The Study of Reverse Water Gas Shift Reaction Activity over Different Interfaces: The Design of Cu-Plate ZnO Model Catalysts

 ,
,
Abstract
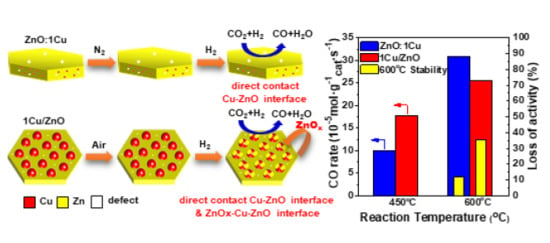
:
1. Introduction
2. Results and Discussion
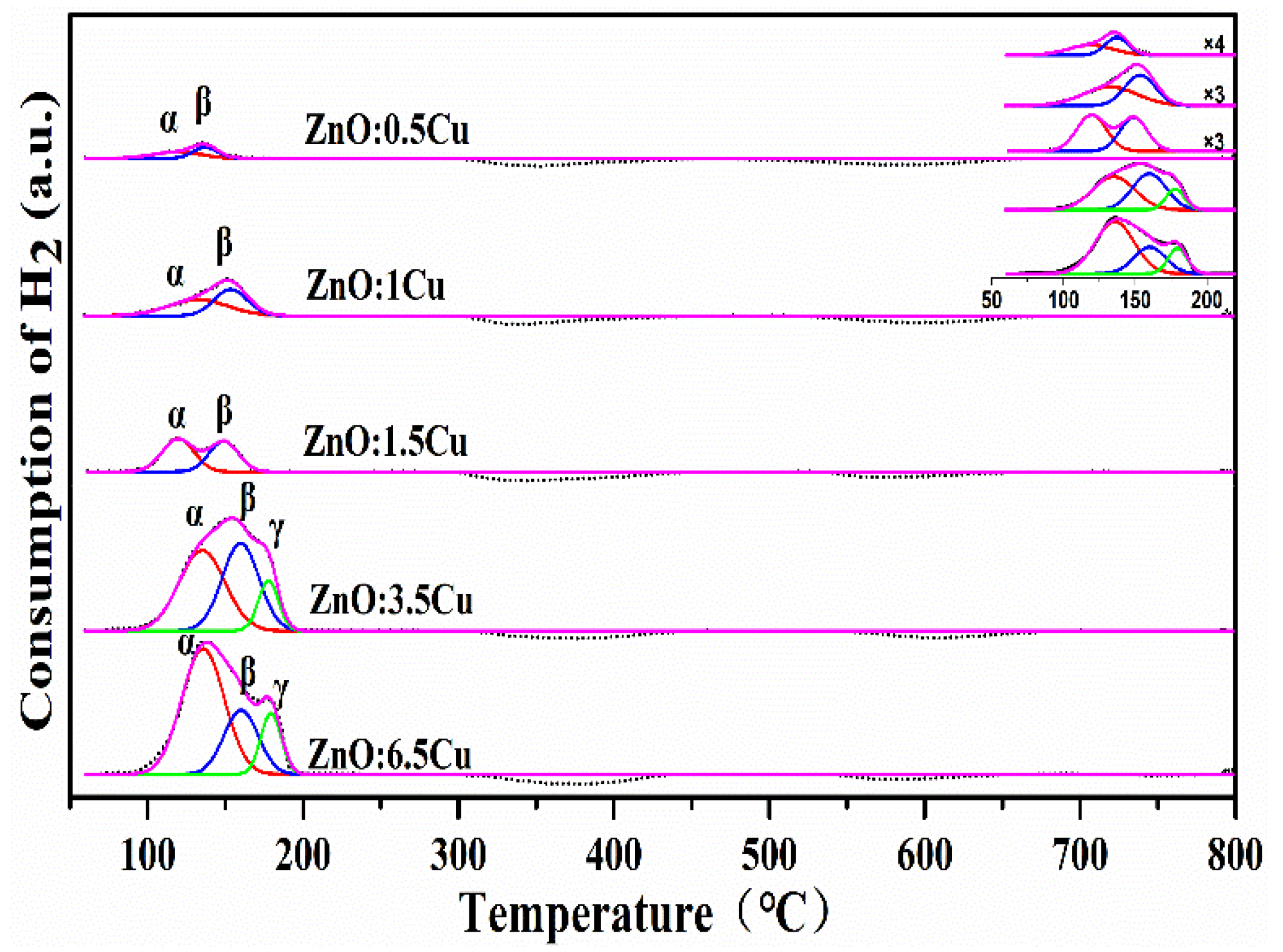
2.1. Catalyst Characterization
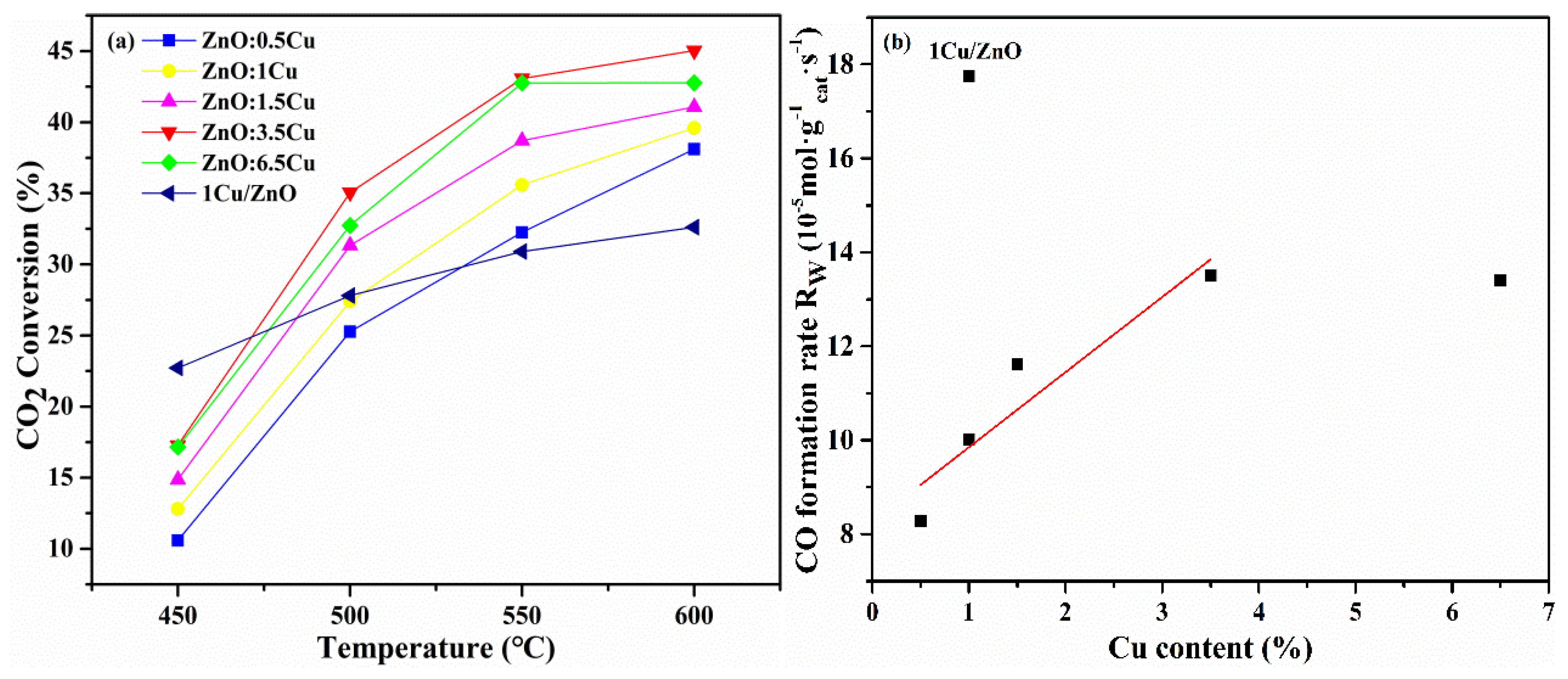
2.2. Catalytic Performance
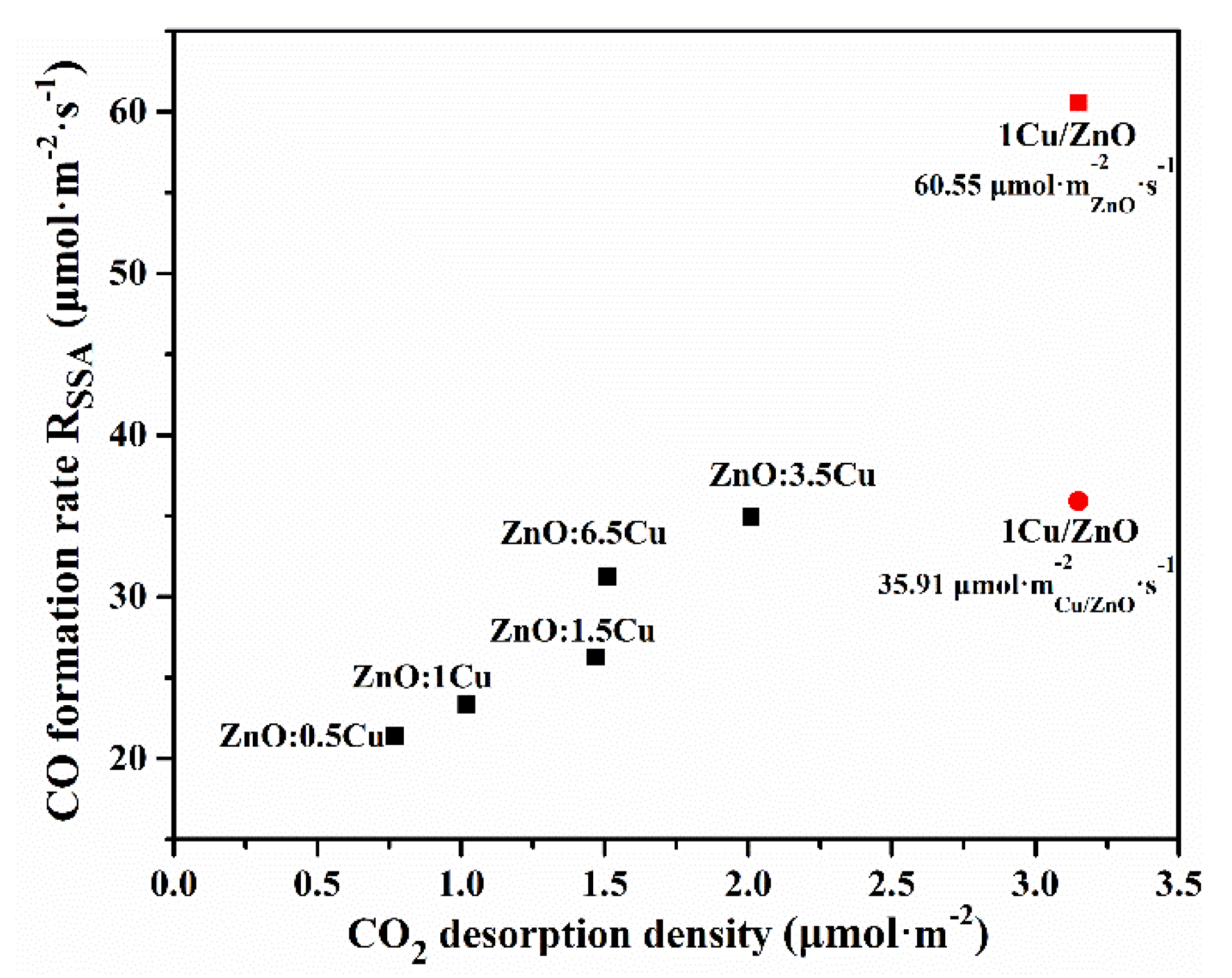
2.3. Discussion
2.3.1. The RWGS Reactivity over Different Cu-ZnO Interfaces
2.3.2. The RWGS Reaction Stability over Different Cu-ZnO Interfaces
3. Materials and Methods
3.1. Catalyst Preparation
3.1.1. Synthesis of Cu Doped Plate ZnO Catalysts
3.1.2. Synthesis of Cu/Plate ZnO Catalysts
3.2. Characterization Technologies
3.3. Catalytic Activity Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Huang, C.-H.; Tan, C.-S. A review: CO2 utilization. Aerosol Air Qual. Res. 2014, 14, 480–499. [Google Scholar] [CrossRef] [Green Version]
- Song, C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Khatib, H. IEA world energy outlook 2011—A comment. Energy Policy 2012, 48, 737–743. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem. Rev. 2013, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, W.; Li, Y.; Chen, J.; Yu, B.; Wang, J.; Zhang, L.; Zhang, J. Energy related CO2 conversion and utilization: Advanced materials/nanomaterials, reaction mechanisms and technologies. Nano Energy 2017, 40, 512–539. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Li, J.; Pippel, E.; Yang, H.; Gao, Z.; Qin, Y. High efficiency Cu-ZnO hydrogenation catalyst: The tailoring of Cu-ZnO interface sites by molecular layer deposition. ACS Catal. 2015, 5, 5567–5573. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Martinez-Suarez, L.; Siemer, N.; Frenzel, J.; Marx, D. Reaction network of methanol synthesis over Cu/ZnO nanocatalysts. ACS Catal. 2015, 5, 4201–4218. [Google Scholar] [CrossRef]
