Metal-Catalyzed Synthesis and Transformations of β-Haloenol Esters


Laboratorio de Compuestos Organometálicos y Catálisis (Unidad Asociada al CSIC), Departamento de Química Orgánica e Inorgánica, Facultad de Química, Universidad de Oviedo, Julián Clavería 8, E-33006 Oviedo, Spain
Catalysts 2020, 10(4), 399; https://doi.org/10.3390/catal10040399
Submission received: 23 March 2020
/
Revised: 3 April 2020
/
Accepted: 3 April 2020
/
Published: 4 April 2020
(This article belongs to the Special Issue Recent Advances in Organometallic Chemistry and Catalysis)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:In the last years there has been an increasing interest in the search for protocols to obtain β-haloenol esters in an efficient and selective manner as they are versatile building blocks in synthetic organic chemistry. In this article, metal-catalyzed transformations allowing the access to both acyclic and cyclic (i.e., haloenol lactones) β-haloenol esters are reviewed. Metal-catalyzed reactions in which these molecules participate as substrates are also discussed.
1. Introduction
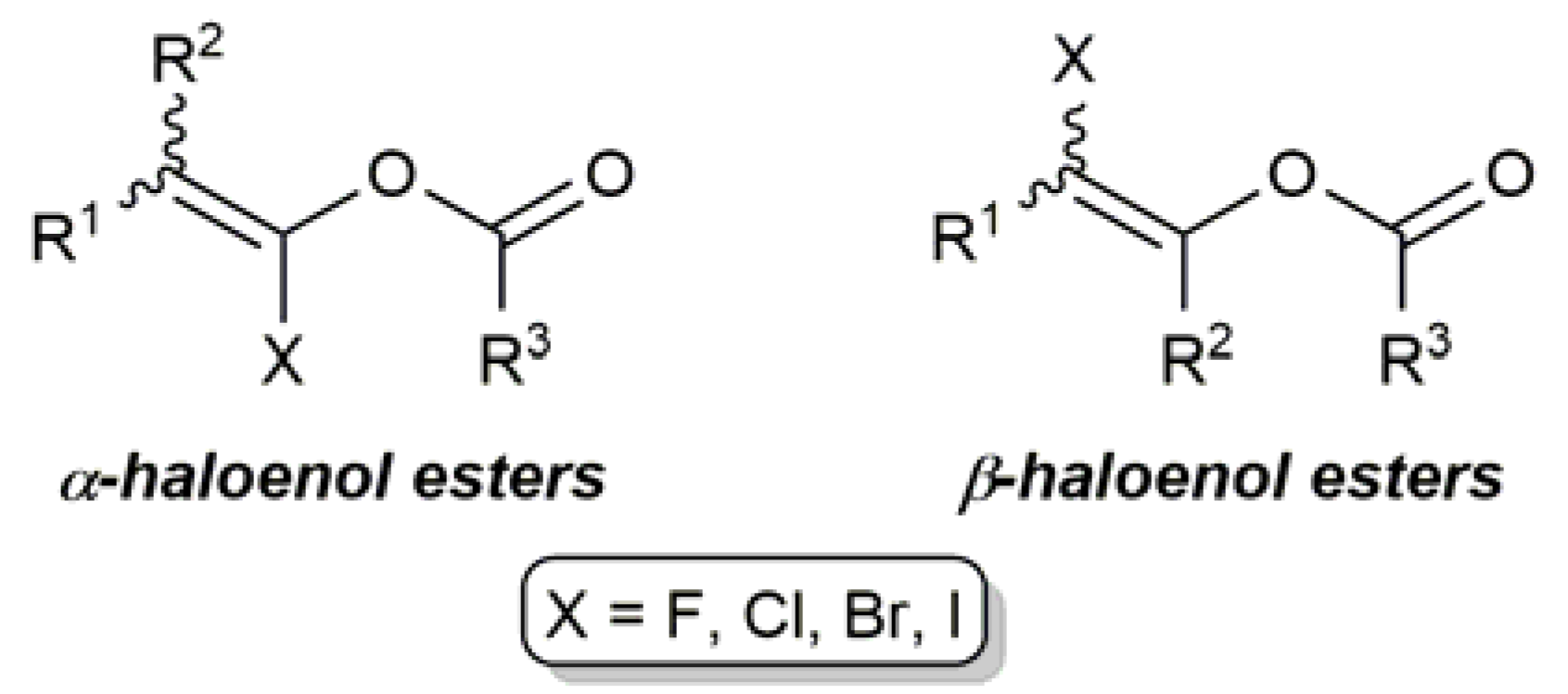
Alkenyl halides (haloalkenes) are a pivotal class of compounds in organic synthesis that can be used in a variety of carbon–carbon and carbon–heteroatom bond-forming reactions. For example, alkenyl halides are widely used as substrates in transition metal-catalyzed cross-coupling reactions [1,2] and can be easily converted, through a metal–halogen exchange, into nucleophiles for 1,2-additions to carbonyl compounds [3]. Enol esters are also relevant olefinic derivatives with multitude of applications in modern organic chemistry [4,5,6]. The introduction of a halogen atom on the C=C bond of the latter leads to functionalized molecules, i.e., α-and β-haloenol esters (Figure 1), in which the reactivities of the haloalkene and enol ester functionalities can be combined and potentially exploited in numerous synthetic ways.
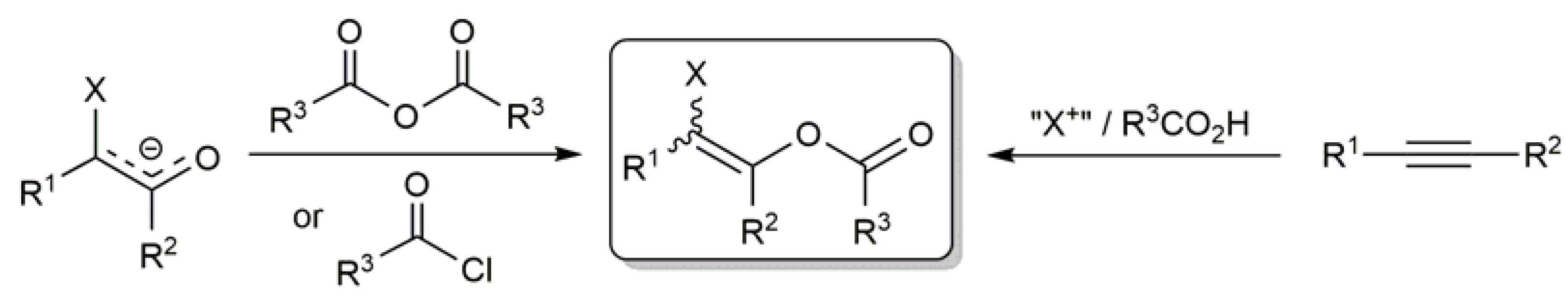
In this context, while the utility of α-haloenol esters remains almost unexplored due to the lack of efficient and general synthetic methods [7,8,9,10], more accessible β-haloenol esters have gained significance in recent years as coupling partners in diverse chemical transformations. Several methodologies can be employed for the preparation β-haloenol esters, the most classical ones involving the acylation of haloenolate anions [11,12,13,14,15] or the haloacyloxylation of alkynes employing the elemental halogens [16,17,18,19,20], bis(pyridine)iodonium tetrafluoroborate (IPy2BF4) [21,22,23,24,25], N-halosuccinimides (NXS; X = Cl, Br, I) [26,27,28], PhI(OAc)2 [29,30,31], trihaloisocyanuric acids [32] or N,N-dibromo-p-toluenesulfonamide (TsNBr2) [33] as the electrophilic halogen source (Scheme 1).
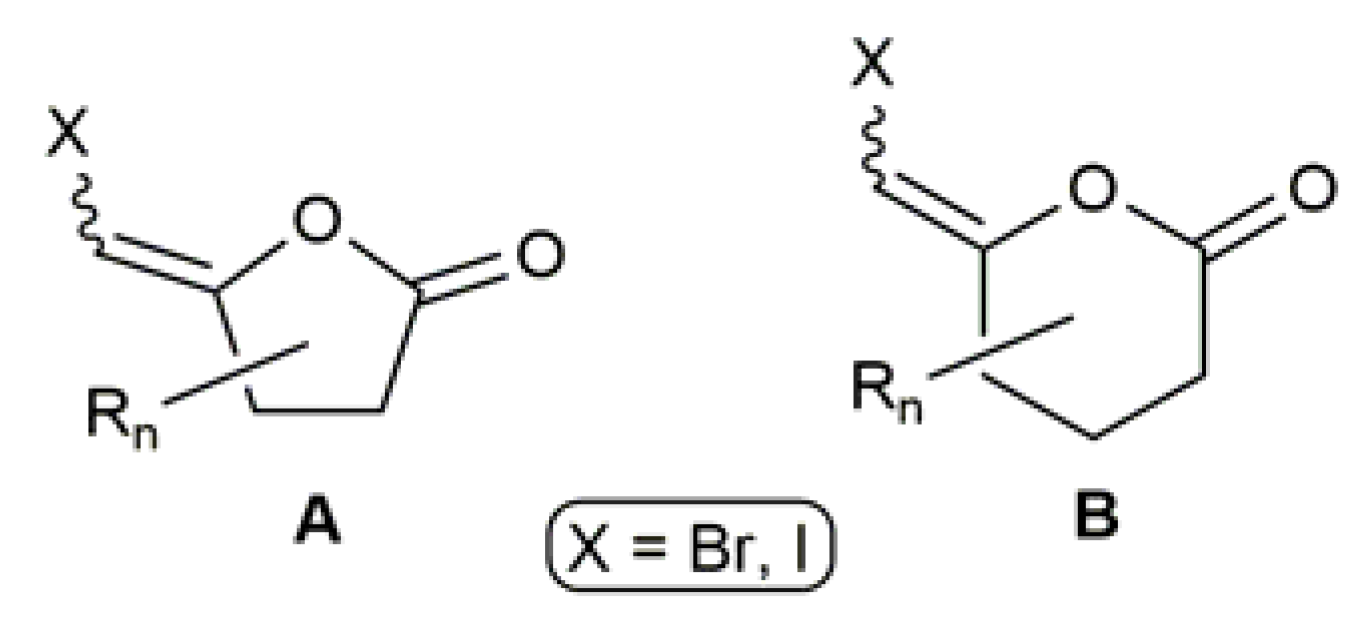
Haloenol lactones of type A and B (see Figure 2) are also a relevant class of compounds due to their biological activity as enzymes inhibitors [34,35,36,37,38], and also because they are a common structural motif in many natural products [39].
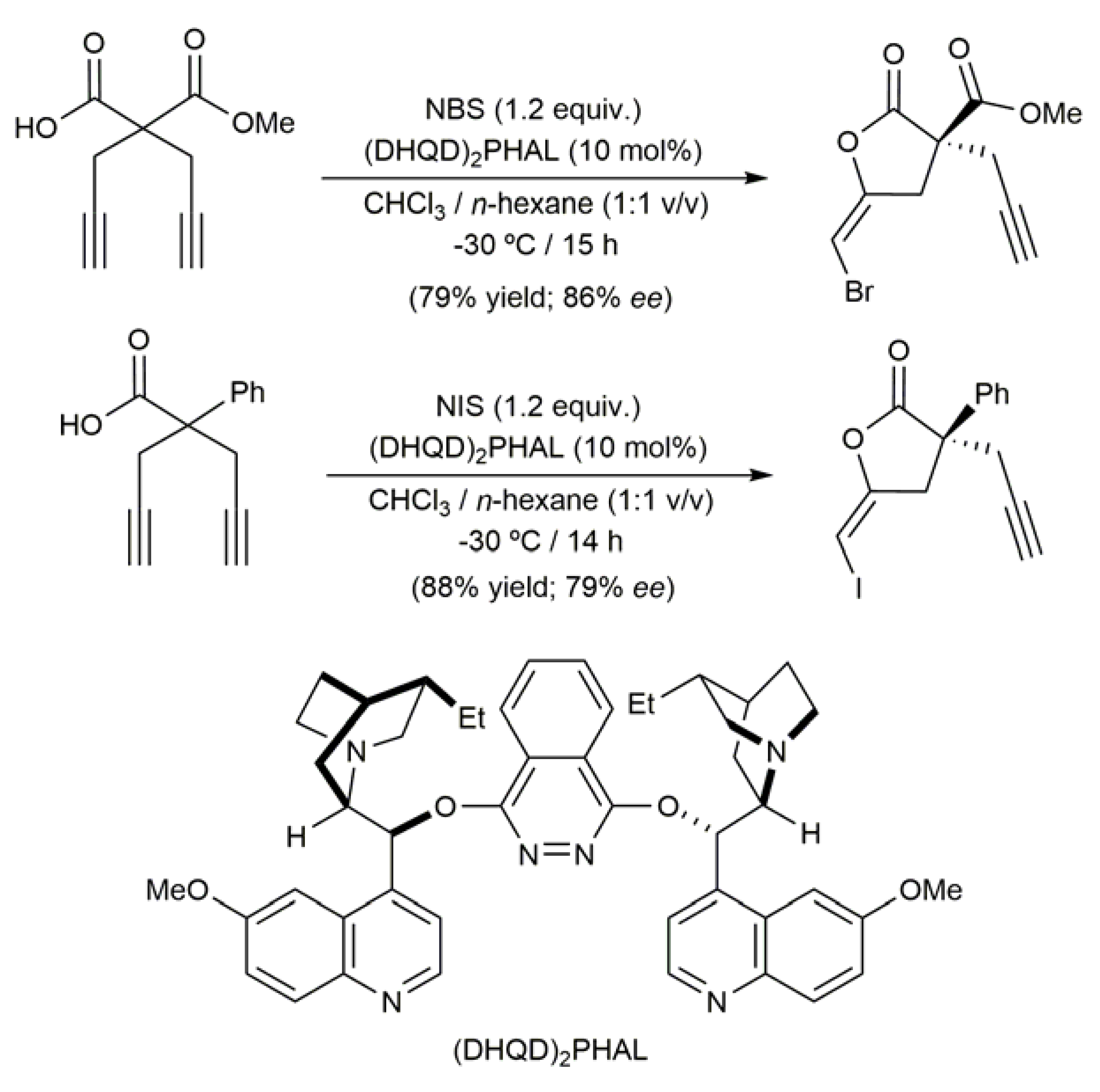
Access to these molecules is usually achieved by halolactonization of the corresponding alkynoic acid using I2, NBS, NIS or related halogenating agents, reactions that deliver the haloenol lactone products as the E isomers exclusively (Scheme 2) [34,35,36,37,38,40,41]. We would like also to highlight here that asymmetric versions of these halolactonization processes have been recently described employing cinchona alkaloid-based organocatalysts [42,43,44]. A couple of illustrative examples are given in Scheme 3. Mechanistic investigations indicated a bifunctional behavior of the organocatalysts, activating simultaneously the halogenating agent and the carboxylic acid unit.
Synthetic approaches to both acyclic and cyclic β-haloenol esters have been expanded in the most recent years by the aid of metal catalysts, employing in most of the cases in situ generated or preformed haloalkynes [45,46,47] as starting materials, and are the subject of the present review article. Thus, the catalytic synthesis of β-haloenol esters will be discussed, as well as the participation of these molecules in metal-catalyzed cross-coupling processes.
2. Metal-Catalyzed Synthesis of β-Haloenol Esters
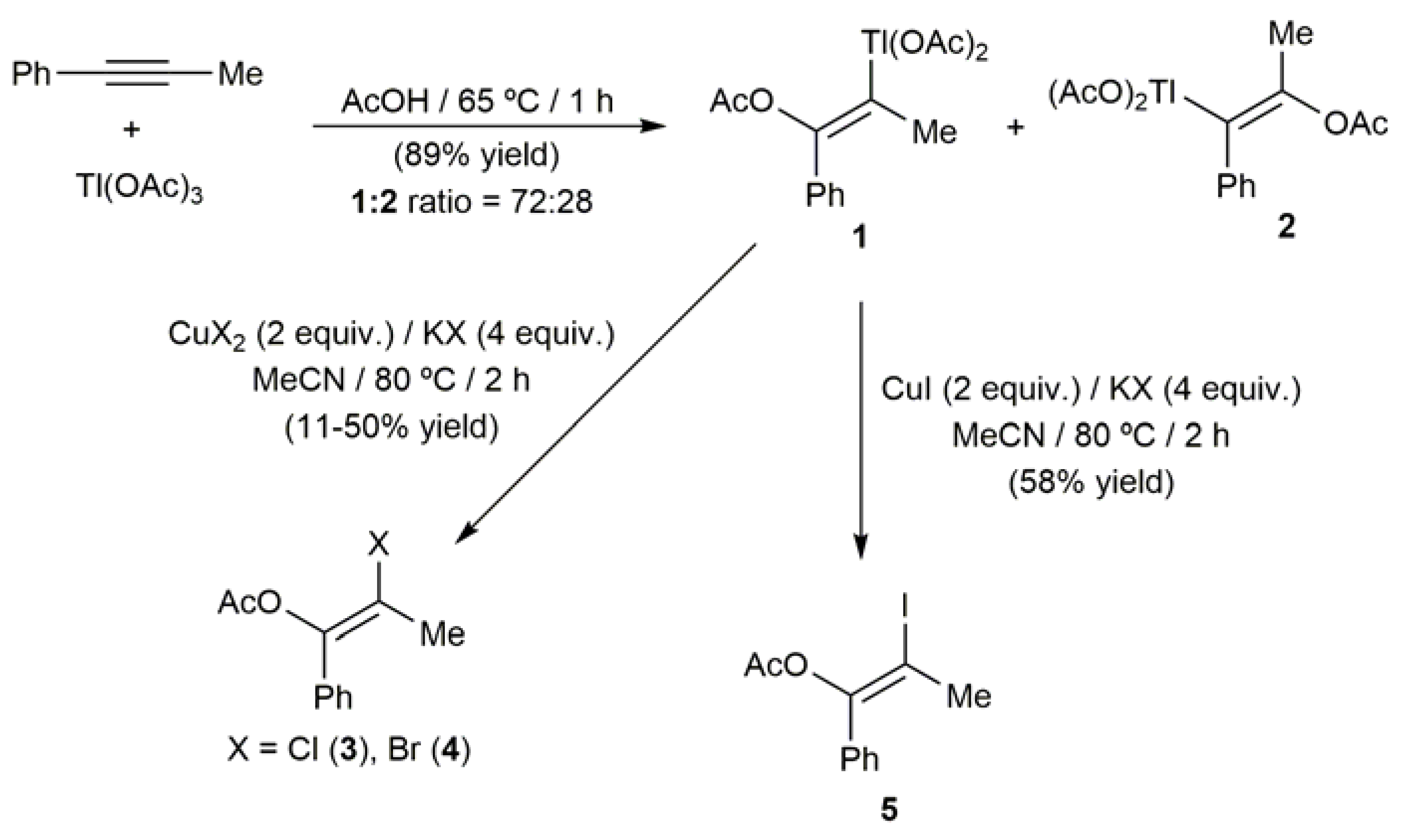
The first metal-mediated route to access β-haloenol esters was described by Ichikawa and co-workers in 1974 (Scheme 4) [48]. They found that 1-propynylbenzene readily undergoes a trans-acetoxythallation, upon treatment of with Tl(OAc)3 in acetic acid, to give a separable mixture of the two isomeric vinylthallium(III) compounds 1 and 2. Subsequent reaction of the major isomer 1 with the corresponding copper(I) or copper(II) halide salt in acetonitrile resulted in the formation of the (E)-β-haloenol acetates 3–5, which were isolated in low yield (11–58%). The halogenodethallation step proceeded in all the cases with complete retention of the C=C configuration.
After this seminal work, more general methodologies based on the use of Hg, Ag, Au and Pd metals have been described.
2.1. Hg-Catalyzed Synthesis of β-Haloenol Esters
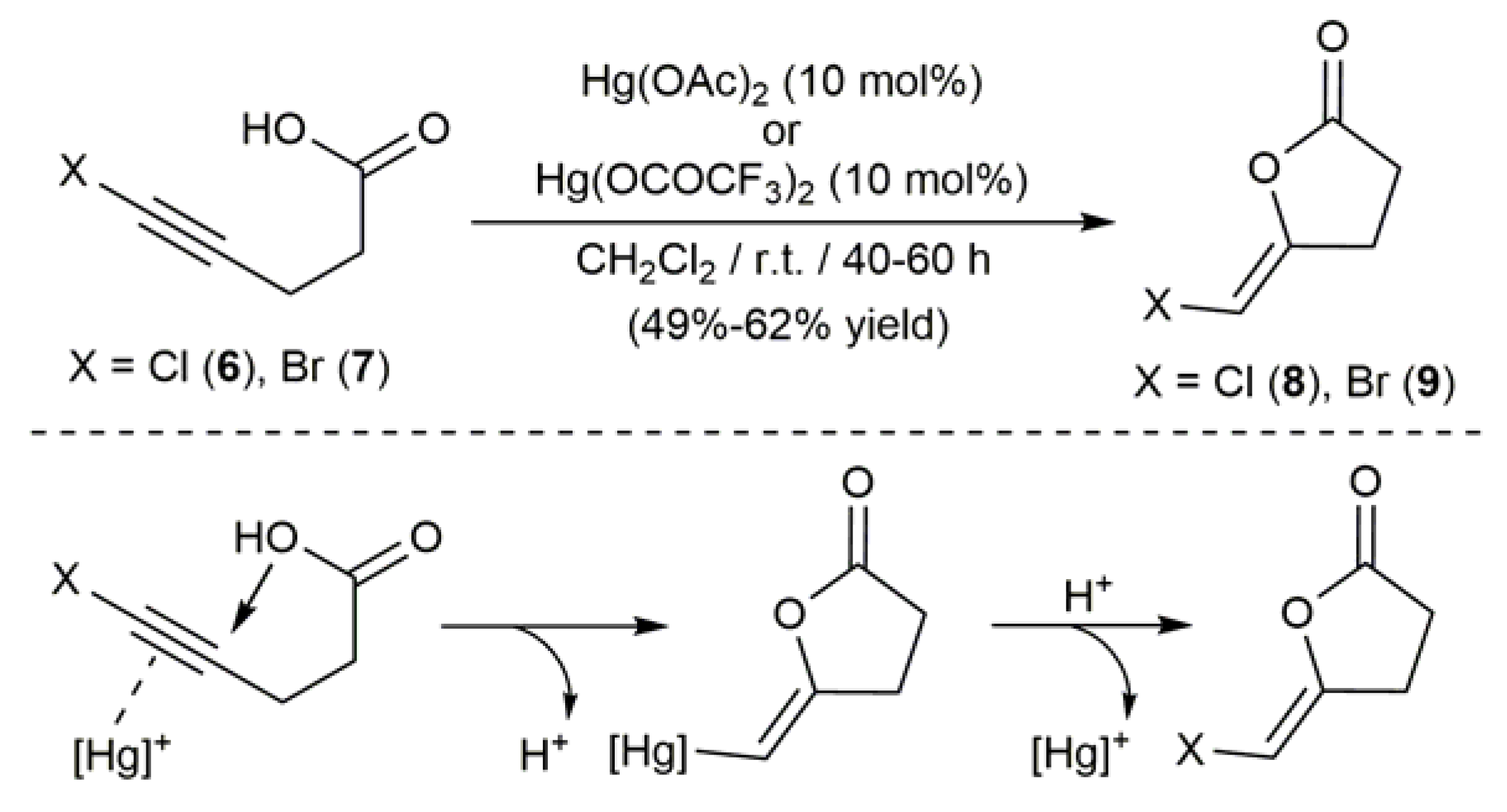
Extending previous studies with non-halogenated molecules [49], Krafft and Katzenellenbogen described in 1981 a synthetic route to haloenol lactones through the mercury-catalyzed cyclization of halogen-substituted alkynoic acids [50]. In particular, they were able to generate compounds 8 and 9 in moderate yield (40–69%) by treatment of dichloromethane solutions of 5-chloro-4-pentynoic acid (6) and 5-bromo-4-pentynoic acid (7), respectively, with 10 mol % of Hg(OAc)2 or Hg(OCOCF3)2 (Scheme 5). The reactions proceed through the anti addition of the carboxylate group to the C≡C bond activated by π-coordination to mercury and, contrary to the halolactonization methods commented above (see Scheme 2), they delivered the haloenol lactone products as the Z isomers exclusively.
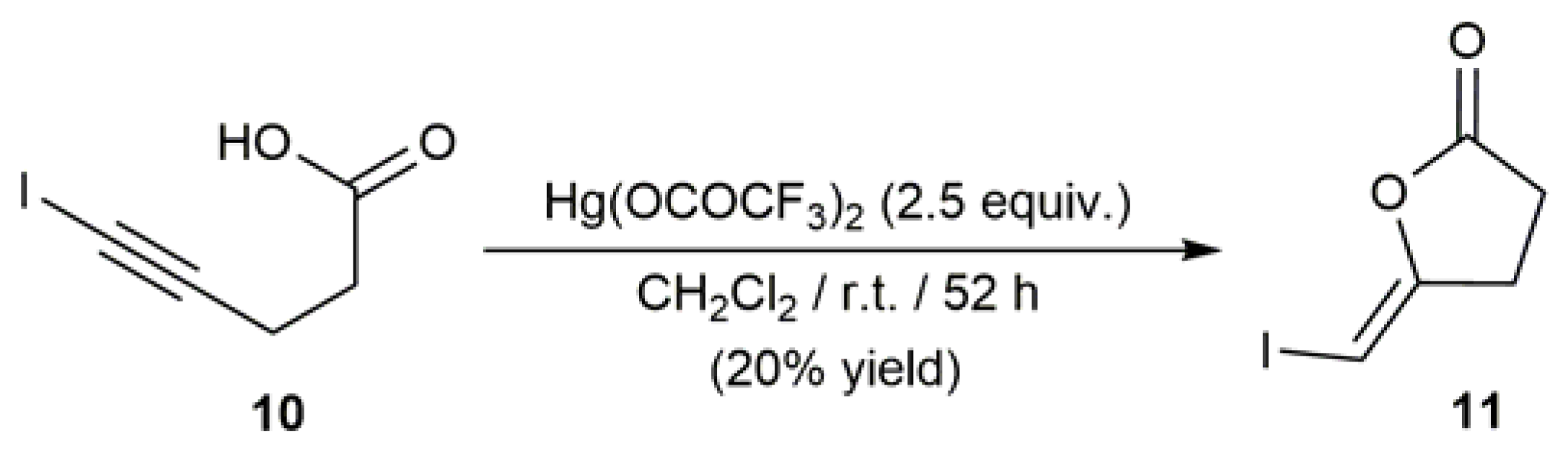
In an independent study, Krantz and co-workers extended this cyclization reaction to 5-iodo-4-pentynoic acid (10). However, the use of an excess of mercury(II) trifluoroacetate was in this case needed, and the (Z)-iodoenol lactone 11 could only be obtained in low yield (Scheme 6) [51].
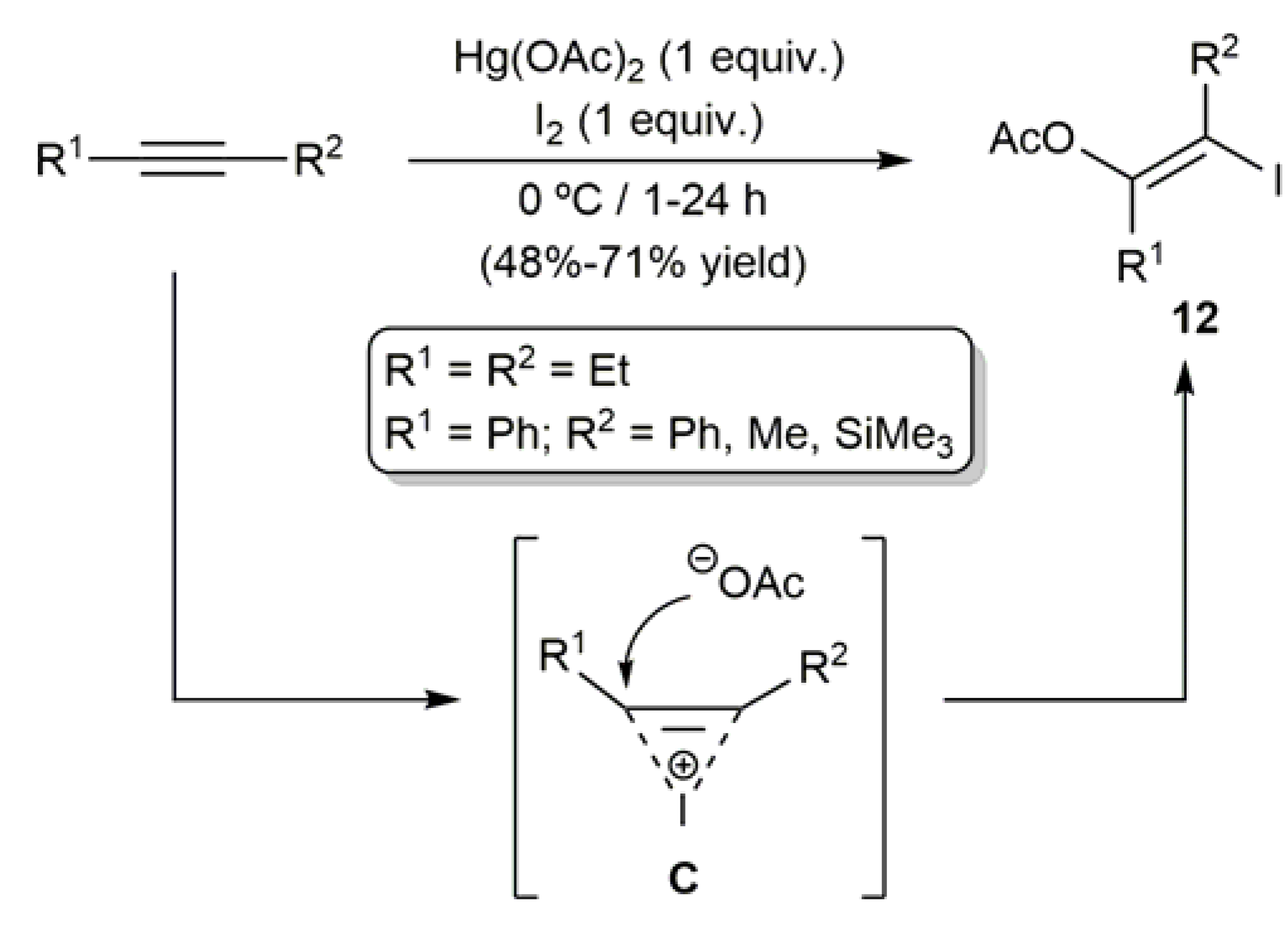
