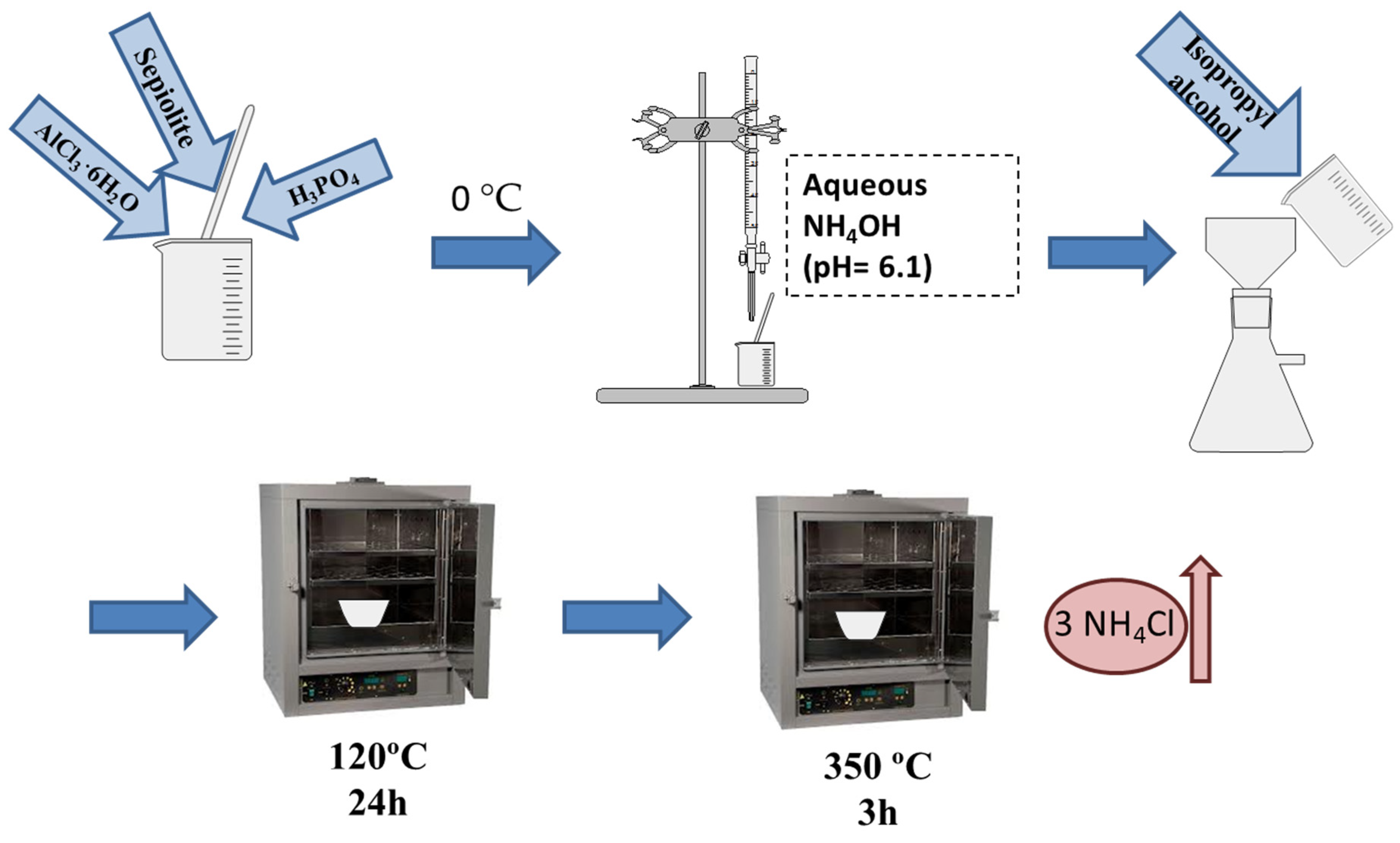
2.1. Characterization of the Support
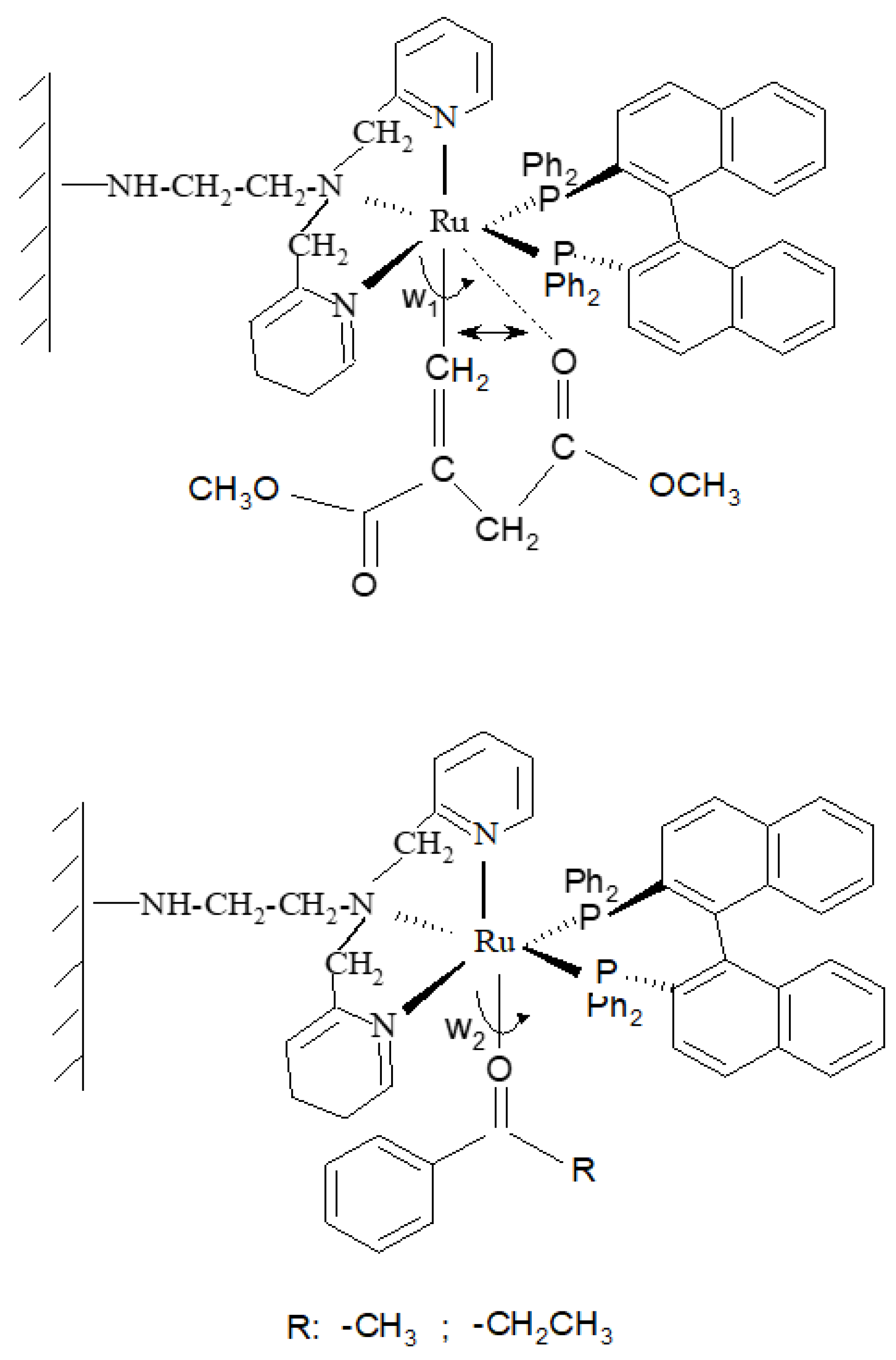
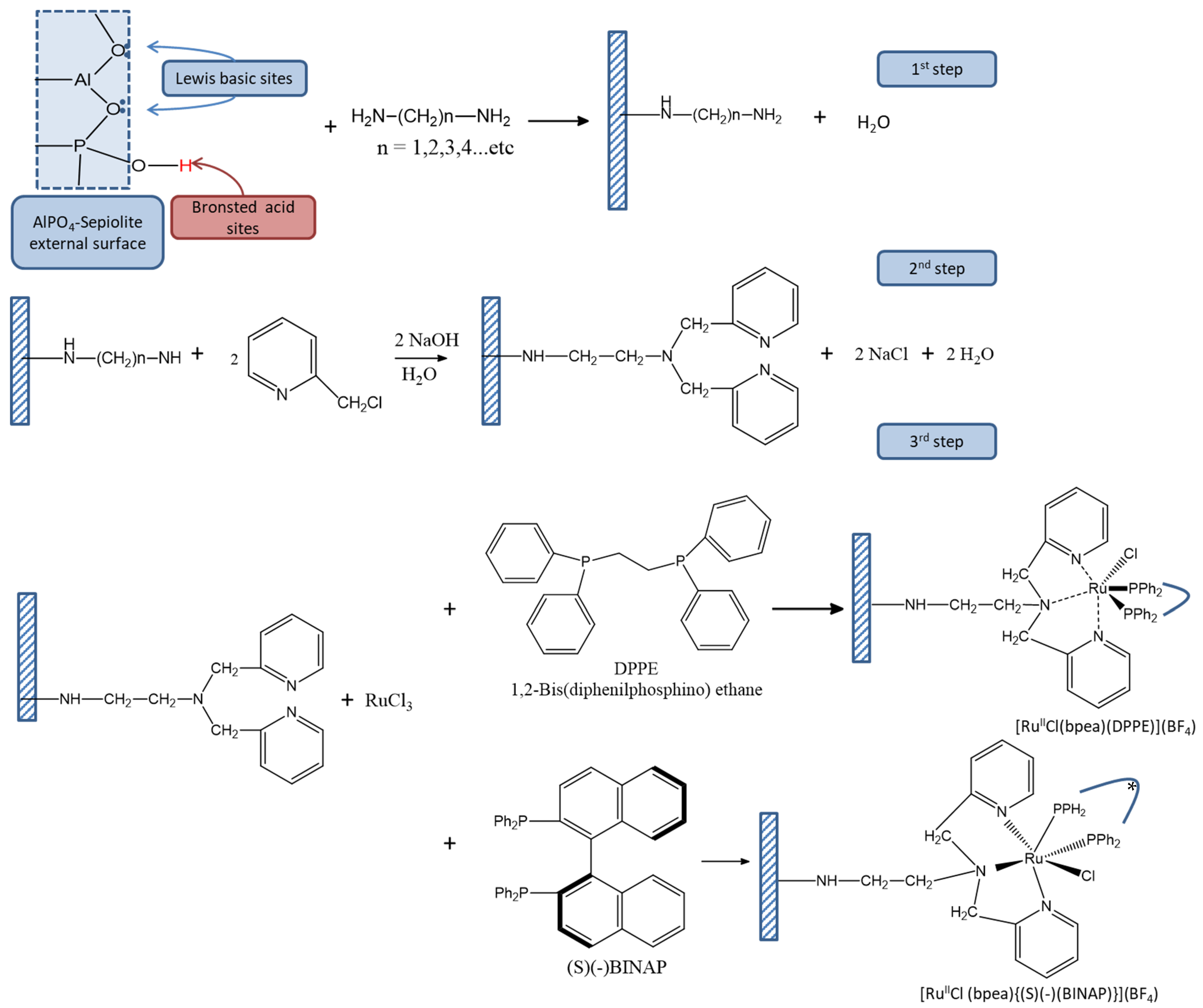
2.2. Synthesis of the Covalently Attached Ru Coordination Complexes
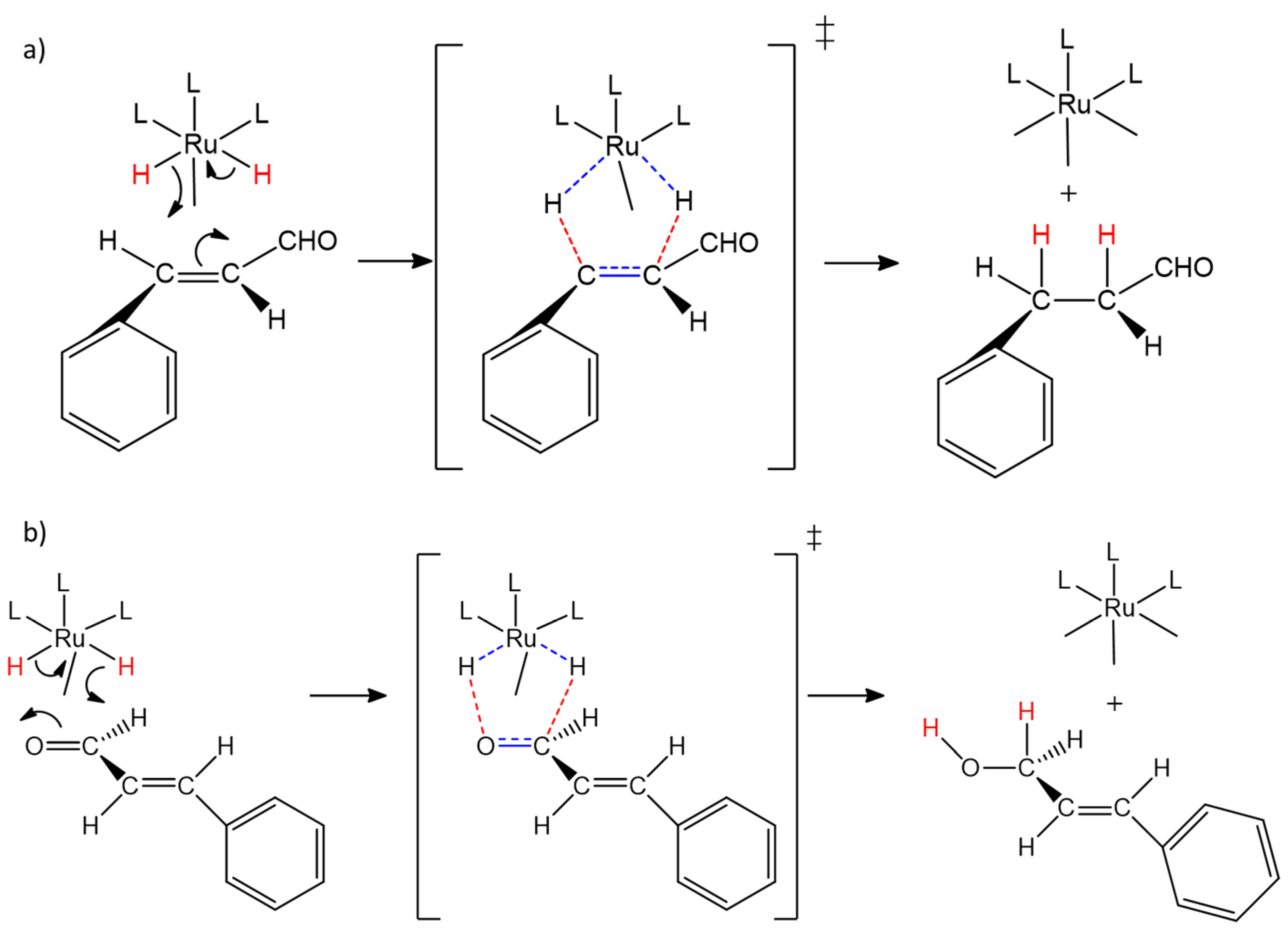
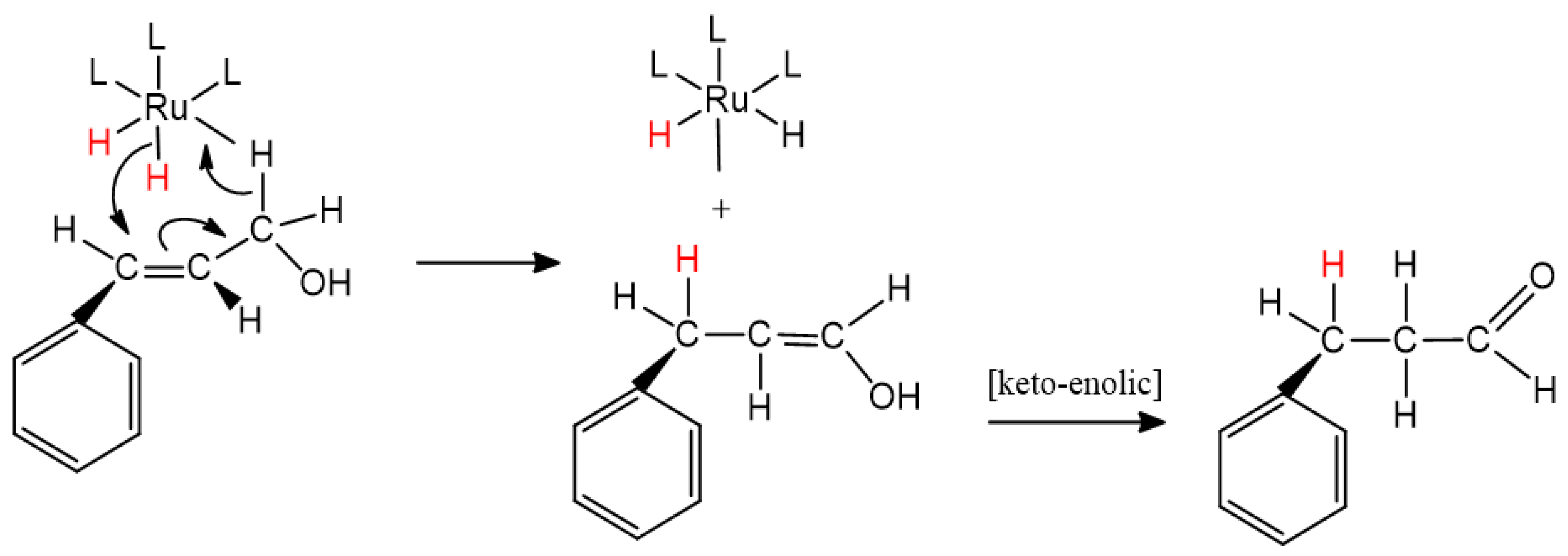
2.3. Catalytic Behavior of [RuIICl(bpea)(DPPE)](BF4) Complex, 2 and of the Covalently Immobilized Complex [RuIICl(bpea)(DPPE)](BF4), AlPO4-Sepiolite@2
2.4. Catalytic Behavior of the Covalently Immobilized Complex [RuIICl(bpea){(S)(-)(BINAP)}](BF4), AlPO4-Sepiolite@1
3. Materials and Methods
3.1. Inorganic Support Synthesis and Surface Characterization
3.2. Synthesis and Characterization of the Homogeneous Complexes (RuIICl(bpea){(S)(-)(BINAP)}](BF4) 1 and (RuIICl(bpea)(DPPE)](BF4) 2
3.3. Covalent Immobilization of the Complexes [RuIICl(bpea){(S)(-)(BINAP)}](BF4) and [RuIICl (bpea)(DPPE)](BF4) on AlPO4-Sepiolite Supports, (AlPO4-Sepiolite@1 and AlPO4-Sepiolite@2)
3.4. Catalysts Characterization and Hydrogenation Experiments
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campelo, J.M.; Jaraba, M.; Luna, D.; Luque, R.; Marinas, A.J.M.; Romero, A.A.; Navio, J.A.; Macias, M. Effect of Phosphate Precursor and Organic Additives on the Structural and Catalytic Properties of Amorphous Mesoporous AlPO4 Materials. Chem. Mater. 2003, 15, 3352–3364. [Google Scholar] [CrossRef]
- Luna, D.; Posadillo, A.; Caballero, V.; Verdugo, C.; Bautista, F.M.; Romero, A.A.; Sancho, E.D.; Luna, C.; Calero, J. New Biofuel Integrating Glycerol into Its Composition through the Use of Covalent Immobilized Pig Pancreatic Lipase. Int. J. Mol. Sci. 2012, 13, 10091–10112. [Google Scholar] [CrossRef] [Green Version]
