Characteristics and Behavior of Different Catalysts Used for Water Decontamination in Photooxidation and Ozonation Processes

 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Catalysts Used in Pollutant Photooxidation
2.1. Activated Carbons
2.1.1. Effect of Gamma Irradiation on the Textural and Chemical Properties of ACs
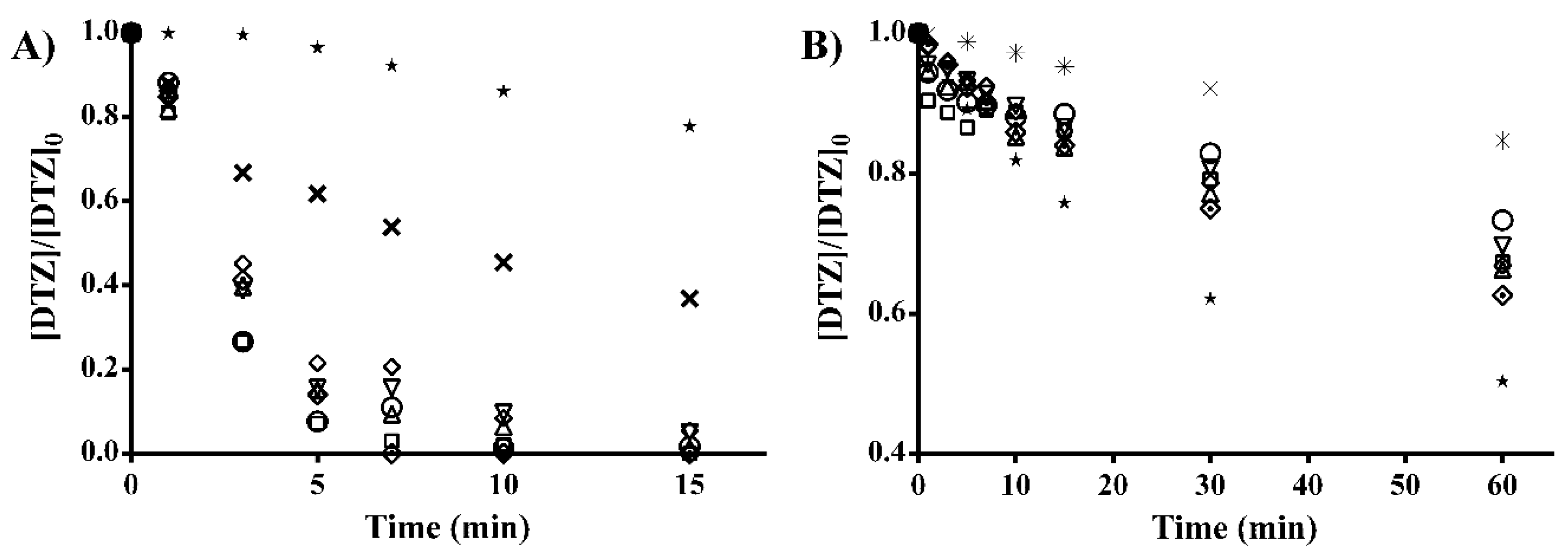
2.1.2. Effect of the Surface Chemistry of ACs on DTZ Degradation by UV/AC System