Barluenga and co-workers achieved also the stereoselective synthesis of the acyclic (Z)-β-iodoenol acetates 12 through a difunctionalization reaction of the corresponding internal alkynes by means of the Hg(OAc)2/I2 combination (Scheme 7) [52]. The formation of a cationic intermediate of type C, which undergoes the attack of the acetate anion, was proposed by the authors as the most likely reaction pathway.
2.2. Ag-Catalyzed Synthesis of β-Haloenol Esters
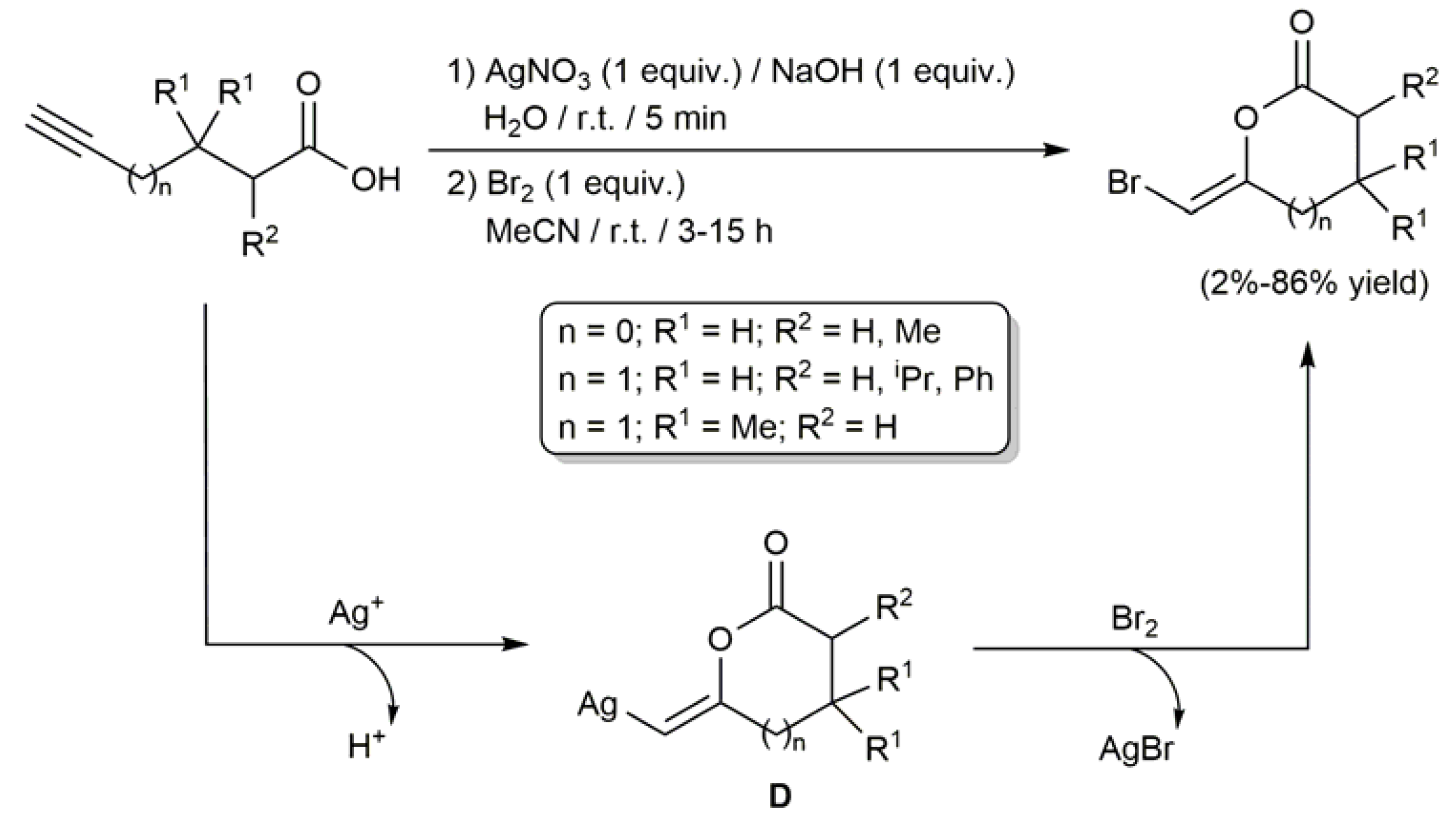
In 1991, the group of Katzenellenbogen described a two-steps protocol for the selective Z-bromoenol lactonization of akynoic acids employing stoichiometric amounts of AgNO3 and Br2 (Scheme 8) [53]. The process involves the initial silver-mediated cyclization of the substrates to generate the metallated lactones D, which subsequently undergo an Ag/Br exchange upon treatment with Br2. Both 5- and 6-membered ring lactones could be synthesized employing this methodology, whose efficiency was found to be conditioned by the substitution pattern of the starting alkynoic acids. In particular, the presence of substituents in α and β position with respect to the carboxylate group (R1 and R2) was key to obtain the products in high yields, those substrates unsubstituted in these positions leading to very poor results (2–5% yield). Additionally, of note is the fact that the lactonization process resulted ineffective with akynoic acids featuring an internal C≡C bond or when I2 was employed as the electrophile.
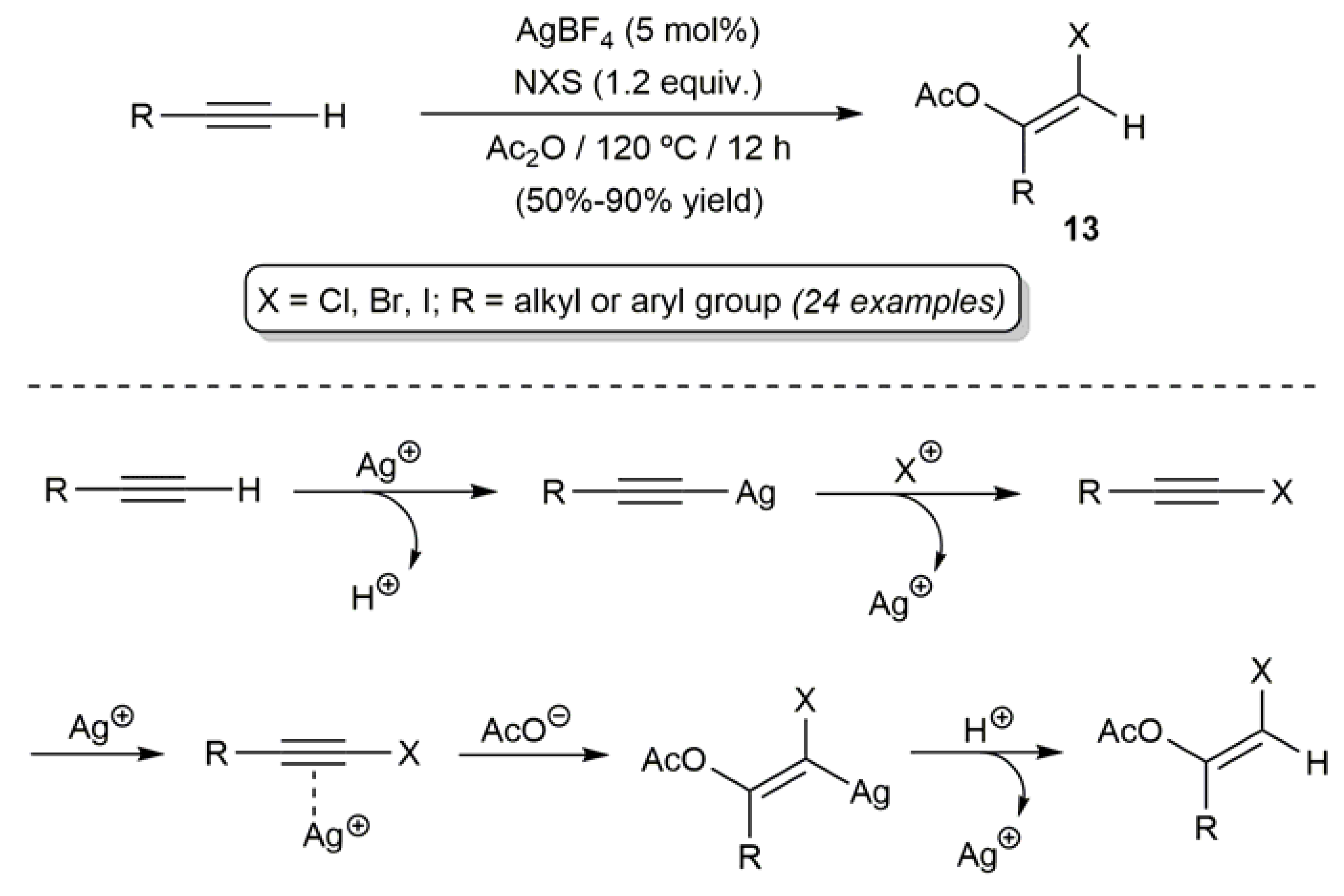
The first general and truly catalytic protocol to obtain acyclic β-haloenol esters came to light only in 2010 and it was developed by Jiang’s group [54]. As shown in Scheme 9, they were able to synthesize a large variety of (Z)-haloenol acetates 13 through an AgBF4-catalyzed difunctionalization reaction of terminal alkynes with N-halosuccinimides (NXS) and acetic anhydride. In the reactions, which were performed at 120 °C with 5 mol % of AgBF4 and employing directly Ac2O as the solvent, the silver(I) cation plays a dual role. Thus, it first acts a σ-activator allowing the in situ generation of the corresponding haloalkynes RC≡CX, and subsequently as a π-activator facilitating the nucleophilic attack of the acetate anion to the C≡C bond. The process featured an exquisite regio- and stereoselectivity, and tolerated the presence of common functional groups in the alkyne substrates.
2.3. Au-Catalyzed Synthesis of β-Haloenol Esters
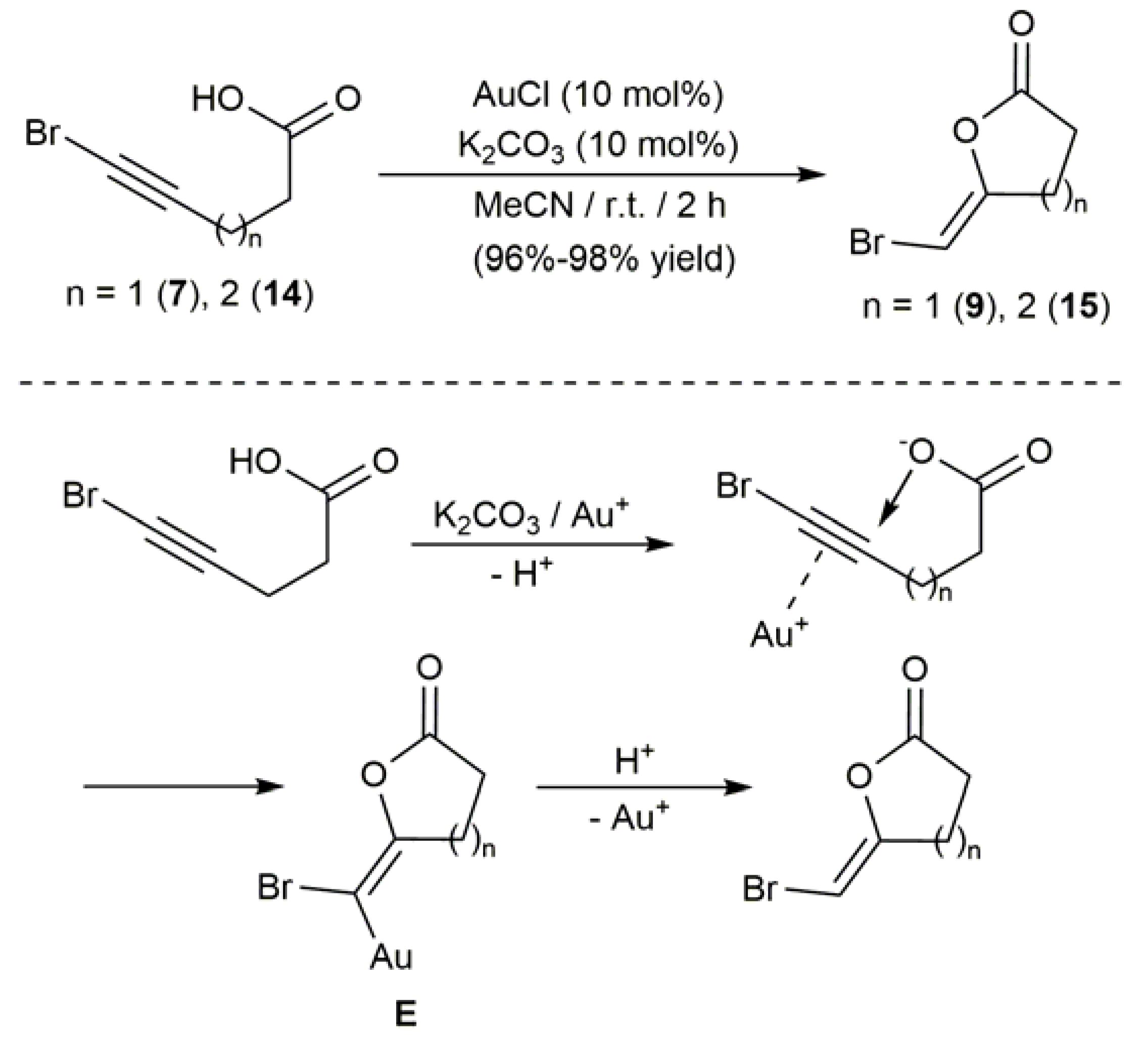
In 2006, and almost simultaneously, the groups of Michelet and Pale demonstrated that both terminal and internal alkynoic acids could be efficiently cyclized under mild conditions employing AuCl as a catalyst [55,56]. 5-Bromo-4-pentynoic acid (7) and 6-bromo-5-hexynoic acid (14) made part of the substrates studied by Pale and, from them, the Z-bromoenol lactones 9 and 15, respectively, could be synthesized in excellent yield (Scheme 10) [56,57]. The cycloisomerization reactions, which were performed at room temperature with 10 mol % of AuCl in combination with K2CO3 (10 mol %), involve the intramolecular exo-dig anti-addition of the carboxylate anion generated by deprotonation with the K2CO3 base to the C≡C bond, which is activated by π-coordination to the Au+ cation. Final protonolysis of the gold-carbon bond in the metallated intermediate E liberates the enol-lactone products.
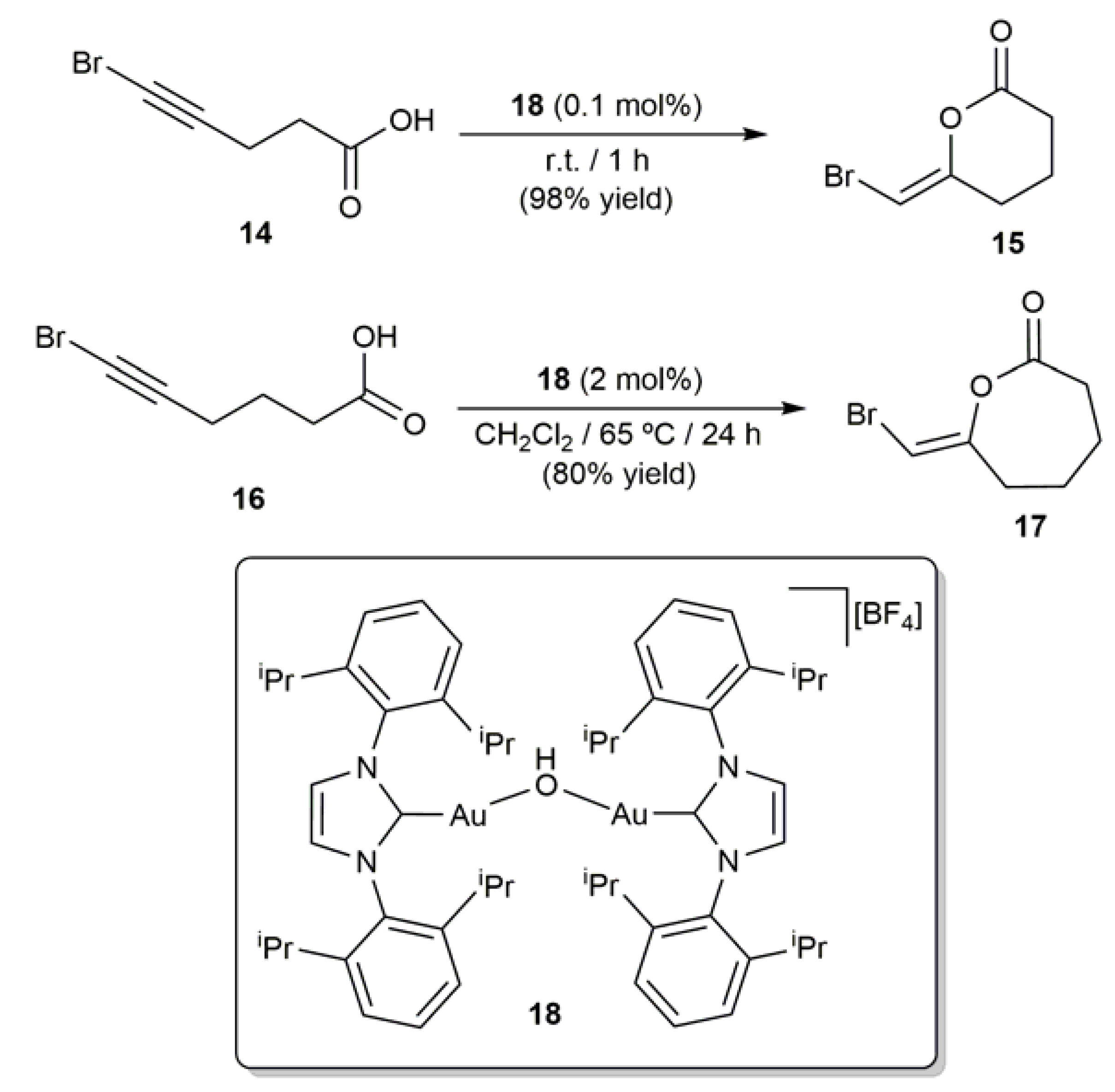
More recently, Nolan and coworkers reported the regio- and stereoselective cyclization of 6-bromo-5-hexynoic acid (14) and 7-bromo-6-heptynoic acid (16) into lactones 15 and 17 employing catalytic amounts of the hydroxo-bridged dinuclear gold(I) complex [{Au(IPr)}2(µ-OH)][BF4] (18; IPr = N,N’-bis(2,6-di-iso-propylphenyl)imidazole-2-ylidene) (Scheme 11) [58]. The addition of an external base was in this case not needed, the bridging OH ligand facilitating the generation of the carboxylate anion. It is also worthy of note that, while the conversion of 14 into 15 proceeded rapidly at r.t. with only 0.1 mol % of 18, the generation of the ε-alkylidene-lactone 17 from 16 resulted in being more demanding and an increase of the Au loading, temperature and reaction time was required.
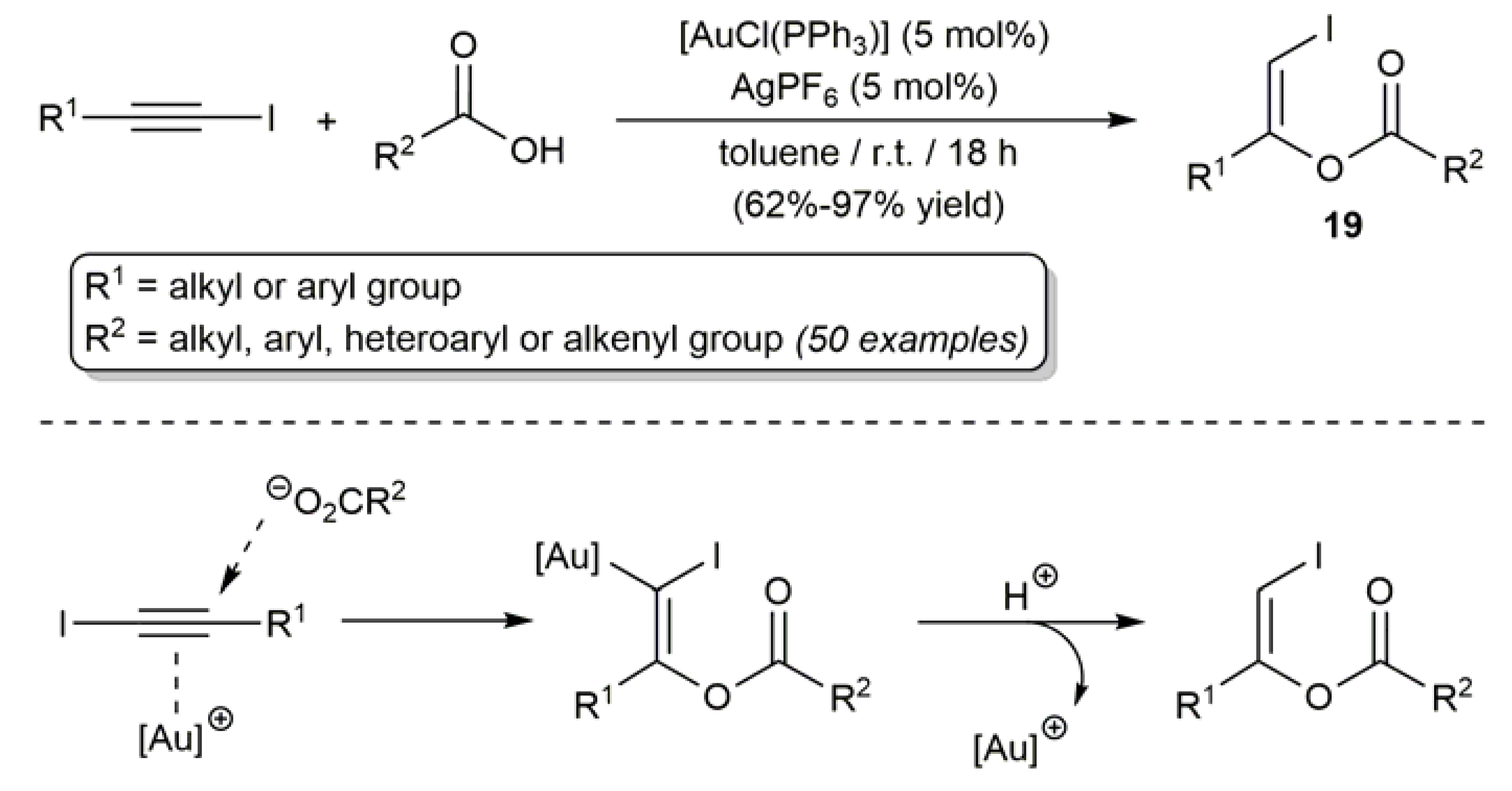
Making use of a catalytic system composed of the gold(I) complex [AuCl(PPh3)] and the chloride abstractor AgPF6, a broad scope procedure for the preparation of acyclic (Z)-β-iodoenol esters was also recently developed by Cadierno and co-workers (Scheme 12) [59,60,61]. The process, which proceeds under mild conditions and tolerates the presence of several functional groups in the substrates, involves the intermolecular addition of carboxylic acids to iodoalkynes, the latter being activated towards the carboxylate anion attack by π-coordination to the in situ generated gold(I) cation [Au(PPh3)]+. As expected, the carboxylate anion adds selectively to the more electrophilic C-2 carbon of the iodoalkyne [45,46,47] and, as usually observed in the chemistry of π-alkyne-gold complexes [62], the addition takes places in an anti fashion, thus affording the olefinic products 19 in a complete regio- and stereoselective manner. With a couple of representative examples, i.e., the addition of acetic acid to 1-(chloroethynyl)-4-methylbenzene and 1-bromooct-1-yne, the authors also demonstrated the applicability of this procedure for the synthesis of related (Z)-β-chloroenol and (Z)-β-bromoenol esters [60].
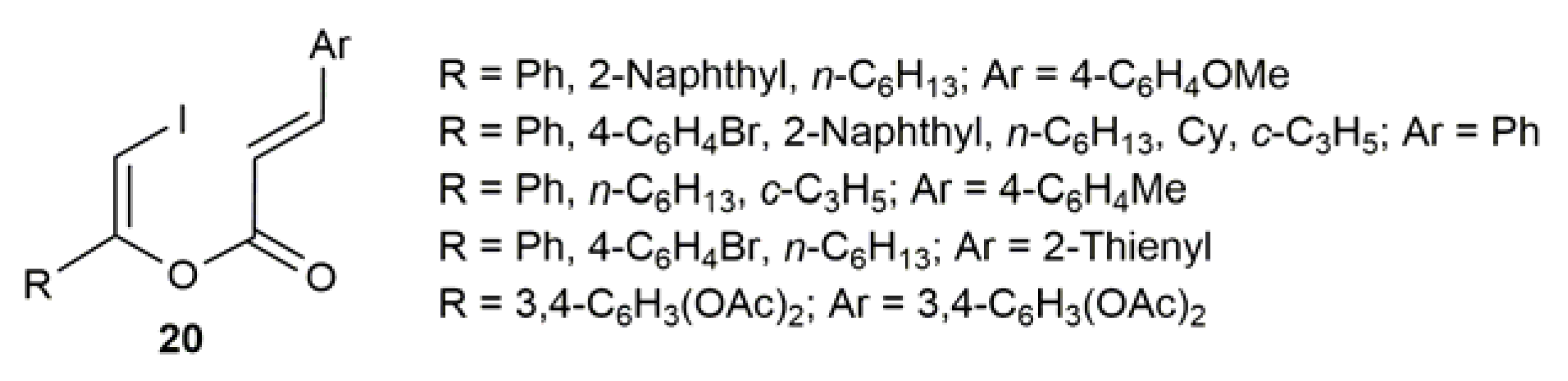
Following the same gold-catalyzed protocol, Muthusamy and Pansare synthesized later a large family of (Z)-β-iodoenol cinnamates 20 starting from the corresponding aromatic or aliphatic iodoalkyne and cinnamic acid (Figure 3) [63].
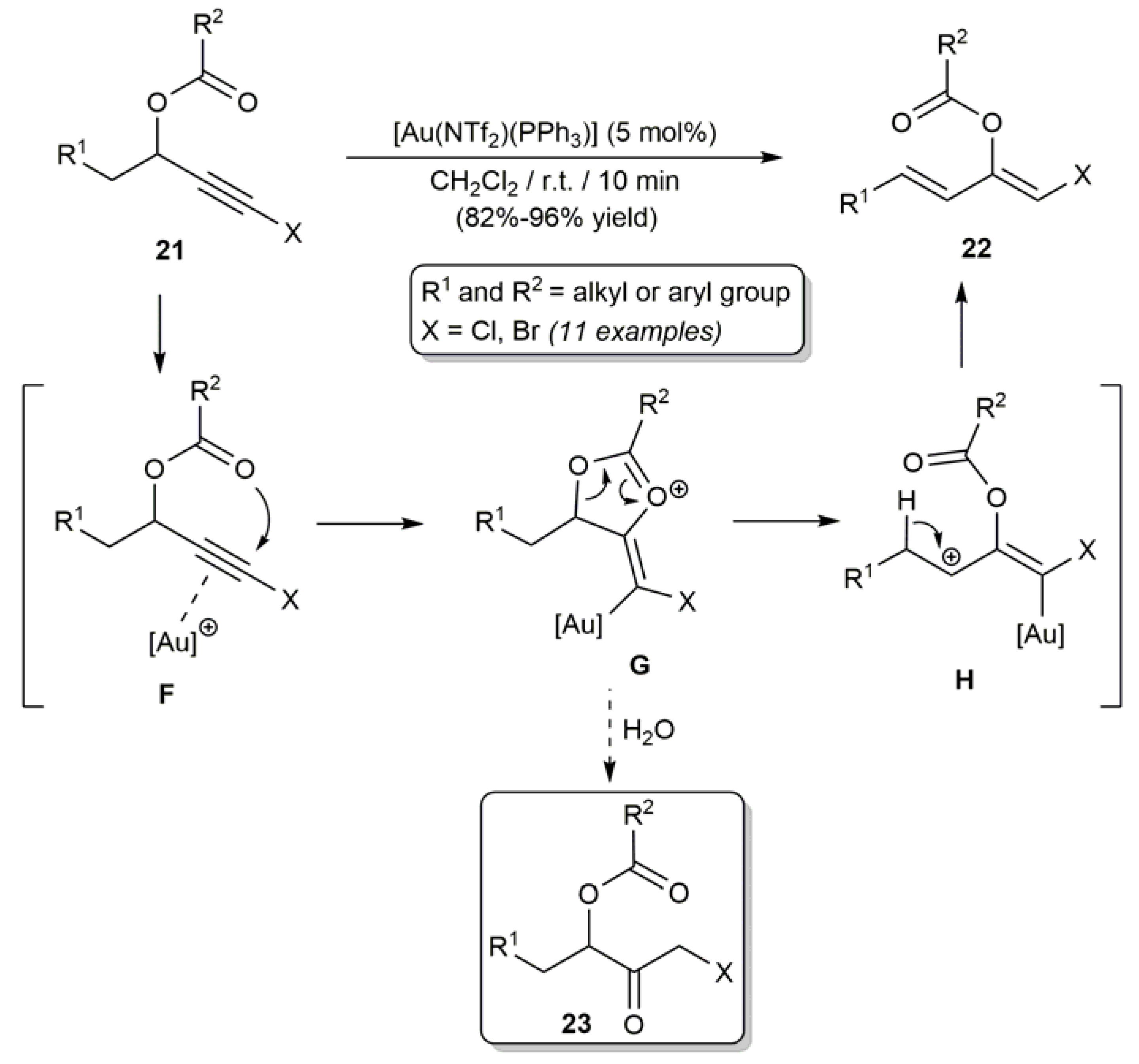
On the other hand, Zhang and coworkers reported the efficient synthesis of different β-haloenol esters of type 22 by rearrangement of the corresponding halo-substituted propargylic carboxylates 21 (Scheme 13) [64]. The process, which is catalyzed by the gold(I) complex [Au(NTf2)(PPh3)] (NTf2 = bis(trifluoromethane)sulfonamide) under mild conditions (r.t.), involves the initial activation of the C≡C bond of the substrates by the cationic gold species [Au(PPh3)]+ (intermediate F), followed by 1,2-migration of the carboxylate group via the cyclic intermediate G [64,65]. The allyl cation H thus generated evolves into the final products 22 by deprotonation and protodeauration. It is important to note that the use of rigorously anhydrous conditions is mandatory for the rearrangement process to proceed selectively since, in the presence of water, hydrolysis of intermediate G readily takes place leading to the corresponding α-halomethyl ketones 23 [64,66,67]. In line with this, we would like to mention that efficient protocols for the hydration of haloalkynes RC≡CX into α-halomethyl ketones RC(=O)CH2X employing catalytic systems composed of AgF/CF3CO2H [68], In(OTf)3/CH3CO2H [69] and Cu(OAc)2·H2O/CF3CO2H [70] have been described, in which hydrolysis of a β-haloenol acetate or trifluoroacetate intermediate (RC(O2CR’)=CHX; R’ = CH3 or CF3) has been proposed as the most likely reaction pathway.
