Highly Active CO2 Fixation into Cyclic Carbonates Catalyzed by Tetranuclear Aluminum Benzodiimidazole-Diylidene Adducts



Abstract
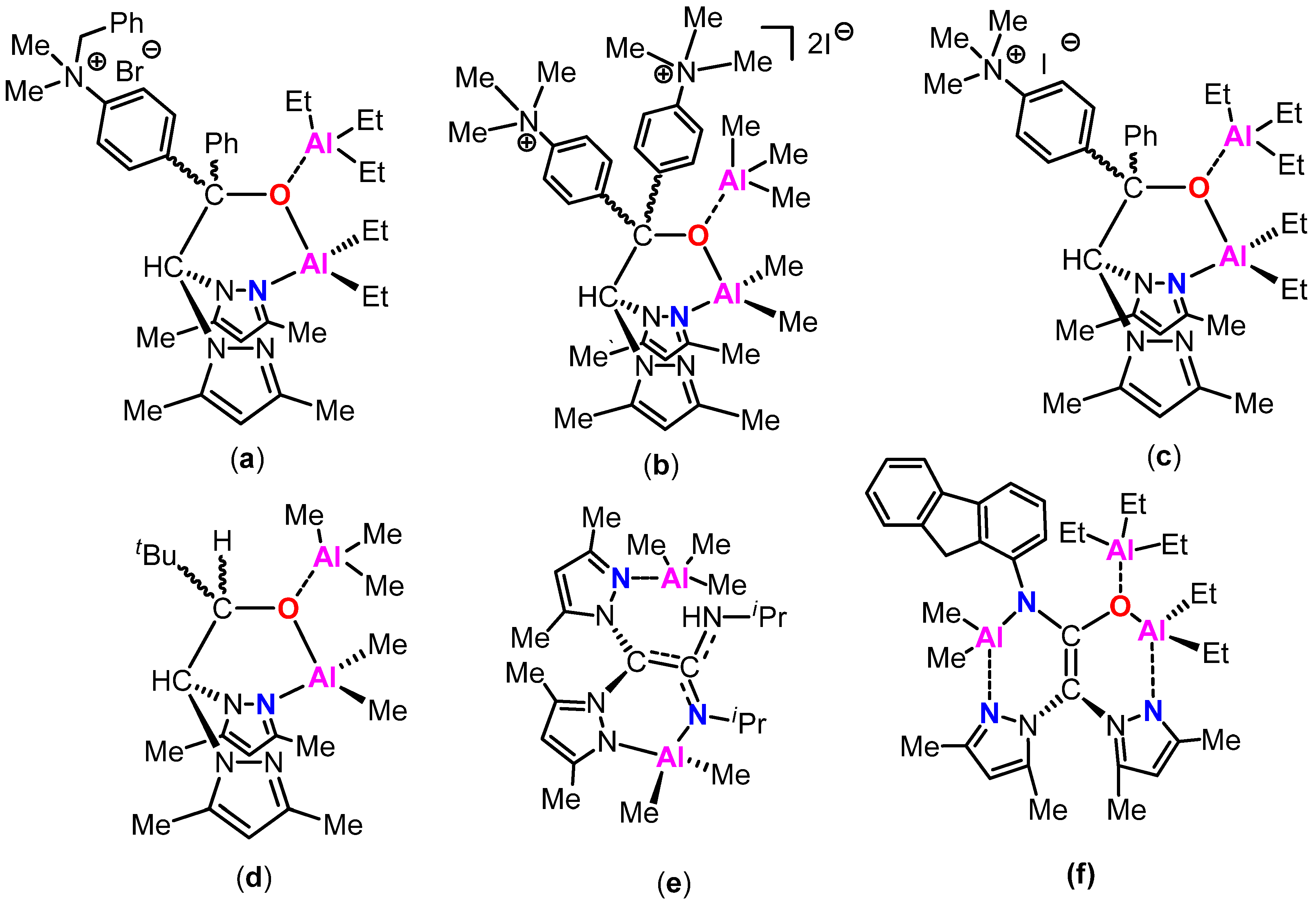
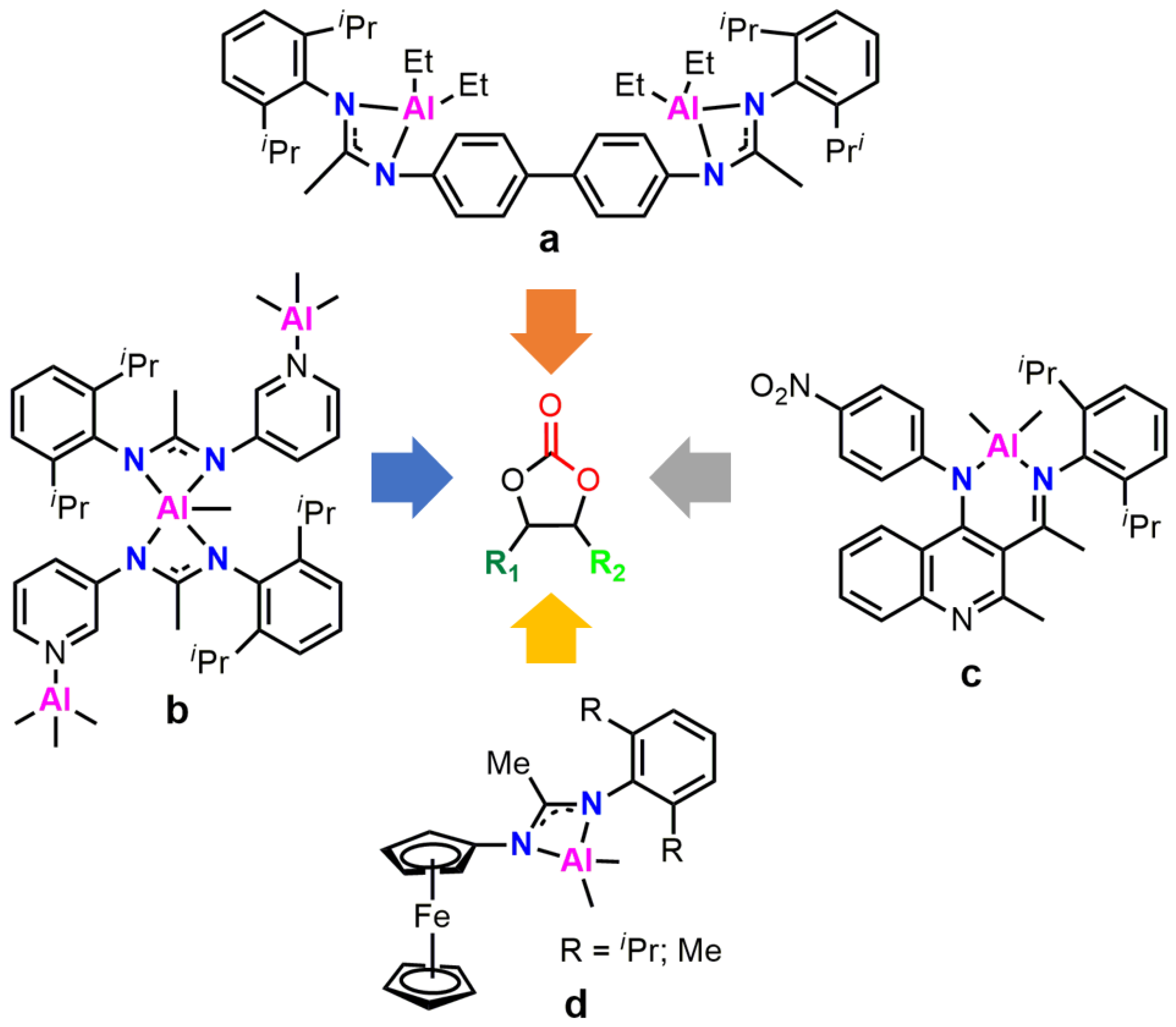
:1. Introduction
2. Results and Discussion
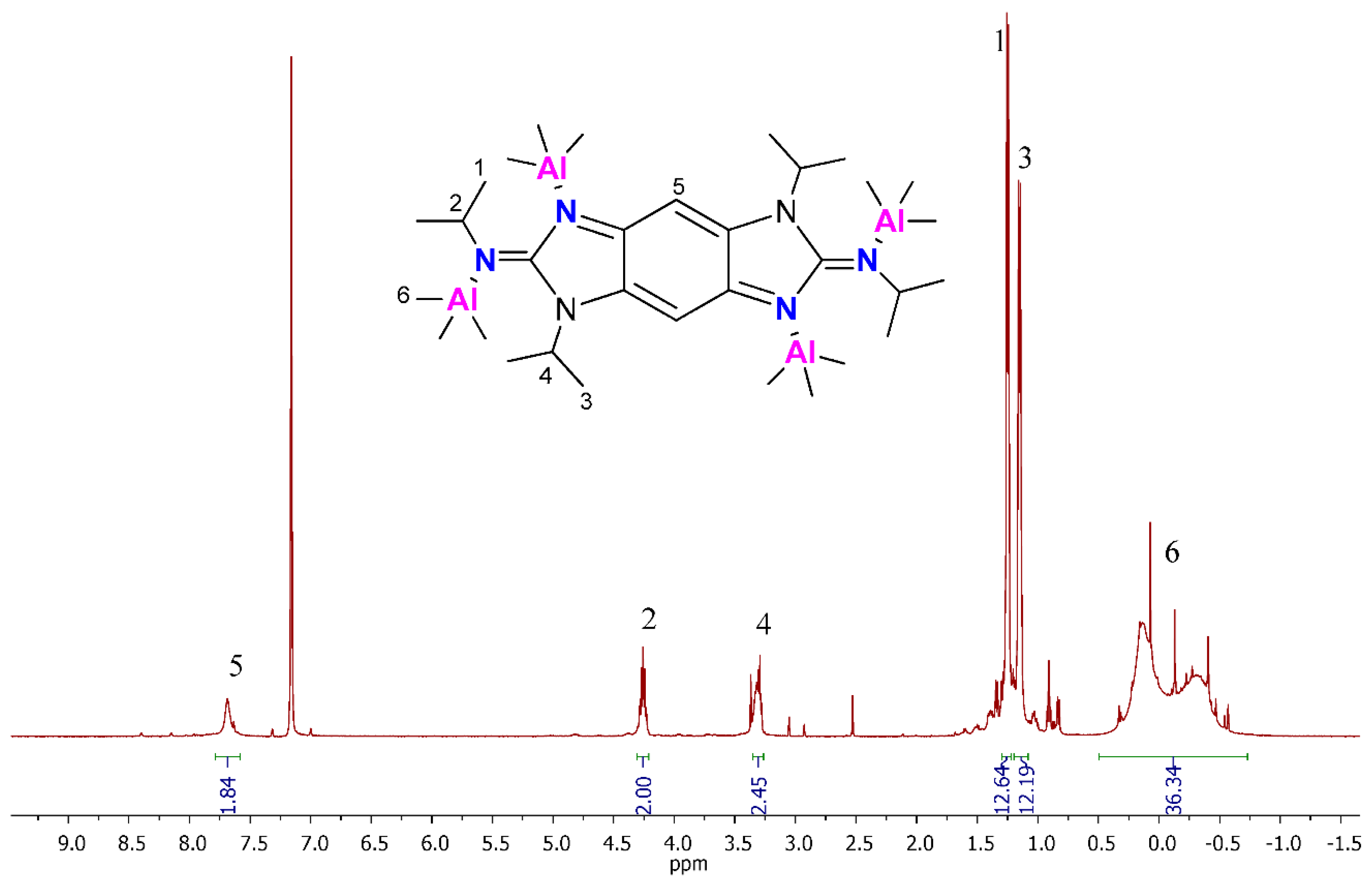
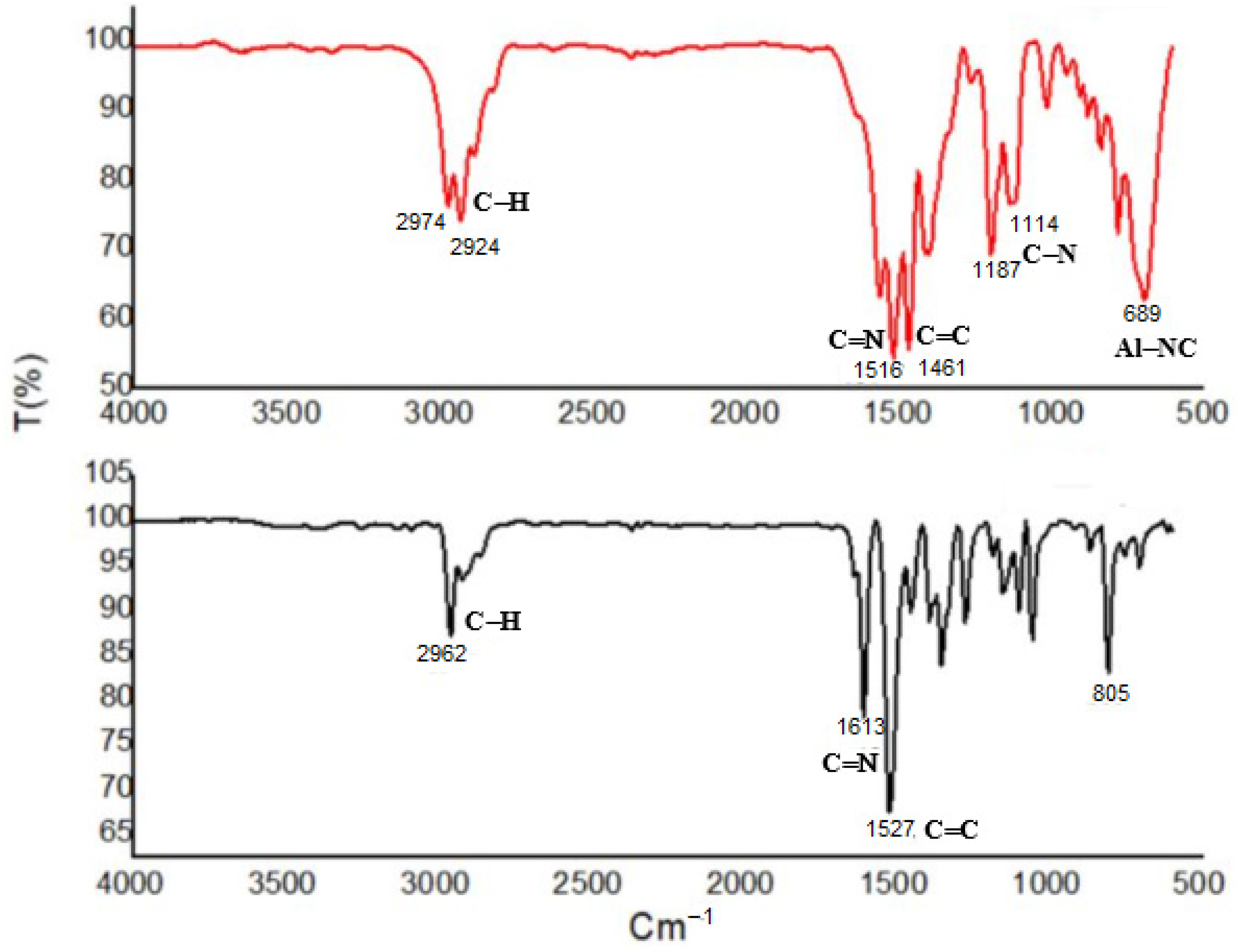
2.1. Tetranuclear Alkyl Aluminum Adduct Characterization
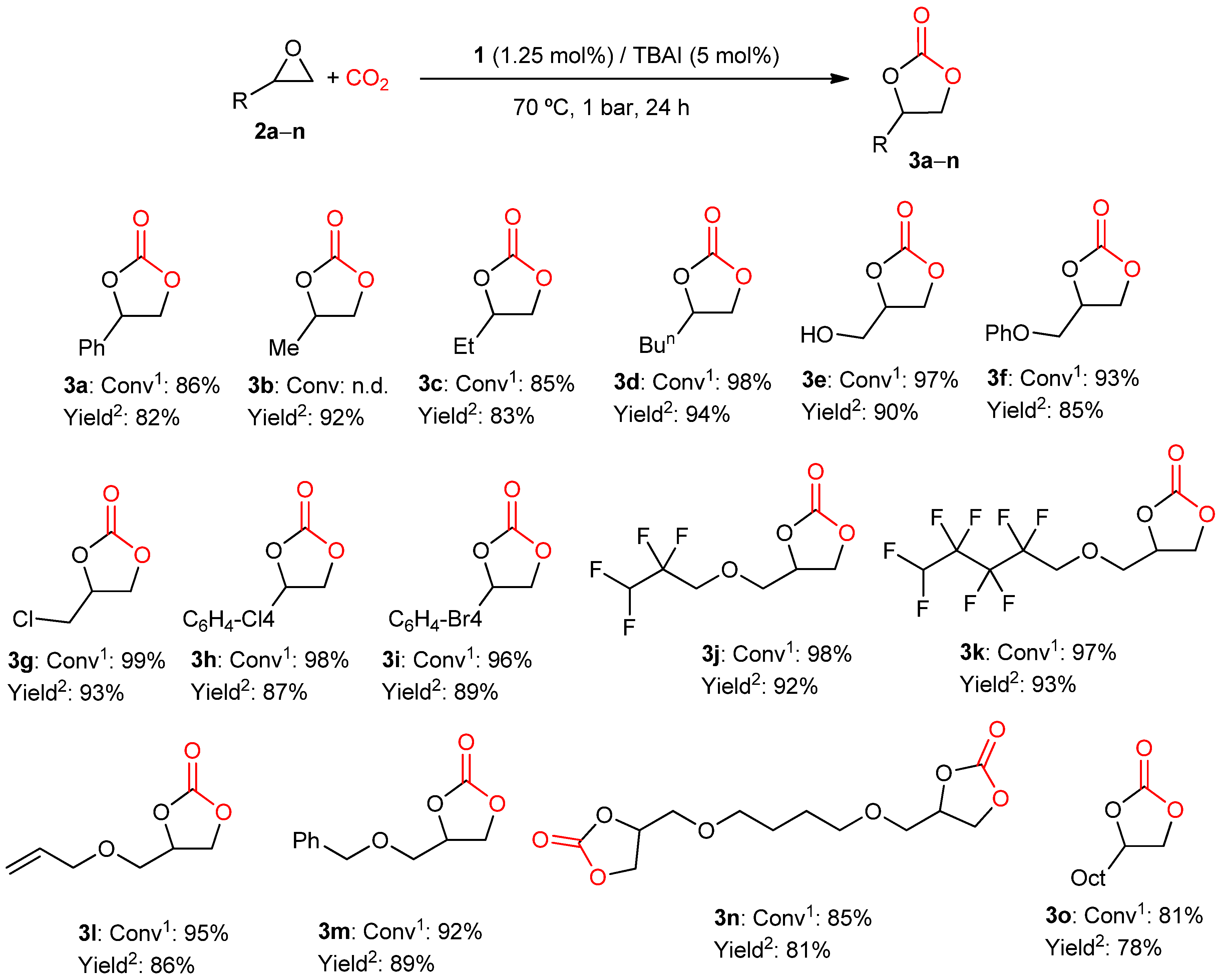
2.2. Catalytic Studies
2.3. Catalysis Comparison
3. Materials and Methods
3.1. Materials and Equipment
3.2. Catalyst Preparation and Characterization
3.3. General Procedure for Catalyst Screening at 1 bar Pressure
3.4. General Method for Preparation of Cyclic Carbonates 3a–o
3.5. NMR Data for Cyclic Carbonates 3a–o
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- James, J.A.; Sung, S.; Jeong, H.; Broesicke, O.A.; French, S.P.; Li, D.; Crittenden, J.C. Impacts of Combined Cooling, Heating and Power Systems, and Rainwater Harvesting on Water Demand, Carbon Dioxide, and NO x Emissions for Atlanta. Environ. Sci. Technol. 2018, 52, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.-W.; Zhou, Z.-H.; He, L.-N. Efficient, Selective and Sustainable Catalysis of Carbon Dioxide. Green Chem. 2017, 19, 3707–3728. [Google Scholar] [CrossRef]
- Dabral, S.; Schaub, T. The Use of Carbon Dioxide (CO2) as a Building Block in Organic Synthesis from an Industrial Perspective. Adv. Synth. Catal. 2019, 361, 223–246. [Google Scholar] [CrossRef] [Green Version]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Bui, M.; Adjiman, C.S.; Bardow, A.; Anthony, E.J.; Boston, A.; Brown, S.; Fennell, P.S.; Fuss, S.; Galindo, A.; Hackett, L.A.; et al. Carbon Capture and Storage (CCS): The Way Forward. Energy Environ. Sci. 2018, 11, 1062–1176. [Google Scholar] [CrossRef] [Green Version]
- Reis Machado, A.S.; Nunes da Ponte, M. CO2 Capture and Electrochemical Conversion. Curr. Opin. Green Sustain. Chem. 2018, 11, 86–90. [Google Scholar] [CrossRef]
- Fang, C.; Lu, C.; Liu, M.; Zhu, Y.; Fu, Y.; Lin, B.-L. Selective Formylation and Methylation of Amines Using Carbon Dioxide and Hydrosilane Catalyzed by Alkali-Metal Carbonates. ACS Catal. 2016, 6, 7876–7881. [Google Scholar] [CrossRef]
- Tundo, P.; Selva, M. The chemistry of dimethyl carbonate. Acc. Chem. Res. 2002, 35, 706–716. [Google Scholar] [CrossRef]
- Jacquet, O.; Das Neves Gomes, C.; Ephritikhine, M.; Cantat, T. Recycling of Carbon and Silicon Wastes: Room Temperature Formylation of N−H Bonds Using Carbon Dioxide and Polymethylhydrosiloxane. J. Am. Chem. Soc. 2012, 134, 2934–2937. [Google Scholar] [CrossRef]
