1. Introduction
Immobilized lipases are currently used in many processes, most of them related to the pharmaceutical industry due to their enantioselectivity. Among many others, typical examples are the enantiomeric esterification of captopryl (antihypertensive) or the aryl propionic analgesics such as (S)-naproxen. The immobilization of lipases to lead solid biocatalysts is essential for their applications as it allows their stabilization and easy recovery from the liquid reaction media and subsequent reuse.
The discovery of permanent porosity in certain thermally-stable coordination polymers, the so-called metal-organic frameworks (MOFs) [
1,
2,
3,
4], at the end of the last century, probably gave rise to the greatest ever revolution in the field of porous materials. MOFs are nanoporous coordination polymers, generally but not always crystalline, formed by either isolated metal ions or metal clusters linked by multidentate (at least, bidentate) organic ligands given rise to structures extended along two or three spatial dimensions [
5]. Indeed, more than 90,000 MOF materials have been described in two decades [
6,
7], involving a diverse selection of metals and numerous organic linkers. The extraordinary versatility of MOFs is not only structural or compositional but also in terms of synthesis methods [
8,
9], which contrasts with the almost monotonous hydrothermal procedures used in the synthesis of inorganic microporous materials.
Amongst the numerous potential applications of MOFs, their use as enzyme supports has awakened the interest of the scientific community in the past few years [
10,
11,
12,
13,
14,
15,
16,
17], particularly since the intrinsic pore size of MOFs does not have to be necessarily larger than the enzyme size [
18,
19,
20,
21,
22,
23,
24].
Owing to the initial pore size limitation (only enzymes of a certain dimension could be encapsulated within a MOF) as reported by Ma et al. [
25,
26,
27,
28], other methodologies were developed in an attempt to universalize the enzymatic immobilization of MOFs. By taking advantage of: (i) The favourable chemical affinity between the MOF materials and enzymes [
18], (ii) the possibility of preparing the MOFs supports under enzymatically compatible conditions, which enables the enzyme immobilization within or onto the surface of the MOF material in one step [
18,
19,
20,
21,
22], or (iii) the chance of using the enzyme as the organic linker for the MOF material [
24], therefore many methodologies of preparing MOF-supported solid biocatalysts were rapidly and simultaneously developed by different research groups.
The synthesis of some carboxylate-based MOF materials, which are the most common MOFs to support enzymes, can be readily carried out in aqueous solutions at room temperature by employing a deprotonating agent to solubilize the insoluble carboxylate acid [
29,
30,
31,
32,
33]. In addition, some of the so-formed MOFs can be crystallized in the nanometre scale, which could have important advantages in particular applications such as heterogeneous catalysis. Moreover, they possess very high external surface areas [
29] that favour their interaction with any macromolecules (for instance, enzymes). This fact, together with the natural chemical affinity of the enzyme for the MOF [
18], makes the in situ approach for enzyme immobilization onto MOF-based supports a very efficient method [
19,
20,
21,
22,
23,
24]. Furthermore, immobilization is presumably non-covalent, so minor activity loss against that of the free enzyme could be reached.
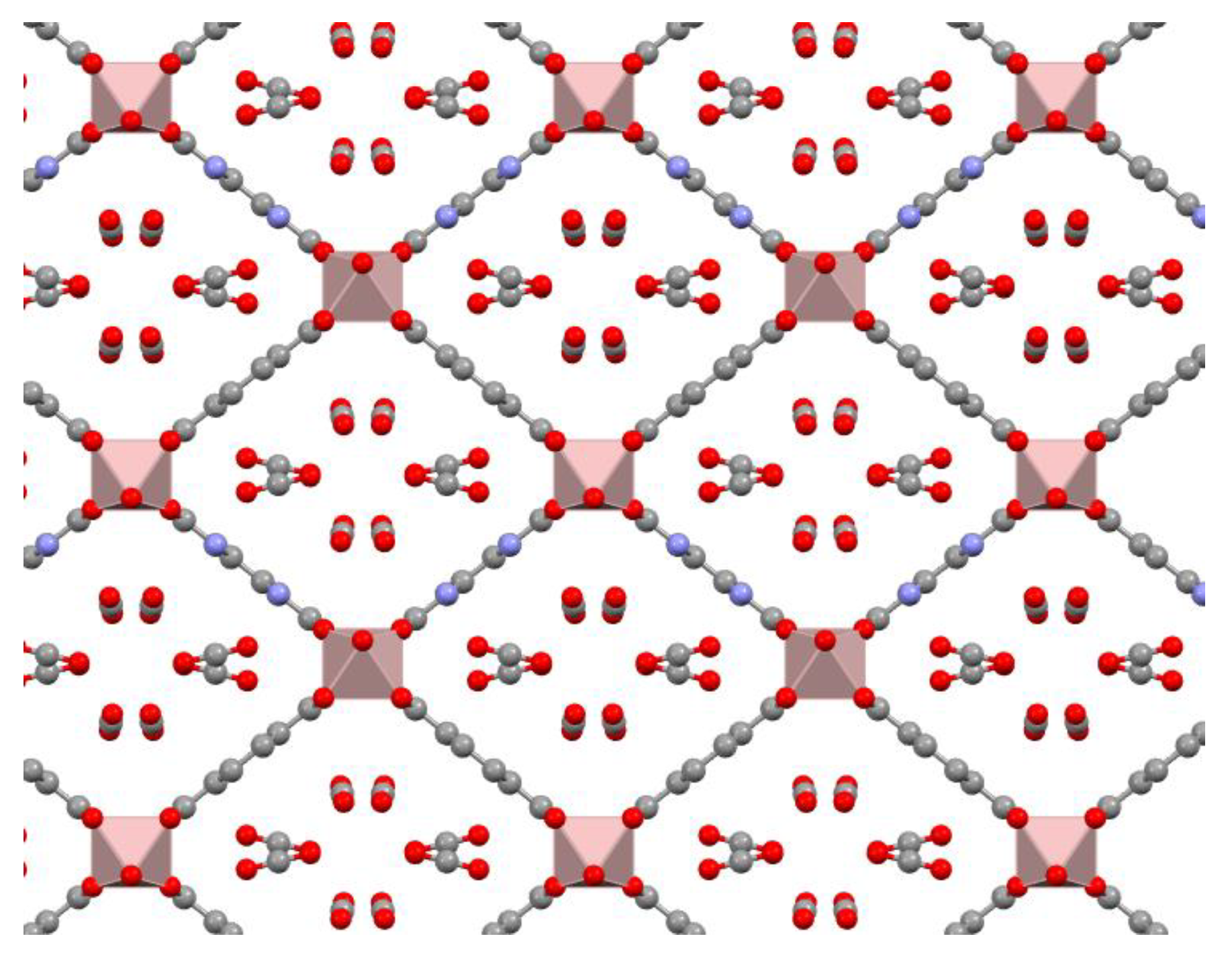
One of the nanocrystalline MOF materials able to be prepared under these sustainable conditions is the X-MIL-53(Al), where X denotes an organic functional group (such as -H, -NH
2 or -NO
2) [
29,
30]. The structure of the as-prepared NH
2-functionalized MIL-53(Al) shown in
Figure A1, evidences that their pores, which are of limited sized and filled by a protonated organic linker cannot play any role in the encapsulation of comparatively much larger lipase CaL B. NH
2-functionalized MIL-53(Al) was already identified as an acceptable in situ support for the enzyme β-glucosidase [
20,
21], whose immobilization is rather a challenge due to its large dimensions. In that study, it was confirmed that the presence of the enzyme in the synthesis medium altered the chemistry of the MOF material, which was formed together with the protonated organic linker. Nevertheless, the phase responsible for the β-glucosidase immobilization was undoubtedly the MOF-based one.