2.4. Pd-Catalyzed Synthesis of β-Haloenol Esters
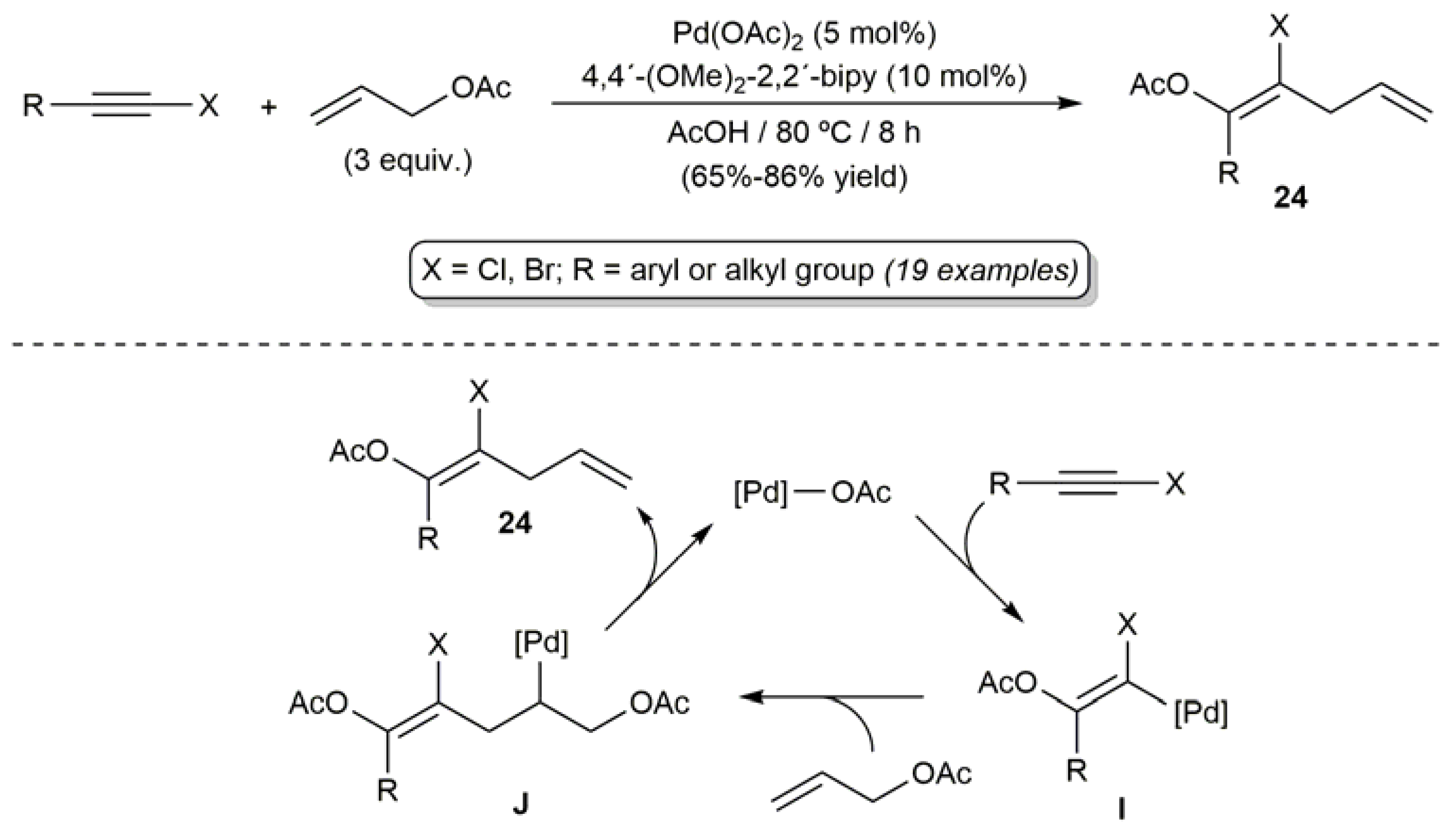
In 2011, an efficient approach to (Z)-β-haloenol acetates 24 was developed by Zhu and coworkers by coupling haloalkynes with allyl acetate, employing a catalytic system composed of Pd(OAc)2 and the bidentate ligand 4,4’-dimethoxy-2,2’-bipyridine (Scheme 14) [71]. The regio- and stereoselectivity of the process was exquisite, the formation of byproducts being in no case observed. A reaction pathway involving the initial acetoxypalladation of the alkyne, followed by insertion of the allyl acetate molecule into the Pd-C bond of the resulting alkenyl-palladium intermediate I, was proposed by the authors. A final β-elimination step in the alkyl-palladium species J furnished the (Z)-β-haloenol acetate products 24. Both chloro- and bromoalkynes participated in the reaction, but the process resulted inoperative with iodoalkynes due to their decomposition under the reaction conditions employed. Negative results were also obtained when substituted allyl acetates, such as 1-phenylallyl acetate, 2-methylallyl acetate or cinnamyl acetate, were used as the olefinic coupling partners.
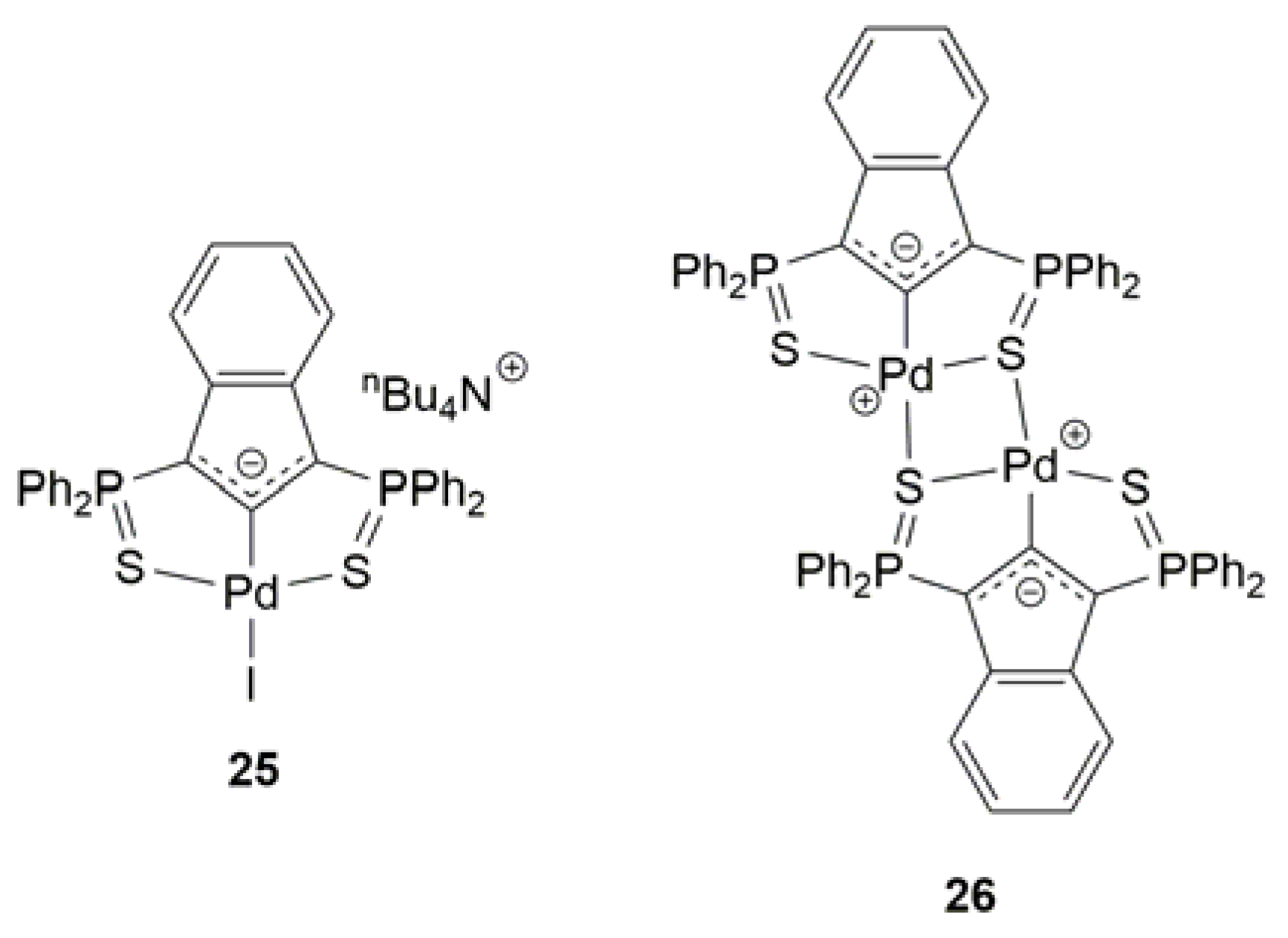
On the other hand, in the context of their studies on the cycloisomerization of alkynoic acids with indenediide-based palladium pincer catalysts, the group of Martin-Vaca and Bourissou also reported the efficient and selective conversion of 6-bromo-5-hexynoic acid (14) into the corresponding lactone 15 (see Scheme 11) employing complexes 25 and 26 (5 mol % of Pd at 90 °C; see Figure 4) [72,73]. Analogously to the case of hydroxo-gold complex [{Au(IPr)}2(µ-OH)][BF4] (18), a reaction mechanism involving the intramolecular exo-dig anti-addition of the carboxylate anion on the π-activated C≡C bond of substrate was proposed, with the electron-rich indenediide ligand being in this case responsible for the deprotonation of the carboxylic acid unit.
3. Metal-Catalyzed Transformations of β-Haloenol Esters
As commented in the introduction of this article, haloalkenes are widely employed in synthetic organic chemistry for the construction of polysubstituted olefins through transition-metal catalyzed cross-coupling reactions [1,2]. In this section, the participation of β-haloenol esters in metal-catalyzed transformations is discussed.
3.1. Acyclic β-Haloenol Esters
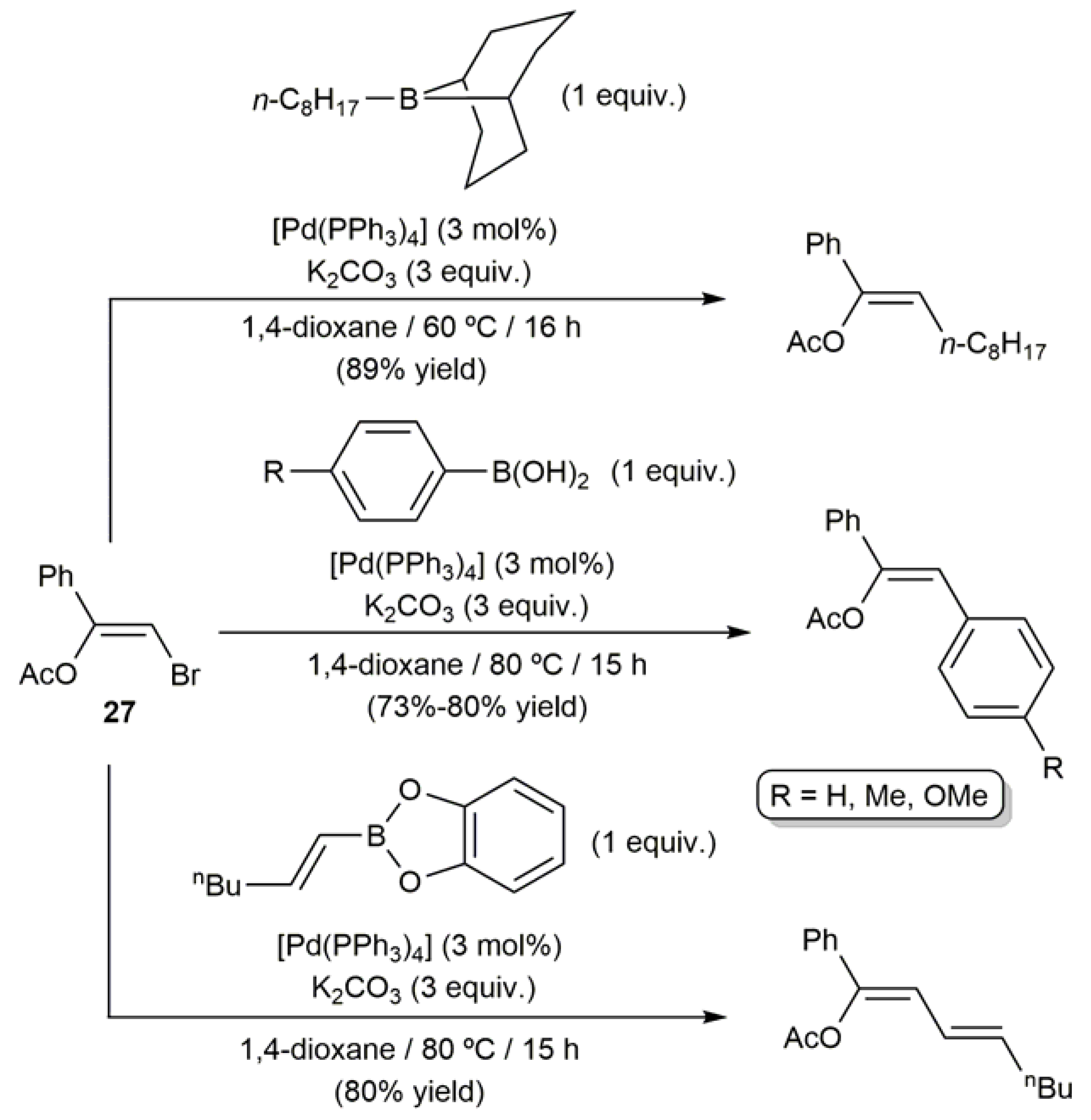
In the context of their studies on the palladium-catalyzed coupling of haloalkenes with organoboron compounds, Suzuki and Miyaura reported in 1992 the first cross-coupling reactions involving a β-haloenol ester [74]. Thus, as shown in Scheme 15, the treatment of the bromoenol acetate 27 with different alkyl-, aryl- and alkenyl-boron reagents in the presence of catalytic amounts of [Pd(PPh3)4] and a base cleanly afforded the corresponding trisubstituted olefins, which were isolated in high yields (73–89%) and with complete retention of the stereochemistry of the starting C=C bond.
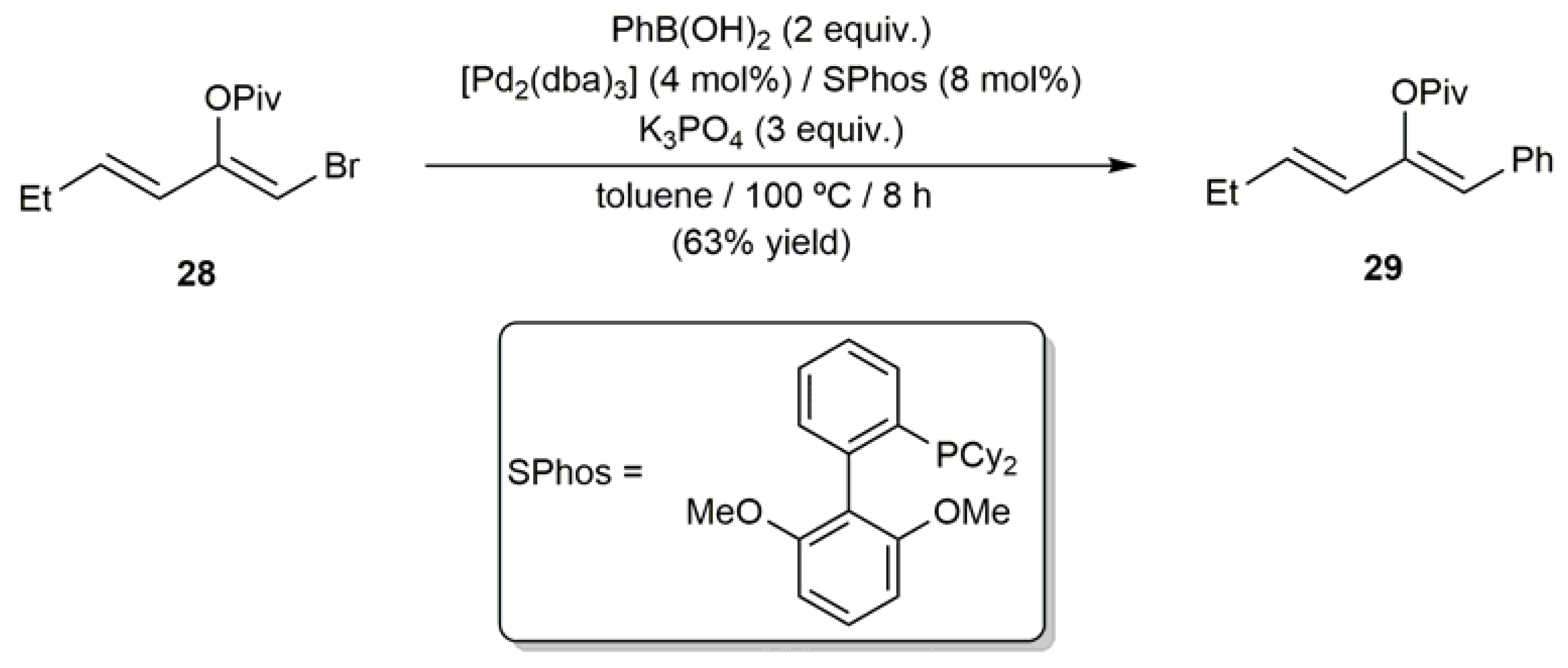
After this seminal contribution, several works reporting the use of β-haloenol esters as substrates in Suzuki–Miyaura type reactions have appeared in the literature. For example, Zhang and coworkers described the preparation of the aryldiene 29 through the cross-coupling of 28 with phenylboronic acid catalyzed by the [Pd2(dba)3]/SPhos system (dba = dibenzylideneacetone; see Scheme 16) [64].
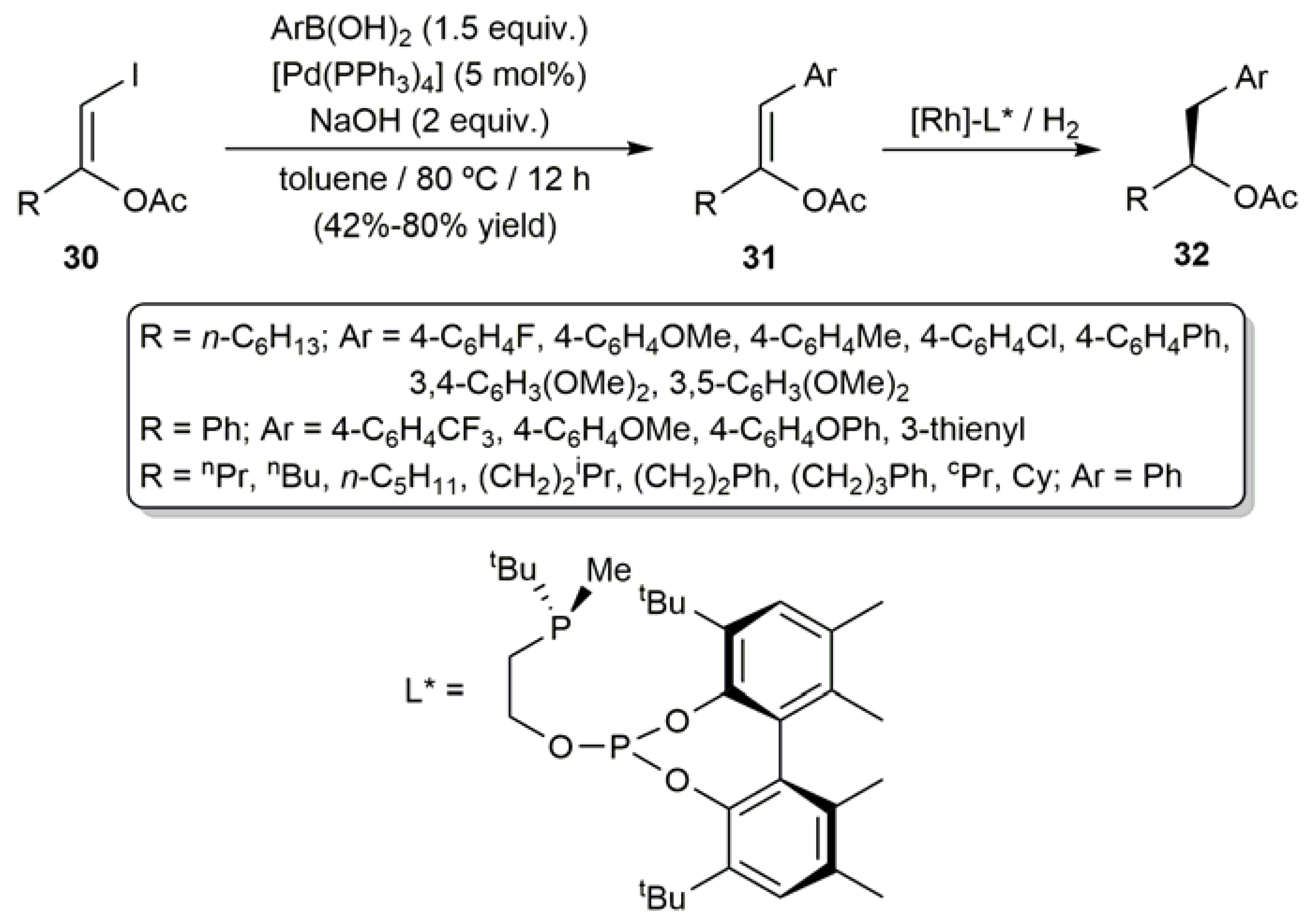
In a wider scope study, Cadierno, Pizzano and coworkers employed the (Z)-β-iodoenol acetates 30 as starting materials for the synthesis of several (Z)-1-substituted-2-arylvinyl acetates 31, via [Pd(PPh3)4]-catalyzed coupling of 30 with aromatic boronic acids (Scheme 17) [59,61]. Remarkably, the asymmetric hydrogenation of compounds 31 to afford the corresponding chiral homobenzylic esters 32, of interest as precursors of synthetically useful chiral alcohols by deacylation, could be successfully accomplished by the same authors employing rhodium(I) catalysts containing optically pure bidentate phosphine-phosphite ligands (ee up to 98%) [59,61].
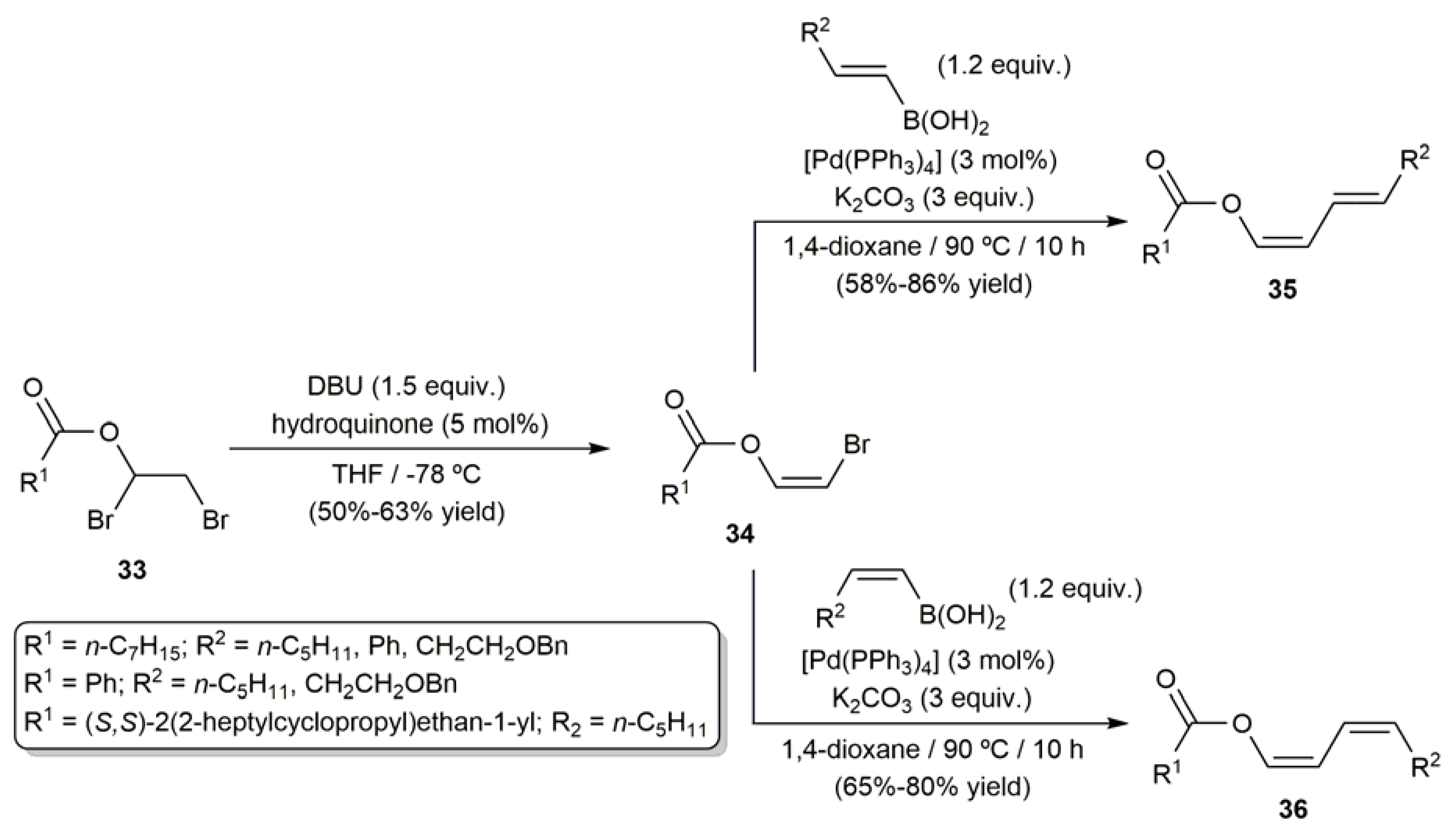
As shown in Scheme 18, the Suzuki–Miyaura coupling of (Z)-2-bromovinyl esters 34, generated by stereocontrolled dehydrobromination of 1,2-dibromoethyl esters 33 with DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) and a catalytic amount of hydroquinone, with stereodefined alkenylboronic acids provided also an efficient route for the selective construction of (Z,E)- and (Z,Z)-conjugated dienyl esters (35 and 36, respectively), molecules of enormous interest since they can be employed as precursors for the preparation of several natural products through Diels–Alder reactions [75].
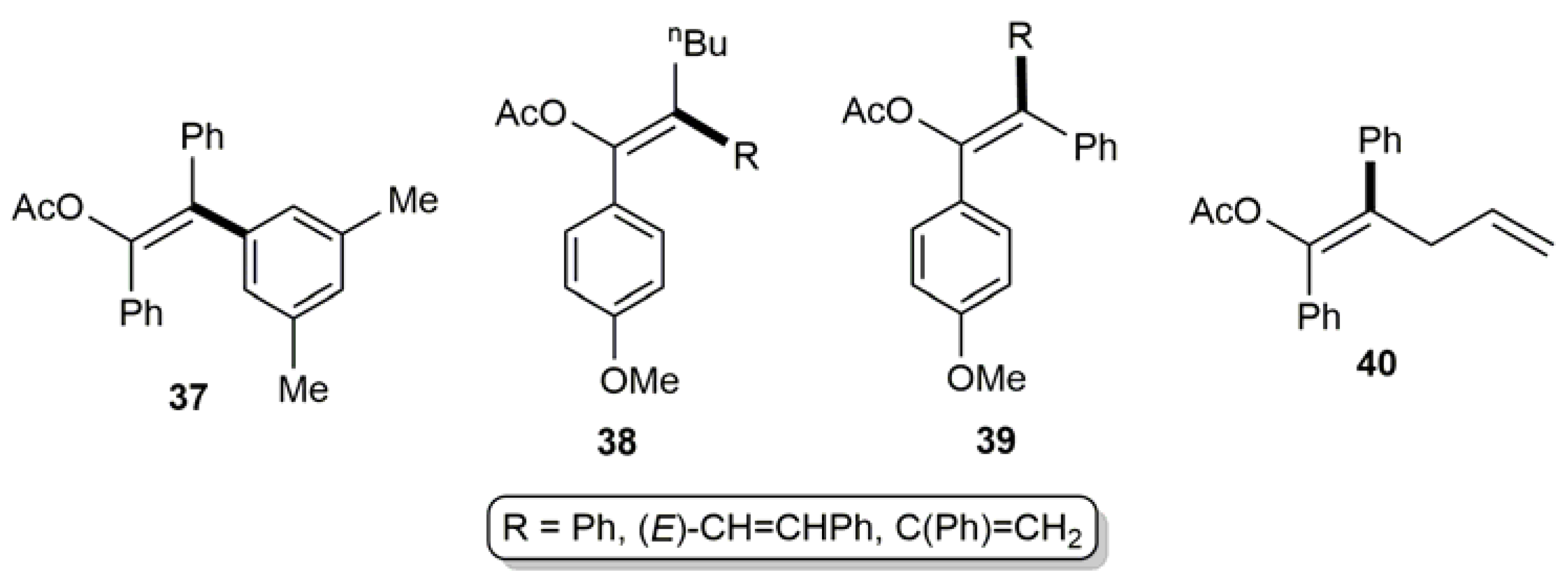
In addition, the tetrasubstituted olefins 37–40 (see Figure 5) were also synthesized in high yields (72–99%) by Suzuki–Miyaura coupling of the corresponding β-haloenol acetates with boronic acids (the C–C bond formed is highlighted in bold) [28,31,71].
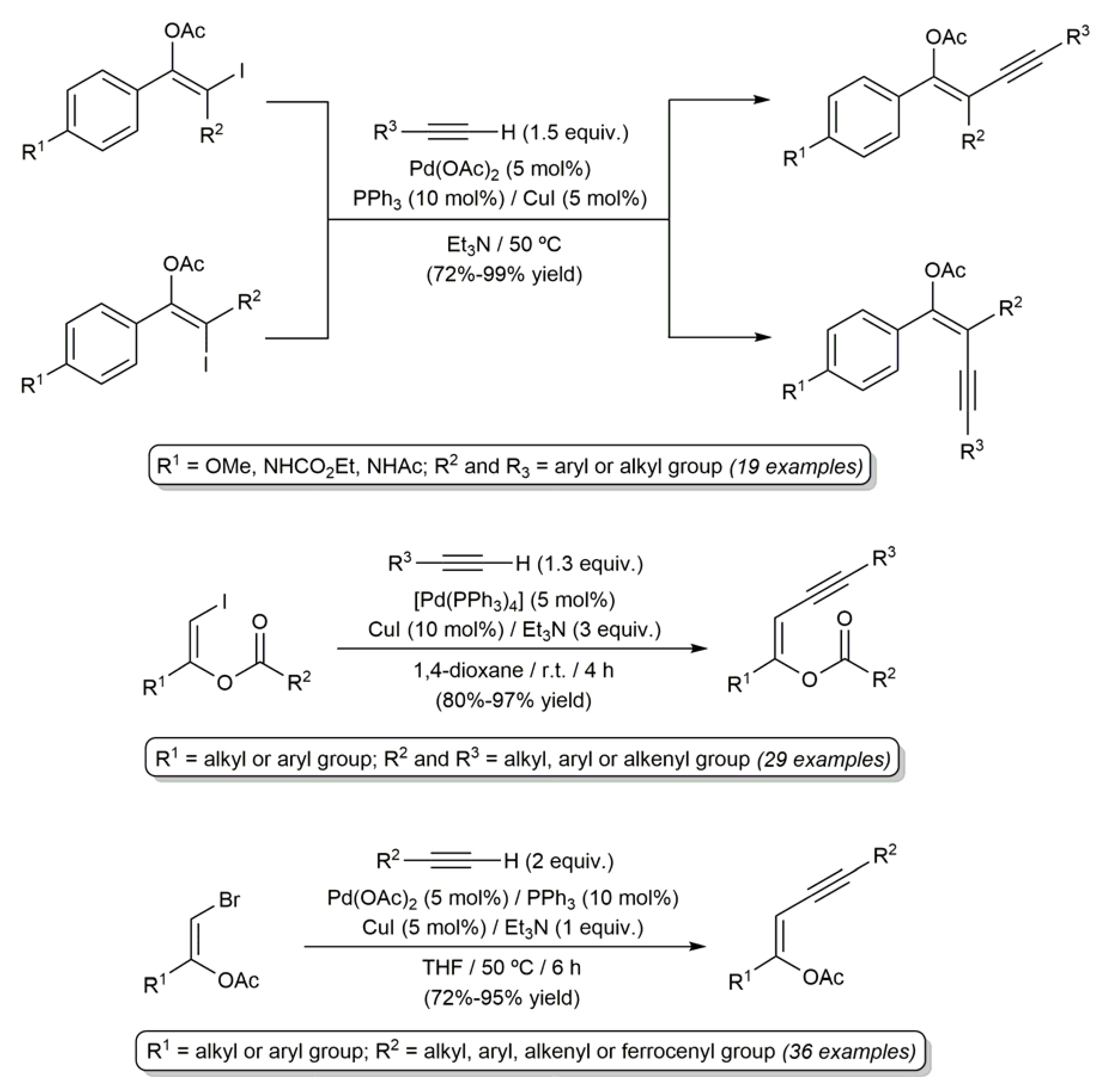
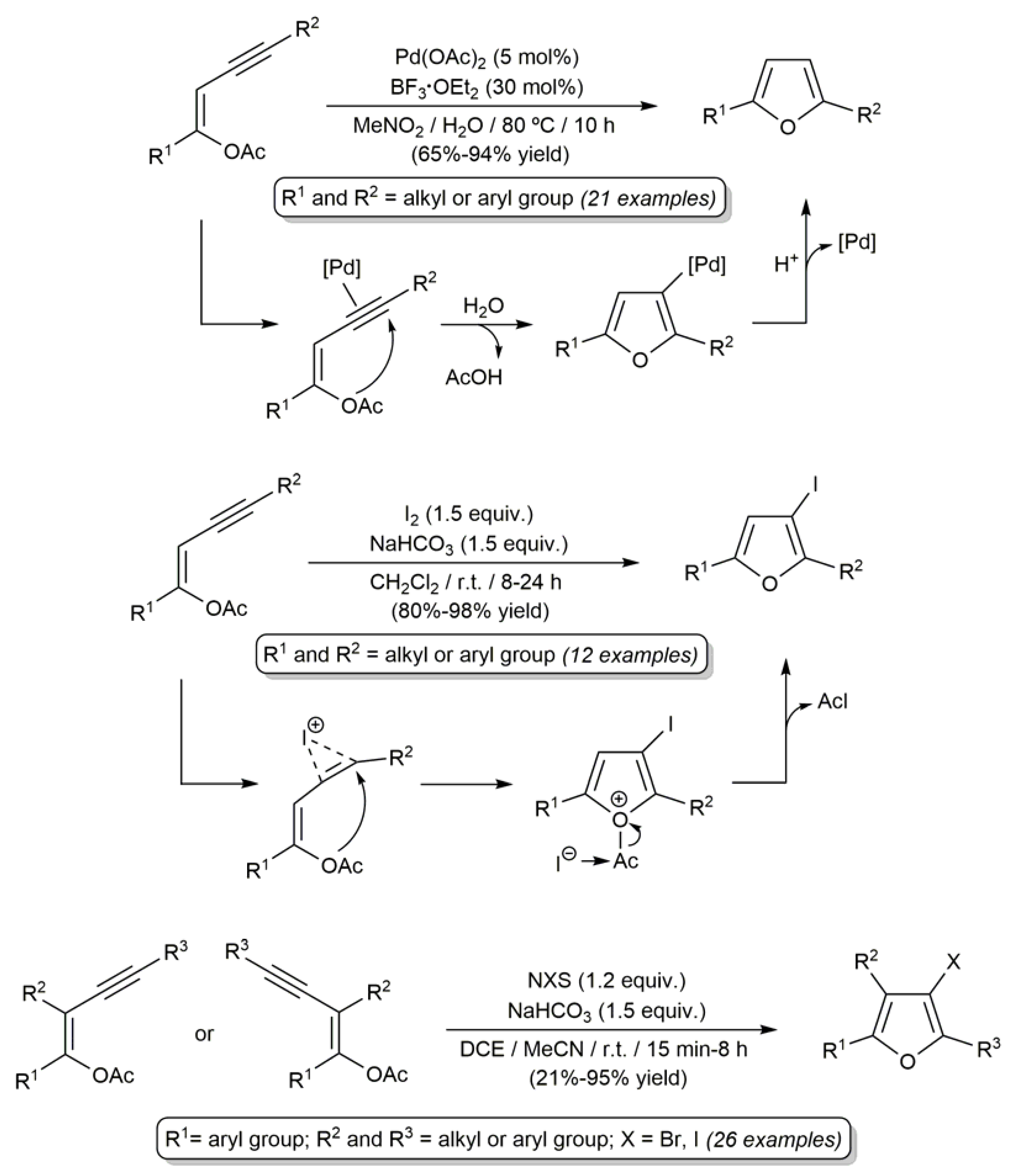
Probably, the reactions of β-haloenol esters most widely studied to date are the palladium-catalyzed Sonogashira-type couplings with terminal alkynes [30,54,60,71,76,77,78]. In this regard, a large number of (Z)- and (E)-β-haloenol esters (both chlorides, bromides and iodides) have been successfully coupled with alipahic and aromatic alkynes, 1,3-enynes or propargylic alcohols to generate the corresponding enynyl ester products in high yields and with complete preservation of the C=C bond stereochemistry of the starting olefins (the results obtained in references [60,76,77] are depicted in Scheme 19).
The main interest in these Sonogashira-type reactions is that the enynyl ester products can be employed as starting materials for the generation of polysubstituted furans via metal-catalyzed [76,77] or halogen-induced electrophilic cyclization reactions [30,79,80,81]. Illustrative examples are shown in Scheme 20.
