Understanding the Effect of Multiple Domain Deletion in DNA Polymerase I from Geobacillus Sp. Strain SK72


Abstract
:1. Introduction
2. Results and Discussion
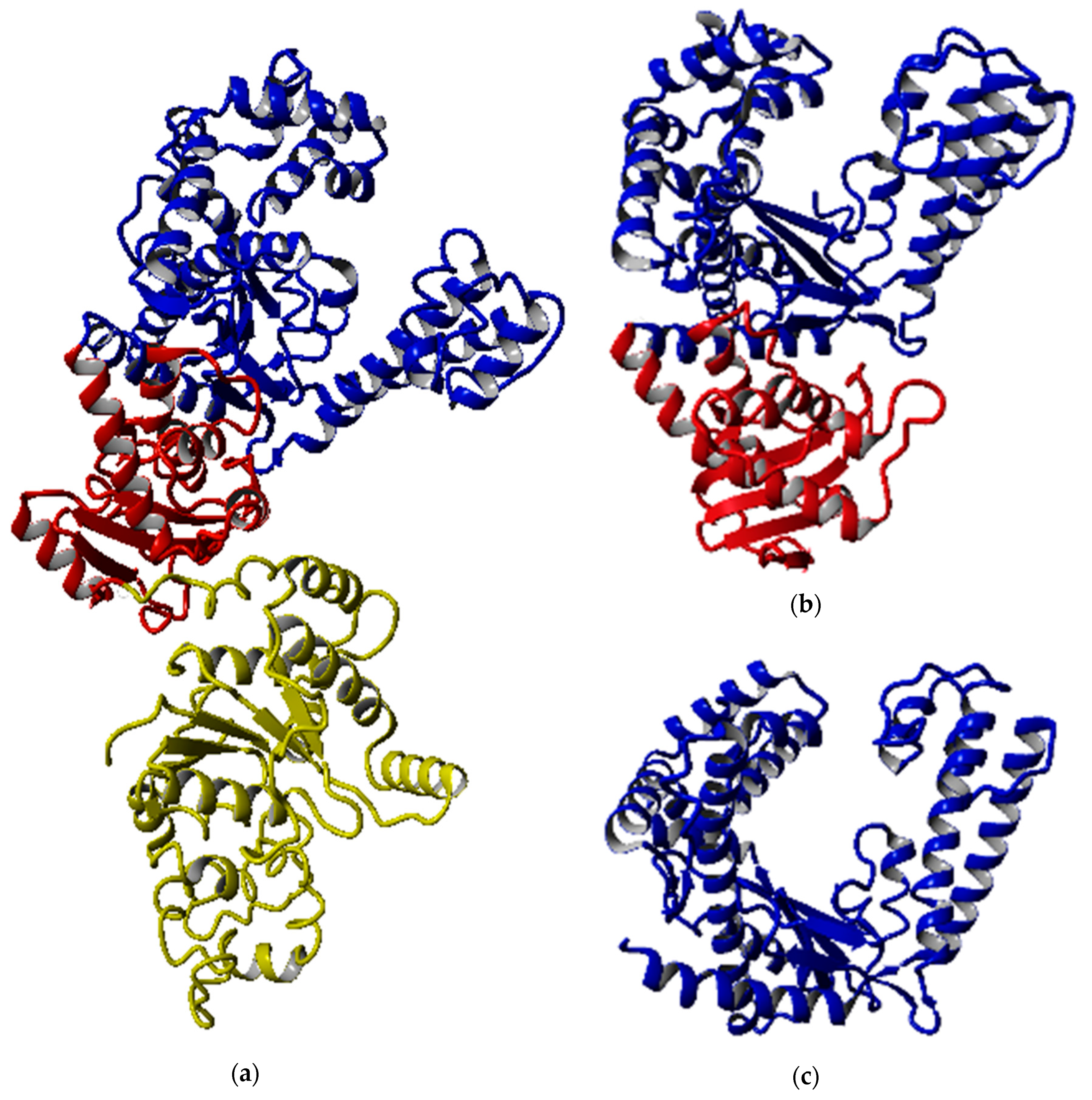
2.1. Conserved Domain and Structure Analyses
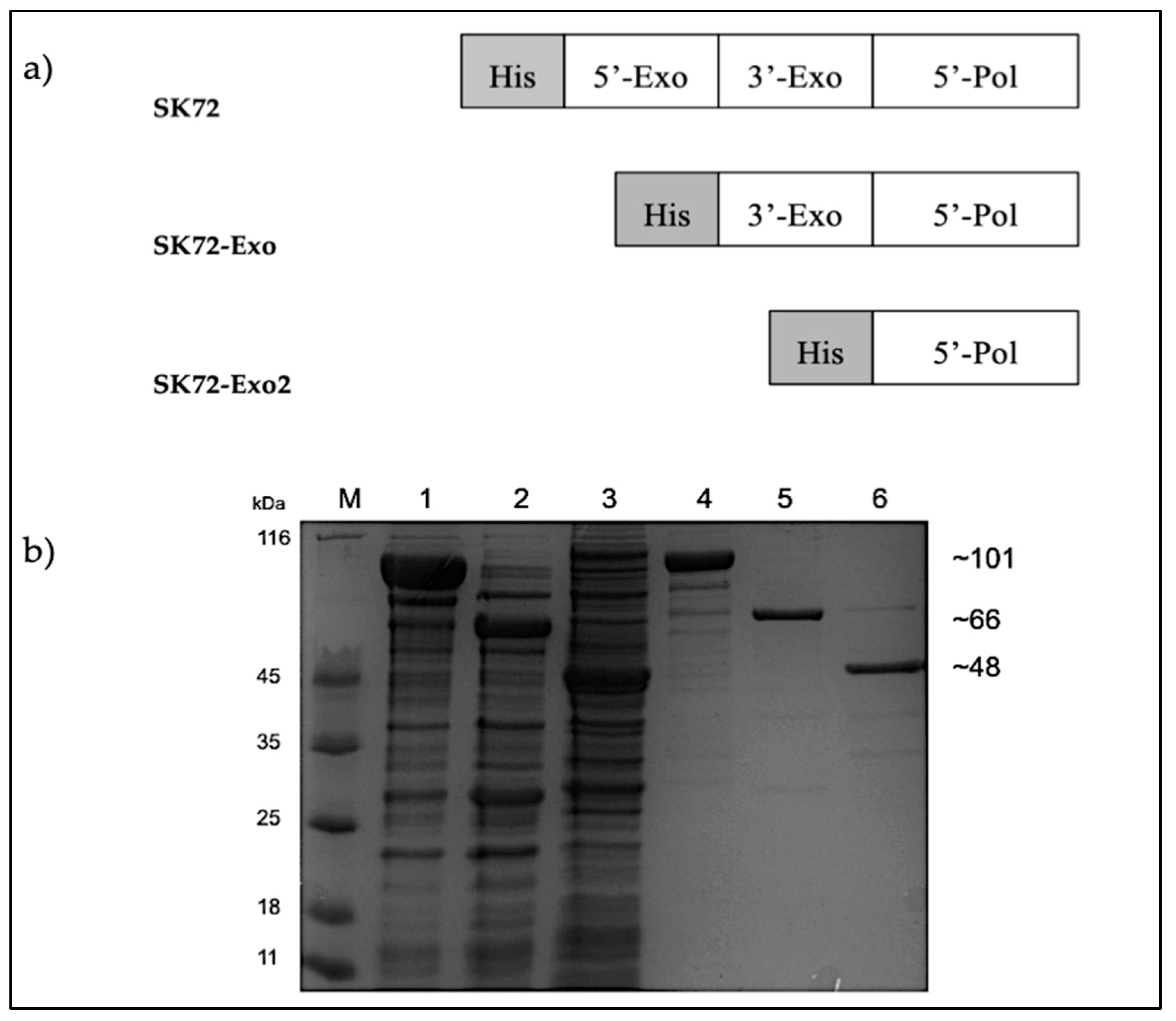
2.2. Cloning, Overexpression, and Purification of SK72 DNA Polymerase I and Its Variants
2.3. Characterization of SK72 DNA Polymerase I and Its Variants
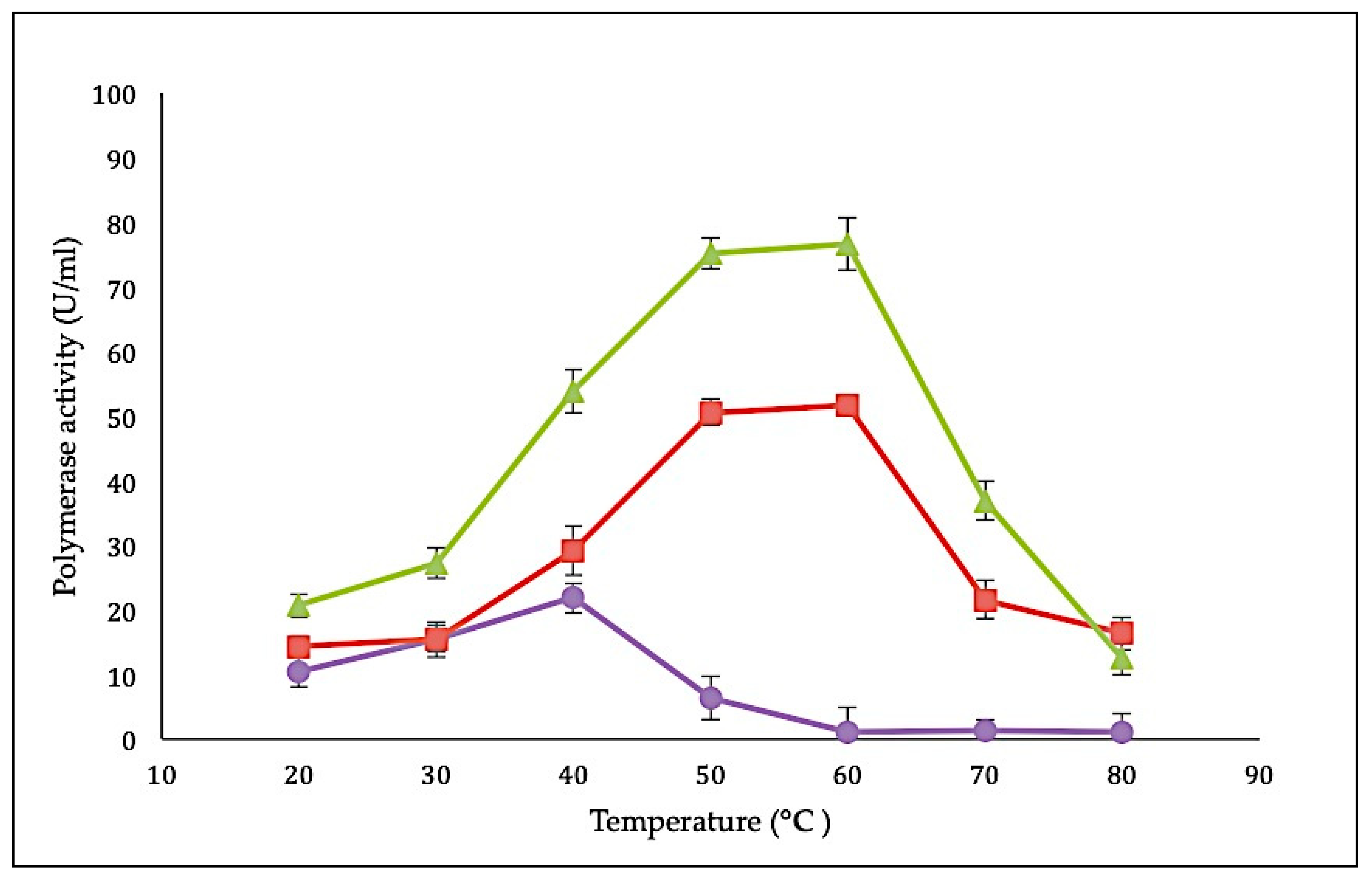
2.3.1. Effect of Temperature on SK72 Polymerase Activity and Its Variants
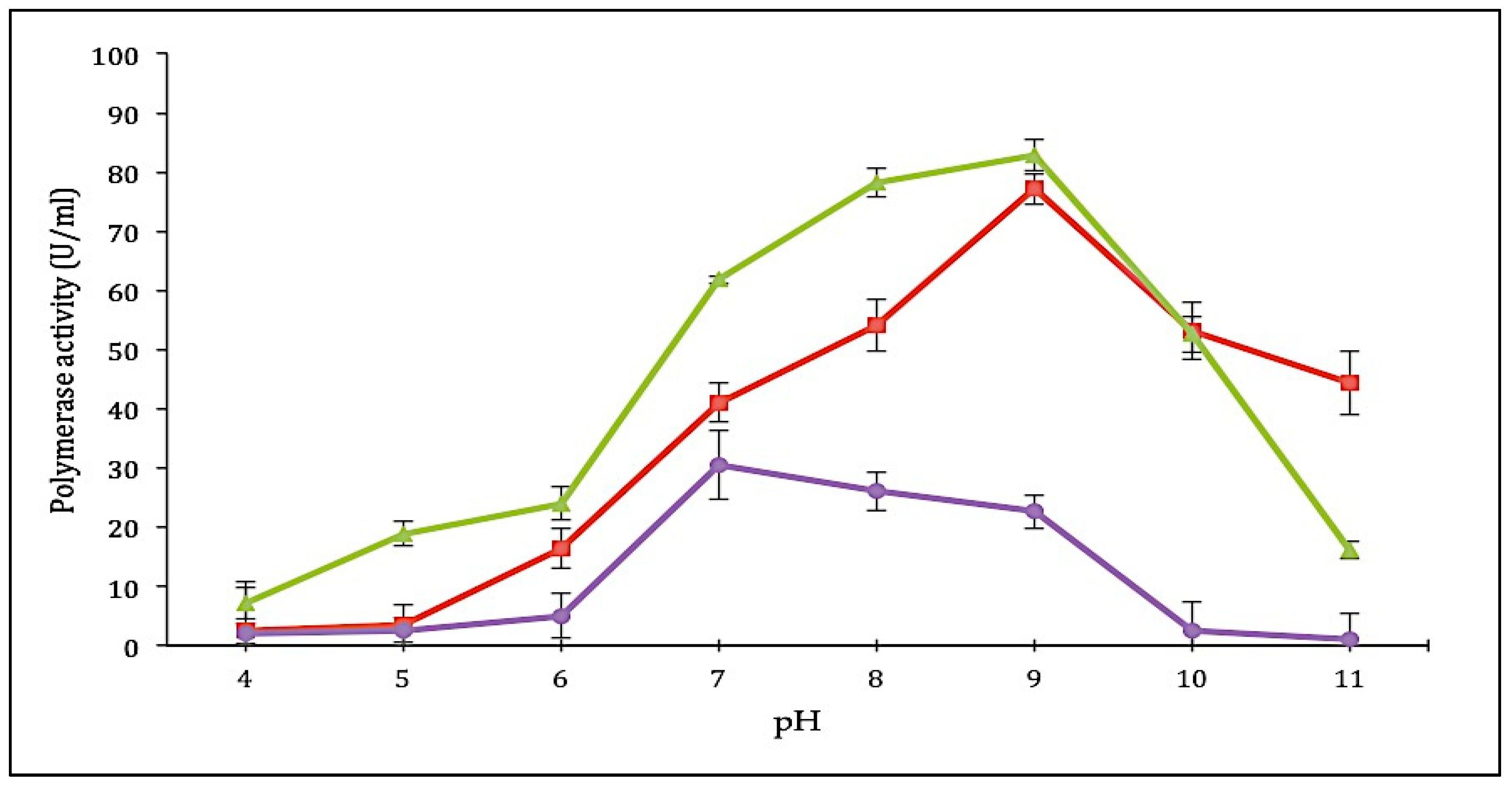
2.3.2. Effect of pH on SK72 Polymerase Activity and Its Variants
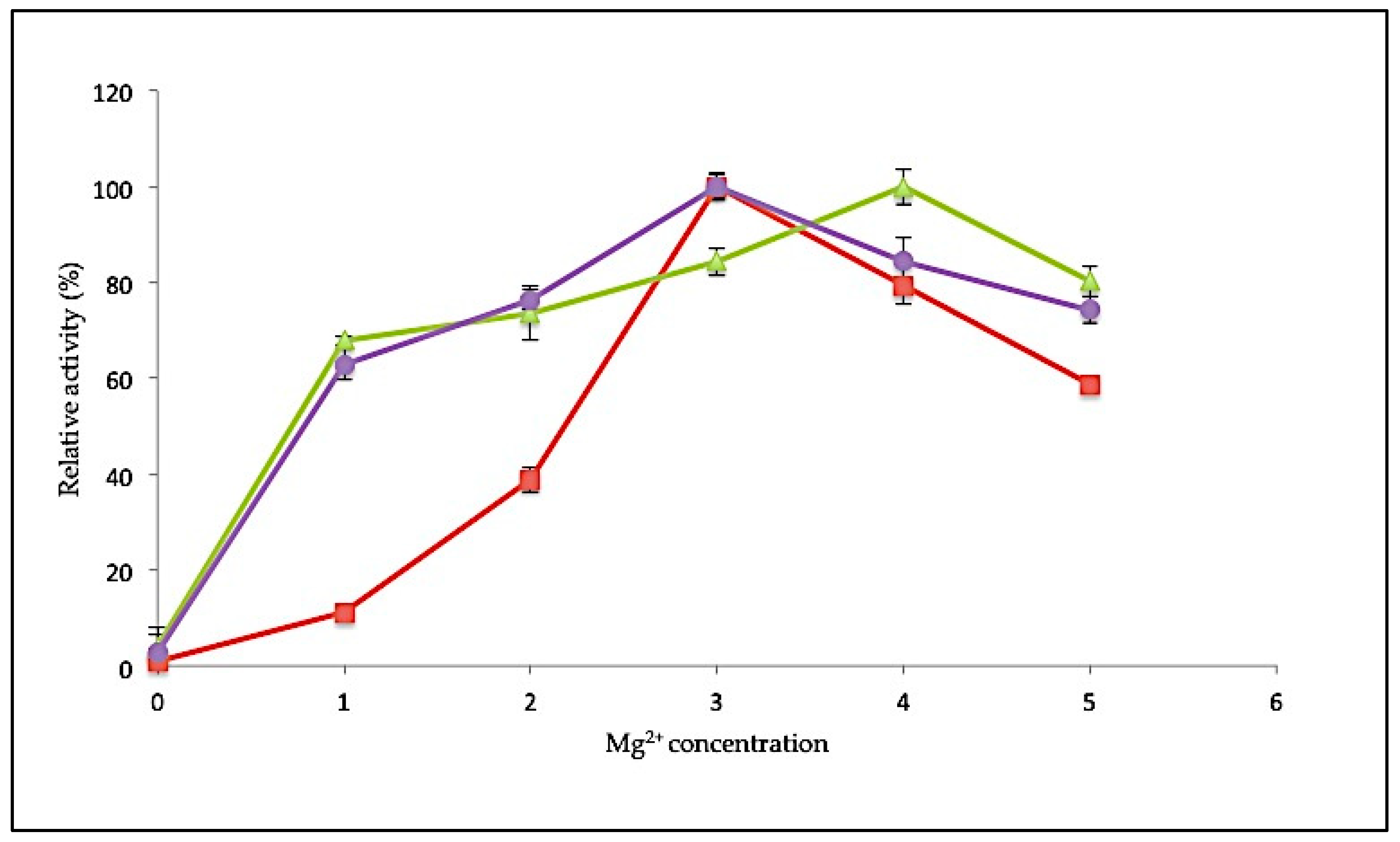
2.3.3. Effect of MgCl2 Concentration on SK72 Polymerase Activity and Its Variants
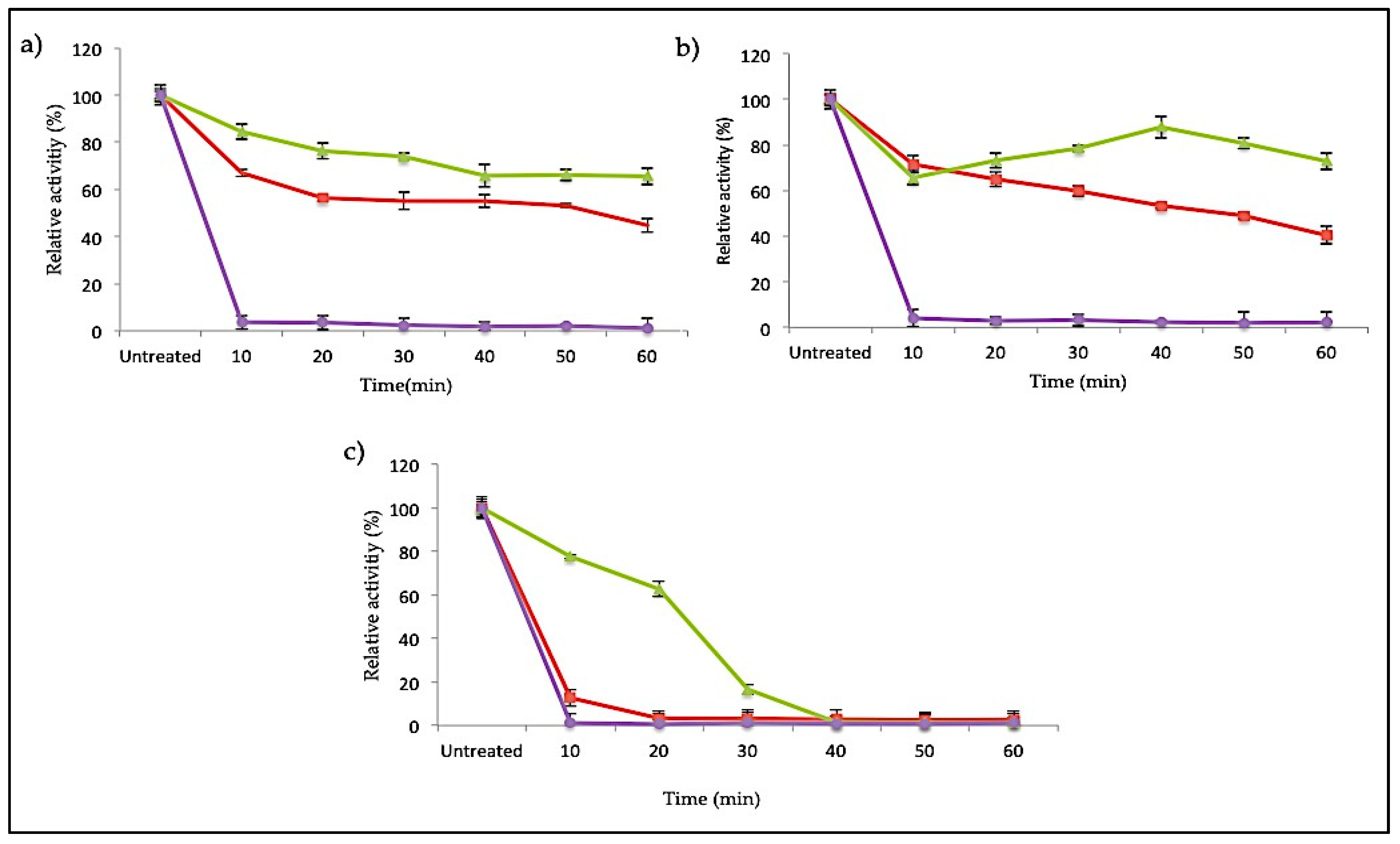
2.3.4. Thermostability of SK72 Polymerase Activity and Its Variants
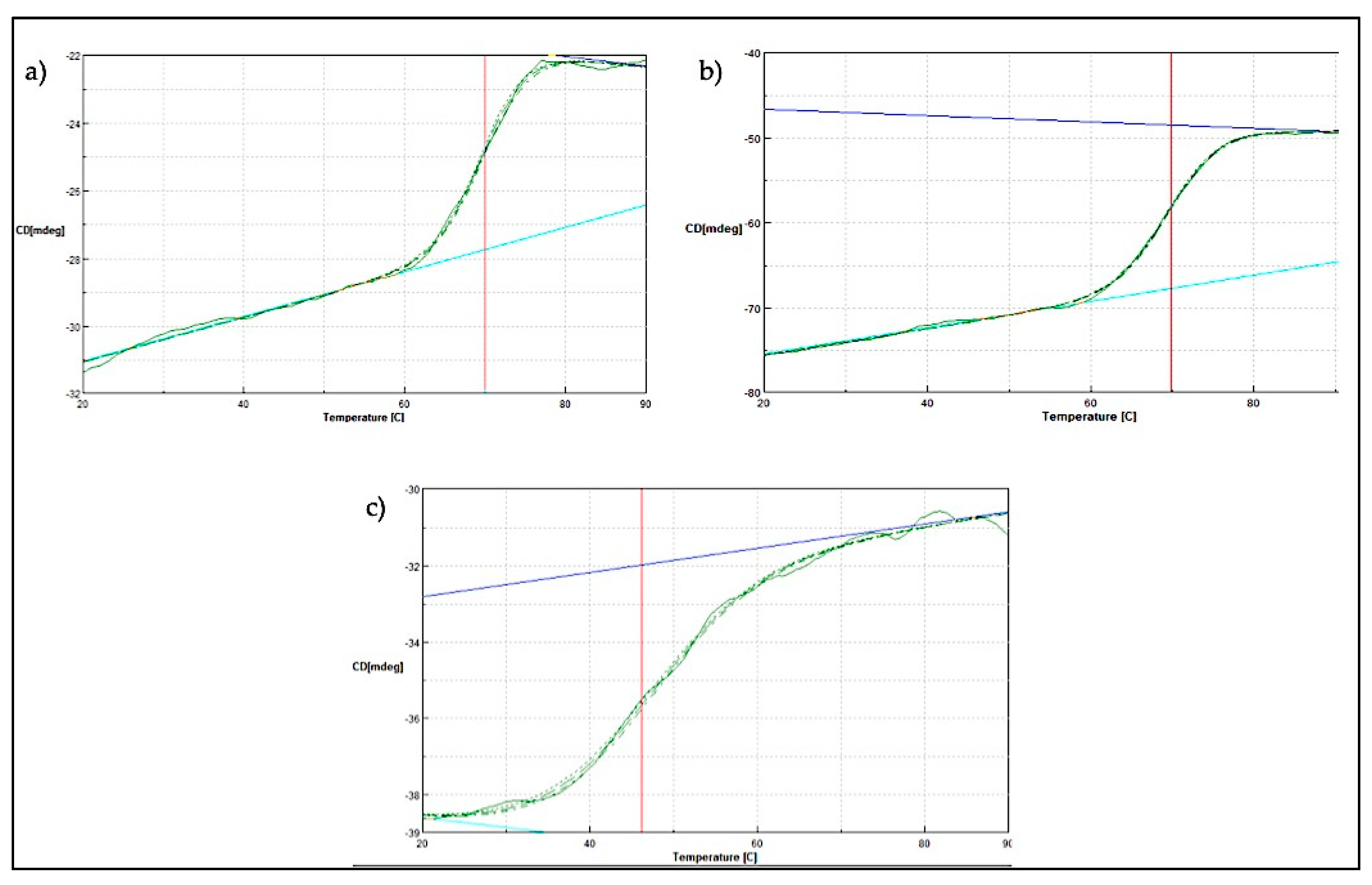
2.4. Thermal Denaturation Analysis of SK72 DNA Polymerase I and Its Variants
2.5. Secondary Structure Analysis of SK72 DNA Polymerase I and Its Variants
3. Materials and Methods
3.1. Strains, Vectors, and Growth Medium
3.2. Isolation of SK72 DNA Polymerase I Gene
3.3. Cloning of SK72 DNA Polymerase I
3.4. Nucleotide Sequencing and Amino Acid Analysis
3.5. Domains Analysis of SK72 DNA Polymerase and Its Variants
3.6. Construction of SK72 Pol and Its Variants
3.7. Expression and Purification of SK72 Polymerase I and Its Variants
3.8. Polymerase Activity Assay
3.9. Thermal Stability, Optimal Temperature and pH, and MgCl2 Concentration Analyses