In this study, the validity of this in situ one-step methodology for enzyme immobilization onto NH
2-MIL-53(Al) is extended to lipase CaLB (from
Candida antarctica), which indeed is one of the most widely investigated enzymes in different immobilization processes [
34,
35,
36]. Apart from our own studies using nanocrystalline NH
2-MIL-53(Al) supports for enzymes [
20,
21], the MIL-53(Al) family has proved to be efficient in the immobilization of: (i) Laccase on NH
2-MIL-53(Al), using our one-step room-temperature methodology [
37]; (ii) laccase on meso-MIL-53(Al) by post-synthesis methods [
38]; and (iii) luciferase on non-functionalized MIL-53(Al), also by post-synthesis methodology [
39]. We also found excellent results for lipase in situ immobilization on the semi-crystalline support Fe-BTC [
23], suggesting that this enzyme could be easily immobilized onto MOF supports. In spite of the fact that Fe-BTC resulted in an excellent support for lipase, it is worth investigating any other MOF-based support for this enzyme such as the NH
2-MIL-53(Al) considered in this work. On one hand, this support has a crystalline-enough structure that allows to properly understand the lipase-MOF interaction and, on the other hand, it is basically inert from a catalytic point of view. In addition, employing NH
2-MIL-53(Al) support for lipase immobilization allows extending the state-of-the-art over a previous immobilization approach [
21,
22,
23]. Different in-solution deprotonating agents such as sodium hydroxide (NaOH), triethylamine (TEA), or ammonium hydroxide (NH
4OH) are studied here for generating lipase-based solid biocatalysts with NH
2-MIL-53(Al) as the crystalline support. The efficiency of the lipase immobilization as well as the activity of the resultant biocatalyst is compared with their counterparts containing the enzyme β-glucosidase.
2. Results and Discussion
2.1. In Situ Immobilization of Lipase on NH2-MIL-53(Al)
The choice of both NH
2-MIL-53(Al) as the support and the in situ approach as the main strategy for the enzyme lipase CaLB immobilization for this work was based on previous results achieved by systematic studies over the immobilization of β-glucosidase performed in our laboratory [
20,
21]. In these studies, parameters such as the nature of the linker deprotonating agent (NaOH, ammonia or trimethylamine) or synthesis time (1–48 h) strongly influenced the phase formed, the enzyme immobilization efficiency, and enzymatic activity [
20,
21]. As a result, these parameters have been methodically investigated for the immobilization of the enzyme lipase CaLB (
Table 1). As shown in
Table 1, on the one hand, the percentage of immobilized lipase (Enz. immob. (%)) was obtained via the Bradford assay by giving the differential enzyme concentration between the initial enzyme concentration offered to the synthesis media and the final concentration present in the supernatant after filtering and recovering of the resultant biocatalyst. As it can be seen, in all cases a percentage higher than 85% was obtained with respect to the enzyme present in the synthesis media. It is worth noting that this method does not consider if the enzyme keeps its activity during the immobilization process. On the other hand, the hydrolysis of
p-NPA was used as a routine test for measuring spectrophotometrically at 348 nm and 25 °C the nonspecific esterase activity during the process of in situ lipase immobilization in order to calculate the enzyme loading (mg·g
−1, column 3). The enzyme concentration was calculated as the difference between the activity of suspension solution in the synthesis media and the remaining activity present in the supernatant after filtering and recovering of the resultant biocatalyst.
The enzyme loading in these biocatalysts is around 20 mg·g
−1, and the variations of the loading found amongst the different biocatalysts was due to the differences in the yield of the MOF support. For a given deprotonating agent (NaOH, TEA, or NH
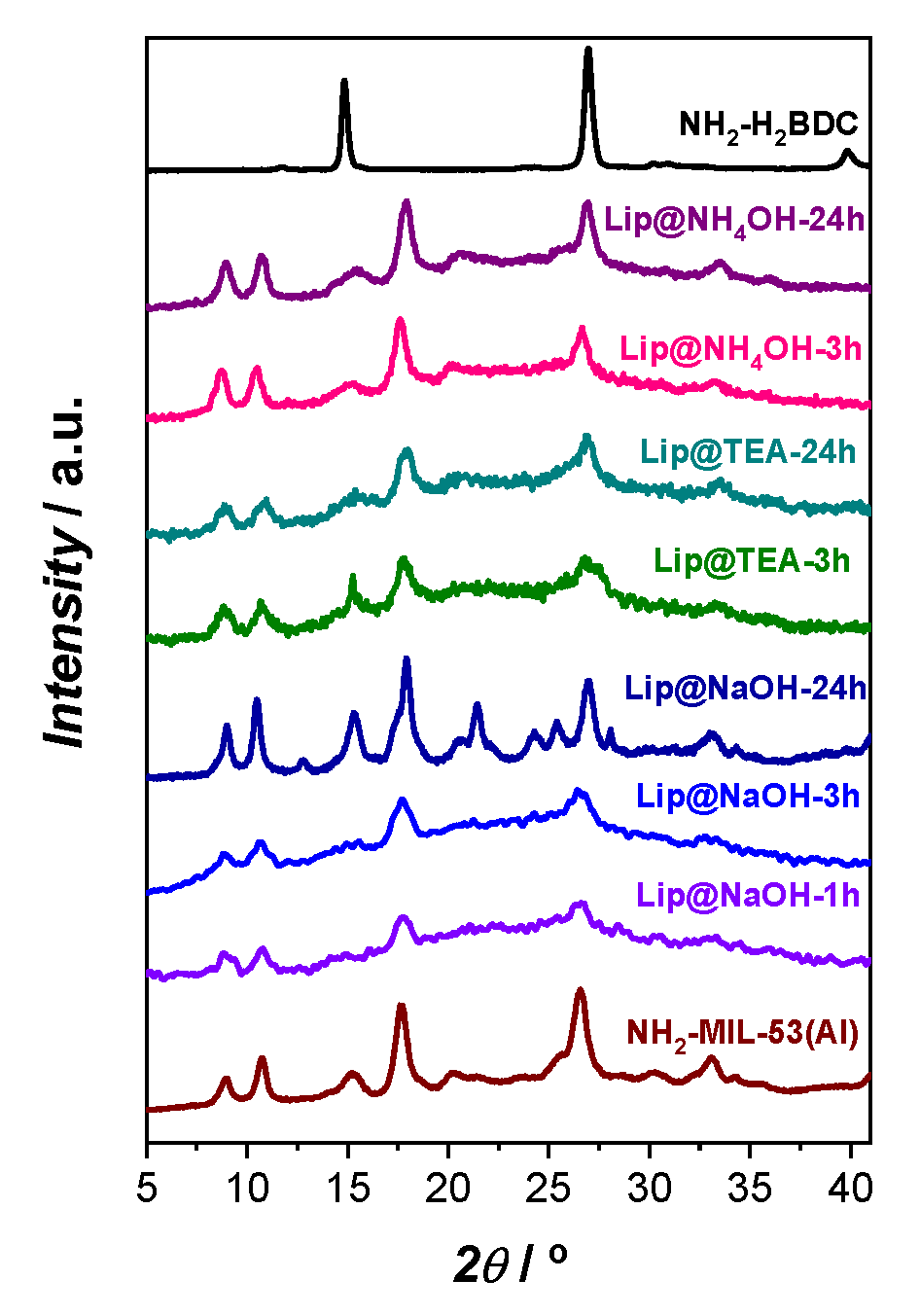
4OH), the percentage of immobilized enzyme was increased or at least maintained with the synthesis time. On the contrary, the specific catalytic activity (activity per mg of immobilized enzyme) was shown to slowly decrease with time. This could be the result of the enzyme having been exposed for a prolonged period of time to the synthesis media designed for the MOF formation or because the first immobilized lipase molecules could become more inaccessible to the substrates if the support particles continue growing over time. Supporting this last possibility, the crystalline order of the samples inferred by powder X-ray diffraction (PXRD) characterization seems to improve with the extended reaction synthesis time (
Figure 1), suggesting a better (and probably more hindered) integration of the enzyme within the MOF.