2.1.3. Effect of the Type of Radiation in the Heterogeneous Photocatalysis Process Using ACs
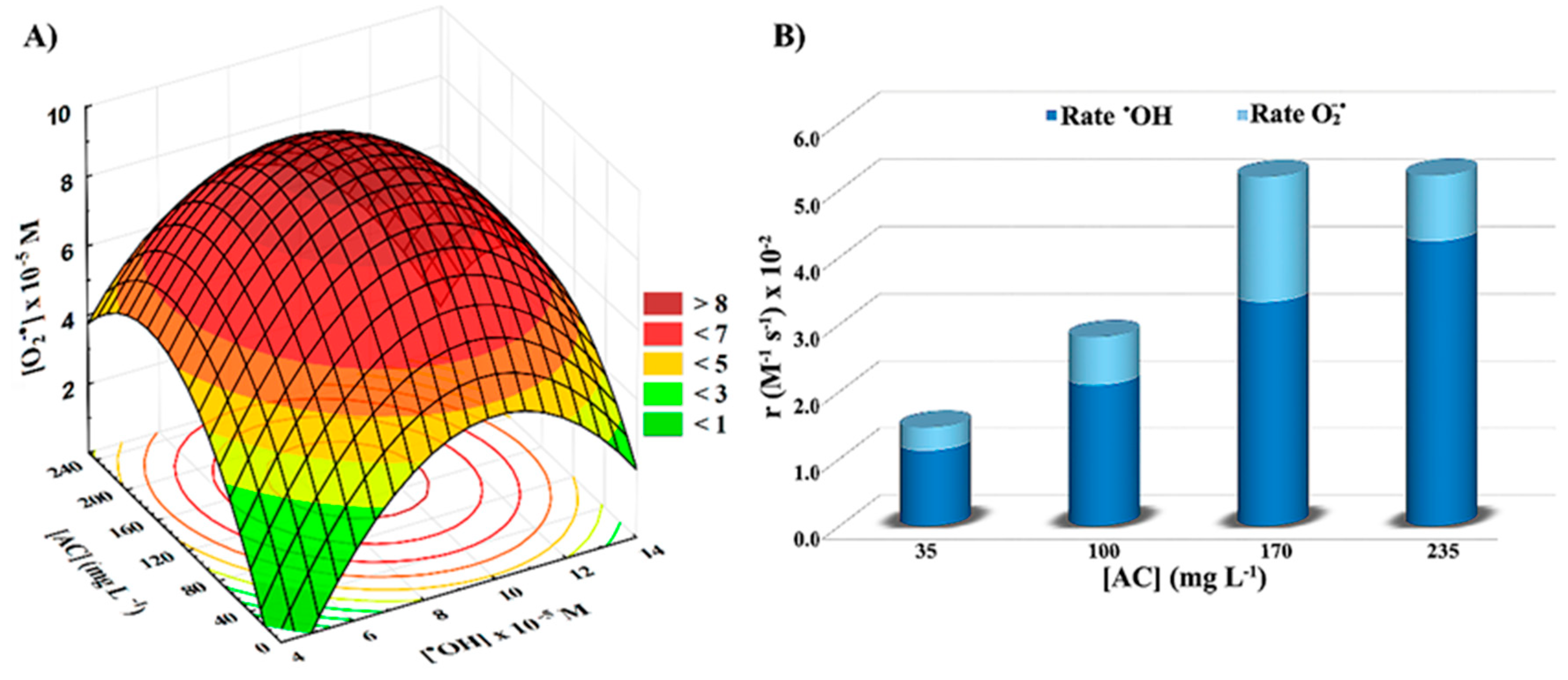
2.1.4. Effect of AC Surface Chemistry on the Photogeneration of Radical Species in UV/AC and Solar/AC Systems
2.2. Metal Carbon Aerogels
2.3. Iron-Doped Silica Xerogels
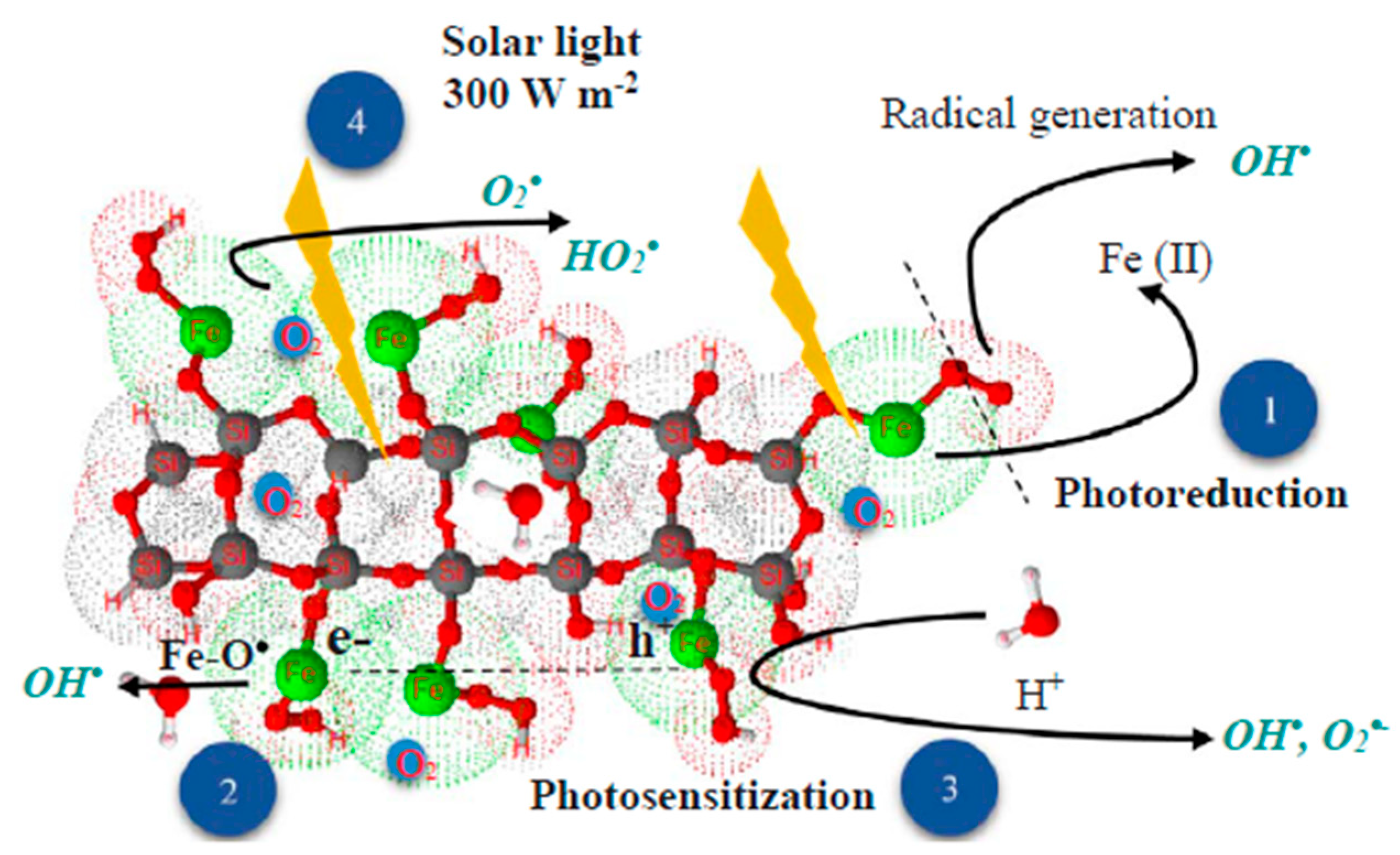
- (1)
- Photoredox reaction between OH groups on the catalyst surface and the iron.