- Luna, C.; Sancho, E.; Luna, D.; Caballero, V.; Calero, J.; Posadillo, A.; Verdugo, C.; Bautista, F.M.; Romero, A.A. Biofuel that Keeps Glycerol as Monoglyceride by 1,3-Selective Ethanolysis with Pig Pancreatic Lipase Covalently Immobilized on AlPO4 Support. Energies 2013, 6, 3879–3900. [Google Scholar] [CrossRef] [Green Version]
- Calero, J.; Luna, D.; Luna, C.; Bautista, F.M.; Hurtado, B.; Romero, A.A.; Posadillo, A.; Estevez, R. Rhizomucor miehei Lipase Supported on Inorganic Solids, as Biocatalyst for the Synthesis of Biofuels: Improving the Experimental Conditions by Response Surface Methodology. Energies 2019, 12, 831. [Google Scholar] [CrossRef] [Green Version]
- Bautista, F.; Bravo, M.; Campelo, J.; Garcia, A.; Luna, D.; Marinas, J.; Romero, A.A. Covalent immobilization of acid phosphatase on amorphous AlPO4 support. J. Mol. Catal. B Enzym. 1999, 6, 473–481. [Google Scholar] [CrossRef]
- Bautista, F.; Campelo, J.; García, A.; Jurado, A.; Luna, D.; Marinas, J.; Romero, A.A. Properties of a glucose oxidase covalently immobilized on amorphous AlPO4 support. J. Mol. Catal. B Enzym. 2001, 11, 567–577. [Google Scholar] [CrossRef]
- Bautista, F.M.; Caballero, V.; Campelo, J.M.; Luna, D.; Marinas, J.M.; Romero, A.A.; Romero, I.; Serrano, I.; Llobet, A. Heterogeneization of a new Ru(II) homogeneous asymmetric hydrogenation catalyst containing BINAP and the N-tridentate bpea ligand, through covalent attachment on amorphous AlPO4 support. Top. Catal. 2006, 40, 193–205. [Google Scholar] [CrossRef]
- Caballero, V.; Bautista, F.M.; Campelo, J.M.; Luna, D.; Luque, R.; Marinas, J.M.; Romero, A.A.; Romero, I.; Rodríguez, M.; Serrano, I.; et al. Efficient hydrogenation of alkenes using a highly active and reusable immobilised Ru complex on AlPO4. J. Mol. Catal. A Chem. 2009, 308, 41–45. [Google Scholar] [CrossRef]
- Deshmukh, A.; Kinage, A.; Kumar, R.; Meijboom, R. Heterogenized Ru(II) phenanthroline complex for chemoselective hydrogenation of diketones under biphasic aqueous medium. J. Mol. Catal. A Chem. 2010, 333, 114–120. [Google Scholar] [CrossRef]