- Galván, C.Á.; Schumann, J.; Behrens, M.; Fierro, J.L.G.; Schlögl, R.; Frei, E. Reverse water-gas shift reaction at the Cu/ZnO interface: Influence of the Cu/Zn ratio on structure-activity correlations. Appl. Catal. B 2016, 195, 104–111. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Li, K.; Zheng, Y.; Xie, Z.; Na, W.; Chen, J.G.; Wang, H. Strong Evidence of the Role of H2O in Affecting Methanol Selectivity from CO2 Hydrogenation over Cu-ZnO-ZrO2. Chem 2020, 6, 419–430. [Google Scholar] [CrossRef]
- Lloyd, L.; Ridler, D.; Twigg, M. Catalyst Handbook; CRC Press: Boca Raton, FL, USA, 1989; p. 283. [Google Scholar]
- Lunkenbein, T.; Schumann, J.; Behrens, M.; Schlögl, R.; Willinger, M.G. Formation of a ZnO overlayer in industrial Cu/ZnO/Al2O3 catalysts induced by strong metal–support interactions. Angew. Chem. Int. Ed. 2015, 54, 4544–4548. [Google Scholar] [CrossRef] [PubMed]
- Le Valant, A.; Comminges, C.; Tisseraud, C.; Canaff, C.; Pinard, L.; Pouilloux, Y. The Cu–ZnO synergy in methanol synthesis from CO2, Part 1: Origin of active site explained by experimental studies and a sphere contact quantification model on Cu+ZnO mechanical mixtures. J. Catal. 2015, 324, 41–49. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Belin, T.; Ahouari, H.; Soualah, A.; Pouilloux, Y.; Le Valant, A. The Cu–ZnO synergy in methanol synthesis from CO2, Part 2: Origin of the methanol and CO selectivities explained by experimental studies and a sphere contact quantification model in randomly packed binary mixtures on Cu–ZnO coprecipitate catalysts. J. Catal. 2015, 330, 533–544. [Google Scholar] [CrossRef]
- Natesakhawat, S.; Lekse, J.W.; Baltrus, J.P.; Ohodnicki, P.R., Jr.; Howard, B.H.; Deng, X.; Matranga, C. Active sites and structure–activity relationships of copper-based catalysts for carbon dioxide hydrogenation to methanol. ACS Catal. 2012, 2, 1667–1676. [Google Scholar] [CrossRef]