- (2)
- Formation of Fe(III)−O• due to the presence of Fe(III), which may induce the generation of HO• and H• radicals.
- (3)
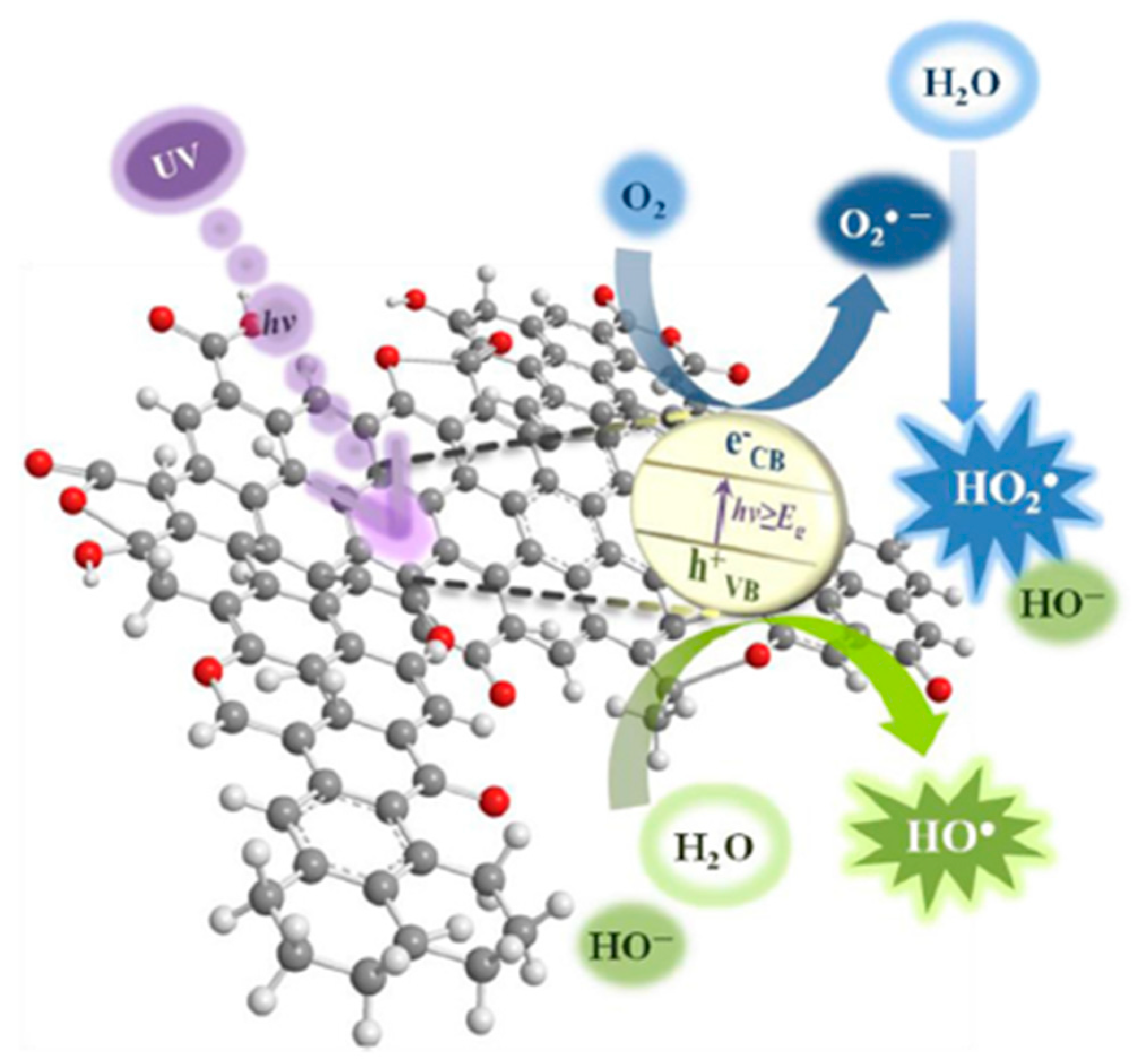
- Generation of electron–hole pairs (e−-h+) by photons from the solar light that reaches the catalyst (reaction (31)); contact of the photogenerated electrons with pollutant or O2 molecules would reduce them to O2•− radicals, which react with water molecules and produce more radical species to enhance TNZ degradation (reactions (32) and (33)) [115].
- (4)
- O2 reduction by photogenerated electrons (e−CB) on the XGS-Fe(III) surface via the conduction band, which can form O2•− for action in reactions (34) and (35), contributing to TNZ degradation [116].