- Sisodiya, S.; Lazar, A.; Shylesh, S.; Wang, L.; Thiel, W.R.; Singh, A.P. Covalently anchored ruthenium–phosphine complex on mesoporous organosilica: Catalytic applications in hydrogenation reactions. Catal. Commun. 2012, 25, 22–27. [Google Scholar] [CrossRef]
- Gorbunov, D.; Safronova, D.; Kardasheva, Y.; Maximov, A.; Rosenberg, E.; Karakhanov, E. New Heterogeneous Rh-Containing Catalysts Immobilized on a Hybrid Organic–Inorganic Surface for Hydroformylation of Unsaturated Compounds. ACS Appl. Mater. Interfaces 2018, 10, 26566–26575. [Google Scholar] [CrossRef]
- Ren, J.; Lan, P.C.; Chen, M.; Zhang, W.; Ma, S. Heterogenization of Trinuclear Palladium Complex into an Anionic Metal–Organic Framework through Postsynthetic Cation Exchange. Organometallics 2019, 38, 3460–3465. [Google Scholar] [CrossRef]
- Pederzolli, F.R.S.; Wolke, S.I.; Da Rosa, R.G. Recyclable rhodium-Cp′-heterogenized catalysts for hydrogenation of olefins. J. Catal. 2018, 360, 201–212. [Google Scholar] [CrossRef]
- Mayakrishnan, G.; Soo, K.I.; Min, C.I. Stepwise Construction of Ru(II)Center Containing Chiral Thiourea Ligand on Graphene Oxide: First Efficient, Reusable, and Stable Catalyst for Asymmetric Transfer Hydrogenation of Ketones. Catalysts 2020, 10, 175. [Google Scholar] [CrossRef] [Green Version]
- Huebner, S.; De Vries, J.G.; Farina, V. Why Does Industry Not Use Immobilized Transition Metal Complexes as Catalysts? Adv. Synth. Catal. 2016, 358, 3–25. [Google Scholar] [CrossRef]
- Calsolaro, F.; Martina, K.; Borfecchia, E.; Chávez-Rivas, F.; Cravotto, G.; Berlier, G. β-Cyclodextrin-Silica Hybrid: A Spatially Controllable Anchoring Strategy for Cu(II)/Cu(I) Complex Immobilization. Catalysts 2020, 10, 1118. [Google Scholar] [CrossRef]