- Wesselbaum, S.; Moha, V.; Meuresch, M.; Brosinski, S.; Thenert, K.M.; Kothe, J.; vom Stein, T.; Englert, U.; Hölscher, M.; Klankermayer, J.; et al. Hydrogenation of carbon dioxide to methanol using a homogeneous ruthenium−Triphos catalyst: From mechanistic investigations to multiphase catalysis. Chem. Sci. 2015, 6, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Sun, Y.; Han, B. Green Carbon Science: Scientific Basis for Integrating Carbon Resource Processing, Utilization, and Recycling. Angew. Chem. Int. Ed. 2013, 52, 9620–9633. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable Polymers from Renewable Resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Martínez, F.; de Sarasa-Buchaca, M.M.; Martínez, J.; Tejeda, J.; Fernández-Baeza, J.; Alonso-Moreno, C.; Rodríguez, A.M.; Castro-Osma, J.A.; Lara-Sánchez, A. Bimetallic Zinc Catalysts for Ring-Opening Copolymerization Processes. Inorg. Chem. 2020, 59, 8412–8423. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Ye, Y.; Peng, H.; Zhou, X.; Xie, X.; Wang, X.; Wang, F. A One-Step Route to CO2-Based Block Copolymers by Simultaneous ROCOP of CO2/Epoxides and RAFT Polymerization of Vinyl Monomers. Angew. Chem. Int. Ed. 2018, 57, 3593–3597. [Google Scholar] [CrossRef]
- Kozak, C.M.; Ambrose, K.; Anderson, T.S. Copolymerization of carbon dioxide and epoxides by metal coordination complexes. Coord. Chem. Rev. 2018, 376, 565–587. [Google Scholar] [CrossRef]
- Wang, Y.; Darensbourg, D.J. Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. [Google Scholar] [CrossRef]
- Lopes, E.J.C.; Ribeiro, A.P.C.; Martins, L.M.D.R.S. New Trends in the Conversion of CO2 to Cyclic Carbonates. Catalysts 2020, 10, 479. [Google Scholar] [CrossRef]
- Akimana, E.; Wang, J.; Likhanova, N.V.; Chaemchuen, S.; Verpoort, F. MIL-101(Cr) for CO2 Conversion into Cyclic Carbonates, Under Solvent and Co-Catalyst Free Mild Reaction Conditions. Catalysts 2020, 10, 453. [Google Scholar] [CrossRef]
- Natongchai, W.; Pornpraprom, S.; D’ Elia, V. Synthesis of Bio-based Cyclic Carbonates Using a Bio-based Hydrogen Bond Donor: Application of Ascorbic Acid to the Cycloaddition of CO2 to Oleochemicals. Asian J. Org. Chem. 2020, 9, 801–810. [Google Scholar] [CrossRef]
- Della Monica, F.; Kleij, A.W. Mechanistic guidelines in nonreductive conversion of CO2: The case of cyclic carbonates. Catal. Sci. Technol. 2020, 10, 3483–3501. [Google Scholar] [CrossRef]
- Fiorani, G.; Guo, W.; Kleij, A.W. Sustainable conversion of carbon dioxide: The advent of organocatalysis. Green Chem. 2015, 17, 1375–1389. [Google Scholar] [CrossRef]
- Martínez, J.; Castro-Osma, J.A.; Alonso-Moreno, C.; Rodríguez-Diéguez, A.; North, M.; Otero, A.; Lara-Sánchez, A. One-Component Aluminum(heteroscorpionate) Catalysts for the Formation of Cyclic Carbonates from Epoxides and Carbon Dioxide. ChemSusChem 2017, 10, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Martínez, F.; de Sarasa Buchaca, M.M.; Martínez, J.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Rodríguez-Diéguez, A.; Castro-Osma, J.A.; Lara-Sánchez, A. Synthesis of Bio-Derived Cyclic Carbonates from Renewable Resources. ACS Sustain. Chem. Eng. 2019, 7, 20126–20138. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, Y.; Wang, K.; Fu, X.; Chen, F.; Jing, H. Chiral oligomers of spiro-salencobalt(III)X for catalytic asymmetric cycloaddition of epoxides with CO2. Catal. Commun. 2016, 81, 50–53. [Google Scholar] [CrossRef]
