
DMF as CO Surrogate in Carbonylation Reactions: Principles and Application to the Synthesis of Heterocycles



Abstract
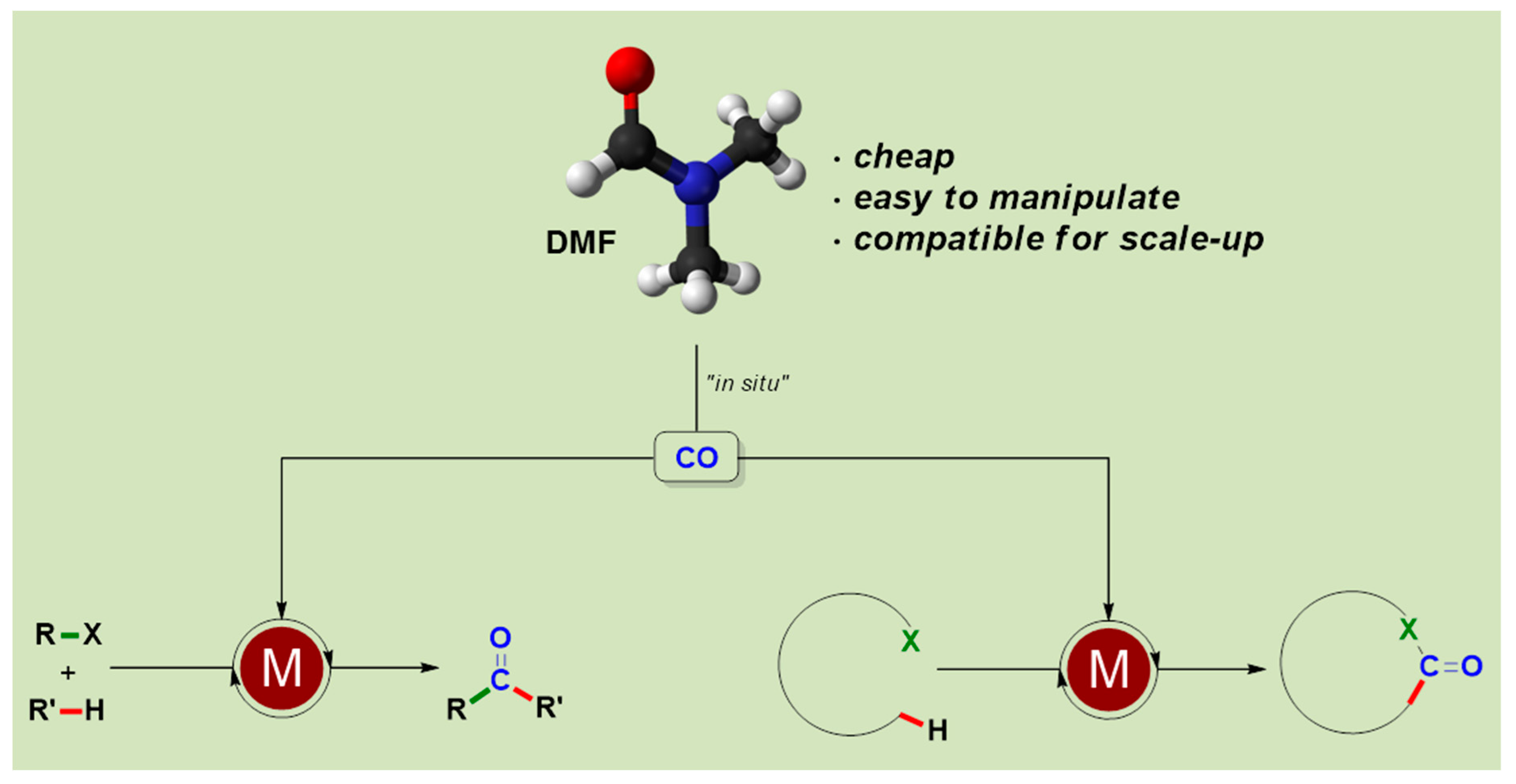
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Transition Metal-Catalyzed Carbonylation Reactions Involving DMF as a CO Surrogate
2.1. Palladium-Catalyzed Carbonylation Involving DMF as a CO Surrogate
2.2. Nickel-Catalyzed Carbonylation Involving DMF as a CO Surrogate
3. Synthesis of Heterocycles by Transition Metal-Catalyzed Carbonylation Reactions Involving DMF as a CO Surrogate
3.1. Synthesis of Heterocycles by Palladium-Catalyzed Carbonylation Involving DMF as a CO Surrogate
3.2. Synthesis of Heterocycles by Nickel-Catalyzed Carbonylation Involving DMF as a CO Surrogate
3.3. Critical Aspects and Perspectives in the Synthesis of Heterocycles by Transition Metal-Catalyzed Carbonylation Reactions Involving DMF as a CO Surrogate
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Semchyshyn, H.M. Reactive Carbonyl Species In Vivo: Generation and Dual Biological Effects. Sci. World J. 2014, 2014, 417842. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zajaczkowski, W.; Velpula, G.; Jänsch, D.; Graf, R.; Marszalek, T.; Parekh, S.H.; Zagranyarski, Y.; Mali, K.; Wagner, M.; et al. Transformation from helical to layered supramolecular organization of asymmetric perylene diimides via multiple intermolecular hydrogen bonding. Chem. Sci. 2020, 11, 4960–4968. [Google Scholar] [CrossRef]
- Hassan Omar, O.; Falcone, M.; Operamolla, A.; Albano, G. Impact of chirality on the aggregation modes of L-phenylalanine- and D-glucose-decorated phenylene–thiophene oligomers. New J. Chem. 2021, 45, 12016–12023. [Google Scholar] [CrossRef]
- Schoenberg, A.; Bartoletti, I.; Heck, R.F. Palladium-catalyzed carboalkoxylation of aryl, benzyl, and vinylic halides. J. Org. Chem. 1974, 39, 3318–3326. [Google Scholar] [CrossRef]
- Wu, X.-F.; Neumann, H.; Beller, M. Palladium-catalyzed carbonylative coupling reactions between Ar–X and carbon nucleophiles. Chem. Soc. Rev. 2011, 40, 4986–5009. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.-B.; Qi, X.; Wu, X.-F. Recent Achievements in Carbonylation Reactions: A Personal Account. Synlett 2017, 28, 175–194. [Google Scholar]
- Dekleva, T.W.; Forster, D. The rhodium-catalyzed carbonylation of linear primary alcohols. J. Am. Chem. Soc. 1985, 107, 3565–3567. [Google Scholar] [CrossRef]
- Haynes, A.; Maitlis, P.M.; Morris, G.E.; Sunley, G.J.; Adams, H.; Badger, P.W.; Bowers, C.M.; Cook, D.B.; Elliott, P.I.P.; Ghaffar, T.; et al. Promotion of Iridium-Catalyzed Methanol Carbonylation: Mechanistic Studies of the Cativa Process. J. Am. Chem. Soc. 2004, 126, 2847–2861. [Google Scholar] [CrossRef]