Table 1 also shows the immobilization results of the same enzyme CaLB lipase on the semi-crystalline MOF Fe-BTC under similar conditions (one-step/in situ procedure, at room temperature, in the presence of a base (NaOH) acting as a deprotonating agent of the corresponding organic linker, etc.) [
23]. Using such MOF material as support, both high enzyme immobilization efficiency and high specific catalytic activity of the resultant biocatalysts were also reached, being even higher under certain conditions with respect to the catalysts of the present study. Nevertheless, it must be considered that both MOF supports are complementary in certain aspects: (i) Unlike the semi-amorphous nature of MOF Fe-BTC material, the NH
2-MIL-53(Al) material is crystalline, which both allowed a more precise study and a better understanding of the possible interaction MOF-enzyme; on the contrary, the structure of semi-amorphous Fe-BTC could not be solved, and the presence, concentration, and/or location of the defects or metal centers interacting with the enzyme could be random and therefore, hard to control from an experimental point of view; (ii) the support NH
2-MIL-53(Al) is basically inert in terms of catalysis, as their amino groups have no basic character because they are directly bonded to an aromatic ring, whereas Fe-BTC has already shown its (photo)catalytic potential in various types of reactions [
32,
41,
42], which could potentially be masking the intrinsic biocatalytic activity carried out by the supported enzyme. In summary, the study of both systems, Lip@NH
2-MIL-53(Al) and Lip@Fe-BTC, is somehow complementary and convenient for extending the state-of-the-art with respect to said systems, and it should not be seen as a strict competition.
In order to detect the crystalline phases present in the biocatalyst, a powder X-ray diffraction (PXRD) characterization technique was employed. In this regard, in our previous work [
21], it was reported that solid biocatalysts containing β-glucosidase supported on the same MOF material than the herein employed (NH
2-MIL-53(Al)) using the same procedure. That approach showed the presence of the protonated linker NH
2-H
2BDC. However, as can be seen in
Figure 1, in this case the only crystalline phase detected by PXRD characterization in the biocatalysts Lip@NH
2-MIL-53(Al) was NH
2-MIL-53(Al). A lower resolution in the PXRD pattern of the biocatalysts in comparison with that of the enzyme-free support is presumably due to the “dilution” of the crystalline phase of the MOF support in the biocatalyst enzyme@support. No trace of crystallized protonated linker NH
2-H
2BDC was detected by PXRD at any synthesis time irrespectively of the deprotonating agent used (
Figure 1). Although the most intense reflections of the XRD pattern of NH
2-H
2BDC are close to certain reflections of NH
2-MIL-53(Al), the presence of the former is quite evident in the diffractogram when both phases coexist [
21]. However, it is worth noting that in the case of using NaOH as a deprotonating agent, the greater the synthesis time, the sharper the intensity of the signal peaks is obtained for the PXRD pattern, obtaining the maximum intensity after 24 h of synthesis (Lip@NaOH-24h). Consequently, among all the tested synthesis times, 24 h was the time that the system took to rearrange itself to accommodate the greatest amount of crystallinity. This behaviour neither was replicated with TEA, nor with NH
4OH deprotonating agents.
This difference in PXRD results indicate that the enzymes play an active role in the formation of the biocatalysts. The higher proportion of basic amino-acids groups in lipase (isoelectric point of 6.0) than in β-glucosidase (isoelectric point of 4.3) could avoid the precipitation of the 2-amino-terephthalic acid, which would tend to be soluble. It must be noted that in the absence of any enzyme, the co-precipitation of the MOF X-MIL-53(Al) material and the corresponding linker is the most common scenario with short reaction times [
29].
Since the enzyme immobilization occurs within/onto the MOF phase and not on the protonated linker [
21], a high percentage of immobilized lipase was reached from the first moment of the synthesis (approx. 1 h). The compromise between enzyme immobilization efficiency and the specific enzymatic activity, which severely increases and decreases respectively with synthesis time for in situ immobilization of β-glucosidase [
21], is not found in the case of lipase immobilization.
2.2. Post-Synthesis Immobilization of Lipase on NH2-MIL-53(Al) and MIL-53(Al) Materials
A potential advantage of the enzyme immobilization over these nanocrystalline sustainable MOF supports is that, apart from the in situ approach, the post-synthesis (two-step) methodology can be investigated in parallel. The comparison between both methodologies can provide valuable information about (i) the chemical affinity between the external surface area of the MOF and the enzyme, (ii) the accessibility of the enzymes to the pores, (iii) the reason behind the possible enzyme leaching, and (iv) the potential to directly compare and rate both approaches in terms of catalytic and specific activities of CaLB lipase.
Unlike the in situ approach, the post-synthesis strategy failed to attain lipase immobilization at a suitable level. Attempts to immobilize CaLB lipase on previously synthesized NH2-MIL-53(Al) and MIL-53(Al) supports were unsuccessful. Immobilization yields were not only negligible but also hard to reproduce.
Their respective catalytic activities in p-nitrophenyl acetate (p-NPA) hydrolysis were: 4.85 units per gram (U·g−1) for Lip#NH2-MIL-53(Al) and 7.19 U·g−1 for Lip#MIL-53(Al), which corresponds to a retained activity relative to the activity of free enzyme of 7.6 and 4.2%, respectively.
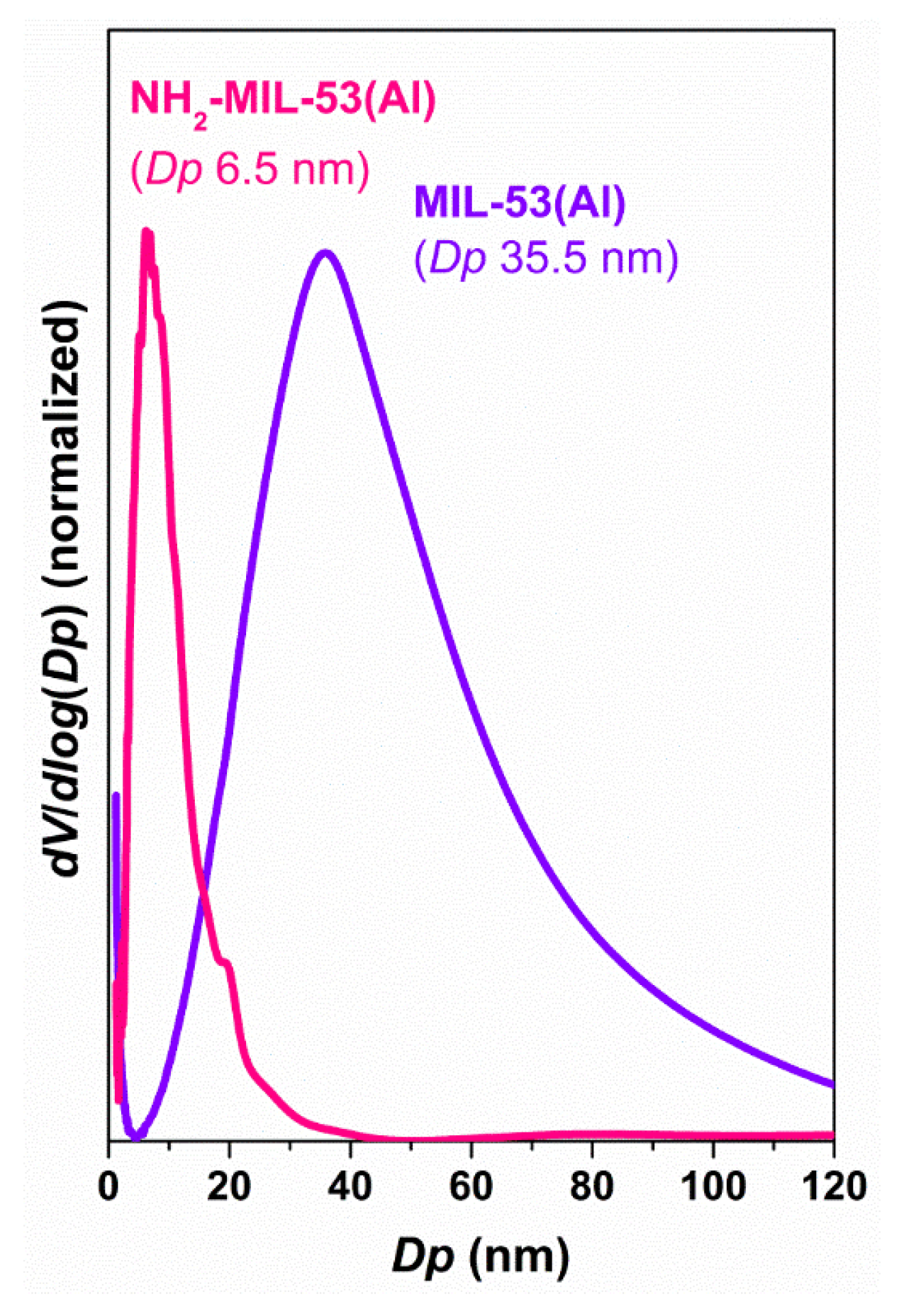
The reduced dimensions of the inter-crystalline space, especially in the case of NH
2-MIL-53(Al) (
Figure A2 shows the pore size distribution), is probably the reason behind these poor results. The nanocrystallinity of this sample (
Figure A3) could favour the interaction enzyme-MOF but especially when the sample offers accessible mesoporosity. It is proposed that the enzyme can only bind the external surface of the material since diffusion throughout the extremely narrow network might be prevented. This also may serve to explain the low reproducibility of the
post-synthesis immobilization yields attained. The size of the particles may vary greatly and therefore the available surface for the enzyme could vary as well.