- Ema, T.; Miyazaki, Y.; Shimonishi, J.; Maeda, C.; Hasegawa, J. Bifunctional Porphyrin Catalysts for the Synthesis of Cyclic Carbonates from Epoxides and CO2: Structural Optimization and Mechanistic Study. J. Am. Chem. Soc. 2014, 136, 15270–15279. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; Lamb, K.; North, M. Cr(salophen) Complex Catalyzed Cyclic Carbonate Synthesis at Ambient Temperature and Pressure. ACS Catal. 2016, 6, 5012–5025. [Google Scholar] [CrossRef]
- Longwitz, L.; Steinbauer, J.; Spannenberg, A.; Werner, T. Calcium-Based Catalytic System for the Synthesis of Bio-Derived Cyclic Carbonates under Mild Conditions. ACS Catal. 2018, 8, 665–672. [Google Scholar] [CrossRef]
- Claver, C.; Yeamin, M.B.; Reguero, M.; Masdeu-Bultó, A.M. Recent advances in the use of catalysts based on natural products for the conversion of CO2 into cyclic carbonates. Green Chem. 2020, 22, 7665–7706. [Google Scholar] [CrossRef]
- Singh Dhankhar, S.; Ugale, B.; Nagaraja, C.M. Co-Catalyst-Free Chemical Fixation of CO2 into Cyclic Carbonates by using Metal-Organic Frameworks as Efficient Heterogeneous Catalysts. Chem. Asian J. 2020, 15, 2403–2427. [Google Scholar] [CrossRef]
- Pal, T.K.; De, D.; Bharadwaj, P.K. Metal–organic frameworks for the chemical fixation of CO2 into cyclic carbonates. Coord. Chem. Rev. 2020, 408, 213173. [Google Scholar] [CrossRef]
- Prasad, D.; Patil, K.N.; Chaudhari, N.K.; Kim, H.; Nagaraja, B.M.; Jadhav, A.H. Paving way for sustainable earth-abundant metal based catalysts for chemical fixation of CO2 into epoxides for cyclic carbonate formation. Catal. Rev. 2020. [Google Scholar] [CrossRef]
- Ma, J.; Liu, J.; Zhang, Z.; Han, B. Mechanisms of ethylene glycol carbonylation with carbon dioxide. Comput. Theor. Chem. 2012, 992, 103–109. [Google Scholar] [CrossRef]
- Hirose, T.M.; Shimizu, S.; Qu, S.; Shitara, H.; Kodama, K.; Wang, L. Economical synthesis of cyclic carbonates from carbon dioxide and halohydrins using K2CO3. RSC Adv. 2016, 6, 69040–69044. [Google Scholar] [CrossRef]
- Hu, J.; Ma, J.; Zhu, Q.; Qian, Q.; Han, H.; Mei, Q.; Han, B. Zinc(II)-catalyzed reactions of carbon dioxide and propargylic alcohols to carbonates at room temperature. Green Chem. 2016, 18, 382–385. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R.; Young, C. Synthesis of Cyclic Carbonates from Epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Lawrenson, S.B.; Arav, R.; North, M. The greening of peptide synthesis. Green Chem. 2017, 19, 1685–1691. [Google Scholar] [CrossRef] [Green Version]
- Sathish, M.; Sreeram, K.J.; Rao, J.R.; Nair, B.U. Cyclic Carbonate: A Recyclable Medium for Zero Discharge Tanning. ACS Sustain. Chem. Eng. 2016, 4, 1032–1040. [Google Scholar] [CrossRef]