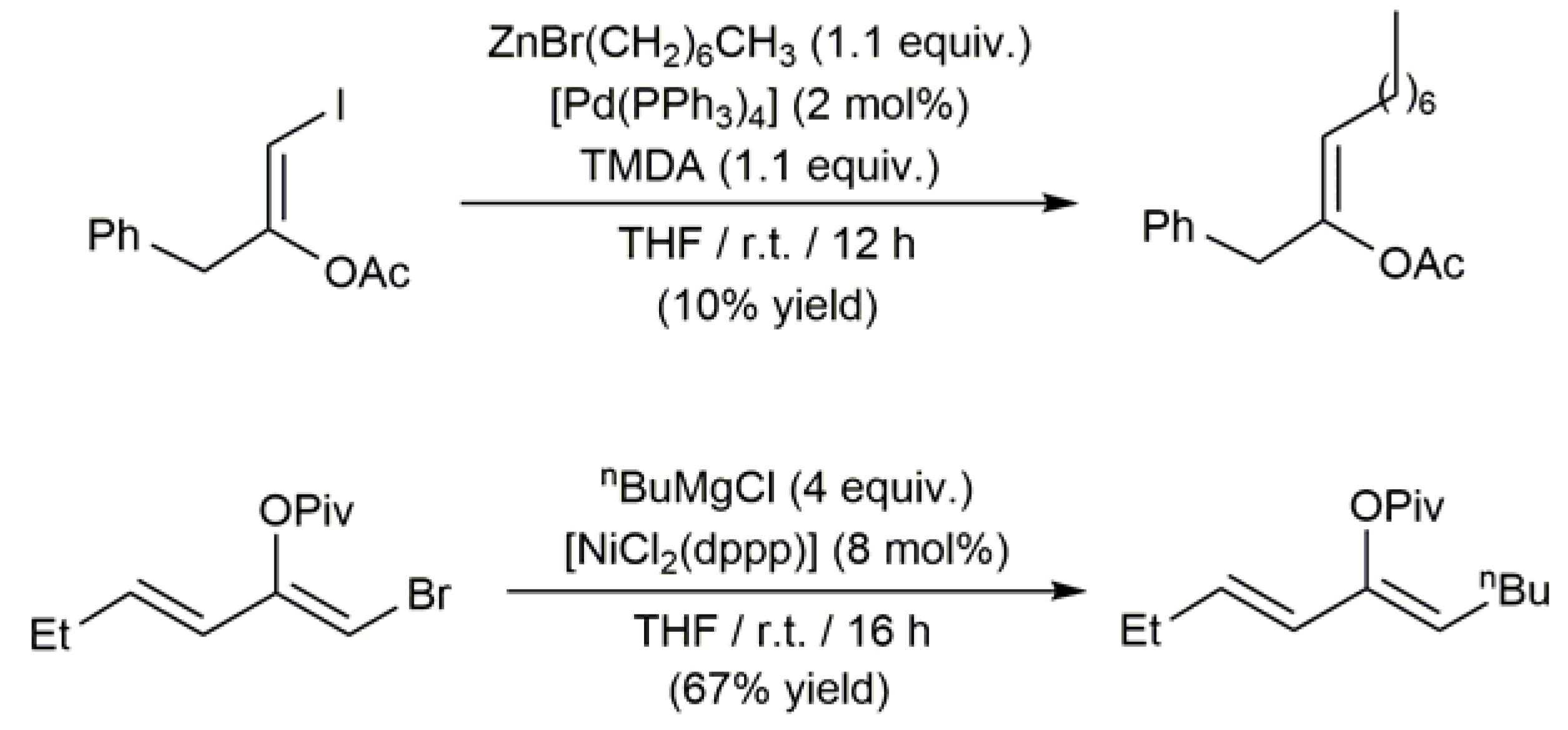
Contrary to the case of the Suzuki-Miyaura and Sonogashira reactions, the participation of β-haloenol esters in Negishi- and Kumada-Corriu-type cross-coupling processes remains almost unexplored. In fact, only the examples depicted in Scheme 21 can be currently found in the literature [61,64].
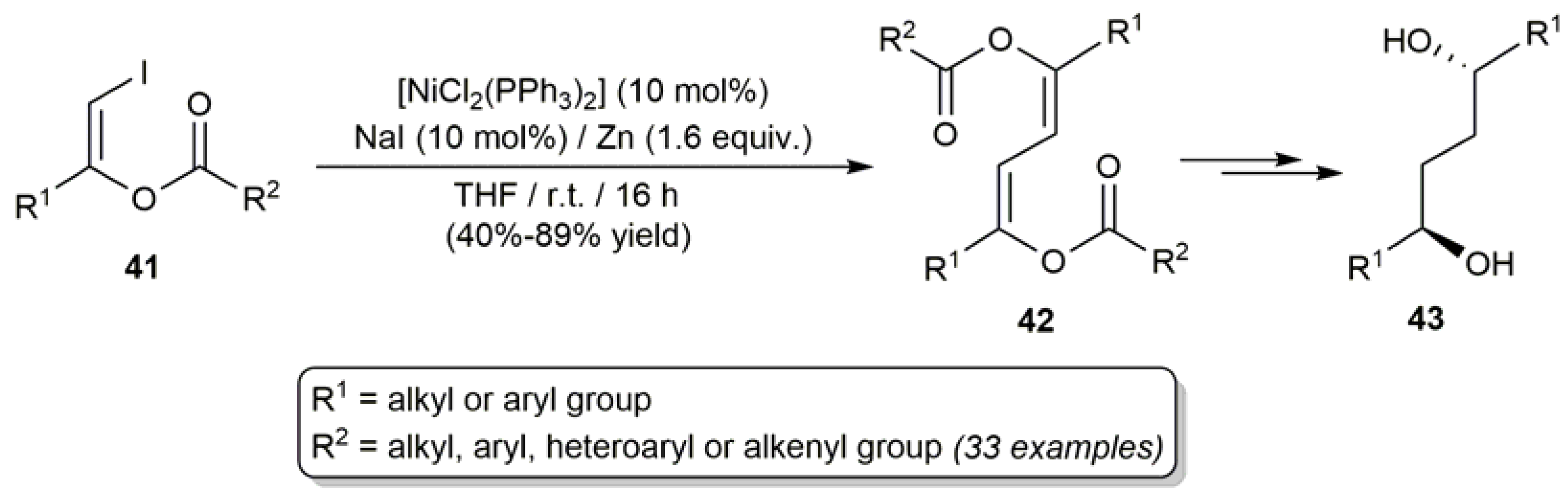
On the other hand, homocoupling reactions of alkenyl halides have been extensively studied during the last decades since they provide a straightforward access to conjugated 1,3-dienes and polyenes [82]. In this context, taking advantage of previous works by Takagi and coworkers with non-functionalized alkenyl halides [83,84,85], an efficient and broad scope protocol for the homocoupling of (Z)-β-iodoenol esters 41 was developed by Francos and Cadierno (Scheme 22) [86]. The process, which is catalyzed by nickel(0) species generated in situ by combining [NiCl2(PPh3)2] with NaI and Zn dust, afforded the buta-1,3-diene-1,4-diyl diester products 42 as the corresponding ZZ-isomers exclusively. It is also worth noting that 1,3-dienes 42 proved to be useful precursors for the preparation of synthetically relevant C2 symmetric 1,4-diols 43, through a rhodium-catalyzed asymmetric hydrogenation of the two C=C bonds of 42 and a subsequent base-promoted deacylation reaction [87].
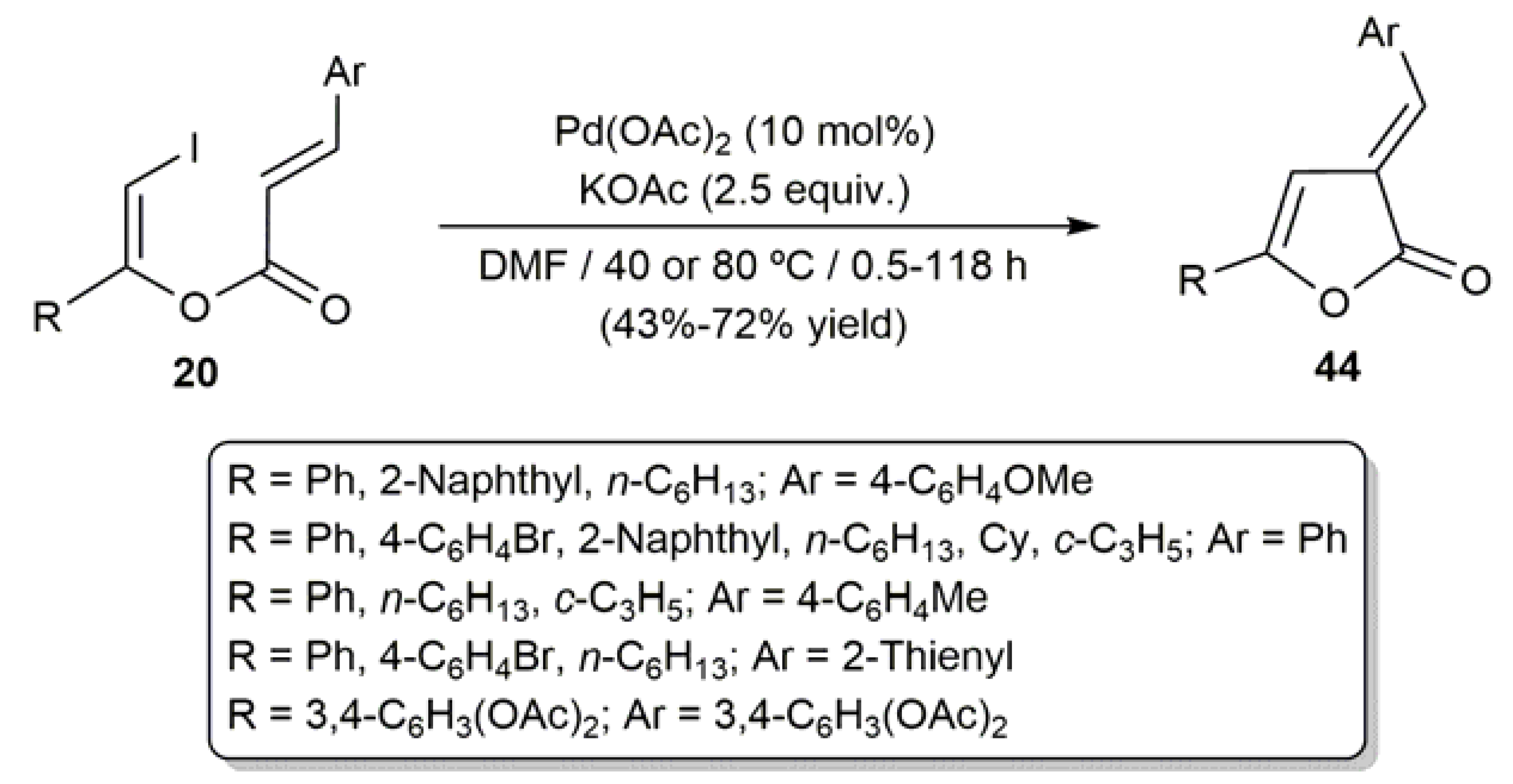
Acyclic β-haloenol esters have also been employed as starting materials for the generation of heterocyclic systems. In this context, the intramolecular palladium-catalyzed Mizoroki–Heck coupling of the (Z)-β-iodoenol cinnamates 20 allowed the preparation of furanones 44 in moderate to high yields (Scheme 23) [63].
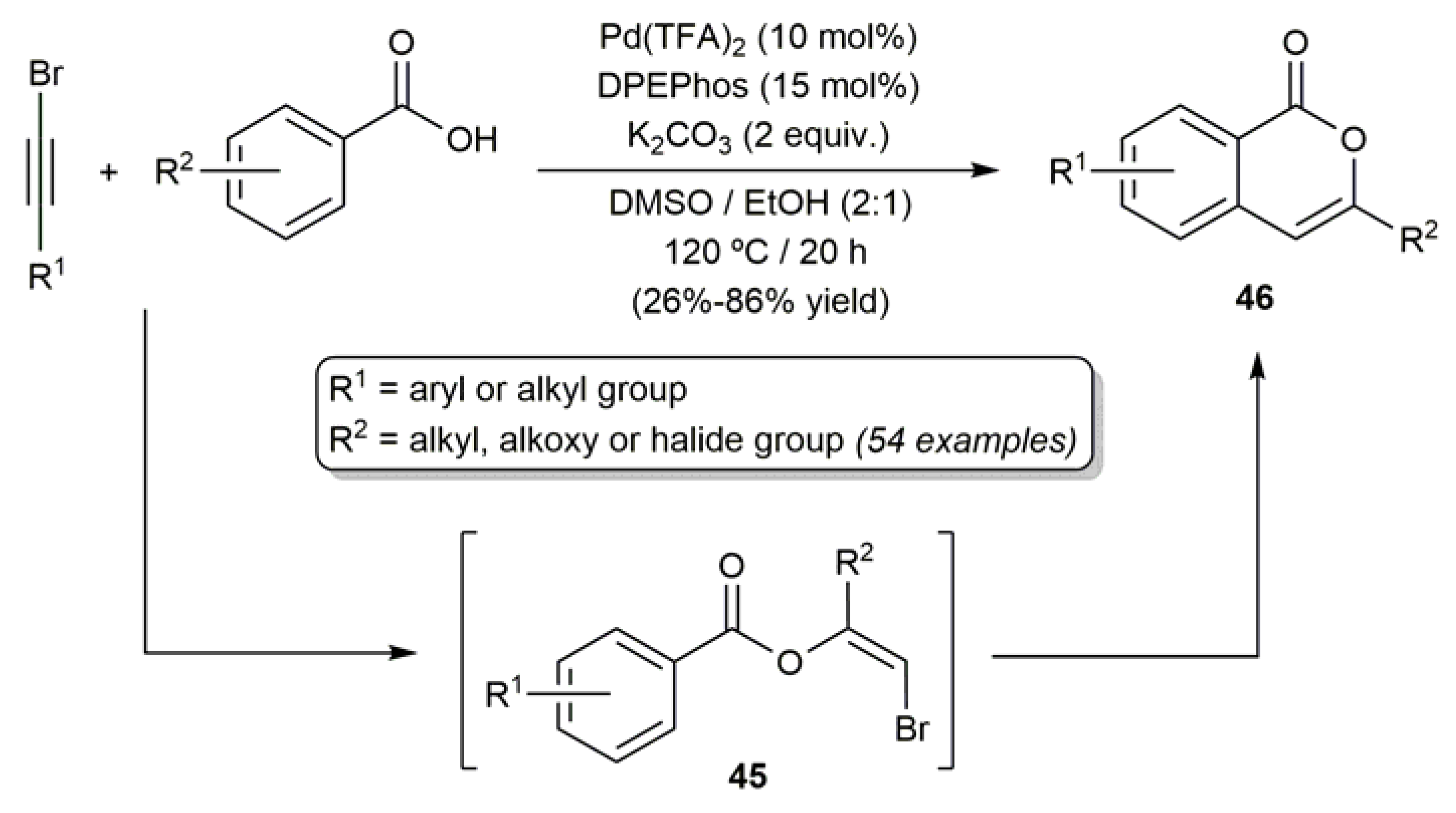
A broad scope protocol for the synthesis of 3-substituted isocoumarins 46 by coupling of bromoalkynes with benzoic acids, catalyzed by palladium(II) trifluoroacetate in combination with the diphosphine ligand DPEPhos (bis[2-(diphenylphosphino)phenyl] ether) and K2CO3, was described by Wu, Jiang and co-workers (Scheme 24) [88]. A reaction pathway involving the initial anti addition of the acid to the alkyne, and subsequent oxidative annulation of the resulting β-bromoenol benzoates 45, was proposed by the authors.
(Z)-β-Halenol acetates proved to be also useful starting materials for the preparation of symmetrical 2,5-disubstituted pyrazines 47 (Scheme 25) [89]. Although the formation of compounds 47 was initially observed in the reactions of the haloenol acetates with an ammonia solution under palladium/copper-catalyzed conditions, a more detailed investigation showed that no metal sources are really needed for the reaction to proceed, and that any source of ammonia can be employed. In this regard, employing ammonium formate, and performing the reaction in DMF at 120 °C, a wide range of 2,5-disubstituted pyrazines could be accessed in moderate to excellent yields starting from both aromatic and aliphatic (Z)-β-iodoenol or (Z)-β-bromoenol acetates (Scheme 25). The regioselectivity of the process was excellent, the formation of the corresponding 2,6-disubtituted regioisomers being not observed under these optimized conditions. According to the authors, α-halomethyl ketones could be involved as intermediates.
3.2. Cyclic β-Haloenol Esters
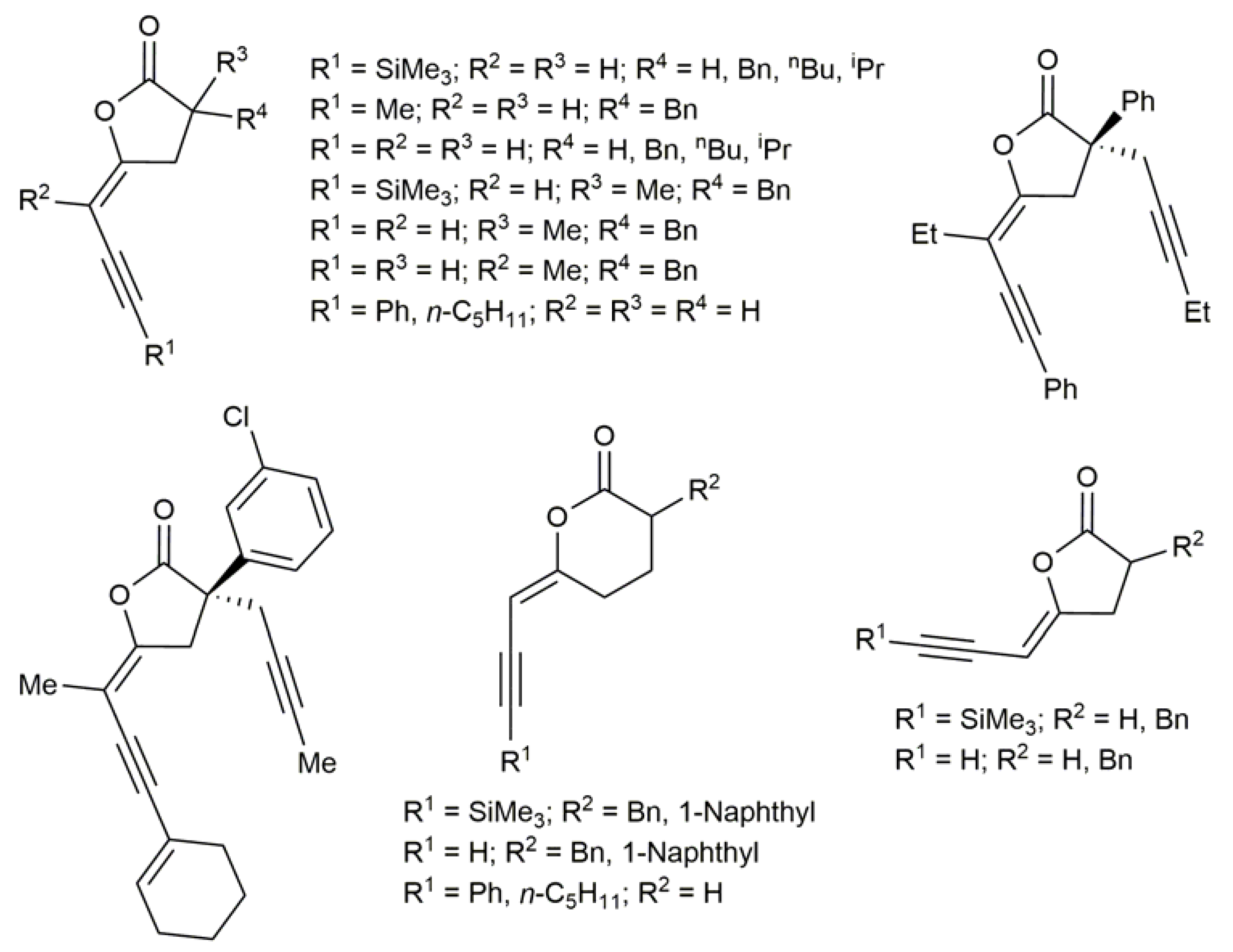
Although to a much lesser extent, the participation of haloenol lactones in metal-catalyzed cross-coupling reactions has also been described. In this regard, several 5- and 6-membered ring ynenol lactones, including some optically pure representatives, could be accessed from the corresponding iodoenol lactones via classical Pd-catalyzed Sonogashira coupling reactions (Figure 6) [41,44,90,91]. Interestingly, these compounds featured biological activity as enzymes inhibitors [44,90,91].
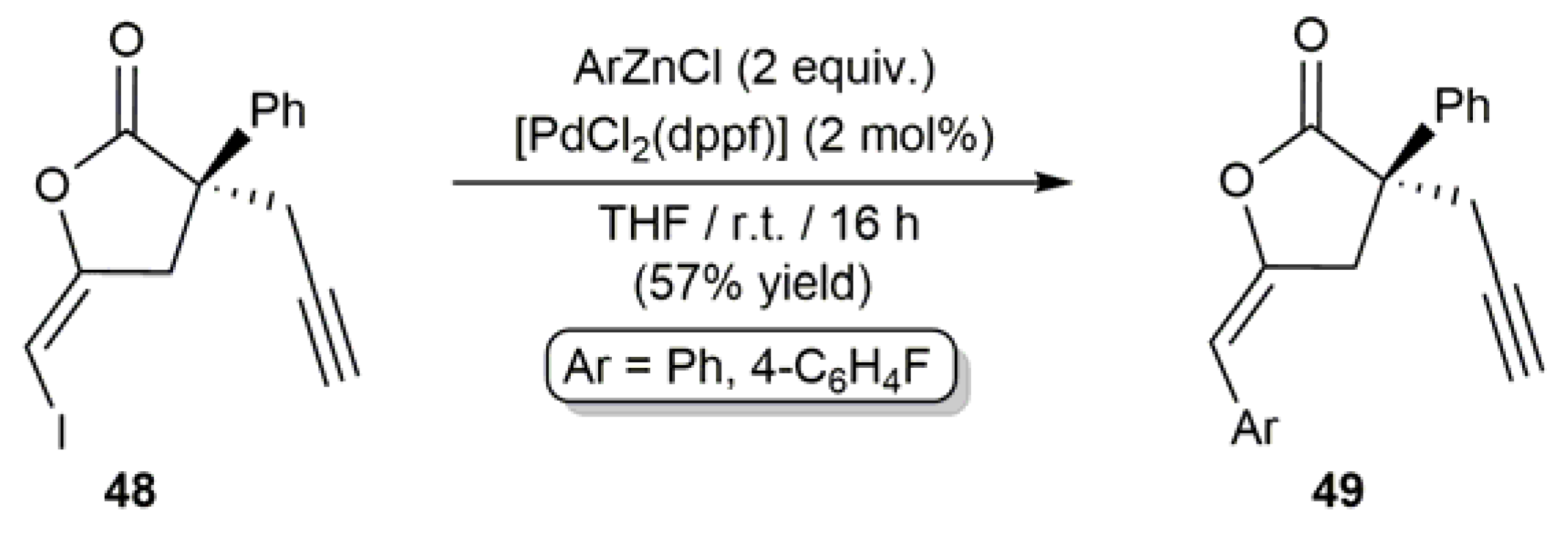
Additionally, Hennecke and coworkers reported the synthesis of the arylenol lactones 49 by Negishi coupling of the iodoenol derivative 48 with the corresponding arylzinc chloride catalyzed by [PdCl2(dppf)] (dppf = 1,1’-bis(diphenylphosphino)ferrocene) (Scheme 26) [44]. The use of [PdCl2(dppf)] as catalyst proved to be crucial, since other palladium sources, i.e., PEPPSI-type catalysts or the [Pd2(dba)3]/trisfurylphosphine combination, led to the extensive dehalogenation of 48. Additionally, of note is the fact that attempts to generate compounds 49 through Pd-catalyzed Suzuki–Miyaura coupling of 48 with aryl boronic acids failed, due to the incompatibility of the iodoenol moiety with the basic conditions required in this particular cross-coupling process [44].
4. Conclusions
In this contribution we summarized the catalytic protocols currently known for the preparation of β-haloenol esters and haloenol lactones, a particular class of haloalkenes, which are gaining significance as coupling partners in diverse chemical transformations of synthetic interest. Metal-catalyzed reactions in which these functionalized olefins participate as substrates, mainly palladium-catalyzed C–C cross-coupling processes, were also discussed. Most of the works herein presented have been published during the last decade, demonstrating the current interest on the use of these molecules as building blocks in organic synthesis. Although there is already a body of work in the field, it still remains open and new synthetic approaches to these compounds and applications can be expected in the near future.
Funding
Financial support from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO project CTQ2016-75986-P) is gratefully acknowledged.
Conflicts of Interest
The author declares no conflict of interest.
References
- De Meijer, A.; Diederich, F. (Eds.) Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef]
- Schlosser, M. (Ed.) Organometallics in Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Jeschke, J.; Gäbler, C.; Lang, H. Regioselective formation of enol esters from the ruthenium-catalyzed Markovnikov addition of carboxylic acids to alkynes. J. Org. Chem. 2016, 81, 476–484. [Google Scholar] [CrossRef]
- Francos, J.; Cadierno, V. Metal-catalyzed intra- and intermolecular addition of carboxylic acids to alkynes in aqueous media: A review. Catalysts 2017, 7, 328. [Google Scholar] [CrossRef] [Green Version]