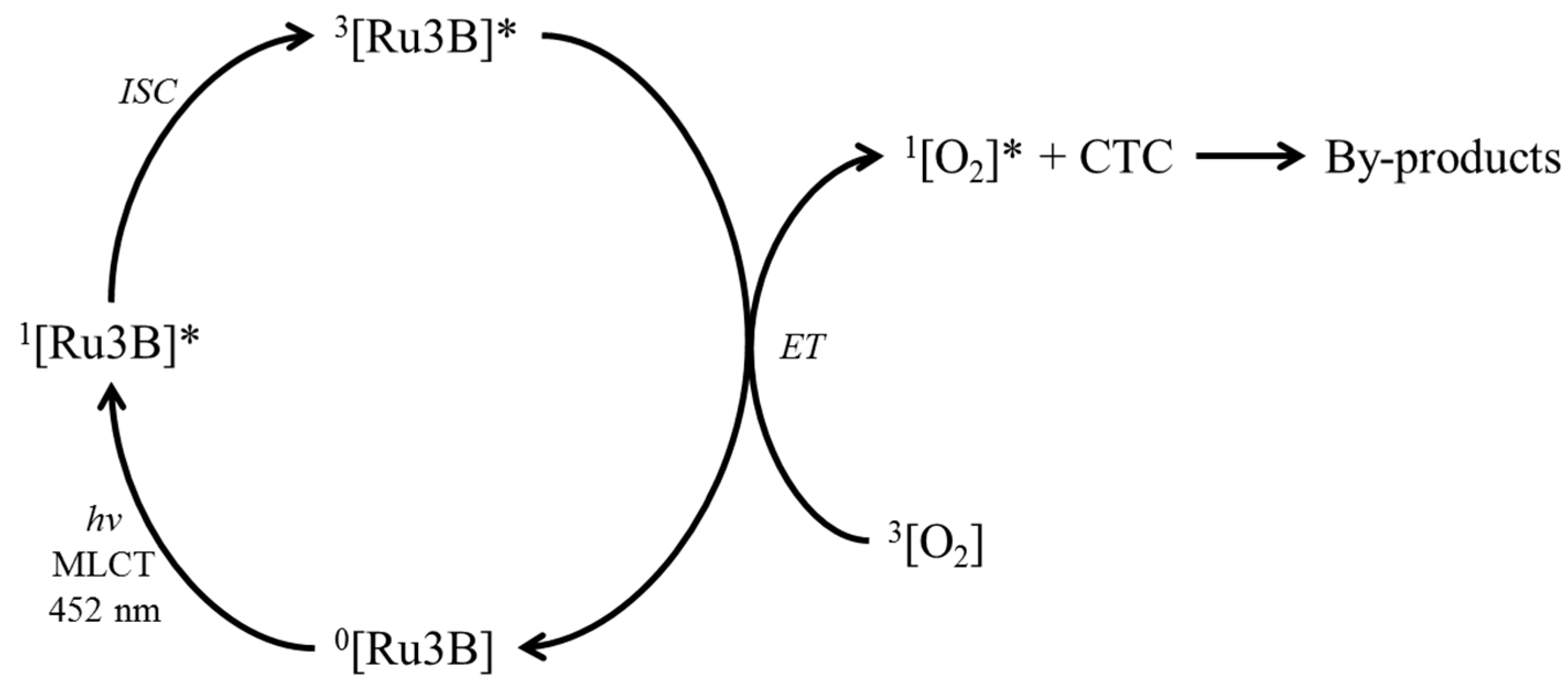
2.4. Ruthenium Metal Complexes
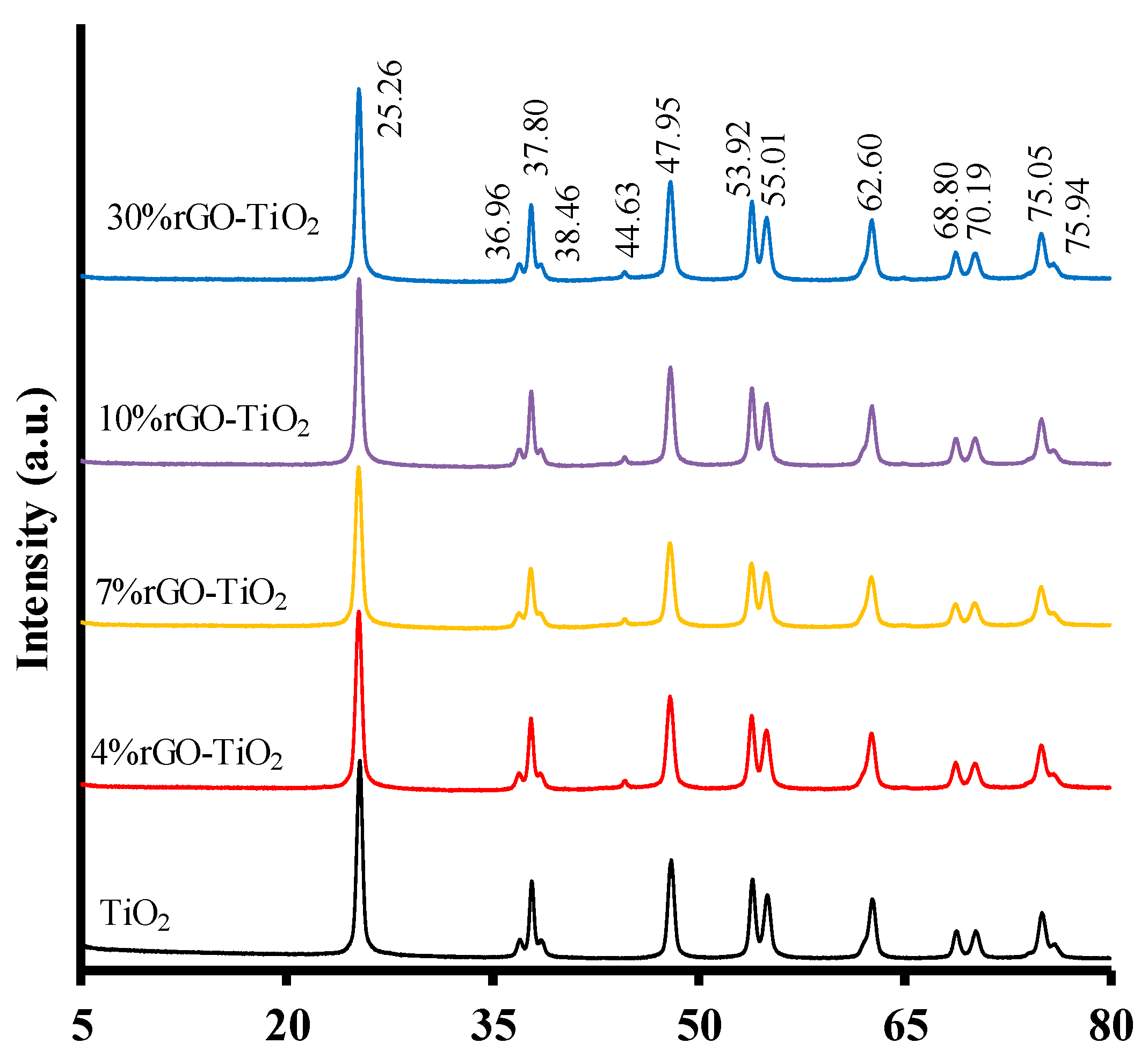
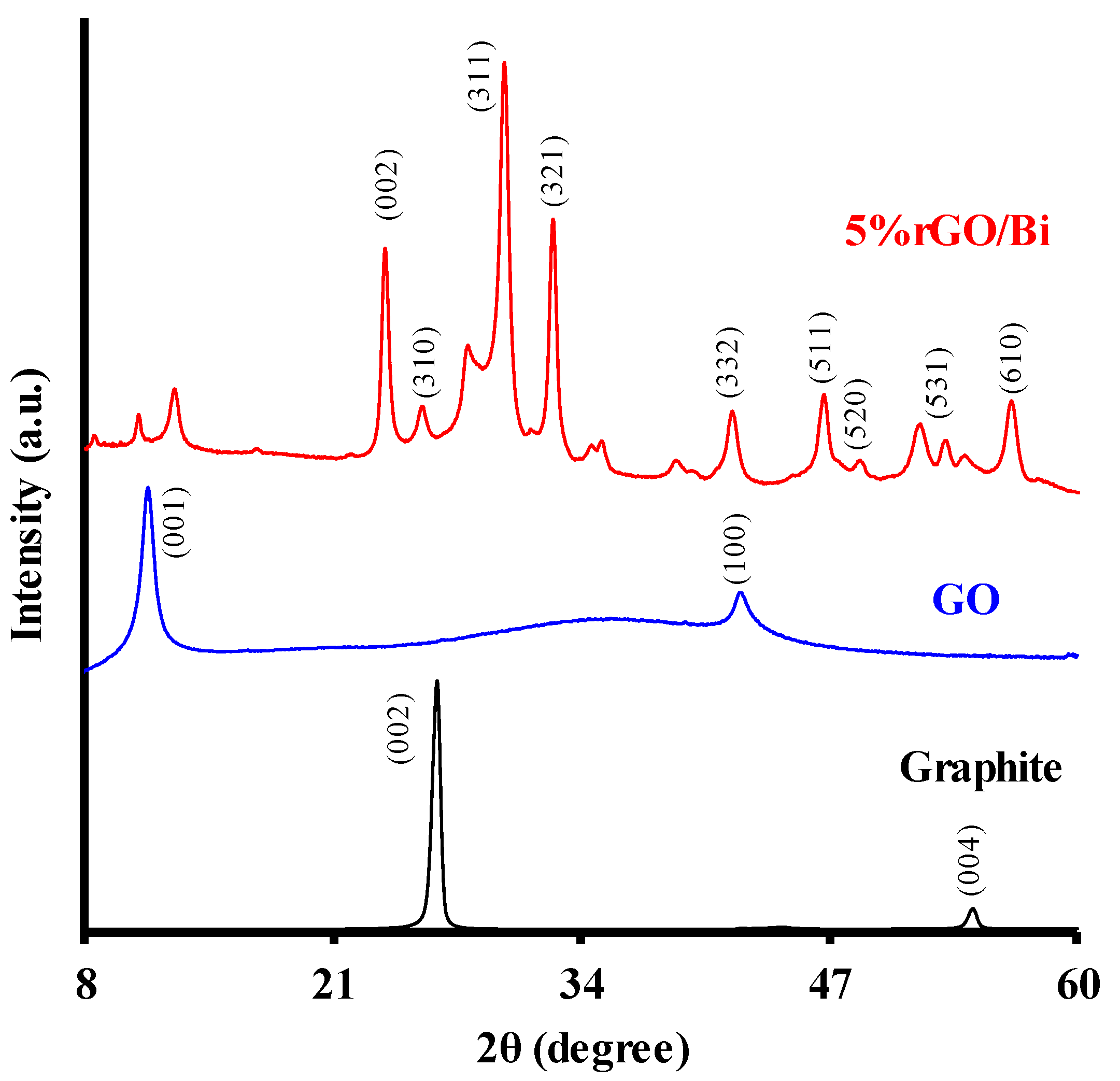
2.5. rGO Metal Oxide Composites
3. Catalysts Used in Pollutant Ozonation
3.1. Activated Carbons


3.2. Metal-Doped Carbon Aerogels
3.3. Basic Treated Zeolites
4. Recent Results Related to Photocatalysis and Catalyzed Ozonation for Water Treatment Using Similar Catalysts to Those Studied in Our Project
5. Conclusions
6. Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Andreozzi, R.; Caprio, V.; Insola, A.; Marotta, R. Advanced oxidation processes (AOP) for water purification and recovery. Catal. Today 1999, 53, 51–59. [Google Scholar] [CrossRef]
- Brillas, E.; Mur, E.; Sauleda, R.; Sanchez, L.; Peral, J.; Domenech, X.; Casado, J. Aniline mineralization by AOP’s: Anodic oxidation, photocatalysis, electro-Fenton and photoelectro-Fenton processes. Appl. Catal. B Environ. 1998, 16, 16–31. [Google Scholar] [CrossRef]
- Matatov, M.Y.I.; Sheintuch, M. Catalytic abatement of water pollutants. Ind. Eng. Chem. Res. 1998, 37, 309–326. [Google Scholar] [CrossRef]
- Niu, L.; Wei, T.; Li, Q.; Zhang, G.; Xian, G.; Long, Z.; Ren, Z. Ce-based catalysts used in advanced oxidation processes for organic wastewater treatment: A review. J. Environ. Sci. 2020, 96, 109–116. [Google Scholar] [CrossRef]
- Singer, P.C.; Reckhow, D.A. Water Quality & Treatment: A Handbook of Community Water Supplies; McGraw-Hill: New York, NY, USA, 1999. [Google Scholar]
- Pitochelli, A. Biocides: Useful applications for the use of chlorine dioxide in water treatment. Ultrapure Water 2006, 23, 40–42. [Google Scholar]
- Zou, L.Y.; Li, Y.; Hung, Y.S. Wet air oxidation for waste treatment. Handb. Environ. Eng. 2007, 5, 575–610. [Google Scholar]
- Von Sonntag, C. The basics of oxidants in water treatment. Part A: OH radical reactions. Water. Sci. Technol. 2007, 55, 19–23. [Google Scholar] [CrossRef]
- Peter, A.; Von Gunten, U. Oxidation kinetics of selected taste and odor compounds during ozonation of drinking water. Environ. Sci. Technol. 2007, 41, 626–631. [Google Scholar] [CrossRef]
- Barry, T.I.; Stone, F.S. The reactions of oxygen at dark and irradiated zinc oxide surface. Proc. R. Soc. A 1960, 255, 124–144. [Google Scholar]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Henderson, M.A. A surface science perspective on TiO2 photocatalysis. Surf. Sci. Rep. 2011, 66, 185–297. [Google Scholar] [CrossRef]
- Serpone, N.; Pelizzetti, E. Photocatalysis: Fundamental and Applications; Wiley: New York, NY, USA, 1989. [Google Scholar]
- Zu, D.; Song, H.; Wang, Y.; Chao, Z.; Li, Z.; Wang, G.; Shen, Y.; Li, C.; Ma, J. One-pot in-situ hydrothermal synthesis of CdS/Nb2O5/Nb2C heterojunction for enhanced visible-light-driven photodegradation. Appl. Catal. B Environ. 2020, 277, 119140. [Google Scholar] [CrossRef]