- Witoon, T.; Kachaban, N.; Donphai, W.; Kidkhunthod, P.; Faungnawakij, K.; Chareonpanich, M.; Limtrakul, J. Tuning of catalytic CO2 hydrogenation by changing composition of CuO–ZnO–ZrO2 catalysts. Energy Convers. Manag. 2016, 118, 21–31. [Google Scholar] [CrossRef]
- Liao, F.; Huang, Y.; Ge, J.; Zheng, W.; Tedsree, K.; Collier, P.; Hong, X.; Tsang, S.C. Morphology-Dependent Interactions of ZnO with Cu Nanoparticles at the Materials’ Interface in Selective Hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2011, 50, 2162–2165. [Google Scholar] [CrossRef]
- Grunwaldt, J.-D.; Molenbroek, A.; Topsøe, N.-Y.; Topsøe, H.; Clausen, B. In situ investigations of structural changes in Cu/ZnO catalysts. J. Catal. 2000, 194, 452–460. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef]
- Li, C.; Yuan, X.; Fujimoto, K. Development of highly stable catalyst for methanol synthesis from carbon dioxide. Appl. Catal. A 2014, 469, 306–311. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Pronier, S.; Pouilloux, Y.; Le Valant, A. The Cu–ZnO synergy in methanol synthesis Part 3: Impact of the composition of a selective Cu@ ZnOx core–shell catalyst on methanol rate explained by experimental studies and a concentric spheres model. J. Catal. 2016, 343, 106–114. [Google Scholar] [CrossRef]
- Kuld, S.; Thorhauge, M.; Falsig, H.; Elkjær, C.F.; Helveg, S.; Chorkendorff, I.; Sehested, J. Quantifying the promotion of Cu catalysts by ZnO for methanol synthesis. Science 2016, 352, 969–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattel, S.; Ramírez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, C.T.; Daube, K.A.; White, J. Cu/ZnO (0001) and ZnOx/Cu (111): Model catalysts for methanol synthesis. Surf. Sci. 1987, 182, 458–476. [Google Scholar] [CrossRef]