- González-Liste, P.J.; Francos, J.; García-Garrido, S.E.; Cadierno, V. The intermolecular hydro-oxycarbonylation of internal alkynes: Current state of the art. Arkivoc 2018, 2, 17–39. [Google Scholar]
- Takai, K.; Kokumai, R.; Nobunaka, T. Reactions of coordinated geminal dichromium reagents with aldehydes: Stereoselective formation of (Z)-2-chloroalk-2-en-1-ols. Chem. Commun. 2001, 1128–1129. [Google Scholar] [CrossRef]
- Bejot, R.; Tisserand, S.; Reddy, L.M.; Barma, D.K.; Baati, R.; Falck, J.R.; Mioskowski, C. Stereoselective transformations of trihalomethylcarbinols induced by chromous chloride. Angew. Chem. Int. Ed. 2005, 44, 2008–2011. [Google Scholar] [CrossRef]
- Bejot, R.; Anjaiah, S.; Falck, J.R.; Mioskowski, C. α-Haloenol acetates: Versatile reactants for oxetan-2-one, azetidin-2-one and isoxazolidin-5-one synthesis. Eur. J. Org. Chem. 2007, 101–107. [Google Scholar] [CrossRef]
- Kashinath, D.; Mioskowski, C.; Falck, J.R.; Goli, M.; Meunier, S.; Baati, R.; Wagner, A. Highly stereoselective synthesis of (Z,E)-1-halo-1,3-dienol esters via rearrangement of Fischer chromium chloro-carbenes using microwave irradiation. Org. Biomol. Chem. 2009, 7, 1771–1774. [Google Scholar] [CrossRef]
- Joshi, G.C.; Chambers, W.D.; Warnhoff, E.W. Camphor enol and homoenol acetates. Tetrahedron Lett. 1967, 8, 3613–3617. [Google Scholar] [CrossRef]
- Kowalski, C.J.; O’Dowd, M.L.; Burke, M.C.; Fields, K.W. α-Ketodianions. New reactive intermediates. J. Am. Chem. Soc. 1980, 102, 5411–5412. [Google Scholar] [CrossRef]
- Kowalski, C.J.; Haque, M.S.; Fields, K.W. Ester homologation via α-bromo α-keto dianion rearrangement. J. Am. Chem. Soc. 1985, 107, 1429–1430. [Google Scholar] [CrossRef]
- Kowalski, C.J.; Haque, M.S. Bromomethyl ketones and enolates: Alternative products from ester homologation reactions. J. Org. Chem. 1985, 50, 5140–5142. [Google Scholar] [CrossRef]
- Kowalski, C.J.; Reddy, R.E. Ester homologation revisited: A reliable, higher yielding and better understood procedure. J. Org. Chem. 1992, 57, 7194–7208. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, M.A.; González, J.M.; Campos, P.J. A new electrophilic addition to acetylenes. Synthesis of 1,2-iodofunctionalized olefins. Tetrahedron Lett. 1986, 27, 3303–3306. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, M.A.; Campos, P.J. Synthesis of 2-functionalized 1,1-diiodo-1-alkenes. Generation and reactions of 1-iodo-1-lithio-1-alkenes and 1,1-dilithio-1-alkenes. J. Am. Chem. Soc. 1988, 110, 5567–5568. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, M.A.; Campos, P.J. Synthesis of 2-functionalized 1-chloro-1-iodo-1-alkenes from 1-chloro-1-alkynes and IPy2BF4. Tetrahedron Lett. 1990, 31, 2751–2754. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, M.A.; Campos, P.J. Electrophilic additions of positive iodine to alkynes through an iodonium mechanism. J. Org. Chem. 1990, 55, 3104–3106. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, M.A.; Campos, P.J. Stereoselective synthesis of 2-functionalized 1-bromo-1-iodo-1-alkenes by electrophilic iodination of 1-bromo-1-alkynes. Synthesis 1992, 270–272. [Google Scholar] [CrossRef]
- Pincock, J.A.; Yates, K. Kinetics and mechanism of electrophilic bromination of acetylenes. Can. J. Chem. 1970, 48, 3332–3348. [Google Scholar] [CrossRef]
- Ogata, Y.; Urasaki, I. The reaction of tolan with a mixture of iodine and peracetic acid. J. Org. Chem. 1971, 36, 2164–2168. [Google Scholar] [CrossRef]
- Yates, K.; Go, T.A. Vinyl cation intermediates in electrophilic additions to triple bonds. 2. Chlorination of alkylacetylenes. J. Org. Chem. 1980, 45, 2385–2391. [Google Scholar] [CrossRef]
- Heasley, G.E.; Codding, C.; Sheehy, J.; Gering, K.; Heasley, V.L.; Shellhamer, D.F.; Rempel, T. Chlorination of 1-hexyne and 3-hexyne in acetic acid and methanol. J. Org. Chem. 1985, 50, 1773–1776. [Google Scholar] [CrossRef]
- Hokamp, T.; Storm, A.T.; Yusubov, M.; Wirth, T. Iodine monoacetate for efficient oxyiodinations of alkenes and alkynes. Synlett 2018, 29, 415–418. [Google Scholar]
- Jovtscheff, A.; Spassov, S.L. Heterolytische anlagerung an acetylen-verbindungen I: Stereochemie und kinetik der anlagerung im system diphenylacetylen-N-brom-succinimid-essigsäure-wasser. Monatsh. Chem. 1967, 98, 2272–2281. [Google Scholar] [CrossRef]
- Jovtscheff, A.; Spassov, S.L. Heterolytische anlagerungen an acetylen-verbindungen III: Über den mechanismus der umsetzung von acetylen-verbindungen mit N-bromsuccinimid in eisessig oder waβriger essigsäure. Monatsh. Chem. 1969, 100, 328–341. [Google Scholar] [CrossRef]
- Okamoto, N.; Miwa, Y.; Minami, H.; Takeda, K.; Yanada, R. Regio- and stereoselective multisubstituted enol ester synthesis. J. Org. Chem. 2011, 76, 9133–9138. [Google Scholar] [CrossRef] [PubMed]
- Merkushev, E.B.; Karpitskaya, L.G.; Novosel’tseva, G.I.; Raida, V.S. Conjugated iodoacetoxylation of triple bonds. Izv. Akad. Nauk Ser. Khim. 1978, 1153–1154. [Google Scholar] [CrossRef]
- Xia, X.-F.; Gu, Z.; Liu, W.; Wang, N.; Wang, H.; Xia, Y.; Gao, H.; Liu, X. Selective oxygenation of alkynes: A direct approach to diketones and vinyl acetate. Org. Biomol. Chem. 2014, 12, 9909–9913. [Google Scholar] [CrossRef]
- Priebbenow, D.L.; Gable, R.W.; Baell, J. Regio- and stereoselective iodoacyloxylations of alkynes. J. Org. Chem. 2015, 80, 4412–4418. [Google Scholar] [CrossRef]
- Crespo, L.T.C.; Nogueira, G.P.; de Mattos, M.C.S.; Esteves, P.M. Reaction of trihaloisocyanuric acids with alkynes: An efficient methodology for the preparation of β-haloenol acetates. Arkivoc 2018, 2, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Chawla, R.; Singh, A.K.; Yadav, L.D.S. Catalyst- and metal-free rapid functionalizations of alkynes using TsNBr2. Synlett 2013, 24, 1558–1562. [Google Scholar]
- Daniels, S.B.; Cooney, E.; Sofia, M.J.; Chakravarty, P.K.; Katzenellenbogen, J.A. Haloenol lactones: Potent enzyme-activated irreversible inhibitors for α-chymotrypsin. J. Biol. Chem. 1983, 258, 15046–15053. [Google Scholar] [PubMed]
- Sofia, M.J.; Katzenellenbogen, J.A. Enol lactone inhibitors of serine proteases. The effect of regiochemistry on the inactivation behavior of phenyl-substituted (halomethylene)tetra- and -dihydrofuranones and (halomethylene)tetrahydropyranones toward α-chymotrypsin: Stable acyl enzyme intermediate. J. Med. Chem. 1986, 29, 230–238. [Google Scholar] [PubMed]
- Rai, R.; Katzenellenbogen, J.A. Guanidinophenyl-substituted enol lactones as selective, mechanism-based inhibitors of trypsin-like serine proteases. J. Med. Chem. 1992, 35, 4150–4159. [Google Scholar] [CrossRef]