- Morimoto, T.; Kakiuchi, K. Evolution of Carbonylation Catalysis: No Need for Carbon Monoxide. Angew. Chem. Int. Ed. 2004, 43, 5580–5588. [Google Scholar] [CrossRef]
- Wu, L.; Liu, Q.; Jackstell, R.; Beller, M. Carbonylations of Alkenes with CO Surrogates. Angew. Chem. Int. Ed. 2014, 53, 6310–6320. [Google Scholar] [CrossRef]
- Gautam, P.; Bhanage, B.M. Recent advances in the transition metal catalyzed carbonylation of alkynes, arenes and aryl halides using CO surrogates. Catal. Sci. Technol. 2015, 5, 4663–4702. [Google Scholar] [CrossRef]
- Chen, J.; Natte, K.; Spannenberg, A.; Neumann, H.; Beller, M.; Wu, X.-F. Palladium-Catalyzed Carbonylative [3+2+1] Annulation of N-Aryl-Pyridine-2-Amines with Internal Alkynes by C–H Activation: Facile Synthesis of 2-Quinolinones. Chem. Eur. J. 2014, 20, 14189–14193. [Google Scholar] [CrossRef]
- Cao, J.; Zheng, Z.-J.; Xu, Z.; Xu, L.-W. Transition-metal-catalyzed transfer carbonylation with HCOOH or HCHO as non-gaseous C1 source. Coord. Chem. Rev. 2017, 336, 43–53. [Google Scholar] [CrossRef]
- Li, H.; Neumann, H.; Beller, M.; Wu, X.-F. Aryl Formate as Bifunctional Reagent: Applications in Palladium-Catalyzed Carbonylative Coupling Reactions Using In Situ Generated CO. Angew. Chem. Int. Ed. 2014, 53, 3183–3186. [Google Scholar] [CrossRef] [PubMed]
- Natte, K.; Dumrath, A.; Neumann, H.; Beller, M. Palladium-Catalyzed Carbonylations of Aryl Bromides using Paraformaldehyde: Synthesis of Aldehydes and Esters. Angew. Chem. Int. Ed. 2014, 53, 10090–10094. [Google Scholar] [CrossRef] [PubMed]
- Shil, A.K.; Kumar, S.; Reddy, C.B.; Dadhwal, S.; Thakur, V.; Das, P. Supported Palladium Nanoparticle-Catalyzed Carboxylation of Aryl Halides, Alkenylsilanes, and Organoboronic Acids Employing Oxalic Acid as the C1 Source. Org. Lett. 2015, 17, 5352–5355. [Google Scholar] [CrossRef]
- Lescot, C.; Nielsen, D.U.; Makarov, I.S.; Lindhardt, A.T.; Daasbjerg, K.; Skrydstrup, T. Efficient Fluoride-Catalyzed Conversion of CO2 to CO at Room Temperature. J. Am. Chem. Soc. 2014, 136, 6142–6147. [Google Scholar] [CrossRef]
- Mondal, K.; Halder, P.; Gopalan, G.; Sasikumar, P.; Radhakrishnan, K.V.; Das, P. Chloroform as a CO surrogate: Applications and recent developments. Org. Biomol. Chem. 2019, 17, 5212–5222. [Google Scholar] [CrossRef]
- Albano, G.; Aronica, L.A. Potentiality and Synthesis of O- and N-Heterocycles: Pd-Catalyzed Cyclocarbonylative Sonogashira Coupling as a Valuable Route to Phthalans, Isochromans, and Isoindolines. Eur. J. Org. Chem. 2017, 2017, 7204–7221. [Google Scholar] [CrossRef]
- Aronica, L.A.; Albano, G.; Giannotti, L.; Meucci, E. Synthesis of N-Heteroaromatic Compounds through Cyclocarbonylative Sonogashira Reactions. Eur. J. Org. Chem. 2017, 2017, 955–963. [Google Scholar] [CrossRef]