- Palomino, R.M.; Ramírez, P.J.; Liu, Z.; Hamlyn, R.; Waluyo, I.; Mahapatra, M.; Orozco, I.; Hunt, A.; Simonovis, J.P.; Senanayake, S.D. Hydrogenation of CO2 on ZnO/Cu (100) and ZnO/Cu (111) Catalysts: Role of Copper Structure and Metal–Oxide Interface in Methanol Synthesis. J. Phys. Chem. B 2017, 122, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wen, J.; Sun, Y.; Zhang, M.; Bao, Y.; Zhang, Y.; Liang, L.; Fu, M.; Wu, J.; Ye, D.; et al. CO2 hydrogenation to methanol over Cu/ZnO plate model catalyst: Effects of reducing gas induced Cu nanoparticle morphology. Chem. Eng. J. 2019, 374, 221–230. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, C.; Chen, L.; Zhang, Y.; Fu, M.; Wu, J.; Ye, D. Active site structure study of Cu/Plate ZnO model catalysts for CO2 hydrogenation to methanol under the real reaction conditions. J. CO2 Util. 2020, 37, 55–64. [Google Scholar] [CrossRef]
- Chen, L. Controllable Synthesis Method of Novel Disc-Shaped Zinc Oxide Doped with Transition Metals or Rare Earth Metals. Patent ZL 201610559571.8, 18 June 2019. [Google Scholar]
- Bohle, D.S.; Spina, C.J. Controlled Co (II) doping of zinc oxide nanocrystals. J. Phys. Chem. C 2010, 114, 18139–18145. [Google Scholar] [CrossRef]
- Chakraborti, D.; Narayan, J.; Prater, J. Room temperature ferromagnetism in Zn1−xCuxO thin films. Appl. Phys. Lett. 2007, 90, 062504. [Google Scholar] [CrossRef]
- Xu, H.; Liu, Y.; Xu, C.; Liu, Y.; Shao, C.; Mu, R. Structural, optical, and magnetic properties of Mn-doped ZnO thin film. J. Chem. Phys. 2006, 124, 074707. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Leung, Y.H.; Djurišić, A.B.; Liu, Z.T.; Xie, M.H.; Shi, S.L.; Xu, S.J.; Chan, W.K. Different origins of visible luminescence in ZnO nanostructures fabricated by the chemical and evaporation methods. Appl. Phys. Lett. 2004, 85, 1601–1603. [Google Scholar] [CrossRef] [Green Version]
- Kakazey, N.; Sreckovic, T.; Ristic, M. Electronic paramagnetic resonance investigation of the evolution of defects in zinc oxide during tribophysical activation. J. Mater. Sci. 1997, 32, 4619–4622. [Google Scholar] [CrossRef]