Furthermore, it is important to note that the experimental conditions used for the post-synthesis approach have been optimized to retain enzymatic activity, thus enabling catalytic activity to be measured despite the low enzyme loading of the resultant biocatalysts. In comparison, lipase was in situ immobilized under conditions designed for the formation of the MOF material. Therefore, it is presumable that there is still much room to improve the experimental conditions in order to optimize the specific activity in the in situ strategy.
2.3. Certifying the Presence of the Enzyme in the Biocatalysts
Apart from the evidence inferred by the catalytic activity tests performed, the presence of the enzyme lipase CaLB on the biocatalysts was also studied by different physico-chemical and biological characterization techniques.
Table 2 shows the results of the chemical analyses of the organic elements C, H, N, and S.
Although elemental analysis is a quantitative technique, since the linker of the MOF also contains elements C, N, and H, this technique cannot be used as definitive proof of the presence of the enzyme within the MOF. Since sulphur (S) is not present in the MOF but is present in the enzyme, this could potentially be used as a means of confirming the enzyme loading of the MOF material. All the biocatalysts contain a certain amount of S except that of the enzyme-free NH
2-MIL-53(Al), which indeed suggests the presence of lipase in every solid biocatalyst. In addition, the S content generally increases with the synthesis time for a given deprotonating agent, which is in good agreement with the trend to increase the enzyme immobilization shown in
Table 1.
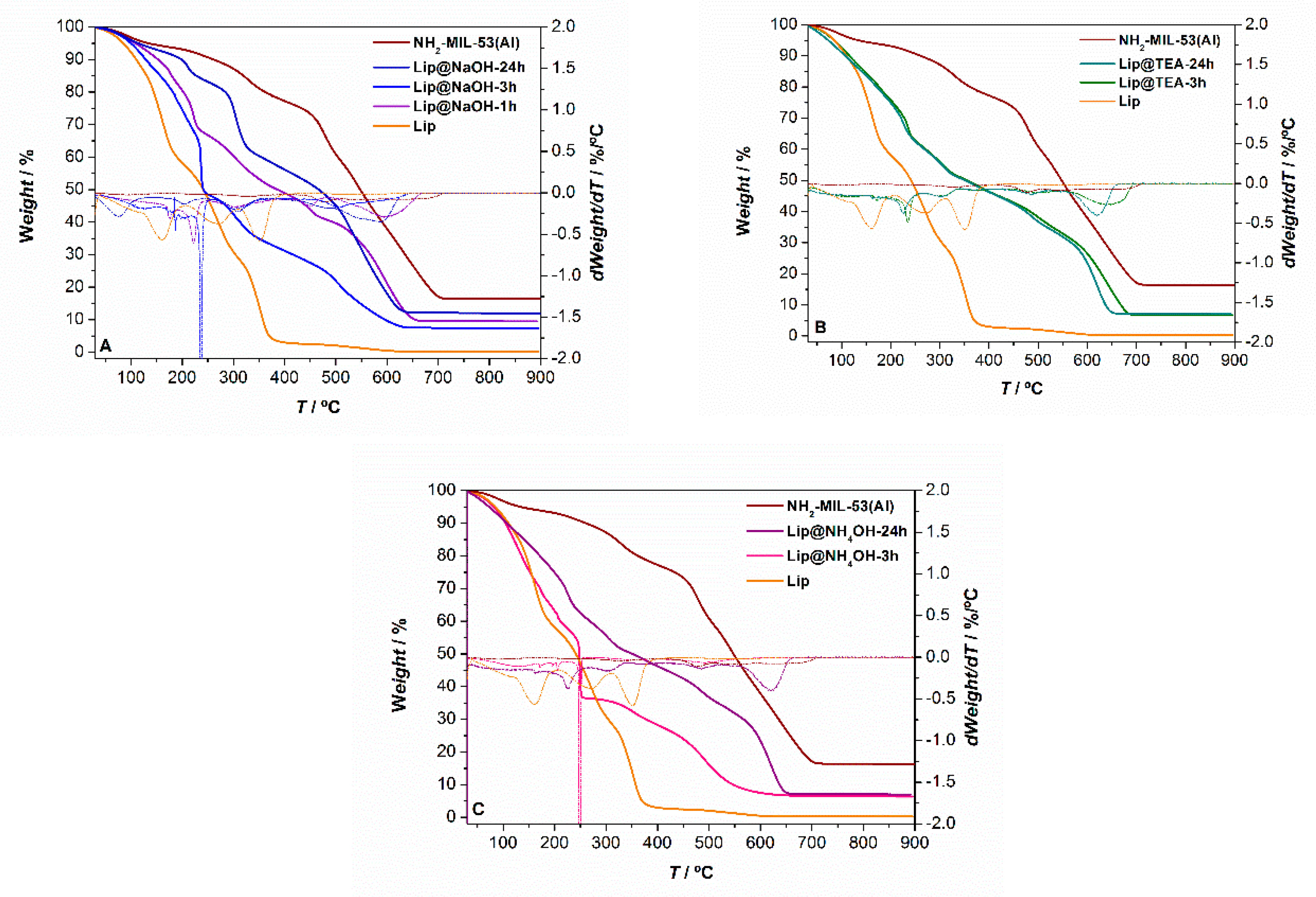
The biocatalysts were also characterized by thermogravimetric analysis (TGA) in order to try to identify the enzymes taking advantage of its different thermal stability compared with the rest of components of the biocatalyst. In this regard,
Figure 2 shows the TGA curves of all Lip@NH
2-MIL-53(Al) samples included in
Table 1. The TGA curves of the enzyme-free MOF and the free lipase CaLB have been included to aid in the interpretation of the mass losses. Both biocatalysts prepared using TEA as a deprotonating agent under different synthesis times, have practically the same TGA curves. Under 3 h of synthesis time the three biocatalysts (one with NaOH, one with TEA, and one with NH
4OH) present an abrupt weight loss near 250 °C, which might be attributed to the presence of the enzyme. The magnitude of these mass losses is higher than 10%, much higher than the immobilized enzyme (
Table 1), suggesting that this mass loss may be ascribed to the enzyme molecules encapsulated by the MOF as well as the enzyme located on the surface of the MOF. This behaviour has already been detected in other in situ immobilizations of enzymes with different MOF materials [
23]. The TGA curves of the biocatalysts prepared at longer times (24 h) with any of these three deprotonating agents exhibit similar mass losses but not as noticeable, and they occur at higher temperatures (>300 °C) which might hint at a higher lipase encapsulation in the MOF compared to on the surface.