- Wu, Z.; Minhas, G.S.; Wen, D.; Jiang, H.; Chen, K.; Zimniak, P.; Zheng, J. Design, synthesis, and structure-activity relationships of haloenol lactones: Site-directed and isozyme-selective glutathione S-transferase inhibitors. J. Med. Chem. 2004, 47, 3282–3294. [Google Scholar] [CrossRef]
- Mock, J.N.; Taliaferro, J.P.; Lu, X.; Patel, S.K.; Cummings, B.S.; Long, T.E. Haloenol pyranones and morpholinones as antineoplastic agents. Bioorg. Med. Chem. Lett. 2012, 22, 4854–4858. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, L.; Albrecht, A.; Janecki, T. α-Alkylidene-γ- and δ-lactones and lactams. In Natural Lactones and Lactams: Synthesis, Occurrence and Biological Activity; Janecki, T., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 147–192. [Google Scholar]
- Ranganathan, S.; Muraleedharan, K.M.; Vaish, N.K.; Jayaraman, N. Halo- and selenolactonisation: The two major strategies for cyclofunctionalisation. Tetrahedron 2004, 60, 5273–5308. [Google Scholar] [CrossRef]
- Halder, J.; Das, D.; Nanda, S. A distinctive transformation based diversity oriented synthesis of small ring carbocycles and heterocycles from biocatalytically derived enantiopure α-substituted-β-hydroxyesters. Org. Biomol. Chem. 2018, 16, 2549–2575. [Google Scholar] [CrossRef]
- Wilking, M.; Mück-Lichtenfeld, C.; Daniliuc, C.G.; Hennecke, U. Enantioselective, desymmetrizing bromolactonization of alkynes. J. Am. Chem. Soc. 2013, 135, 8133–8136. [Google Scholar] [CrossRef]
- Wilking, M.; Daniliuc, C.G.; Hennecke, U. Monomeric cinchona alkaloid-based catalysts for highly enantioselective bromolactonisation of alkynes. Chem. Eur. J. 2016, 22, 18601–18607. [Google Scholar] [CrossRef]
- Fricke, C.; Wilking, M.; Daniliuc, C.G.; Hennecke, U. An enantioselective iodolactonization/cross-coupling protocol for the synthesis of highly substituted enol lactones. Eur. J. Org. Chem. 2018, 3158–3166. [Google Scholar] [CrossRef]
- Wu, W.; Jiang, H. Haloalkynes: A powerful and versatile building block in organic synthesis. Acc. Chem. Res. 2014, 47, 2483–2504. [Google Scholar] [CrossRef] [PubMed]
- Petko, D.; Koh, S.; Tam, W. Transition metal-catalyzed reactions of alkynyl halides. Curr. Org. Synth. 2019, 16, 546–582. [Google Scholar] [CrossRef] [PubMed]
- Cadierno, V. Metal-catalyzed hydrofunctionalization reactions of haloalkynes. Eur. J. Inorg. Chem. 2020, 886–898. [Google Scholar] [CrossRef]
- Uemura, S.; Tara, H.; Okano, M.; Ichikawa, K. Acetoxythallation of acetylenes and the proto- and halogenodethallation of the products. Bull. Chem. Soc. Jpn. 1974, 47, 2663–2671. [Google Scholar] [CrossRef] [Green Version]
- Amos, R.A.; Katzenellenbogen, J.A. An efficient synthesis of γ-methylene-γ-butyrolactone (α’-angelicalactone). Application to the synthesis of deoxyobtusilactone and deoxyisoobtusilactone. J. Org. Chem. 1978, 43, 560–564. [Google Scholar] [CrossRef]
- Krafft, G.A.; Katzenellenbogen, J.A. Synthesis of halo enol lactones. Mechanism-based inactivators of serine proteases. J. Am. Chem. Soc. 1981, 103, 5459–5466. [Google Scholar] [CrossRef]
- Spencer, R.W.; Tam, F.T.; Thomas, E.; Robinson, V.J.; Krantz, A. Ynenol lactones: Synthesis and investigation of reactions relevant to their inactivation of serine proteases. J. Am. Chem. Soc. 1986, 108, 5589–5597. [Google Scholar] [CrossRef]
- Barluenga, J.; Martínez-Gallo, J.M.; Nájera, C.; Yus, M. Stereoselective bifunctionalization of alkynes by means of the mercury(II) salt-iodine combination. J. Chem. Soc. Perkin Trans. 1 1987, 1017–1019. [Google Scholar] [CrossRef]
- Dai, W.; Katzenellenbogen, J.A. Stereoselective Z- and E-bromo enol lactonization of alkynois acids. J. Org. Chem. 1991, 56, 6893–6896. [Google Scholar] [CrossRef]
- Chen, Z.; Li, J.; Jiang, H.; Zhu, S.; Li, Y.; Qi, C. Silver-catalyzed difunctionalization of terminal alkynes: Highly regio- and stereoselective synthesis of (Z)-β-haloenol acetates. Org. Lett. 2010, 12, 3262–3265. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Toullec, P.Y.; Antoniotti, S.; Brancour, C.; Genêt, J.-P.; Michelet, V. Room temperature Au(I)-catalyzed exo-selective cycloisomerization of acetylenic acids: An entry to functionalized γ-lactones. J. Am. Chem. Soc. 2006, 128, 3112–3113. [Google Scholar] [CrossRef] [PubMed]
- Harkat, H.; Weibel, J.-M.; Pale, P. A mild access to γ- and δ-alkylidene lactones through gold catalysis. Tetrahedron Lett. 2006, 47, 6273–6276. [Google Scholar] [CrossRef]
- Harkat, H.; Dembelé, A.Y.; Weibel, J.-M.; Blanc, A.; Pale, P. Cyclization of alkynoic acids with gold catalysts: A surprising dichotomy between AuI and AuIII. Tetrahedron 2009, 65, 1871–1879. [Google Scholar] [CrossRef]
- Gasperini, D.; Maggi, L.; Dupuy, S.; Veenboer, R.M.P.; Cordes, D.B.; Slawin, A.M.Z.; Nolan, S.P. Gold(I)-catalysed cyclisation of alkynoic acids: Towards an efficient and eco-friendly synthesis of γ-,δ- and ε-lactones. Adv. Synth. Catal. 2016, 358, 3857–3862. [Google Scholar] [CrossRef]
- González-Liste, P.J.; León, F.; Arribas, I.; Rubio, M.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Highly stereoselective synthesis and hydrogenation of (Z)-1-alkyl-2-arylvinyl acetate: A wide scope procedure for the preparation of chiral homobenzylic esters. ACS Catal. 2016, 6, 3056–3060. [Google Scholar] [CrossRef] [Green Version]
- González-Liste, P.J.; Francos, J.; García-Garrido, S.E.; Cadierno, V. Gold-catalyzed regio- and stereoselective addition of carboxylic acids to iodoalkynes: Access to (Z)-β-iodoenol esters and 1,4-disubstituted (Z)-enynyl esters. J. Org. Chem. 2017, 82, 1507–1516. [Google Scholar] [CrossRef]
- León, F.; González-Liste, P.J.; García-Garrido, S.E.; Arribas, I.; Rubio, M.; Cadierno, V.; Pizzano, A. Broad scope synthesis of esters precursors of nonfunctionalized chiral alcohols based on the asymmetric hydrogenation of α,β-dialkyl-, α,β-diaryl-, and α-alkyl-β-aryl-vinyl esters. J. Org. Chem. 2017, 82, 5852–5867. [Google Scholar] [CrossRef] [Green Version]