- Yao, K.; Zheng, J.P.; Liang, R. Ethylene carbonate-free fluoroethylene carbonate-based electrolyte works better for freestanding Si-based composite paper anodes for Li-ion batteries. J. Power Sources 2018, 381, 164–170. [Google Scholar] [CrossRef]
- Chai, J.; Liu, Z.; Zhang, J.; Sun, J.; Tian, Z.; Ji, Y.; Tang, K.; Zhou, X.; Cui, G. A Superior Polymer Electrolyte with Rigid Cyclic Carbonate Backbone for Rechargeable Lithium Ion Batteries. ACS Appl. Mater. Interfaces 2017, 9, 17897–17905. [Google Scholar] [CrossRef]
- Guo, W.; Martínez-Rodríguez, L.; Kuniyil, R.; Martin, E.; Escudero-Adán, E.C.; Maseras, F.; Kleij, A.W. Stereoselective and Versatile Preparation of Tri- and Tetrasubstituted Allylic Amine Scaffolds under Mild Conditions. J. Am. Chem. Soc. 2016, 138, 11970–11978. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Seidi, F.; Crespy, D.; D’Elia, V. Polymers Based on Cyclic Carbonates as Trait d’Union Between Polymer Chemistry and Sustainable CO2 Utilization. ChemSusChem 2019, 12, 724–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphuis, A.J.; Picchioni, F.; Pescarmona, P.P. CO2-Fixation into Cyclic and Polymeric Carbonates: Principles and Applications. Green Chem. 2019, 21, 406–448. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.-C.; Tsui, C.-H.; Tsai, C.-Y.; Ko, B.-T. Highly active bimetallic nickel catalysts for alternating copolymerization of carbon dioxide with epoxides. Polym. Chem. 2020, 11, 3225–3236. [Google Scholar] [CrossRef]
- Kuznetsova, S.A.; Rulev, Y.A.; Larionov, V.A.; Smol’yakov, A.F.; Zubavichus, Y.V.; Maleev, V.I.; Li, H.; North, M.; Saghyan, A.S.; Belokon, Y.N. Self-Assembled Ionic Composites of Negatively Charged Zn(salen) Complexes and Triphenylmethane Derived Polycations as Recyclable Catalysts for the Addition of Carbon Dioxide to Epoxides. ChemCatChem 2019, 11, 511–519. [Google Scholar] [CrossRef]
- Jayakumar, S.; Li, H.; Tao, L.; Li, C.; Liu, L.; Chen, J.; Yang, Q. Cationic Zn-Porphyrin Immobilized in Mesoporous Silicas as Bifunctional Catalyst for CO2 Cycloaddition Reaction under Cocatalyst Free Conditions. ACS Sustain. Chem. Eng. 2018, 6, 9237–9245. [Google Scholar] [CrossRef]
- Martínez, J.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Castro-Osma, J.A.; Lara-Sánchez, A.; Otero, A. An Efficient and Versatile Lanthanum Heteroscorpionate Catalyst for Carbon Dioxide Fixation into Cyclic Carbonates. ChemSusChem 2017, 10, 2886–2890. [Google Scholar] [CrossRef]
- Zhao, Z.; Qin, J.; Zhang, C.; Wang, Y.; Yuan, D.; Yao, Y. Recyclable Single-Component Rare-Earth Metal Catalysts for Cycloaddition of CO2 and Epoxides at Atmospheric Pressure. Inorg. Chem. 2017, 56, 4568–4575. [Google Scholar] [CrossRef]