- Albano, G.; Giuntini, S.; Aronica, L.A. Synthesis of 3-Alkylideneisoindolin-1-ones via Sonogashira Cyclocarbonylative Reactions of 2-Ethynylbenzamides. J. Org. Chem. 2020, 85, 10022–10034. [Google Scholar] [CrossRef]
- Albano, G.; Evangelisti, C.; Aronica, L.A. Palladium Nanoparticles Supported on Smopex-234® as Valuable Catalysts for the Synthesis of Heterocycles. Catalysts 2021, 11, 706. [Google Scholar] [CrossRef]
- Albano, G.; Morelli, M.; Aronica, L.A. Synthesis of Functionalised 3-Isochromanones by Silylcarbocyclisation/Desilylation Reactions. Eur. J. Org. Chem. 2017, 2017, 3473–3480. [Google Scholar] [CrossRef]
- Albano, G.; Morelli, M.; Lissia, M.; Aronica, L.A. Synthesis of Functionalised Indoline and Isoquinoline Derivatives through a Silylcarbocyclisation/Desilylation Sequence. ChemistrySelect 2019, 4, 2505–2511. [Google Scholar] [CrossRef]
- Albano, G.; Aronica, L.A. From Alkynes to Heterocycles through Metal-Promoted Silylformylation and Silylcarbocyclization Reactions. Catalysts 2020, 10, 1012. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, L.-C.; Wu, X.-F. Carbonylative synthesis of heterocycles involving diverse CO surrogates. Chem. Commun. 2020, 56, 6016–6030. [Google Scholar] [CrossRef]
- Ding, S.; Jiao, N. N,N-Dimethylformamide: A Multipurpose Building Block. Angew. Chem. Int. Ed. 2012, 51, 9226–9237. [Google Scholar] [CrossRef]
- Wan, Y.; Alterman, M.; Larhed, M.; Hallberg, A. Dimethylformamide as a Carbon Monoxide Source in Fast Palladium-Catalyzed Aminocarbonylations of Aryl Bromides. J. Org. Chem. 2002, 67, 6232–6235. [Google Scholar] [CrossRef]
- Heravi, M.M.; Ghavidel, M.; Mohammadkhani, L. Beyond a solvent: Triple roles of dimethylformamide in organic chemistry. RSC Adv. 2018, 8, 27832–27862. [Google Scholar] [CrossRef] [Green Version]
- Hosoi, K.; Nozaki, K.; Hiyama, T. Carbon Monoxide Free Aminocarbonylation of Aryl and Alkenyl Iodides Using DMF as an Amide Source. Org. Lett. 2002, 4, 2849–2851. [Google Scholar] [CrossRef]
- Vilsmeier, A.; Haack, A. Über die Einwirkung von Halogenphosphor auf Alkyl-formanilide. Eine neue Methode zur Darstellung sekundärer und tertiärer p-Alkylamino-benzaldehyde. Ber. Dtsch. Chem. Ges. 1927, 60, 119–122. [Google Scholar] [CrossRef]
- Tambade, P.J.; Patil, Y.P.; Bhanushali, M.J.; Bhanage, B.M. Pd/C: An efficient, heterogeneous and reusable catalyst for carbon monoxide-free aminocarbonylation of aryl iodides. Tetrahedron Lett. 2008, 49, 2221–2224. [Google Scholar] [CrossRef]
- Sawant, D.N.; Wagh, Y.S.; Bhatte, K.D.; Bhanage, B.M. Palladium-Catalyzed Carbon-Monoxide-Free Aminocarbonylation of Aryl Halides Using N-Substituted Formamides as an Amide Source. J. Org. Chem. 2011, 76, 5489–5494. [Google Scholar] [CrossRef]