- Czech, B.; Zygmunt, P.; Kadirova, Z.C.; Yubuta, K.; Hojamberdiev, M. Effective photocatalytic removal of selected pharmaceuticals and personal care products by elsmoreite/tungsten oxide@ZnS photocatalyst. J. Environ. Manag. 2020, 270, 110870. [Google Scholar] [CrossRef]
- Matos, J.; Laine, J.; Herrmann, J.M. Synergy effect in the photocatalytic degradation of phenol on a suspended mixture of titania and activated carbon. Appl. Catal. B Environ. 1998, 18, 281–291. [Google Scholar] [CrossRef]
- Leary, R.; Westwood, A. Carbonaceous nanomaterials for the enhancement of TiO2 photocatalysis. Carbon 2011, 49, 741–772. [Google Scholar] [CrossRef]
- Onkani, S.P.; Diagboya, P.N.; Mtunzi, F.M.; Klink, M.J.; Olu-Owolabi, B.I.; Pakade, V. Comparative study of the photocatalytic degradation of 2-chlorophenol under UV irradiation using pristine and Ag-doped species of TiO2, ZnO and ZnS photocatalysts. J. Environ. Manag. 2020, 260, 110145. [Google Scholar] [CrossRef]
- Qiao, S.; Feng, C.; Guo, Y.; Chen, T.; Akram, N.; Zhang, Y.; Wang, W.; Yue, F.; Wang, J. CdS nanoparticles modified Ni@NiO spheres as photocatalyst for oxygen production in water oxidation system and hydrogen production in water reduction system. Chem. Eng. J. 2020, 395, 125068. [Google Scholar] [CrossRef]
- Cao, D.; Wang, Q.; Wu, Y.; Zhu, S.; Jia, Y.; Wang, R. Solvothermal synthesis and enhanced photocatalytic hydrogen production of Bi/Bi2MoO6 co-sensitized TiO2 nanotube arrays. Sep. Purif. Technol. 2020, 250, 117132. [Google Scholar] [CrossRef]
- Faria, J.L.; Wang, W. Carbon materials in photocatalysis. In Carbon Materials for Catalysis; Serp, P., Figueiredo, J.L., Eds.; John Wiley & Sons: New York, NY, USA, 2009; pp. 481–506. [Google Scholar]
- Ania, C.O.; Velasco, L.F.; Valdés-Solís, T. Photochemical behavior of carbon adsorbents. In Novel Carbon Adsorbents; Tascón, J.M.D., Ed.; Elsevier: Oxford, UK, 2012; pp. 521–547. [Google Scholar]
- Velasco, L.F.; Parra, J.B.; Ania, C.O. Role of activated carbon features on the photocatalytic degradation of phenol. Appl. Surf. Sci. 2010, 256, 5254–5258. [Google Scholar] [CrossRef] [Green Version]
- Velasco, L.F.; Fonseca, I.M.; Parra, J.B.; Lima, J.C.; Ania, C.O. Photochemical behaviour of activated carbons under UV irradiation. Carbon 2012, 50, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Velasco, L.F.; Maurino, V.; Laurenti, E.; Ania, C. Light-induced generation of radicals on semiconductor-free carbon photocatalysts. Appl. Catal. A Gen. 2013, 453, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Velasco, L.F.; Maurino, V.; Laurenti, E.; Fonseca, I.M.; Lima, J.C.; Ania, C.O. Photoinduced reactions occurring on activated carbons. A combined photooxidation and ESR study. Appl. Catal. A Gen. 2013, 452, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Araña, J.; Doña-Rodríguez, J.M.; Tello Rendón, E.; Garriga, I.; Cabo, C.; González-Díaz, O.; Herrera-Melián, J.A.; Pérez-Peña, J.; Colón, G.; Navío, J.A. TiO2 activation by using activated carbon as a support: Part I. Surface characterisation and decantability study. Appl. Catal. B Environ. 2003, 44, 161–172. [Google Scholar] [CrossRef]