- Qu, L.; del Rosal, I.; Li, Q.; Wang, Y.; Yuan, D.; Yao, Y.; Maron, L. Efficient CO2 transformation under ambient condition by heterobimetallic rare earth complexes: Experimental and computational evidences of a synergistic effect. J. CO2 Util. 2019, 33, 413–418. [Google Scholar] [CrossRef]
- Alassmy, Y.; Pour, Z.A.; Pescarmona, P.P. Efficient and easily reusable metal-free heterogeneous catalyst beads for the conversion of CO2 into cyclic carbonates in the presence of water as hydrogen bond donor. ACS Sustain. Chem. Eng. 2020, 8, 7993–8003. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; Martínez, J.; de la Cruz-Martínez, F.; Caballero, M.P.; Fernández-Baeza, J.; Rodríguez-López, J.; Otero, A.; Lara-Sánchez, A.; Tejeda, J. Development of hydroxy-containing imidazole organocatalysts for CO2 fixation into cyclic carbonates. Catal. Sci. Technol. 2018, 8, 1981–1987. [Google Scholar] [CrossRef]
- Steinbauer, J.; Kubis, C.; Ludwig, R.; Werner, T. Mechanistic Study on the Addition of CO2 to Epoxides Catalyzed by Ammonium and Phosphonium Salts: A Combined Spectroscopic and Kinetic Approach. ACS Sustain. Chem. Eng. 2018, 6, 10778–10788. [Google Scholar] [CrossRef]
- Mesías-Salazar, A.; Martínez, J.; Rojas, R.S.; Carrillo-Hermosilla, F.; Ramos, A.; Fernández-Galán, R.; Antiñolo, A. Aromatic guanidines as highly active binary catalytic systems for the fixation of CO2 into cyclic carbonates under mild conditions. Catal. Sci. Technol. 2019, 9, 3879–3886. [Google Scholar] [CrossRef]
- Kim, Y.; Hyun, K.; Ahn, D.; Kim, R.; Park, M.H.; Kim, Y. Efficient Aluminum Catalysts for the Chemical Conversion of CO2 into Cyclic Carbonates at Room Temperature and Atmospheric CO2 Pressure. ChemSusChem 2019, 12, 4211–4220. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.; Mousavi, B.; Younus, H.A.; Khattak, Z.A.K.; Suleman, S.; Jan, M.T.; Yu, B.; Chaemchuen, S.; Verpoor, F. ONO pincer type ligand complexes of Al(III) as efficient catalyst for chemical fixation of CO2 to epoxides at atmospheric pressure. J. Catal. 2019, 377, 190–198. [Google Scholar] [CrossRef]
- Carvalho, P.A.; Comerford, J.W.; Lamb, K.J.; North, M.; Reiss, P.S. Influence of mesoporous silica properties on cyclic carbonate synthesis catalysed by supported aluminium(salen) complexes. Adv. Synth. Catal. 2019, 361, 345–354. [Google Scholar] [CrossRef]
- Ng, C.K.; Toh, R.W.; Lin, T.T.; Luo, H.-K.; Hor, T.S.A.; Wu, J. Metal-salen molecular cages as efficient and recyclable heterogeneous catalysts for cycloaddition of CO2 with epoxides under ambient conditions. Chem. Sci. 2019, 10, 1549–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; North, M. A Bimetallic Aluminium(Salphen) Complex for the Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide. ChemSusChem 2017, 10, 74–78. [Google Scholar] [CrossRef]
- Rintjema, J.; Kleij, A.W. Aluminum-Mediated Formation of Cyclic Carbonates: Benchmarking Catalytic Performance Metrics. ChemSusChem 2017, 10, 1274–1282. [Google Scholar] [CrossRef]