3.10. Secondary Structure and Melting Point Estimation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kornberg, A.; Baker, T.A. DNA Replication; W.H. Freeman & Co.: New York, NY, USA, 1992; Volume 3. [Google Scholar]
- Garcia-Diaz, M.; Bebenek, K. Multiple functions of DNA polymerases. Crit. Rev. Plant Sci. 2007, 26, 105–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rittie, L.; Perbal, B. Enzymes used in molecular biology: A useful guide. J. Cell Commun. Signal. 2008, 2, 25–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braithwaite, D.; Ito, J. Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 1993, 21, 787. [Google Scholar] [CrossRef]
- Hubscher, U.; Maga, G.; Spadari, S. Eukaryotic DNA polymerases. Annu. Rev. Biochem. 2002, 71, 133–163. [Google Scholar] [CrossRef] [Green Version]
- Ishino, S.; Ishino, Y. DNA polymerases as useful reagents for biotechnology—The history of developmental research in the field. Front. Microbiol. 2014, 5, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, P.R.; Sethy, K.; Mohapatra, S.; Panda, D. Loop mediated isothermal amplification: An innovative gene amplification technique for animal diseases. Vet. World 2016, 9, 465–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowski, Y.; Gurung, M.K.; Larsen, A.N. Characterization and engineering of a DNA polymerase reveals a single amino-acid substitution in the fingers subdomain to increase strand-displacement activity of A-family prokaryotic DNA polymerases. BMC Mol. Cell Biol. 2019, 20, 31. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A. DNA Polymerases: Structural Diversity and Common Mechanisms. J. Biol. Chem. 1999, 274, 17395–17398. [Google Scholar] [CrossRef] [Green Version]