- Hashmi, A.S.K.; Toste, F.D. (Eds.) Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Muthusamy, G.; Pansare, S.V. Stereoselective synthesis of E-3-(arylmethylidene)-5-(alkyl/aryl)-2(3H)-furanones by sequential hydroacyloxylation-Mizoroki-Heck reactions of iodoalkynes. Org. Biomol. Chem. 2018, 16, 7971–7983. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, B.; Zhang, L. The use of Br/Cl to promote regioselective gold-catalyzed rearrangement of propargylic carboxylates: An efficient synthesis of (1Z, 3E)-1-bromo/cloro-2-carboxy-1,3-dienes. Chem. Commun. 2010, 46, 9179–9181. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Liu, Y.; Hou, C.; Li, Y.; Luan, Z.; Zhao, C.; Ke, Z. Rationalization of the selectivity between 1,3- and 1,2-migration: A DFT study on gold(I)-catalyzed propargylic ester rearrangement. Org. Biomol. Chem. 2016, 14, 3558–3563. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Nayak, S.; Prabagar, B.; Sahoo, A.K. Regioselective hydration of terminal halo-substituted propargyl carboxylates by gold catalyst: Synthesis of α-acyloxy α’-halo ketones. J. Org. Chem. 2014, 79, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, Y.; Zhao, X. Theoretical insight into the Au(I)-catalyzed hydration of halo-substituted propargyl acetate: Dynamic water-assisted mechanism. RSC Adv. 2016, 6, 89836–89846. [Google Scholar]
- Chen, Z.-W.; Ye, D.-N.; Ye, M.; Zhou, Z.-G.; Li, S.-H.; Liu, L.-X. AgF/TFA-promoted highly efficient synthesis of α-haloketones from haloalkynes. Tetrahedron Lett. 2014, 55, 1373–1375. [Google Scholar] [CrossRef]
- Zeng, M.; Huang, R.-X.; Li, W.-Y.; Liu, X.-W.; He, F.-L.; Zhang, Y.-Y.; Xiao, F. In(OTf)3/acid co-catalyzed hydration of 1-haloalkynes to α-halomethyl ketones. Tetrahedron 2016, 72, 3818–3822. [Google Scholar] [CrossRef]
- Zou, H.; He, W.; Dong, Q.; Wang, R.; Yi, N.; Jiang, J.; Pen, D.; He, W. First catalyzed hydration of haloalkynes by a recyclable catalytic system. Eur. J. Org. Chem. 2016, 116–121. [Google Scholar] [CrossRef]
- Chen, X.; Chen, D.; Lu, Z.; Kong, L.; Zhu, G. Palladium-catalyzed coupling of haloalkynes with allyl acetate: A regio- and stereoselective synthesis of (Z)-β-haloenol acetates. J. Org. Chem. 2011, 76, 6338–6343. [Google Scholar] [CrossRef]
- Espinosa-Jalapa, N.A.; Ke, D.; Nebra, N.; Le Goanvic, L.; Mallet-Ladeira, S.; Monot, J.; Martin-Vaca, B.; Bourissou, D. Enhanced catalytic performance of indenediide palladium pincer complexes for cycloisomerization: Efficient synthesis of alkylidene lactams. ACS Catal. 2014, 4, 3605–3611. [Google Scholar] [CrossRef]
- Monot, J.; Brunel, P.; Kefalidis, C.E.; Espinosa-Jalapa, N.A.; Maron, L.; Martin-Vaca, B.; Bourissou, D. A case study of proton shuttling in palladium catalysis. Chem. Sci. 2016, 7, 2179–2187. [Google Scholar] [CrossRef] [Green Version]
- Abe, S.; Miyaura, N.; Suzuki, A. The palladium-catalyzed cross-coupling reaction of enol acetates with 1-alkenyl-, aryl-, or alkylboron compounds; A facile synthesis of ketones and their enol acetates. Bull. Chem. Soc. Jpn. 1992, 65, 2863–2865. [Google Scholar] [CrossRef]
- He, R.; Deng, M.-Z. A novel method for the construction of (Z,E)- or (Z,Z)-conjugated alkadienyl carboxylates. Org. Lett. 2002, 4, 2759–2762. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Huang, G.; Jiang, H.; Huang, H.; Pan, X. Synthesis of 2,5-disubstituted 3-iodofurans via palladium-catalyzed coupling and iodocyclization of terminal alkynes. J. Org. Chem. 2011, 76, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Yanada, R. Multisubstituted furan formation from (Z)- or (E)-enynyl acetates: Tandem reactions accelerated by electron-donating groups on aromatic rings. J. Org. Chem. 2012, 77, 3944–3951. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.W.; Chen, H.-C.; Ye, D.-N.; Hu, Q.-S. (Z)-1,4-Diphenylbut-1-en-3-ynyl acetate. Acta Cryst. 2012, E68, o2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.-W.; Luo, M.-T.; Wen, Y.-L.; Ye, M.; Zhou, Z.-G.; Liu, L.-X. A highly efficient synthesis of 2,5-disubstituted furans from enyne acetates catalyzed by Lewis acid and palladium. Synlett 2014, 25, 2341–2344. [Google Scholar] [CrossRef]
- Lee, H.; Yi, Y.; Jun, C.-H. Copper(II)-promoted, one-pot conversion of 1-alkynes with anhydrides or primary amines to the respective 2,5-disubstituted furans or pyrroles under microwave irradiation conditions. Adv. Synth. Catal. 2015, 357, 3485–3490. [Google Scholar] [CrossRef]
- Okamoto, N.; Sueda, T.; Yanada, R. Tandem reaction of enynyl acetate: Precursor of allenyl ketones. Chem. Pharm. Bull. 2016, 64, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Stefani, H.A.; Guarezemini, A.S.; Cella, R. Homocoupling reactions of alkynes, alkenes and alkyl compounds. Tetrahedron 2010, 66, 7871–7918. [Google Scholar] [CrossRef]
- Takagi, K.; Hayama, N.; Sasaki, K. Ni(0)-trialkylphosphine complexes. Efficient homo-coupling catalyst for aryl, alkenyl, and heteroaromatic halides. Bull. Chem. Soc. Jpn. 1984, 57, 1887–1890. [Google Scholar] [CrossRef] [Green Version]
- Takagi, K.; Mimura, H.; Inokawa, S. The in situ-generated nickel(0)-catalyzed homo-coupling of alkenyl halides with zinc powder. A specific outcome in stereochemistry. Bull. Chem. Soc. Jpn. 1984, 57, 3517–3522. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Nakao, K.; Kobayashi, Y.; Sakai, M.; Uchino, N.; Sakakibara, Y.; Takagi, K. Ni(0)-triphenylphosphine complex-catalyzed homo-coupling of 1-alkenyl halides with zinc powder. Bull. Chem. Soc. Jpn. 1993, 66, 2446–2448. [Google Scholar] [CrossRef]
- Francos, J.; Cadierno, V. Nickel-catalyzed homocoupling of (Z)-β-iodoenol esters: Stereoselective access to (Z,Z)-buta-1,3-diene-1,4-diyl diesters. Synthesis 2019, 51, 3117–3126. [Google Scholar] [CrossRef] [Green Version]
- León, F.; Francos, J.; López-Serrano, J.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Double asymmetric hydrogenation of conjugated dienes: A self-breeding chirality route to C2 symmetric 1,4-diols. Chem. Commun. 2019, 55, 786–789. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Li, J.; Zhu, C.; Wu, W.; Jiang, H. Palladium-catalyzed sequential nucleophilic addition/oxidative annulation of bromoalkynes with benzoic acids to construct functionalized isocoumarins. Org. Lett. 2017, 19, 4440–4443. [Google Scholar] [CrossRef]
- Chen, Z.; Ye, D.; Xu, G.; Ye, M.; Liu, L. Highly efficient synthesis of 2,5-disubstituted pyrazines from (Z)-β-haloenol acetates. Org. Biomol. Chem. 2013, 11, 6699–6702. [Google Scholar] [CrossRef]
- Tam, F.T.; Spencer, R.W.; Thomas, E.M.; Copp, L.J.; Krantz, A. Novel suicide inhibitors of serine proteinases. Inactivation of human leukocyte elastase by ynenol lactones. J. Am. Chem. Soc. 1984, 106, 6849–6851. [Google Scholar] [CrossRef]
- Zupan, L.A.; Weiss, R.H.; Hazen, S.L.; Parnas, B.L.; Aston, K.W.; Lennon, P.J.; Getman, D.P.; Gross, R.W. Structural determinants of haloenol lactone-mediated suicide inhibition of canine myocardial calcium-independent phospholipase A2. J. Med. Chem. 1993, 36, 95–100. [Google Scholar] [CrossRef]
Figure 1.
Generic structures of α- and β-haloenol esters.