- Feng, X.; Li, Z. Photocatalytic promoting dimethylformamide (DMF) decomposition to in-situ generation of self-supplied CO for carbonylative Suzuki reaction. J. Photochem. Photobiol. A 2017, 337, 19–24. [Google Scholar] [CrossRef]
- Tasker, S.Z.; Standley, E.A.; Jamison, T.F. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.K.; Burton, P.M.; Nelson, D.J. Nickel versus Palladium in Cross-Coupling Catalysis: On the Role of Substrate Coordination to Zerovalent Metal Complexes. Synthesis 2020, 52, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.; Jeong, M.; Moon, J.; Jung, H.M.; Lee, S. Aminocarbonylation of Aryl Halides Using a Nickel Phosphite Catalytic System. Org. Lett. 2007, 9, 4615–4618. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Ju, J.; Choe, J.; Song, K.H.; Lee, S. The Scope and Limitation of Nickel-Catalyzed Aminocarbonylation of Aryl Bromides from Formamide Derivatives. J. Org. Chem. 2009, 74, 6358–6361. [Google Scholar] [CrossRef]
- Li, Y.; Tu, D.-H.; Wang, B.; Lu, J.-Y.; Wang, Y.-Y.; Liu, Z.-T.; Liu, Z.-W.; Lu, J. Nickel-catalyzed carbonylation of arylboronic acids with DMF as a CO source. Org. Chem. Front. 2017, 4, 569–572. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, K.; Yan, B.; Li, S.; Du, Y. Hydrothermal Method Using DMF as a Reducing Agent for the Fabrication of PdAg Nanochain Catalysts towards Ethanol Electrooxidation. Catalysts 2016, 6, 103. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Feng, J.-B.; Natte, K.; Wu, X.-F. Palladium-Catalyzed Carbonylative Cyclization of Arenes by C-H Bond Activation with DMF as the Carbonyl Source. Chem. Eur. J. 2015, 21, 16370–16373. [Google Scholar] [CrossRef]
- Chen, J.; Natte, K.; Spannenberg, A.; Neumann, H.; Langer, P.; Beller, M.; Wu, X.-F. Base-Controlled Selectivity in the Synthesis of Linear and Angular Fused Quinazolinones by a Palladium-Catalyzed Carbonylation/Nucleophilic Aromatic Substitution Sequence. Angew. Chem. Int. Ed. 2014, 53, 7579–7583. [Google Scholar] [CrossRef]
- Khan, I.; Ibrar, A.; Abbas, N.; Saeed, A. Recent advances in the structural library of functionalized quinazoline and quinazolinone scaffolds: Synthetic approaches and multifarious applications. Eur. J. Med. Chem. 2014, 76, 193–244. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Zaib, S.; Batool, S.; Abbas, N.; Ashraf, Z.; Iqbal, J.; Saeed, A. Quinazolines and quinazolinones as ubiquitous structural fragments in medicinal chemistry: An update on the development of synthetic methods and pharmacological diversification. Biorg. Med. Chem. 2016, 24, 2361–2381. [Google Scholar] [CrossRef] [PubMed]