- Djurišić, A.B.; Choy, W.C.; Roy, V.A.L.; Leung, Y.H.; Kwong, C.Y.; Cheah, K.W.; Gundu Rao, T.; Chan, W.K.; Fei Lui, H.; Surya, C. Photoluminescence and electron paramagnetic resonance of ZnO tetrapod structures. Adv. Funct. Mater. 2004, 14, 856–864. [Google Scholar] [CrossRef]
- Alnoor, H.; Savoyant, A.; Liu, X.; Pozina, G.; Willander, M.; Nur, O. An effective low-temperature solution synthesis of Co-doped [0001]-oriented ZnO nanorods. J. Appl. Phys. 2017, 121, 215102. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Wang, X.; Qi, G.; Wang, J.; Shen, M.; Li, W. Characterization of copper species over Cu/SAPO-34 in selective catalytic reduction of NOx with ammonia: Relationships between active Cu sites and de-NOx performance at low temperature. J. Catal. 2013, 297, 56–64. [Google Scholar] [CrossRef]
- Li, G.; Dimitrijevic, N.M.; Chen, L.; Rajh, T.; Gray, K.A. Role of surface/interfacial Cu2+ sites in the photocatalytic activity of coupled CuO−TiO2 nanocomposites. J. Phys. Chem. C 2008, 112, 19040–19044. [Google Scholar] [CrossRef]
- He, H.; Guo, J.; Xie, X.; Peng, J. Location and migration of cations in Cu2+-adsorbed montmorillonite. Environ. Int. 2001, 26, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Dietz, R.; Kamimura, H.; Sturge, M.; Yariv, A. Electronic structure of copper impurities in ZnO. Phys. Rev. 1963, 132, 1559. [Google Scholar] [CrossRef] [Green Version]
- Vreugdenhil, W.; Haasnoot, J.G.; Kahn, O.; Thuery, P.; Reedijk, J. A copper (II) dope as a detector for the high-spin. tautm. low-spin transition in the two-dimensional compound [trans-bis (thiocyanato) bis (4, 4’-bi-1, 2, 4-triazole) iron] hydrate. J. Am. Chem. Soc. 1987, 109, 5272–5273. [Google Scholar] [CrossRef]
- Sambasivam, S.; Sathyaseelan, B.; Reddy, D.R.; Reddy, B.; Jayasankar, C. ESR and photoluminescence properties of Cu doped ZnS nanoparticles. Spectrochim. Acta A 2008, 71, 1503–1506. [Google Scholar] [CrossRef]
- Gervasini, A.; Manzoli, M.; Martra, G.; Ponti, A.; Ravasio, N.; Sordelli, L.; Zaccheria, F. Dependence of copper species on the nature of the support for dispersed CuO catalysts. J. Phys. Chem. B 2006, 110, 7851–7861. [Google Scholar] [CrossRef]