- Lovett, S.T. The DNA exonucleases of Escherichia Coli. EcoSal Plus 2011, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Joyce, C.M.; Steitz, T.A. Function and Structure Relationship in DNA Polymerase. Annu. Rev. Biochem. 1994, 63, 777–822. [Google Scholar] [CrossRef]
- Phang, S.M.; Teo, C.Y.; Lo, E.; Wong, V.W. Cloning and complete sequence of the DNA polymerase-encoding gene (BstpolI) and characterisation of the Klenow-like fragment from Bacillus stearothermophilus. Gene 1995, 163, 65–68. [Google Scholar] [CrossRef]
- Kiefer, J.R.; Mao, C.; Hansen, C.J.; Basehore, S.L.; Hogrefe, H.H.; Braman, J.C.; Beese, L.S. Crystal structure of a thermostable Bacillus DNA polymerase l large fragment at 2.1 Å resolution. Structure 1998, 5, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Oscorbin, I.P.; Boyarskikh, U.A.; Filipenko, M.L. Large Fragment of DNA Polymerase I from Geobacillus sp. 777: Cloning and Comparison with DNA Polymerases I in Practical Applications. Mol. Biotechnol. 2015, 57, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Shevelev, I.V.; Hübscher, U. The 3’–5’ exonucleases. Nat. Rev. Mol. Cell Biol. 2002, 3, 364–376. [Google Scholar] [CrossRef]
- Ishino, Y.; Iwasaki, H.; Kato, I.; Shinagawa, H. Amino acid sequence motifs essential to 3’-->5’ exonuclease activity of Escherichia coli DNA polymerase II. J. Biol. Chem. 1994, 269, 14655–14660. [Google Scholar]