- Calvete, M.; Silva, M.; Pereira, M.; Burrows, H. Inorganic helping organic: Recent advances in catalytic heterogeneous oxidations by immobilised tetrapyrrolic macrocycles in micro and mesoporous supports. RSC Adv. 2013, 3, 22774–22789. [Google Scholar] [CrossRef]
- Benzaqui, M.; Semino, R.; Carn, F.; Tavares, S.R.; Menguy, N.; Giménez-Marqués, M.; Bellido, E.; Horcajada, P.; Berthelot, T.; Kuzminova, A.I.; et al. Covalent and Selective Grafting of Polyethylene Glycol Brushes at the Surface of ZIF-8 for the Processing of Membranes for Pervaporation. ACS Sustain. Chem. Eng. 2019, 7, 6629–6639. [Google Scholar] [CrossRef]
- Nikoorazm, M.; Khanmoradi, M. Application of Cu (II)-Guanine Complexes Anchored on SBA-15 and MCM-41 as Efficient Nanocatalysts for One-Pot, Four-Component Domino Synthesis of Phenazine Derivatives and Investigation of Their Antimicrobial Behavior. Catal. Lett. 2020, 150, 2823–2840. [Google Scholar] [CrossRef]
- Sharma, R.K.; Sharma, S.; Dutta, S.; Zboril, R.; Gawande, M.B. Silica-nanosphere-based organic–inorganic hybrid nanomaterials: Synthesis, functionalization and applications in catalysis. Green Chem. 2015, 17, 3207–3230. [Google Scholar] [CrossRef]
- Harish, N.; Kathyayini, N.; Bindhu, B.; Nagaraju, N. Investigation on the nature of catalytically active sites in non-crystalline aluminophosphates from 27Al and 31P MAS-NMR studies. J. NonCryst. Solids 2019, 522, 119577. [Google Scholar]
- Luna, D.; Bautista, F.M.; Garcia, A.; Campelo, J.M.; Marinas, J.M.; Romero, A.A.; Llobet, A.; Romero, I.; Serrano, I. Method for the Chemical Binding of Homogeneous Catalysts to Inorganic Solids Supports, Products Thus Obtained and Application of Same. International Patent Application No. PCT/ES2004/000187; International Publication No. WO 2004/096442 A1, 11 November 2004. [Google Scholar]