Scheme 1.
Synthetic routes commonly employed in the preparation of β-haloenol esters.

Figure 2.
Generic structures of haloenol lactones featuring biological activity.

Scheme 2.
The E-halolactonization of alkynoic acids.

Scheme 3.
Enantioselective halolactonization of alkynoic acids employing an organocatalyst.

Scheme 4.
Thallium-mediated synthesis of the (E)-β-haloenol acetates 3–5 from 1-propynylbenzene.

Scheme 5.
Mercury-catalyzed cyclization of the alkynoic acids 6 and 7.

Scheme 6.
Mercury-mediated cyclization of 5-iodo-4-pentynoic acid.

Scheme 7.
Synthesis of acyclic (E)-β-iodoenol acetates by bifunctionalization of internal alkynes.

Scheme 8.
Silver-mediated Z-bromoenol lactonization of alkynoic acids.

Scheme 9.
Silver-catalyzed synthesis of (Z)-β-haloenol acetates from terminal alkynes.

Scheme 10.
AuCl-catalyzed cycloisomerization of bromo-substituted alkynoic acids.

Scheme 11.
Cycloisomerization of alkynoic acids 14 and 16 catalyzed by [{Au(IPr)}2(µ-OH)][BF4] (18).
Scheme 11.
Cycloisomerization of alkynoic acids 14 and 16 catalyzed by [{Au(IPr)}2(µ-OH)][BF4] (18).

Scheme 12.
Gold-catalyzed intermolecular addition of carboxylic acids to iodoalkynes.

Figure 3.
Structure of the (Z)-β-iodoenol cinnamates 20.

Scheme 13.
Gold(I)-catalyzed rearrangement of halo-substituted propargylic carboxylates.

Scheme 14.
Palladium-catalyzed coupling of haloalkynes with allyl acetate.

Figure 4.
Structure of the palladium(II) pincer complexes 25 and 26.

Scheme 15.
Pd-catalyzed coupling of (Z)-β-bromoenol acetate 27 with organoboron compounds.

Scheme 16.
Synthesis of the aryldiene 29 through Suzuki–Miyaura coupling of 28 with PhB(OH)2.

Scheme 17.
(Z)-β-Iodoenol acetates as starting materials for the preparation of chiral homobenzylic esters.
Scheme 17.
(Z)-β-Iodoenol acetates as starting materials for the preparation of chiral homobenzylic esters.

Scheme 18.
Stereoselective synthesis of conjugated dienyl esters through Suzuki–Miyaura-type cross-coupling reactions.
Scheme 18.
Stereoselective synthesis of conjugated dienyl esters through Suzuki–Miyaura-type cross-coupling reactions.

Figure 5.
Structure of the tetrasubstituted olefins 37–40.

Scheme 19.
Sonogashira-type coupling reactions of β-haloenol esters.

Scheme 20.
Some furan-ring formation reactions employing enynyl acetates as precursors.

Scheme 21.
Negishi- and Kumada-Corriu cross-coupling reactions involving β-haloenol esters.

Scheme 22.
Ni-catalyzed homocoupling of (Z)-β-iodoenol esters.

Scheme 23.
Intramolecular Mizoroki–Heck coupling of (Z)-β-iodoenol cinnamates.

Scheme 24.
Catalytic synthesis of isocoumarins from bromoalkynes and benzoic acids.

Scheme 25.
Synthesis of 2,5-disubstituted pyrazines from (Z)-β-haloenol acetates.

Figure 6.
Ynenol lactones synthesized by Sonogashira coupling of the respective iodoenol lactones with terminal alkynes.
Figure 6.
Ynenol lactones synthesized by Sonogashira coupling of the respective iodoenol lactones with terminal alkynes.

Scheme 26.
Access to arylenol lactones by Negishi coupling.

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cadierno, V. Metal-Catalyzed Synthesis and Transformations of β-Haloenol Esters. Catalysts 2020, 10, 399. https://doi.org/10.3390/catal10040399
AMA Style
Cadierno V. Metal-Catalyzed Synthesis and Transformations of β-Haloenol Esters. Catalysts. 2020; 10(4):399. https://doi.org/10.3390/catal10040399
Chicago/Turabian StyleCadierno, Victorio. 2020. "Metal-Catalyzed Synthesis and Transformations of β-Haloenol Esters" Catalysts 10, no. 4: 399. https://doi.org/10.3390/catal10040399
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.