- De la Cruz-Martínez, F.; Martínez, J.; Gaona, M.A.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Rodríguez, A.M.; Castro-Osma, J.A.; Otero, A.; Lara-Sánchez, A. Bifunctional Aluminum Catalysts for the Chemical Fixation of Carbon Dioxide into Cyclic Carbonates. ACS Sustain. Chem. Eng. 2018, 6, 5322–5332. [Google Scholar] [CrossRef]
- Martínez, J.; de la Cruz-Martínez, F.; Gaona, M.A.; Pinilla-Peñalver, E.; Fernández-Baeza, J.; Rodríguez, A.M.; Castro-Osma, J.A.; Otero, A.; Lara-Sánchez, A. Influence of the Counterion on the Synthesis of Cyclic Carbonates Catalyzed by Bifunctional Aluminum Complexes. Inorg. Chem. 2019, 58, 3396–3408. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Castro-Osma, J.A.; Earlam, A.; Alonso-Moreno, C.; Otero, A.; Lara-Sánchez, A.; North, M.; Rodríguez-Diéguez, A. Synthesis of Cyclic Carbonates Catalysed by Aluminium Heteroscorpionate Complexes. Chem. Eur. J. 2015, 21, 9850–9862. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Sánchez-Barba, L.F.; Garcés, A.; Fernández-Baeza, J.; Fernández, I.; Lara-Sánchez, A.; Rodríguez, A.M. Bimetallic scorpionate-based helical organoaluminum complexes for efficient carbon dioxide fixation into a variety of cyclic carbonates. Catal. Sci. Technol. 2020, 10, 3265–3278. [Google Scholar] [CrossRef]
- Gaona, M.A.; de la Cruz-Martínez, F.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Alonso-Moreno, C.; Rodríguez, A.M.; Rodríguez-Diéguez, A.; Castro-Osma, J.A.; Otero, A.; Lara-Sánchez, A. Synthesis of Helical Aluminium Catalysts for Cyclic Carbonate Formation. Dalton Trans. 2019, 48, 4218–4227. [Google Scholar] [CrossRef]
- Meléndez, D.O.; Lara-Sánchez, A.; Martínez, J.; Wu, X.; Otero, A.; Castro-Osma, J.A.; North, M.; Rojas, R.S. Amidinate Aluminium Complexes as Catalysts for Carbon Dioxide Fixation into Cyclic Carbonates. ChemCatChem 2018, 10, 2271–2277. [Google Scholar] [CrossRef] [Green Version]
- Rios Yepes, Y.; Quintero, C.; Meléndez, D.O.; Daniliuc, C.G.; Martínez, J.; Rojas, R.S. Cyclic Carbonates from CO2 and Epoxides Catalyzed by Tetra- and Pentacoordinate Amidinate Aluminum Complexes. Organometallics 2019, 38, 469–478. [Google Scholar] [CrossRef]
- Moreno da Costa, D.; Borja, L.; Verdugo, C.; Martinez, J.; Quintero, C.; Jaque, P.; Trofymchuk, O.S.; Daniliuc, C.G.; Cabrera, A.R.; Rojas, R.S. Unexpected intramolecular N-arylcyano-bdiketiminate cyclization in new aminoquinoline derivative complexes of aluminium for CO2 fixation into cyclic carbonates. New J. Chem. 2019, 43, 12059–12068. [Google Scholar] [CrossRef]
- Rios Yepes, R.; Martínez, J.; Rangel-Sánchez, H.; Quintero, C.; Ortega-Alfaro, M.C.; López-Cortés, J.G.; Daniliuc, C.G.; Antiñolo, A.; Ramos, A.; Rojas, R.S. Aluminum complexes with new non-symmetric ferrocenyl amidine ligands and their application in CO2 transformation into cyclic carbonates. Dalton Trans. 2020, 49, 1124–1134. [Google Scholar] [CrossRef]