- Mola, J.; Rodríguez, A.M.; Romero, I.; Llobet, A.; Parella, T.; Poater, A.; Duran, A.M.; Solà, M.; Benet-Buchholz, J. New Ru Complexes Containing the N-Tridentate bpea and Phosphine Ligands: Consequences of Meridional vs. Facial Geometry. Inorg. Chem. 2006, 45, 10520–10529. [Google Scholar] [CrossRef] [PubMed]
- Vaquer, L.; Poater, A.; De Tovar, J.; García-Antón, J.; Solà, M.; Llobet, A.; Sala, X. Ruthenium Complexes with Chiral Bis-Pinene Ligands: An Array of Subtle Structural Diversity. Inorg. Chem. 2013, 52, 4985–4992. [Google Scholar] [CrossRef] [PubMed]
- Mola, J.; Romero, I.; Rodríguez, M.; Bozoglian, F.; Poater, A.; Solà, M.; Parella, T.; Benet-Buchholz, J.; Fontrodona, X.; Llobet, A. Mechanistic Insights into the Chemistry of Ru(II) Complexes Containing Cl and DMSO Ligands. Inorg. Chem. 2007, 46, 10707–10716. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Fukui, S.; Oi, T.; Nagao, H. Synthesis and Characterization of Ruthenium Complexes Having TridentateN-Ethyl-N,N-bis(2-pyridylmethyl)amine Coordinating in a Facial or Meridional Fashion. Bull. Chem. Soc. Jpn. 2008, 81, 1285–1295. [Google Scholar] [CrossRef]
- Poater, A.; Mola, J.; Saliner, A.G.; Romero, I.; Rodríguez, M.; Llobet, A.; Solà, M. Mechanistic theoretical insight of Ru(II) catalysts with a meridional–facial bpea fashion competition. Chem. Phys. Lett. 2008, 458, 200–204. [Google Scholar] [CrossRef]
- Dakkach, M.; Atlamsani, A.; Parella, T.; Fontrodona, X.; Romero, I.; Rodríguez, M. New Aqua N-Heterocyclic Carbene Ru(II) Complexes with Two-Electron Process as Selective Epoxidation Catalysts: An Evaluation of Geometrical and Electronic Effects. Inorg. Chem. 2013, 52, 5077–5087. [Google Scholar] [CrossRef]
- Mola, J.; Pujol, D.; Rodríguez, M.; Romero, I.; Sala, X.; Katz, N.; Parella, T.; Benet-Buchholz, J.; Fontrodona, X.; Llobet, A. Synthesis and Structure of Novel RuII—N≡C—Me Complexes and their Activity Towards Nitrile Hydrolysis: An Examination of Ligand Effects. Aust. J. Chem. 2009, 62, 1675–1683. [Google Scholar] [CrossRef]