- Kivelson, D.; Neiman, R. ESR studies on the bonding in copper complexes. J. Chem. Phys. 1961, 35, 149–155. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; van Bokhoven, J.A. Revisiting copper reduction in zeolites: The impact of autoreduction and sample synthesis procedure. Chem. Commun. 2018, 54, 7447–7450. [Google Scholar] [CrossRef] [PubMed]
- Drmosh, Q.A.; Rao, S.G.; Yamani, Z.H.; Gondal, M.A. Crystalline nanostructured Cu doped ZnO thin films grown at room temperature by pulsed laser deposition technique and their characterization. Appl. Surf. Sci. 2013, 270, 104–108. [Google Scholar] [CrossRef]
- Peng, X.; Xu, J.; Zang, H.; Wang, B.; Wang, Z. Structural and PL properties of Cu-doped ZnO films. J. Lumin. 2008, 128, 297–300. [Google Scholar] [CrossRef]
- Šćepanović, M.; Grujić-Brojčin, M.; Vojisavljević, K.; Bernik, S.; Srećković, T. Raman study of structural disorder in ZnO nanopowders. J. Raman Spectrosc. 2010, 41, 914–921. [Google Scholar] [CrossRef]
- Wang, X.; Xu, J.; Zhang, B.; Yu, H.; Wang, J.; Zhang, X.; Yu, J.; Li, Q. Signature of intrinsic high-temperature ferromagnetism in cobalt-doped zinc oxide nanocrystals. Adv. Mater. 2006, 18, 2476–2480. [Google Scholar] [CrossRef]
- Rajalakshmi, M.; Arora, A.K.; Bendre, B.; Mahamuni, S. Optical phonon confinement in zinc oxide nanoparticles. J. Appl. Phys. 2000, 87, 2445–2448. [Google Scholar] [CrossRef]
- Reddy, G.K.; Gunasekera, K.; Boolchand, P.; Dong, J.; Smirniotis, P.G. High temperature water gas shift reaction over nanocrystalline copper codoped-modified ferrites. J. Phys. Chem. C 2011, 115, 7586–7595. [Google Scholar] [CrossRef]
- Schumann, J.; Eichelbaum, M.; Lunkenbein, T.; Thomas, N.; Alvarez Galvan, M.C.; Schlögl, R.; Behrens, M. Promoting strong metal support interaction: Doping ZnO for enhanced activity of Cu/ZnO: M (M = Al, Ga, Mg) catalysts. ACS Catal. 2015, 5, 3260–3270. [Google Scholar] [CrossRef]