- Horváth, I.T.; Anastas, P.T. Innovations and Green Chemistry. Chem. Rev. 2007, 107, 2169–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Panda, B.; Sarkar, T.K. Gold and Palladium Combined for the Sonogashira Coupling of Aryl and Heteroaryl Halides. Synthesis 2013, 45, 817–829. [Google Scholar]
- Mphahlele, M.J.; Maluleka, M.M. Advances in Metal-Catalyzed Cross-Coupling Reactions of Halogenated Quinazolinones and Their Quinazoline Derivatives. Molecules 2014, 19, 17435–17463. [Google Scholar] [CrossRef] [PubMed]
- Campanati, M.; Fornasari, G.; Vaccari, A. Fundamentals in the preparation of heterogeneous catalysts. Catal. Today 2003, 77, 299–314. [Google Scholar] [CrossRef]
- Copéret, C.; Chabanas, M.; Petroff Saint-Arroman, R.; Basset, J.-M. Homogeneous and Heterogeneous Catalysis: Bridging the Gap through Surface Organometallic Chemistry. Angew. Chem. Int. Ed. 2003, 42, 156–181. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D.; Lu, F.; Aranzaes, J.R. Nanoparticles as Recyclable Catalysts: The Frontier between Homogeneous and Heterogeneous Catalysis. Angew. Chem. Int. Ed. 2005, 44, 7852–7872. [Google Scholar] [CrossRef]
- Albano, G.; Evangelisti, C.; Aronica, L.A. Hydrogenolysis of Benzyl Protected Phenols and Aniline Promoted by Supported Palladium Nanoparticles. ChemistrySelect 2017, 2, 384–388. [Google Scholar] [CrossRef]
- Chen, J.; Natte, K.; Wu, X.-F. Pd/C-catalyzed carbonylative C–H activation with DMF as the CO source. Tetrahedron Lett. 2015, 56, 6413–6416. [Google Scholar] [CrossRef]
- Bhattacharya, T.; Dutta, S.; Maiti, D. Deciphering the Role of Silver in Palladium-Catalyzed C–H Functionalizations. ACS Catal. 2021, 11, 9702–9714. [Google Scholar] [CrossRef]
- Piou, T.; Slutskyy, Y.; Kevin, N.J.; Sun, Z.; Xiao, D.; Kong, J. Direct Arylation of Azoles Enabled by Pd/Cu Dual Catalysis. Org. Lett. 2021, 23, 1996–2001. [Google Scholar] [CrossRef]
- Hamlin, J.E.; Hirai, K.; Millan, A.; Maitlis, P.M. A Simple Practical Test for Distinguishing a Heterogeneous Component in an Homogeneously Catalysed Reaction. J. Mol. Catal. A Chem. 1980, 7, 543–544. [Google Scholar]
- Albano, G.; Interlandi, S.; Evangelisti, C.; Aronica, L.A. Polyvinylpyridine-Supported Palladium Nanoparticles: A Valuable Catalyst for the Synthesis of Alkynyl Ketones via Acyl Sonogashira Reactions. Catal. Lett. 2020, 150, 652–659. [Google Scholar] [CrossRef]
- Nageswar Rao, D.; Rasheed, S.; Das, P. Palladium/Silver Synergistic Catalysis in Direct Aerobic Carbonylation of C(sp2)−H Bonds Using DMF as a Carbon Source: Synthesis of Pyrido-Fused Quinazolinones and Phenanthridinones. Org. Lett. 2016, 18, 3142–3145. [Google Scholar] [CrossRef]
- Aleti, R.R.; Festa, A.A.; Voskressensky, L.G.; Van der Eycken, E.V. Synthetic Strategies in the Preparation of Phenanthridinones. Molecules 2021, 26, 5560. [Google Scholar] [CrossRef]