- Naota, T.; Takaya, H.; Murahashi, S.-I. Ruthenium-Catalyzed Reactions for Organic Synthesis. Chem. Rev. 1998, 98, 2599–2660. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.J.; Xie, J.H.; Li, Y.L.; Liu, S.; Zhou, Q.L. Enantioselective synthesis of chiral β-aryloxy alcohols by ruthenium-catalyzed ketone hydrogenation via dynamic kinetic resolution (DKR). Adv. Synth. Catal. 2010, 352, 81–84. [Google Scholar] [CrossRef]
- Serrano, I.; Rodríguez, M.; Romero, I.; Llobet, A.; Parella, T.; Campelo, J.M.; Luna, D.; Marinas, J.M.; Benet-Buchholz, J. Catalytic Ability of a Cationic Ru(II) Monochloro Complex for the Asymmetric Hydrogenation of Dimethyl Itaconate and Enamides. Inorg. Chem. 2006, 45, 2644–2651. [Google Scholar] [CrossRef]
- Sala, X.; Serrano, I.; Rodríguez, M.; Romero, I.; Llobet, A.; Van Leeuwen, P.W. Ruthenium-catalyzed asymmetric hydrogenation of N-(3,4-dihydro-2-naphthalenyl)-acetamide. Catal. Commun. 2008, 9, 117–119. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Support | SBET (m2/g) | Acidity | Basicity | |
|---|---|---|---|---|
| Pyridine | Dimethylpyridine | Benzoic Acid | ||
| Sepiolite | 233 | 10 | 9 | 115 |
| AlPO4/Sepiolite | 139 | 198 | 128 | 393 |
| Heterogeneous Ru Complexes | Support | Ru (%) |
|---|---|---|
| Ru-(DPPE) (supernatant in synthesis) | - | 0.40 ± 0.01 |
| AlPO4-Sepiolite@2 | AlPO4-Sepiolite | 1.65 ± 0.03 |
| AlPO4-Sepiolite@2 (after 14 successive uses) | AlPO4-Sepiolite | 0.25 ± 0.01 |
| Ru-BINAP (supernatant in synthesis) | +++ - | 0.48 ± 0.02 |
| AlPO4-Sepiolite@1 | AlPO4-Sepiolite | 1.50 ± 0.02 |
| AlPO4-Sepiolite@1 (after 12 successive uses) | AlPO4-Sepiolite | 1.46 ± 0.01 |
| Substrate | t (h) | Conv. (%) | Rate (mmol/h) | TOF (h−1) |
|---|---|---|---|---|
| 1-hexene | 24 | 100 | 5.2 | 86.8 |
| Methyl acetoacetate | 68 | 24 | 0.2 | 3.4 |
| Nº | Substrate | Product | t (h) | Conv. (%) | Rate (μmol/h) | TOF (h−1) |