- Chen, L.; Wang, J.; Valenzuela, M.; Bokhimi, X.; Acosta, D.; Novaro, O. Hydrogen spillover and structural defects in a PdO/zirconia nanophase synthesized through a surfactant-templated route. J. Alloy. Compd. 2006, 417, 220–223. [Google Scholar] [CrossRef]
- Dong, X.; Li, F.; Zhao, N.; Xiao, F.; Wang, J.; Tan, Y. CO2 hydrogenation to methanol over Cu/ZnO/ZrO2 catalysts prepared by precipitation-reduction method. Appl. Catal. B 2016, 191, 8–17. [Google Scholar] [CrossRef]
- Huang, C.; Chen, S.; Fei, X.; Liu, D.; Zhang, Y. Catalytic hydrogenation of CO2 to methanol: Study of synergistic effect on adsorption properties of CO2 and H2 in CuO/ZnO/ZrO2 system. Catalysts 2015, 5, 1846–1861. [Google Scholar] [CrossRef] [Green Version]
- Thang, H.V.; Pacchioni, G. Electronic structure of Al, Ga, In and Cu doped ZnO/Cu (111) bilayer films. Phys. Chem. Chem. Phys. 2019, 21, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Derchi, M.; Hensen, E.J. Promotional effect of transition metal doping on the basicity and activity of calcined hydrotalcite catalysts for glycerol carbonate synthesis. Appl. Catal. B 2014, 144, 135–143. [Google Scholar] [CrossRef]
- Cantrell, D.G.; Gillie, L.J.; Lee, A.F.; Wilson, K. Structure-reactivity correlations in MgAl hydrotalcite catalysts for biodiesel synthesis. Appl. Catal. A 2005, 287, 183–190. [Google Scholar] [CrossRef]
- Di Cosimo, J.; Dıez, V.; Xu, M.; Iglesia, E.; Apesteguıa, C. Structure and surface and catalytic properties of Mg-Al basic oxides. J. Catal. 1998, 178, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Yue, H.; Zhao, Y.; Zhao, S.; Wang, B.; Ma, X.; Gong, J. A copper-phyllosilicate core-sheath nanoreactor for carbon–oxygen hydrogenolysis reactions. Nat. Commun. 2013, 4, 2339. [Google Scholar] [CrossRef]