- Aliotta, J.M.; Pelletier, J.J.; Ware, J.L.; Moran, L.S.; Benner, J.S.; Kong, H. Thermostable Bst DNA polymerase I lacks a 3’- 5’ proofreading exonuclease activity. Genet. Anal. Biomol. Eng. 1996, 12, 185–195. [Google Scholar] [CrossRef]
- Kamarudin, N.H.; Rahman, R.N.; Ali, M.S.; Leow, T.C.; Basri, M.; Salleh, A.B. Unscrambling the effect of C-terminal tail deletion on the stability of a cold-adapted, organic solvent stable lipase from Staphylococcus epidermidis AT2. Mol. Biotechnol. 2014, 56, 747–757. [Google Scholar] [CrossRef]
- Lamers, M.H.; Georgescu, R.E.; Lee, S.G.; O’Donnell, M.; Kuriyan, J. Crystal structure of the catalytic a subunit of E. coli replicative DNA polymerase III. Cell 2006, 126, 881–892. [Google Scholar] [CrossRef] [Green Version]
- Barros, T.; Guenther, J.; Kelch, B.A.; Anaya, J.; Prabhakar, A.; Odonnell, M.; Kuriyan, J.; Lamers, M.H. A structural role for the PHP domain in E. coli DNA polymerase III. BMC Struct. Biol. 2013, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishwanath, S.; Brevern, A.G.; Srinivasan, N. Same but not alike: Structure, flexibility and energetics of domains in multi-domain proteins are influenced by the presence of other domains. PLoS Comput. Biol. 2018, 14, e1006008. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Sinha, S. Thermostability of in vitro evolved Bacillus subtilis lipase A: A network and dynamics perspective. PLoS ONE 2014, 9, e102856. [Google Scholar] [CrossRef] [PubMed]
- Federly, R.G.; Romano, L.J. DNA Polymerase: Structural Homology, Conformational Dynamics, and the Effects of Carcinogenic DNA Adducts. J. Nucleic Acids 2010, 2010, 457176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoso, Y.; Joyce, C.M.; Potapova, O.; Reste, L.L.; Hohlbein, J.; Torella, J.P.; Grindley, N.D.F.; Kapinidis, A.N. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc. Natl. Acad. Sci. USA 2010, 107, 715–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.H. Viral Polymerases. Adv. Exp. Med. Biol. 2012, 726, 267–304. [Google Scholar]
- Miller, B.R.; Beese, L.S.; Parish, C.A.; Wu, E.Y. The Closing Mechanism of DNA Polymerase I at Atomic Resolution. Structure 2015, 23, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- San-Miguel, T.; Perez-Bermudez, P.; Gavidia, I. Production of soluble eukaryotic recombinant proteins in E. coli is favoured in early log-phase cultures induced at low temperature. SpringerPlus 2013, 2, 89. [Google Scholar] [CrossRef] [Green Version]
- Gutierrezgonzalez, M.; Farias, C.; Tello, S.; Perezetcheverry, D.; Romero, A.; Zuniga, R.; Ribeiro, C.H.; Lorenzoferreiro, C.; Molina, M.C. Optimization of culture conditions for the expression of three different insoluble proteins in Escherichia coli. Sci. Rep. 2019, 9, 16850. [Google Scholar] [CrossRef]
- Lee, J.; Cho, S.; Kil, E.; Kwon, S. Characterization and PCR application of a thermostable DNA polymerase from Thermococcus pacificus. Enzym. Microb. Technol. 2010, 47, 147–152. [Google Scholar] [CrossRef]
- Latip, W.; Raja Noor, Z.R.; Rahman, A.; Adam Thean, C.L.; Fairolniza, M.S.; Nor Hafizah, A.K.; Mohd Shukuri, M.A. The effect of N-terminal domain removal towards the biochemical and structural features of a thermotolerant lipase from an antarctic Pseudomonas sp. strain AMS3. Int. J. Mol. Sci. 2018, 19, 560. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Wang, Q.; Jiang, S.; Zhang, G.; Ma, Y. Truncation of the unique N-terminal domain improved the thermos-stability and specific activity of alkaline α-amylase Amy703. Sci. Rep. 2016, 6, 22465. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Tian, C.; Li, A.; Zhang, G.; Ma, Y. Identification and characterization of a novel alkaline [alpha]-amylase Amy703 belonging to a new clade from Bacillus pseudofirmus. J. Ind. Microbiol. Biotechnol. 2014, 41, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ma, T.; Zhang, B.; Yao, N.; Li, M.; Cui, L.; Li, G.; Ma, Z.; Cheng, J. A novel mechanism of protein thermostability: A unique N-terminal domain confers heat resistance to Fe/Mn-SODs. Sci. Rep. 2015, 4, 7284. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Pang, H.; Wang, Z.; Lu, J.; Wei, Y.; Huang, R. Characterization of an Invertase with pH Tolerance and Truncation of Its N-Terminal to Shift Optimum Activity toward Neutral pH. PLoS ONE 2013, 8, e62306. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Subarna, P.; Young, J.Y. Mutation of non-conserved amino acids surrounding catalytic site to shift pH optimum of Bacillus circulans xylanase. J. Mol. Catal. B Enzym. 2008, 55, 130–136. [Google Scholar] [CrossRef]
- Śpibida, M.; Krawczyk, B.; Olszewski, M.; Kur, J. Modified DNA polymerases for PCR troubleshooting. J. Appl. Genet. 