- Keller, N.; Rebmann, G.; Barraud, E.; Zahraa, O.; Keller, V. Macroscopic carbon nanofibers for use as photocatalyst support. Catal. Today 2005, 101, 323–329. [Google Scholar] [CrossRef]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: Are we a step closer to achieving sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef]
- Wang, J.; Huang, H.; Xu, Z.; Kou, J.; Lu, C. The potential of carbon-based materials for photocatalytic application. Curr. Org. Chem. 2014, 18, 1346–1364. [Google Scholar] [CrossRef]
- Chen, J.W.; Hui, C.; Keller, T.; Smith, G. Catalytic ozonation in aqueous system. AIChE Symp. Ser. 1977, 73, 206–212. [Google Scholar]
- Andreozzi, R.; Insola, A.; Caprio, V.; Marotta, R.; Tufano, V. The use of manganese dioxide as a heterogeneous catalyst for oxalic acid ozonation in aqueous solution. Appl. Catal. A Gen. 1996, 138, 75–81. [Google Scholar] [CrossRef]
- Ma, J.; Graham, N.J.D. Preliminary investigation of manganese-catalyzed ozonation for the destruction of atrazine. Ozone Sci. Eng. 1997, 19, 227–236. [Google Scholar] [CrossRef]
- Al Hayek, N.; Legube, B.; Doré, M. Ozonation catalytique (Fe(III)/Al2O3) du phénol y de ses produits d’ozonation. Environ. Technol. 1992, 10, 415–422. [Google Scholar]
- Gracia, R.; Cortés, S.; Sarasa, J.; Ormad, P.; Ovelleiro, J.L. Catalytic ozonation with supported titanium dioxide. The stability of catalyst in water. Ozone Sci. Eng. 2000, 22, 185–193. [Google Scholar] [CrossRef]
- Leitner, N.K.V.; Fu, H. pH effects on catalytic ozonation of carboxylic acids with metal on metal oxides catalysts. Top. Catal. 2005, 33, 249–256. [Google Scholar] [CrossRef]
- Cooper, C.; Burch, R. Mesoporous materials for water treatment processes. Water Res. 1999, 33, 3689–3694. [Google Scholar] [CrossRef]
- McKay, G.; McAleavey, G. Ozonation and carbon adsorption in a three-phase fluidised bed for colour removal from peat water. Chem. Eng. Res. Des. 1988, 66, 532–536. [Google Scholar]
- Lin, S.H.; Lai, C.L. Kinetic characteristics of textile wastewater ozonation in fluidized and fixed activated carbon beds. Water Res. 2000, 34, 763–772. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M. Ozonation of 1,3,6-naphthalenetrisulphonic acid catalysed by activated carbon in aqueous phase. Appl. Catal. B Environ. 2002, 39, 31–329. [Google Scholar] [CrossRef] [Green Version]
- Orge, C.A.; Sousa, J.P.S.; Gonçalves, F.; Freire, C.; Órfão, J.J.M.; Pereira, M.F.R. Development of novel mesoporous carbon materials for the catalytic ozonation of organic pollutants. Catal. Lett. 2009, 132, 1–9. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Ma, J.; Cui, Y.H.; Zhang, B.P. Effect of ozonation pretreatment on the surface properties and catalytic activity of multi-walled carbon nanotube. Appl. Catal. B Environ. 2009, 92, 301–306. [Google Scholar] [CrossRef]