- Prieto, G.; Zečević, J.; Friedrich, H.; De Jong, K.P.; De Jongh, P.E. Towards stable catalysts by controlling collective properties of supported metal nanoparticles. Nat. Mater. 2013, 12, 34. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | ZnO | ZnO:0.5Cu | ZnO:1Cu | ZnO:1.5Cu | ZnO:3.5Cu | ZnO:6.5Cu |
|---|---|---|---|---|---|---|
| (cm−1) | 439 | 436 | 432 | 432 | 428 | 428 |
| E1(LO)/ | 0.09 | 0.13 | 0.22 | 0.27 | 0.45 | 0.53 |
| Samples | CO2 Desorption Amount (μmol·g−1), Temperature | ||||
|---|---|---|---|---|---|
| α, T (°C) | β1, T (°C) | β2, T (°C) | γ, T (°C) | Total (β1 + β2) | |
| ZnO | - | 0.39 (342) | 1.06 (486) | 8.46 (665) | 1.45 |
| ZnO:0.5Cu | 0.40 (82) | 1.65 (371) | 1.37 (427) | 1.03 (648) | 3.02 |
| ZnO:1Cu | 0.36 (83) | 2.73 (363) | 1.64 (416) | 1.74 (656) | 4.37 |
| ZnO:1.5Cu | 0.63 (84) | 3.46 (371) | 3.02 (429) | 1.45 (662) | 6.48 |
| ZnO:3.5Cu | 0.55 (89) | 5.09 (383) | 2.76 (436) | 1.56 (645) | 7.85 |
| ZnO:6.5Cu | 0.58 (89) | 3.18 (382) | 3.31 (431) | 1.95 (661) | 6.49 |
| 1Cu/ZnO | 0.88 (90) | 4.34 (322) | 4.80 (392) | 9.48 (662) | 9.14 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, J.; Huang, C.; Sun, Y.; Liang, L.; Zhang, Y.; Zhang, Y.; Fu, M.; Wu, J.; Chen, L.; Ye, D. The Study of Reverse Water Gas Shift Reaction Activity over Different Interfaces: The Design of Cu-Plate ZnO Model Catalysts. Catalysts 2020, 10, 533. https://doi.org/10.3390/catal10050533
Wen J, Huang C, Sun Y, Liang L, Zhang Y, Zhang Y, Fu M, Wu J, Chen L, Ye D. The Study of Reverse Water Gas Shift Reaction Activity over Different Interfaces: The Design of Cu-Plate ZnO Model Catalysts. Catalysts. 2020; 10(5):533. https://doi.org/10.3390/catal10050533
Chicago/Turabian StyleWen, Jinjun, Chunlei Huang, Yuhai Sun, Long Liang, Yudong Zhang, Yujun Zhang, Mingli Fu, Junliang Wu, Limin Chen, and Daiqi Ye. 2020. "The Study of Reverse Water Gas Shift Reaction Activity over Different Interfaces: The Design of Cu-Plate ZnO Model Catalysts" Catalysts 10, no. 5: 533. https://doi.org/10.3390/catal10050533