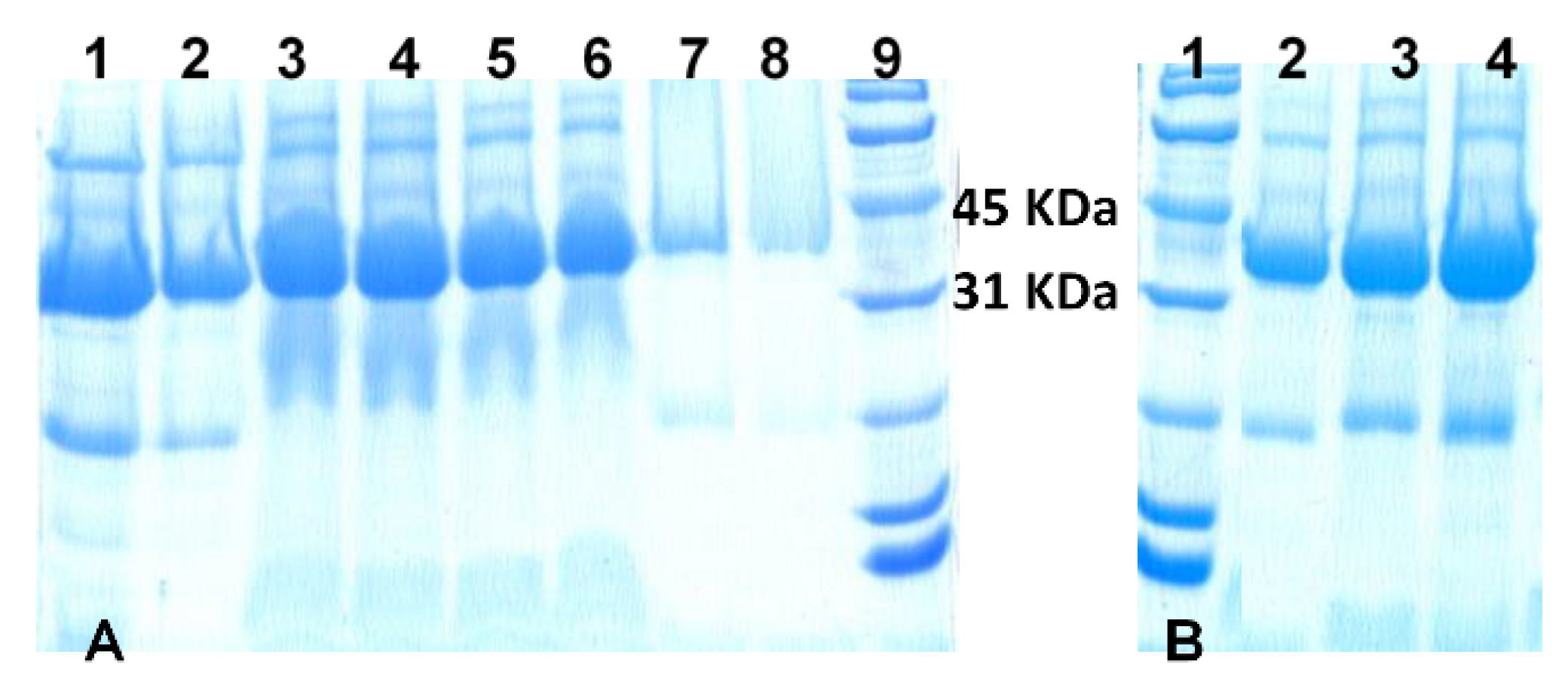
In another effort to detect the presence and/or absence of the enzyme in the biocatalysts, the SDS-PAGE electrophoresis gel technique was employed [
43]. In this sense,
Figure 3 shows the image of the lipase CaLB extract and the in situ and
post-synthesis lipase-containing biocatalysts. All the biocatalysts gave a signal in the electrophoresis gel between 31 and 45 kDa, which was attributed to the enzyme lipase CaLB, whose molecular weight is 33.5 kDa. Thin bands in lanes 7 and 8 in
Figure 3A show the large difference in the immobilization yields of the
post-synthesis biocatalysts. In situ biocatalysts Lip@NH
2-MIL-53(Al) produced much wider bands, due to the higher enzyme loading.
2.4. Enzyme Leaching
To ascertain the amount of enzyme immobilized in the MOF, the enzyme concentration initially present as identified by the Bradford method [
40] was subtracted from the residual concentration of enzyme after filtering (Bradford method,
Table 1). The resultant biocatalysts were tested in the leaching experiment.
Enzyme leaching is a key parameter in the solid biocatalysts formed by the immobilization of enzymes. In this work, the biocatalysts (in situ and
post-synthesis) were suspended in a solution under conditions designed for lipase leaching, which were even more aggressive than the conditions used under the tested catalytic reaction.
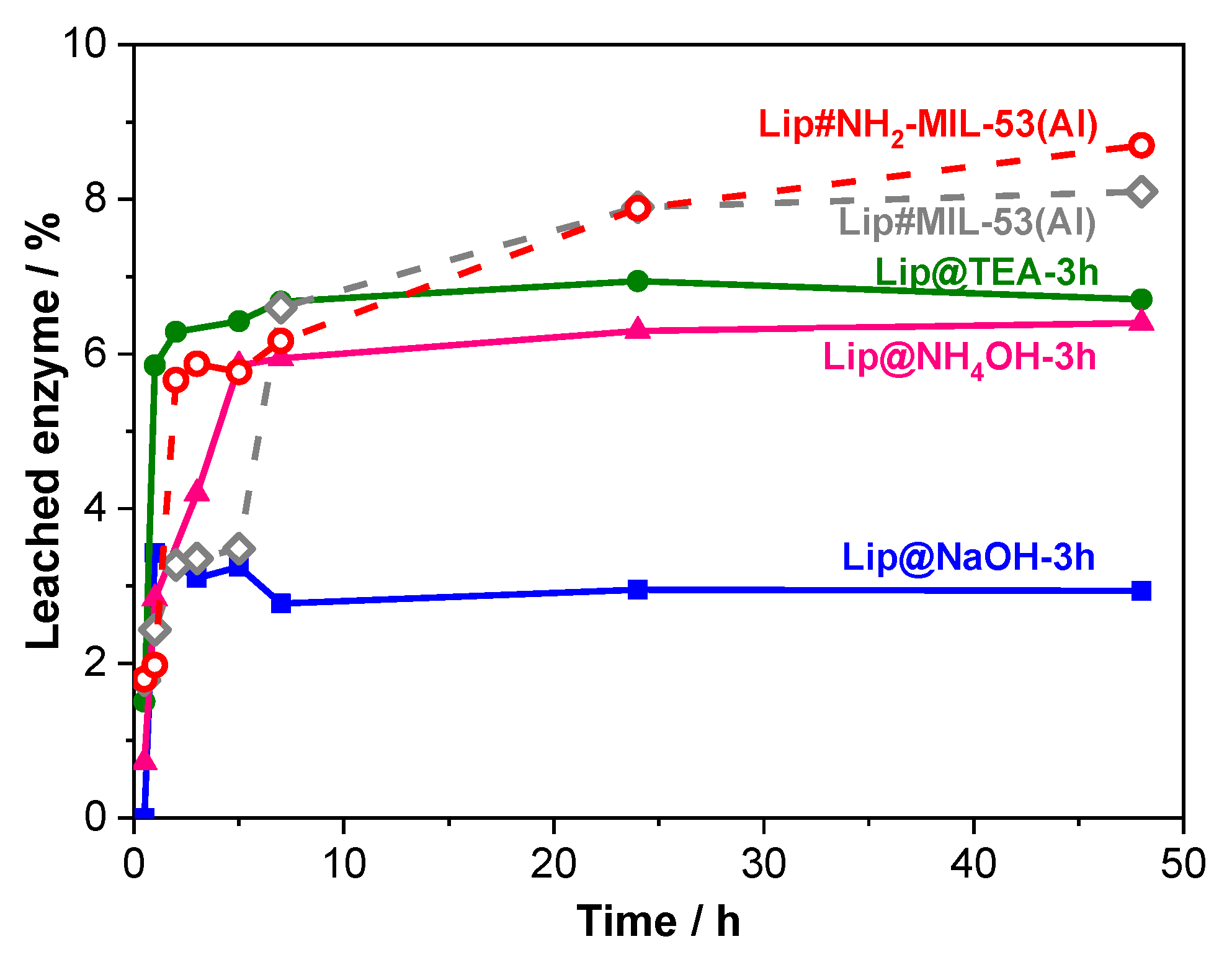
Figure 4 shows the enzyme leaching during 48 h for the main catalysts prepared by either methodology.