- Ma, J.; Sui, M.; Zhang, T.; Guan, C. Effect of pH on MnOx/GAC catalyzed ozonation for degradation of nitrobenzene. Water Res. 2005, 39, 779–786. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; Rivera-Utrilla, J.; von Gunten, U. Metal-doped carbon aerogels as catalysts during ozonation processes in aqueous solutions. Water Res. 2006, 40, 3375–3384. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Ma, J.; Cui, Y.H. Carbon nanotube supported platinum catalysts for the ozonation of oxalic acid in aqueous solutions. Carbon 2008, 46, 890–897. [Google Scholar] [CrossRef]
- Nidheesh, P.V. Graphene-based materials supported advanced oxidation processes for water and wastewater treatment: A review. Environ. Sci. Pollut. Res. 2017, 24, 27047–27069. [Google Scholar] [CrossRef]
- Valdés, H.; Sánchez-Polo, M.; Rivera-Utrilla, J.; Zaror, C.A. Effect of ozone treatment on surface properties of activated carbon. Langmuir 2002, 18, 2111–2116. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; Moncada, M.A.; Rivera-Utrilla, J.; Zaror, C.A. Effect of ozone and ozone/activated carbon treatments on genotoxic activity of naphthalenesulphonic acids. J. Chem. Technol. Biot. 2002, 77, 883–890. [Google Scholar]
- Sánchez-Polo, M.; Rivera-Utrilla, J. Effect of the ozone-carbon reaction on the catalytic activity of activated carbon during degradation of 1,3,6-naphthalenetrisulphonic acid with ozone. Carbon 2003, 41, 303–307. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M. Degradation and removal of naphthalenesulphonic acids by means of adsorption and ozonation catalyzed by activated carbon in water. Water Resour. Res. 2003, 39, 1232. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; Rivera-Utrilla, J. The use of activated carbon and ozone in water treatment to eliminate naphthalenesulphonic acids. Solid Fuel Chem. 2004, 1, 26–34. [Google Scholar]
- Sánchez-Polo, M.; Rivera-Utrilla, J. Ozonation of 1,3,6-naphthalenetrisulfonic acid in presence of heavy metals. J. Chem. Technol. Biot. 2004, 79, 902–909. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M. Ozonation of naphthalenesulphonic acid in aqueous phase in the presence of basic activated carbons. Langmuir 2004, 20, 9217–9222. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; Leyva-Ramos, R.; Rivera-Utrilla, J. Kinetics of 1,3,6-naphthalenetrisulphonic acid oxidation in presence of activated carbon. Carbon 2005, 43, 962–969. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; von Gunten, U.; Rivera-Utrilla, J. Efficiency of activated carbon to transform ozone into ·OH radicals: Influence of operational parameters. Water Res. 2005, 39, 3189–3198. [Google Scholar] [CrossRef]