- Nakamura, M.; Aoyama, A.; Salim, M.T.A.; Okamoto, M.; Baba, M.; Miyachi, H.; Hashimoto, Y.; Aoyama, H. Structural development studies of anti-hepatitis C virus agents with a phenanthridinone skeleton. Biorg. Med. Chem. 2010, 18, 2402–2411. [Google Scholar] [CrossRef]
- Fang, Y.; Tranmer, G.K. Continuous flow photochemistry as an enabling synthetic technology: Synthesis of substituted-6(5H)-phenanthridinones for use as poly(ADP-ribose) polymerase inhibitors. MedChemComm 2016, 7, 720–724. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Mori, S.; Makishima, M.; Fujii, S.; Kagechika, H.; Hashimoto, Y.; Ishikawa, M. Novel Nonsteroidal Progesterone Receptor (PR) Antagonists with a Phenanthridinone Skeleton. ACS Med. Chem. Lett. 2018, 9, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, Y.; Ge, H. Direct Aerobic Carbonylation of C(sp2)–H and C(sp3)–H Bonds through Ni/Cu Synergistic Catalysis with DMF as the Carbonyl Source. J. Am. Chem. Soc. 2015, 137, 4924–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lao, Z.-Q.; Zhong, W.-H.; Lou, Q.-H.; Li, Z.-J.; Meng, X.-B. KI-catalyzed imidation of sp3 C–H bond adjacent to amide nitrogen atom. Org. Biomol. Chem. 2012, 10, 7869–7871. [Google Scholar] [CrossRef]
- Itoh, M.; Hirano, K.; Satoh, T.; Miura, M. Copper-Catalyzed α-Methylenation of Benzylpyridines Using Dimethylacetamide as One-Carbon Source. Org. Lett. 2014, 16, 2050–2053. [Google Scholar] [CrossRef]
- Li, Y.; Xue, D.; Lu, W.; Wang, C.; Liu, Z.-T.; Xiao, J. DMF as Carbon Source: Rh-Catalyzed α-Methylation of Ketones. Org. Lett. 2014, 16, 66–69. [Google Scholar] [CrossRef]
- Kushwaha, N.; Kaushik, D. Recent Advances and Future Prospects of Phthalimide Derivatives. J. App. Pharm. Sci. 2016, 6, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Yue, J.; Ji, X.; Nian, M.; Kang, K.; Qiao, H.; Zheng, X. Research progress in biological activities of succinimide derivatives. Bioorg. Chem. 2021, 108, 104557. [Google Scholar] [CrossRef]























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panda, B.; Albano, G. DMF as CO Surrogate in Carbonylation Reactions: Principles and Application to the Synthesis of Heterocycles. Catalysts 2021, 11, 1531. https://doi.org/10.3390/catal11121531
Panda B, Albano G. DMF as CO Surrogate in Carbonylation Reactions: Principles and Application to the Synthesis of Heterocycles. Catalysts. 2021; 11(12):1531. https://doi.org/10.3390/catal11121531
Chicago/Turabian StylePanda, Biswajit, and Gianluigi Albano. 2021. "DMF as CO Surrogate in Carbonylation Reactions: Principles and Application to the Synthesis of Heterocycles" Catalysts 11, no. 12: 1531. https://doi.org/10.3390/catal11121531