The most remarkable feature of
Figure 4 is that the leaching is below 7% of the in situ immobilized lipase. The total enzyme leaching of these biocatalysts Lip@NH
2-MIL-53(Al) is produced in the first 5 h. After this time, no more enzyme is released in any of these biocatalysts indicating that the remaining enzyme is strongly retained inside the crystalline network of the MOFs. The initial desorption may be due to the presence of weakly (physically) adsorbed enzyme molecules, which are easily released from their associated support.
It is particularly remarkable that the biocatalyst Lip@NH2-MIL-53(Al) prepared by the in situ methodology using NaOH as a deprotonating agent (1 h), scarcely lost 3% of the initially retained lipase providing an additional advantage to this biocatalyst.
The shape of the curves corresponding to lipase leaching of the post-synthesis Lip#NH2-MIL-53(Al) is different. After an initial fast enzyme desorption, similar to that exhibited by the in situ method, the enzyme released does not stop but continues increasing. This indicates that the enzyme molecules may not be retained within the MOF network.
3. Materials and Methods
3.1. Reagents and Chemicals
The organic linkers 2-aminoterephthalic acid (NH2-H2BDC, 98%) and terephthalic acid (H2BDC, 99%) was purchased from Sigma-Aldrich. The metallic precursor aluminum nitrate nonahydrate (Al(NO3)3·9H2O, ≥98%) was acquired from Sigma-Aldrich. Sodium hydroxide (≥98%) and ammonia (29%) were from Panreac whereas triethylamine (≥99%) was acquired from Sigma-Aldrich.
The extract of soluble CaLB L Lipase (Lipozyme CaLB L) from Candida sp. recombinant, expressed in Aspergillus niger was kindly provided by Novozymes. Enzyme substrates 4-nitrophenyl-acetate (p-NPA, ≥98%) and tributyrin (TB) were purchased from Sigma-Aldrich (Madrid, Spain).
Sodium hydroxide (NaOH, ≥98%), acetic acid (99%), and di-sodium hydrogen phosphate dihydrate were purchased from Panreac. Sodium acetate anhydrous (>99%) was acquired from Scharlab. Phosphoric acid (85%) and citric acid was purchased from Sigma-Aldrich. Tri-sodium citrate dehydrate was from Analyticals Carlo Erba (Cornaredo, Milan, Italy).
Commercial protein standard bovine serum albumin (BSA) purchased from Sigma-Aldrich along with Coomassie Brilliant Blue R-250 staining solution acquired from Bio-Rad were used for determination of protein concentration employing the Bradford assay determination method.
The employed reagents for electrophoresis (SDS-PAGE) were: Mercaptoethanol (>99%) purchased from Sigma-Aldrich; tetrabromophenolsulfonphthalein or bromophenol blue indicator and glycerol (>99%) provided by Sigma; tris(hydroxymethyl) aminomethane (Tris base) purchased from Fluka Analytical; sodium dodecyl sulfate (SDS), ammonium persulfate (APS), N,N,N’,N’-tetramethylethylenediamine (TEMED), 40% acrylamide/bis solution, 10x Tris base/glycine/SDS buffer and bio-safeTM coomasie G-250 stain all purchased from Bio-Rad, and finally the SDS molecular weight standard was provided by Bio-Rad.
All reagents and solvents were of analytical grade, and were used as received without any further purification. All solutions were prepared using deionized water obtained from a Milli-Q Plus/Millipore purification system (18 MΩ·cm).
3.2. Characterization of the Supports and Biocatalysts
Powder X-ray diffraction (PXRD) patterns were acquired with a X’PERT Pro PANalytical diffractometer using the Cu-Kα radiation (λ = 1.5406 Å). The data were registered with a two theta step size 0.04° from 4 to 90° and accumulation time of 20 s.
Adsorption-desorption isotherms of the samples were measured on ASAP 2020 or ASAP 2420 devices at −196 °C using nitrogen as adsorption gas. The samples were outgassed at 70 °C for 16 h prior to analysis. Pore diameter distributions were determined from the adsorption branch of the nitrogen isotherms using the BJH (Barrett-Joyner-Halenda) method.
Quantitative determinations of nitrogen and sulphur contents belonging to the enzymes extract and biocatalysts were performed using a LECO CHNS-932 Elemental Analyser with an AD-4 autobalance.
Thermogravimetric analyses (TGA) were carried out using a Perkin-Elmer TGA 7 instrument. Samples were heated in an air flow at atmospheric pressure from 25 to 900 °C at a heating rate of 20 °C∙min−1.
The particle size and morphologies were examined by using a scanning electron microscope (SEM). Images were collected using a XL30S FEG XL SERIES Philips microscope with ECON IV detector. The samples were prepared on a double-sided graphite adhesive tape mounted on the sample holder, and afterwards were metalized by sputtering with a coating of ultra-thin chromium layer before being inserted into the microscope.
3.3. In Situ Immobilization: Lip@NH2-MIL-53(Al) Biocatalysts
The NH2-MIL-53(Al) MOF material was prepared in water at room temperature to compare with the in situ biocatalysts (all together in one-pot synthesis) lipase immobilization methods using three different deprotonating agents (NaOH, TEA, and NH4OH).
The preparation method of the biocatalysts Lip@NH
2-MIL-53(Al) is similar to the one described for the enzyme-free NH
2-MIL-53(Al) MOF material employing room temperature and water [
20,
21]. Slight differences are due to the incorporation of the enzyme extract, in which part of the water quantity used within the organic linker solution was substituted by the enzyme extract solution, and a modification of the addition order was employed to prevent exposure of the enzymes to extreme pH values.