- Mesias-Salazar, A.; Trofymchuk, O.S.; Daniliuc, C.D.; Antiñolo, A.; Carrillo-Hermosilla, F.; Nachtigall, F.M.; Santos, L.S.; Rojas, R.S. Copper (II) as catalyst for intramolecular cyclization and oxidation of (1,4-phenylene)bisguanidines to benzodiimidazole-diylidenes. J. Catal. 2020, 382, 150–154. [Google Scholar] [CrossRef]
- Caló, V.; Nacci, A.; Monopoli, A.; Fanizzi, A. Cyclic Carbonate Formation from Carbon Dioxide and Oxiranes in Tetrabutylammonium Halides as Solvents and Catalysts. Org. Lett. 2002, 4, 2561–2563. [Google Scholar] [CrossRef]
- Chang, T.; Jing, H.; Jin, L.; Qiu, W. Quaternary onium tribromide catalyzed cyclic carbonate synthesis from carbon dioxide and epoxides. J. Mol. Catal. A Chem. 2007, 264, 241–247. [Google Scholar] [CrossRef]
- Valerio, O.; Misra, M.; Mohanty, A.K. Poly(glycerol-co-diacids) Polyesters: From Glycerol Biorefinery to Sustainable Engineering Applications, A Review. ACS Sustain. Chem. Eng. 2018, 6, 5681–5693. [Google Scholar] [CrossRef]
- Lee, K.W.; Chung, J.W.; Kwak, S.-Y. Highly Branched Polycaprolactone/Glycidol Copolymeric Green Plasticizer by One-Pot Solvent-Free Polymerization. ACS Sustain. Chem. Eng. 2018, 6, 9006–9017. [Google Scholar] [CrossRef]
- Utrata-Wesołek, A.; Zymełka-Miara, I.; Kowalczuk, A.; Trzebicka, B.; Dworak, A. Photocrosslinking of Polyglycidol and Its Derivative: Route to Thermoresponsive Hydrogels. Photochem. Photobiol. 2018, 94, 52–60. [Google Scholar] [CrossRef]
- Steinbauer, J.; Spannenberg, A.; Werner, T. An in situ formed Ca2+—Crown ether complex and its use in CO2-fixation reactions with terminal and internal epoxides. Green Chem. 2017, 19, 3769–3779. [Google Scholar] [CrossRef]
- Yan, G.; Li, X.; Wang, Z.; Guo, H.; Peng, W.; Hu, Q.; Wang, J. Fluorinated solvents for high-voltage electrolyte in lithium-ion battery. J. Solid State Electr. 2017, 21, 1589–1597. [Google Scholar] [CrossRef]
- Nishikawa, D.; Nakajima, T.; Ohzawa, Y.; Koh, M.; Yamauchi, A.; Kagawa, M.; Aoyama, H. Thermal and oxidation stability of organo-fluorine compound-mixed electrolyte solutions for lithium ion batteries. J. Power Sources 2013, 243, 573–580. [Google Scholar] [CrossRef]
- Schimpf, V.; Max, J.B.; Stolz, B.; Heck, B.; Mülhaupt, R. Semicrystalline Non-Isocyanate Polyhydroxyurethanes as Thermoplastics and Thermoplastic Elastomers and Their Use in 3D Printing by Fused Filament Fabrication. Macromolecules 2019, 52, 320–331. [Google Scholar] [CrossRef]
- Blain, M.; Cornille, A.; Boutevin, B.; Auvergne, R.; Benazet, D.; Andrioletti, B.; Caillol, S. Hydrogen bonds prevent obtaining high molar mass PHUs. J. Appl. Polym. Sci. 2017, 134, 44958. [Google Scholar] [CrossRef] [Green Version]
- Clegg, W.; Harrington, R.W.; North, M.; Pasquale, R. Cyclic Carbonates Synthesis Catalysed by Bimetallic Aluminium-Salen Complexes. Chem. Eur. J. 2010, 16, 6828–6843. [Google Scholar] [CrossRef]