|---|---|---|---|---|---|---|
| Supernatant | 1-hexene | n-hexane | 46 | 100 | 320.0 | 2.404 |
| 1 a | 1-hexene | n-hexane | 30 | 100 | 4000.0 | 7.198 |
| 2 b | Methyl acetoacetate | Methyl 3-hydroxybutyrate | 15 | 2.3 | 24.6 | 0.044 |
| 3 | Methyl acetoacetate | Methyl 3-hydroxybutyrate | 72 | 0.16 | 71.0 | 0.128 |
| Methyl acetoacetate | Methyl 3-hydroxybutyrate | 120 | 0.72 | 61.1 | 0.110 | |
| 4 | Acetophenone | Phenylethanol | 72 | 0.4 | 14.7 | 0.026 |
| Acetophenone | Phenylethanol | 120 | 4.4 | 11.7 | 0.021 | |
| 5 | Methyl acetoacetate | Methyl 3-hydroxybutyrate | 42 | 6.7 | 51.1 | 0.092 |
| 6 | Ethyl pyruvate | Ethyl lactate | 44 | 13.5 | 98.3 | 0.176 |
| 7 | Benzaldehyde | Benzyl alcohol | 18 | 1.6 | 29.3 | 0.052 |
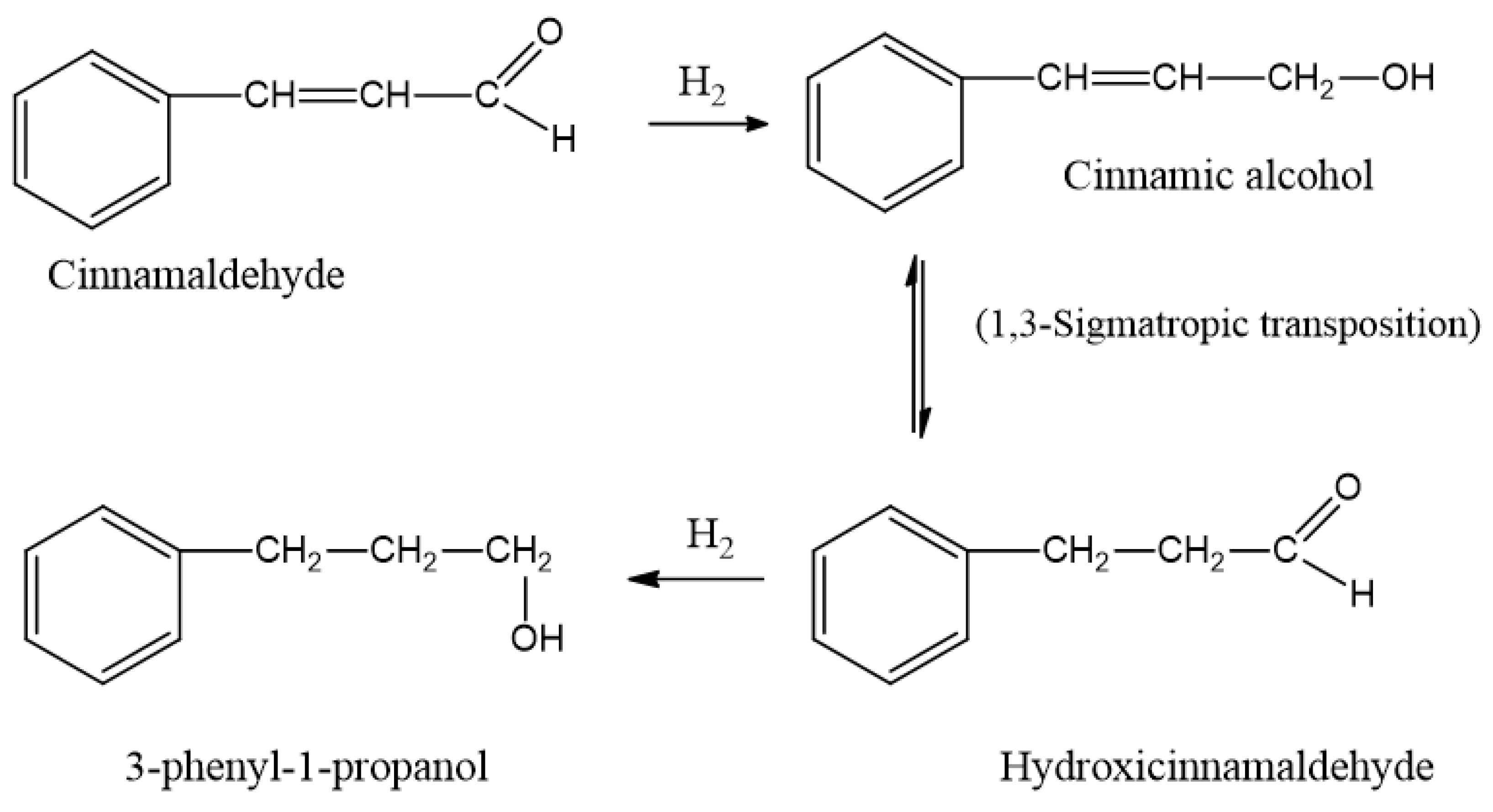
| 8 | Trans cinnamaldehyde | Hydrocinnamaldehyde | 140 | 21.1 | 48.3 | 0.088 |
| Cinnamic alcohol | 140 | 25 | 57.2 | 0.104 | ||
| 9 | Hydrocinnamaldehyde | 3-phenyl-1-propanol | 21 | 0.7 | 10.5 | 0.020 |
| 10 | Cinnamic alcohol | Hydrocinnamaldehyde | 40 | 92 | 736.6 | 1.328 |
| 3-phenyl-1-propanol | 40 | 1.64 | 13.1 | 0.024 | ||
| Cinnamic alcohol | Hydrocinnamaldehyde | 87 | 90 | 331.4 | 0.596 | |
| 3-phenyl-1-propanol | 87 | 10 | 36.8 | 0.068 | ||
| 11 | Geranial | Geraniol | 47 | 59.2 | 403.4 | 0.728 |
| 12 | Furfural | Furfuryl alcohol | 70 | 6.6 | 30.4 | 0.056 |
| 13 | o-hydroxyacetophenone | o-hydroxy-phenylethanol | 40 | 0.2 | 1.60 | 0.003 |
| 14 c | Benzylideneacetone | 4-phenyl-3-buten-2-ol | 72 | 100 | 444.4 | 0.800 |
| Nº | Substrate | t (h) | T (°C) | Conv. (%) | Rate (μmol/h) | TOF (h−1) | ee (%) |
|---|---|---|---|---|---|---|---|
| Supern | Acetophenone | 70 | 50 | 0.7 | 3.2 | 0.0054 | 6.7 |
| 1 | Acetophenone | 168 | 50 | 68.0 | 129.5 | 0.218 | 6.4 |
| 2 | Acetophenone | 165 | 45 | 63.0 | 122.2 | 0.206 | 5.2 |
| 3 | Acetophenone | 216 | 40 | 71.0 | 105.2 | 0.177 | 3.2 |