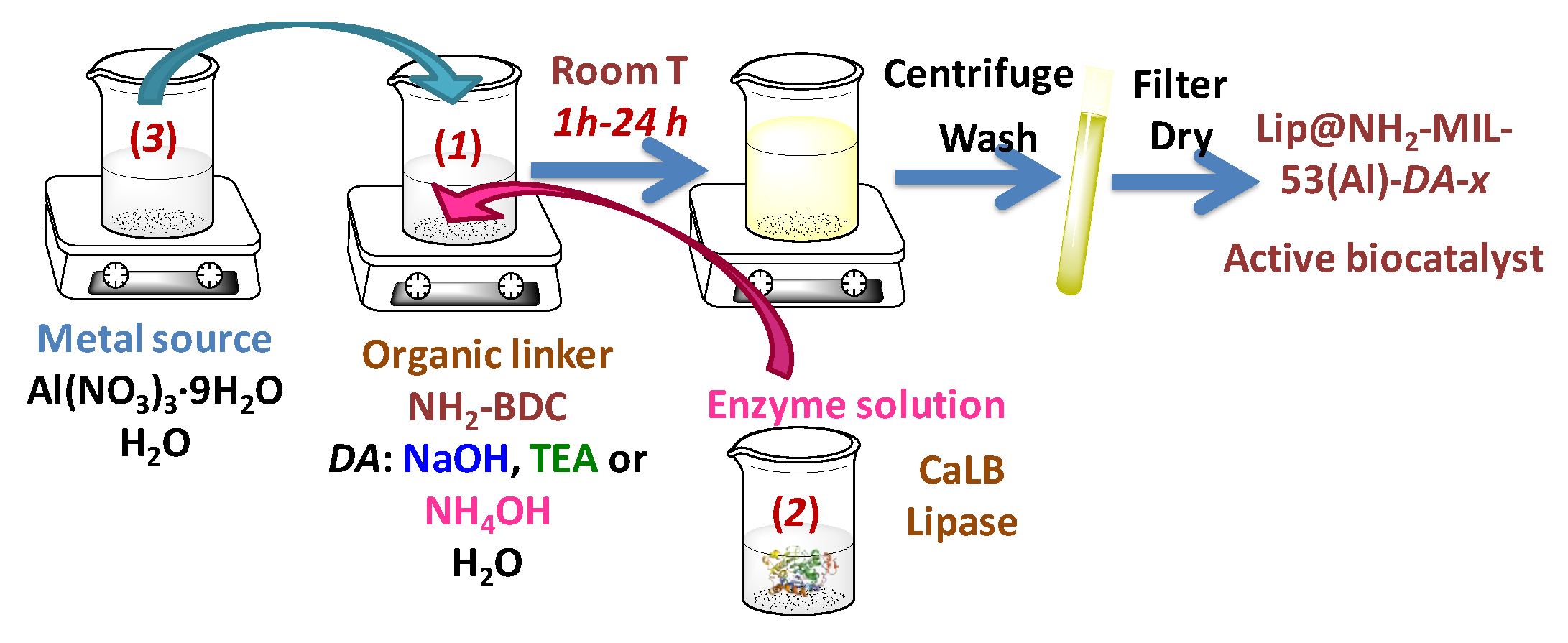
In a typical synthesis procedure, two clear aqueous solutions were separately prepared (
Scheme 1). The organic linker solution (solution 1) was prepared by dissolving 0.482 g of 2-aminoterephtalic acid (NH
2-H
2BDC) in 13.2 g deionized water, assisted by one of the following three deprotonating agents (DA): Sodium hydroxide solution (NaOH 1.04 M, 5.30 g), triethylamine (TEA, 0.539 g), or ammonium hydroxide solution (NH
4OH 29 wt%, 0.644 g). The amount of water was adjusted by taking into account the water content contained in the deprotonating agent source for the case of NaOH and NH
4OH. In the absence of these agents, the linker is not soluble, whereas in the presence of any of them the mixture becomes a clear solution after some time (ca. 10 min NaOH, 3 h TEA and NH
4OH). The resultant pH value of this solution was near neutral (around 6), and it was higher for stronger bases used as deprotonating agents.
The enzymatic solution (solution 2) was formed by 5 mL of a CaLB lipase extract with a concentration of 5.9 mg protein∙mL−1 (measured by the Bradford assay). The metallic source solution (solution 3) was prepared by dissolving 2.0 g of Al(NO3)3·9H2O in 6.0 g Milli-Q grade water reaching a pH around 2.0.
The lipase extract (solution 2) was added over the linker solution (solution 1), inducing a slight pH decrease (ca. 0.5 units) for all mixtures. The metal solution (solution 3) was gently added dropwise into the linker-enzyme solution mixture under slow magnetic stirring in order to prevent pH variations. This procedure resulted in an instant appearance of a yellowish precipitate, which is considered to be the solid biocatalyst MOF material. The pH of the resultant suspension was acidic, slightly above 3. This addition order was selected to minimize enzyme exposure and subsequent denaturing and/or deactivation in extreme acidic media provided by solution 3.
The resultant suspension was maintained under permanent stirring at 25 °C for different reaction times. The obtained solid was then vacuum filtered, washed with Milli-Q grade water and finally dried under a continuous nitrogen flow. The resultant biocatalysts were weighed, gently ground to a powder, and stored at 4 °C. The solid biocatalysts were named as Lip@NH2-MIL-53(Al)-DA-x, where DA can be NaOH, TEA or NH4OH, and x is the synthesis time employed in each sample (1, 3, and 24 h for Lip@NH2-MIL-53(Al)-NaOH biocatalysts, as well as 3 and 24 h for Lip@NH2-MIL-53(Al)-TEA and Lip@NH2-MIL-53(Al)-NH4OH biocatalysts, respectively). During the process of lipase immobilization, aliquots were withdrawn every half an hour, and the enzymatic activity of the blank, suspension and supernatant (after separating it by centrifugation) were assayed spectrophotometrically by the hydrolysis of p-nitrophenyl acetate (p-NPA) conducted by lipase.
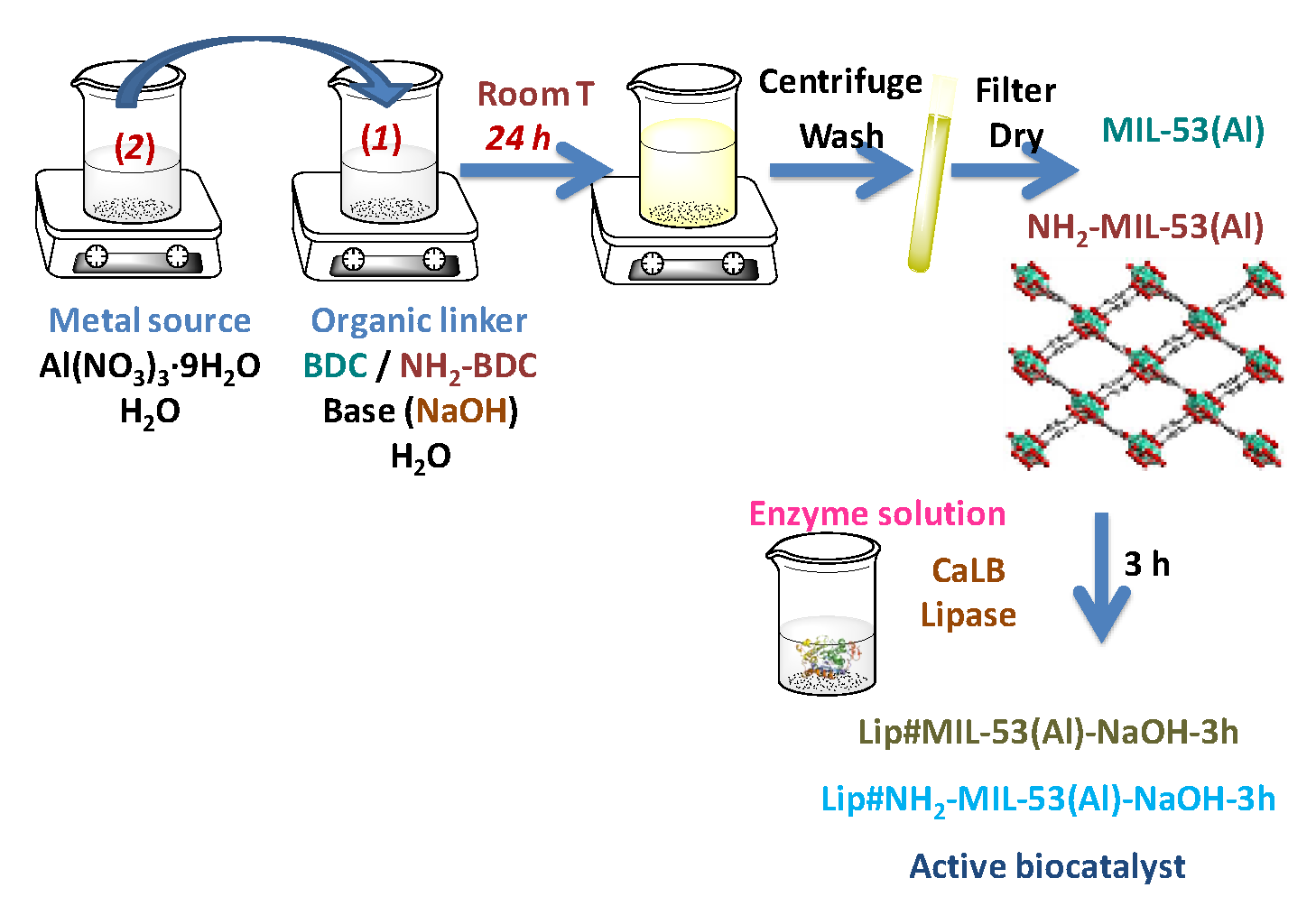
3.4. Post-Synthesis Immobilization: Lip#MIL-53(Al) and Lip#NH2-MIL-53(Al) Biocatalysts
Enzyme-free MOF materials, MIL-53(Al) and NH
2-MIL-53(Al) were prepared in water at room temperature according to the methods described [
20,
21]. These MOF materials were used as supports for the
post-synthesis immobilization of lipase (immobilization after the synthesis of the material) (
Scheme 2).