2017, 58, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawyer, F.C.; Stoffel, S.; Saiki, R.K.; Chang, S.Y.; Landre, P.A.; Ambramson, R.D.; Gelfand, D.H. High-level Expression, Purification, and Enzymatic Characterization of Full-length Thermus aquaticus DNA Polymerase and a Truncated Form Deficient in 5’ to 3’ Exonuclease Activity. PCR Methods Appl. 1993, 2, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Li, S.; Zhao, H.; Bian, L.; Chen, L.; Zhang, Z.; Zhong, X.; Ma, L.; Yu, X. Expression and Characterization of the RKOD DNA Polymerase in Pichia pastoris. PLoS ONE 2015, 10, e0131757. [Google Scholar] [CrossRef]
- Xie, P.; Sayers, J.R. A Model for Transition of 59-Nuclease Domain of DNA Polymerase I from Inert to Active Modes. PLoS ONE 2011, 6, e16213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Liu, Z.; Cui, W.; Zhou, L.; Tian, Y.; Zhou, Z. Enhanced Thermal Stability and Hydrolytic Ability of Bacillus subtilis Aminopeptidase by Removing the Thermal Sensitive Domain in the Non-Catalytic Region. PLoS ONE 2014, 9, e92357. [Google Scholar] [CrossRef] [PubMed]
- Tahir, H.M.; Rahman, R.N.Z.R.A.; Leow, A.T.C.; Ali, M.S.M. Expression, Characterisation and Homology Modelling of a Novel Hormone-Sensitive Lipase (HSL)-Like Esterase from Glaciozyma antarctica. Catalysts 2019, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Hu, D.; Li, J.F.; He, Y.; Zhu, T.D.; Wu, M.C. Contribution of Disulfide Bridges to the Thermostability of a Type A Feruloyl Esterase from Aspergillus usamii. PLoS ONE 2015, 10, e0126864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, M.; Blackshields, G.; Brown, N.; Chenna, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Non bonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthy, R.; Bowie, J.U.; Eisenberg, D. Assesment of Protein Models with 3 Dimensional Profile. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Künzli, M.; Schwede, T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, 510–514. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amount of Secondary Structure (%) | |||