- Xu, Y.; Yuan, D.; Wang, Y.; Yao, Y. Aluminum complexes derived from a hexadentate salen-type Schiff base: Synthesis, structure, and catalysis for cyclic carbonate synthesis. Dalton Trans. 2017, 46, 5848–5855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Yan, L.; Liu, Y.; Wu, X.; Wei, X. Potassium organoaluminate: Synthesis, structure, and catalytic activity for the conversion of CO2 into cyclic carbonates. J. Organomet. Chem. 2020, 922, 121365. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
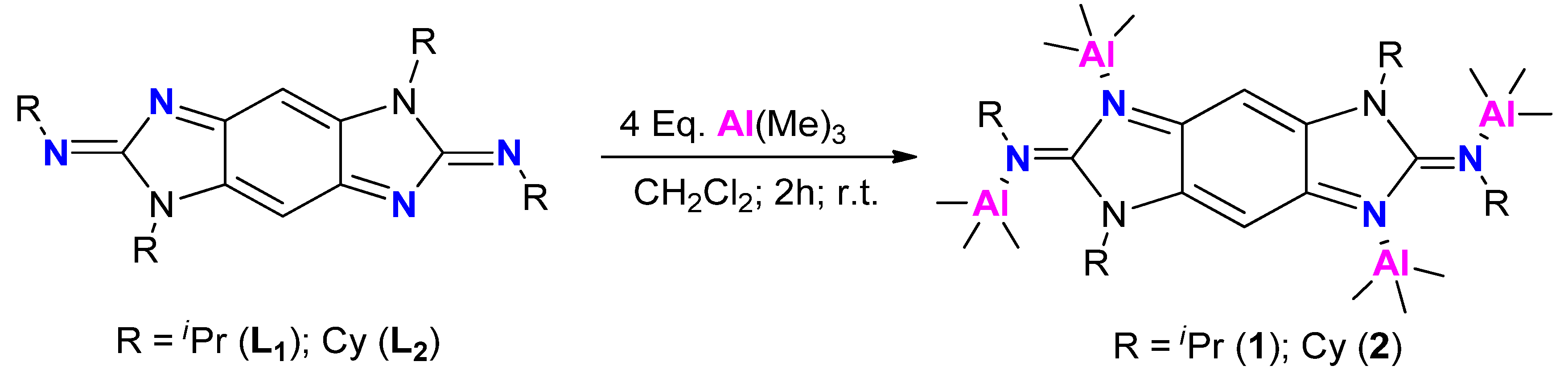{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Co-Catalyst | Temperature (°C) | Conversion 2,3 (%) |
| 1 | 1 | TBAI | 50 | 70 |
| 2 | 1 | TBAB | 50 | 62 |
| 3 | 1 | TBAC | 50 | 46 |
| 4 | 1 | TBAF | 50 | 1 |
| 5 | 1 | TBAI | 70 | 86 |
| 6 | 2 | TBAI | 50 | 42 |
| 7 | 2 | TBAB | 50 | 35 |
| 8 | 2 | TBAC | 50 | 31 |
| 9 | 2 | TBAF | 50 | 2 |
| 10 | 2 | TBAI | 70 | 53 |
| 11 4 | Al(CH3)3 | TBAI | 50 | 20 |
| 12 4 | Al(CH3)3 | TBAI | 70 | 31 |
| 13 5 | Al(CH3)3 | TBAI | 70 | 18 |
| 14 6 | 1 | - | 70 | 0 |
| 15 7 | - | TBAI | 70 | 5 |
| Entry | Epoxide | Catalyst System (mol%) | Reaction Conditions: T (°C), time (h) | Yield (%) 2 | TOF (h−1) 3 | Reference |
|---|---|---|---|---|---|---|
| 1 | 2a | 1/TBAI (1.25/5.0) | 70, 24 | 82 | 2.73 | This work |
| 2 | 2a | Al(Salen)/TBAB (2.5/2.5) | 25, 3 | 57 | 7.65 | [80] |
| 3 | 2a | Al cat/TBAI (2.5/5.0) | 50, 24 | 85 | 1.41 | [66] |
| 4 | 2a | Al cat/TBAI (5.0/5.0) | 25, 24 | 80 | 0.66 | [65] |
| 5 | 2a | Al cat/TBAI (1.5/1.5) | 80, 24 | 90 | 2.5 | [67] |
| 6 | 2a | Al cat/TBAI (5.0/5.0) | 35, 24 | 100 4 | - | [60] |
| 7 | 2g | 1/TBAI (1.25/5.0) | 70, 24 | 93 | 3.10 | This work |
| 8 | 2g | Al(Salen) (0.25) | 100, 24 | 63 | 10.50 | [56] |
| 9 | 2f | 1/TBAI (1.25/5.0) | 70, 24 | 85 | 2.83 | This work |
| 10 | 2f | Al-cage/TBAB (0.33/10.0) | rt, 24 | 54 | 3.40 | [57] |
| 11 | 2f | Al-Schiff/TBAB (0.3/0.9) | 100, 18 | 94 | 17.41 | [81] |
| 12 5 | 2f | Al-K/TBAI (5.0/2.5) | 50, 24 | 73 | 0.61 | [82] |
| 13 | 2f | Al cat/TBAB (5.0/5.0) | 35, 18 | 80 4 | - | [63] |
| 14 | 2f | Al-Fe/TBAI (1.7/1.7) | 80, 24 | 71 | 1.74 | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesías-Salazar, Á.; Yepes, Y.R.; Martínez, J.; Rojas, R.S. Highly Active CO2 Fixation into Cyclic Carbonates Catalyzed by Tetranuclear Aluminum Benzodiimidazole-Diylidene Adducts. Catalysts 2021, 11, 2. https://doi.org/10.3390/catal11010002
Mesías-Salazar Á, Yepes YR, Martínez J, Rojas RS. Highly Active CO2 Fixation into Cyclic Carbonates Catalyzed by Tetranuclear Aluminum Benzodiimidazole-Diylidene Adducts. Catalysts. 2021; 11(1):2. https://doi.org/10.3390/catal11010002
Chicago/Turabian StyleMesías-Salazar, Ángela, Yersica Rios Yepes, Javier Martínez, and René S. Rojas. 2021. "Highly Active CO2 Fixation into Cyclic Carbonates Catalyzed by Tetranuclear Aluminum Benzodiimidazole-Diylidene Adducts" Catalysts 11, no. 1: 2. https://doi.org/10.3390/catal11010002