| 4 | Acetophenone | 150 | 35 | 44.0 | 93.9 | 0.158 | 5.0 |
| 5 | Acetophenone | 189 | 30 | 52.0 | 88.0 | 0.148 | 3.4 |
| 6 | Acetophenone | 168 | 25 | 41.0 | 78.1 | 0.131 | 3.8 |
| 7 | Propiophenone | 168 | 50 | 23.6 | 45.0 | 0.076 | 18.8 |
| 8 | Propiophenone | 144 | 45 | 16.0 | 35.6 | 0.060 | 20.7 |
| 9 | Propiophenone | 143 | 40 | 11.9 | 26.74 | 0.045 | 23.0 |
| 10 | Propiophenone | 220 | 35 | 13.6 | 19.78 | 0.033 | 20.8 |
| 11 | Propiophenone | 144 | 30 | 5.6 | 12.4 | 0.021 | 23.0 |
| 12 a | Propiophenone | 166 | 25 | 4.9 | 9.4 | 0.016 | 20.8 |
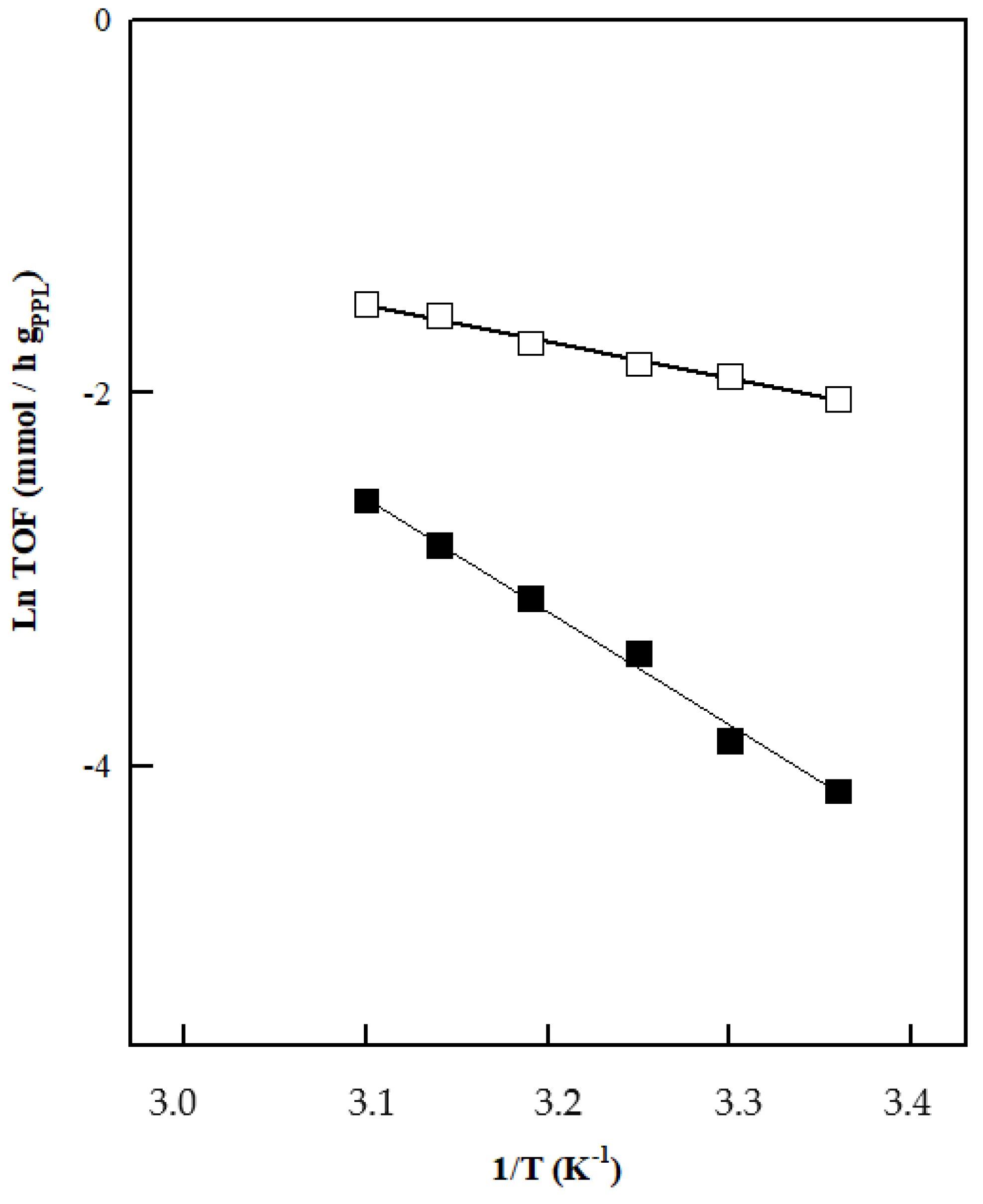
| Substrate | Solvent | Ea (Kcal/mol) | lnA (h−1) | r2 |
|---|---|---|---|---|
| Acetophenone | MeOH | 3.94 ± 0.10 | 4.62 ± 0.33 | 0.99 |
| Propiophenone | MeOH | 12.13 ± 0.26 | 16.37 ± 0.84 | 0.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caballero, V.; Estevez, R.; Luna, D.; Bautista, F.M.; Romero, A.A.; Aguado-Deblas, L.; Hidalgo-Carrillo, J.; Romero, I. Hydrogenation of α,β-Unsaturated Carbonyl Compounds over Covalently Heterogenized Ru(II) Diphosphine Complexes on AlPO4-Sepiolite Supports. Catalysts 2021, 11, 289. https://doi.org/10.3390/catal11020289
Caballero V, Estevez R, Luna D, Bautista FM, Romero AA, Aguado-Deblas L, Hidalgo-Carrillo J, Romero I. Hydrogenation of α,β-Unsaturated Carbonyl Compounds over Covalently Heterogenized Ru(II) Diphosphine Complexes on AlPO4-Sepiolite Supports. Catalysts. 2021; 11(2):289. https://doi.org/10.3390/catal11020289
Chicago/Turabian StyleCaballero, Verónica, Rafael Estevez, Diego Luna, Felipa M. Bautista, Antonio A. Romero, Laura Aguado-Deblas, Jesús Hidalgo-Carrillo, and Isabel Romero. 2021. "Hydrogenation of α,β-Unsaturated Carbonyl Compounds over Covalently Heterogenized Ru(II) Diphosphine Complexes on AlPO4-Sepiolite Supports" Catalysts 11, no. 2: 289. https://doi.org/10.3390/catal11020289