Post-synthesis immobilization of lipase on either of the two MOF materials (MIL-53(Al), NH
2-MIL-53(Al)) was carried out by suspending 50.0 mg of the MOF material in 10 mL of lipase aqueous solution (0.25 mg·mL
−1) in 50 mM acetic acid/sodium acetate buffer at pH 5.0 (
Scheme 2). The suspension was kept under mild stirring conditions for 3 h (no further enzyme molecules were immobilized after 3 h). During the process of lipase immobilization, aliquots were withdrawn following the same sequence as in the in situ procedure. The decrease in activity of the supernatant to a minimum and constant value indicated the end of the immobilization process. The amount of enzyme immobilized onto the support was calculated by two methods: The difference between the respective activities of the blank or suspension and the supernatant. The second method measured the protein content of the supernatant before and after the immobilization process by the Bradford assay [
40]. The suspension was then vacuum filtered and the resultant solid biocatalysts were washed with the same acetic acid/sodium acetate buffer used for immobilization. No protein was detected in the washing residues (measured by the said Bradford assay). The solid samples were then dried under a vacuum and were stored at 4 °C for further analysis.
3.5. Determination of Enzymatic Activity of the Lipase Extract and Heterogeneous Lipase-Containing Biocatalysts
Hydrolysis of
p-NPA was used as a routine test for measuring spectrophotometrically the nonspecific esterase activity during the process of lipase immobilization and to calculate the enzyme loading (mg·g
−1) [
36]. The activity tests were conducted in triplicate and the average value was calculated for the initial enzyme solution (or blank solution), for the suspension solution (heterogeneous biocatalyst + enzyme solution), and for the supernatant (after centrifugation of the suspension solution). Such spectrophotometrically measurements were conducted in a Diode array UV-Vis spectrophotometer (Agilent 8453) equipped with a constant stirring device and temperature controller. The reaction mixture within the sample cuvette contained 1.9 mL of a previously prepared 0.4 mM
p-NPA aqueous solution (pH 7.0). A 50 µL aliquote of the enzyme solution (blank, suspension or supernatant) was then added in the reaction mixture, and afterwards the increase ratio of absorbance per minute due to the release of
p-nitrophenol product coming from
p-NPA hydrolysis was continuously monitored during 5 min at 348 nm and 25 °C. The molar absorption coefficient was taken as ε
348 nm = 5150 M
−1·cm
−1. A unit of lipase activity (U) was defined as the amount of enzyme required to hydrolize 1 µmol of
p-NPA per minute at 25 °C.
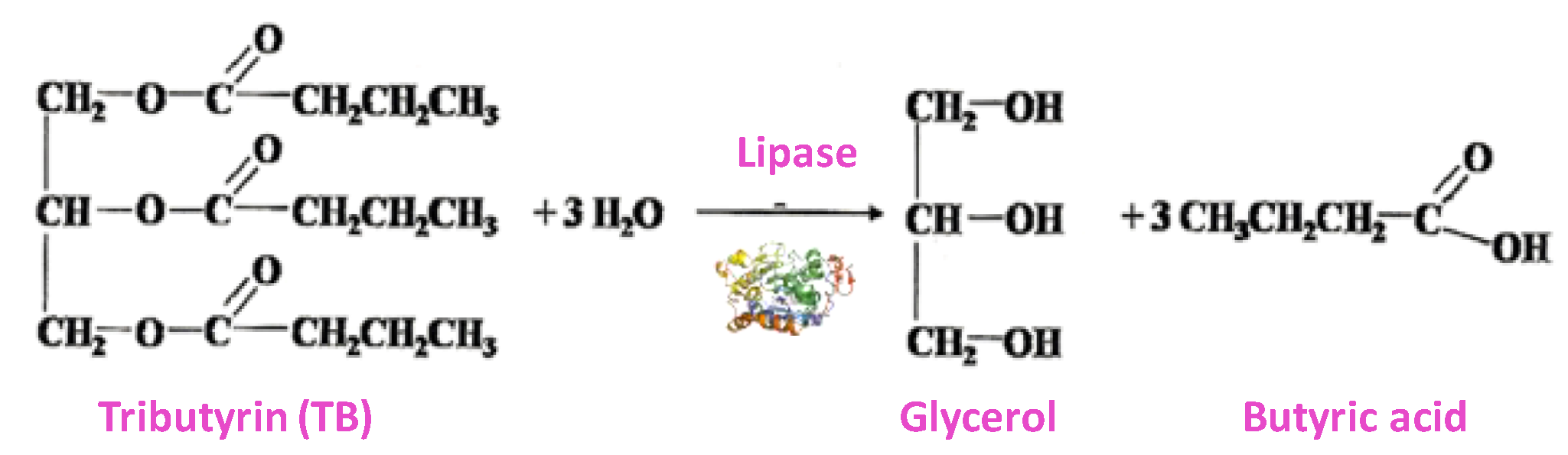
In order to determine the catalytic activity of the Lipase (Lip) heterogeneous biocatalyst, tributyrin hydrolysis was used as an activity test [
36] (
Scheme 3). This method is even more specific than the hydrolysis of
p-NPA. This assay measures the lipase activity by butyric acid release, and it was monitored titrimetrically in a DL-50 pH-state Mettler Toledo, using a 100 mM sodium hydroxide aqueous solution. Then, 48.5 mL of potassium phosphate buffer (10 mM, pH 7.0) were incubated in a thermostated vessel at 25 °C and under stirring. After that, 1.47 mL of tributyrin was added and the pH-state was stated to keep the pH at 7.0. When the pH stabilized, 10 mg of the catalyst were added and the consumption of NaOH was determined. One lipase unit corresponds to consumption of 1 µmol NaOH/min. All measurements were performed in triplicate and their average rate value was taken for plotting and discussion.
3.6. Leaching Tests
In order to infer the resistance of the enzyme to leach from the support, a leaching test was performed. It consisted of subjecting the solid biocatalysts to conditions that presumably favoured the enzyme release by re-suspending the biocatalyst in a high dilution and low ionic strength buffer solution. Consequently, the biocatalysts were incubated along the time in a 50 mM phosphoric acid/trisodium citrate buffer solution at pH 5.0 with a content of 1.25 mg of solid per mL of buffer solution. These incubation periods were 0.5, 1, 2, 3, 5, 7, 24, and 48 h. Enzyme leaching was calculated by monitoring the appearance of protein in the supernatant employing the Bradford assay [
40].
3.7. Electrophoresis Tests
In order to indirectly detect in a qualitative way the presence of lipase enzyme inside the solid supports after 48 h of leaching tests by means of SDS-PAGE electrophoresis, and provided the fact that solid samples are not suitable for this technique, first of all an enzyme denaturalization was conducted by employing a rupture buffer solution obtained according to Laemmli protocol [
41] containing: 240 µL sodium dodecyl sulphate 20%, 500 µL Tris/HCl buffer 2.5 M pH 6.8, 1960 µL Milli-Q water, 2000 µL glycerol, 800 µL bromophenol blue 0.05%, and 1100 µL mercaptoethanol, and then boiled for 10 min. This denaturalization aimed to transform the enzyme tertiary structure (including the split of disulphide bonds) into a random coil chain with the sole purpose of allowing it to leak from the solid framework. As a result, a solution was obtained in which after centrifugation a supernatant could be distinguished presumably containing said protein chains. Afterwards, the withdrawal of this supernatant could be easily performed by centrifugation and then analyzed using a 12% SDS-PAGE electrophoresis gel previously prepared in-house.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}