|---|---|---|---|
| SK72 | SK72-Exo | SK72-Exo2 | |
| Helix | 47.0 | 49.1 | 45.0 |
| Sheet | 18.5 | 12.9 | 5.5 |
| Turn | 11.4 | 17.8 | 19.6 |
| Coil | 23.1 | 20.3 | 29.9 |
| Primer Name | Sequence (5′-3′) | Restriction Site |
|---|---|---|
| SK72-F1 | ATCCCATATGATGCGCCTGAAAAAAAAACTGGTGCTGATTG | NdeI |
| SK72-R1 | TGCGTCGACTCATTTGGCATCATACCAAGTAC | SalI |
| SK72-Exo-F2 | GAAACATATGGCCAAAATGGCGTTTACGCTGGC | NdeI |
| SK72-Exo2-F3 | GCTTCATATGAATGAACAGGATCGGCTGCTGGTG | NdeI |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadrawi, W.H.; Norazman, A.; Mohd Shariff, F.; Mohamad Ali, M.S.; Raja Abd Rahman, R.N.Z. Understanding the Effect of Multiple Domain Deletion in DNA Polymerase I from Geobacillus Sp. Strain SK72. Catalysts 2020, 10, 936. https://doi.org/10.3390/catal10080936
Hadrawi WH, Norazman A, Mohd Shariff F, Mohamad Ali MS, Raja Abd Rahman RNZ. Understanding the Effect of Multiple Domain Deletion in DNA Polymerase I from Geobacillus Sp. Strain SK72. Catalysts. 2020; 10(8):936. https://doi.org/10.3390/catal10080936
Chicago/Turabian StyleHadrawi, Waqiyuddin Hilmi, Anas Norazman, Fairolniza Mohd Shariff, Mohd Shukuri Mohamad Ali, and Raja Noor Zaliha Raja Abd Rahman. 2020. "Understanding the Effect of Multiple Domain Deletion in DNA Polymerase I from Geobacillus Sp. Strain SK72" Catalysts 10, no. 8: 936. https://doi.org/10.3390/catal10080936