- Méndez-Díaz, J.; Sánchez-Polo, M.; Rivera-Utrilla, J.; Bautista-Toledo, I.; Ferro-García, M.A. Ozonation in aqueous phase of sodium dodecylbenzenesulphonate in the presence of powdered activated carbon. Carbon 2005, 43, 3031–3034. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Méndez-Díaz, J.; Sánchez-Polo, M.; Ferro-García, M.A.; Bautista-Toledo, I. Removal of the surfactant sodium dodecylbenzenesulphonte from water by simultaneous use of ozone and powdered activated carbon: Comparison with system based on O3 and O3/H2O2. Water Res. 2006, 40, 1717–1725. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; Salhi, E.; Rivera-Utrilla, J.; von Gunten, U. Combination of ozone with activated carbon as an alternative to conventional advanced oxidation processes. Ozone Sci. Eng. 2006, 28, 237–245. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; Rivera-Utrilla, J. Ozonation of naphthalenetrisulphonic acid in the presence of activated carbons prepared from petroleum coke. Appl. Catal. B Environ. 2006, 67, 113–120. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; Rivera-Utrilla, J. Photooxidation of naphthalenesulphonic acids in presence of transition metal-doped carbon aerogels. Appl. Catal. B Environ. 2006, 69, 93–100. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; Rivera-Utrilla, J.; Méndez-Díaz, J.; López-Peñalver, J. Metal-doped carbon aerogels: New materials for water treatment. Ind. Eng. Chem. Res. 2008, 47, 6001–6005. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; Rivera-Utrilla, J.; Prados-Joya, G.; Ferro-García, M.A.; Bautista-Toledo, I. Removal of pharmaceutical compounds, nitroimidazoles, from waters by using the ozone/carbon system. Water Res. 2008, 42, 4163–4171. [Google Scholar]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Méndez-Díaz, J.D. Ozone decomposition by catalysts and its application in water treatment: An overview. In Ozone Depletion, Chemistry and Impacts; Bakker, S.H., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2008; pp. 17–51. [Google Scholar]
- Sánchez-Polo, M.; Mendez-Díaz, J.D.; Rivera-Utrilla, J.; Bautista-Toledo, I.; Ferro-García, M.A. Influence of presence of tannic acid on removal of sodium dodecylbenzenesulphonate by O3 and advanced oxidation processes. J. Chem. Technol. Biotechnol. 2009, 84, 367–375. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Prados-Joya, G.; Ferro-García, M.A.; Bautista-Toledo, I. Removal of tinidazole from water by using ozone and activated carbon in dynamic regime. J. Hazard. Mater. 2010, 174, 880–886. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Gómez-Serrano, V.; Álvarez, P.M.; Alvim-Ferraz, M.C.M.; Dias, J.M. Activated carbon modifications to enhance its water treatment applications. An overview. J. Hazard. Mater. 2011, 187, 1–23. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Bautista-Toledo, M.I.; Méndez-Díaz, J.D. Enhanced oxidation of sodium dodecylbenzenesulfonate aqueous solution using ozonation catalyzed by base treated zeolite. Chem. Eng. J. 2012, 180, 204–209. [Google Scholar] [CrossRef]
- Medellin-Castillo, N.A.; Ocampo-Pérez, R.; Leyva-Ramos, R.; Sanchez-Polo, M.; Rivera-Utrilla, J.; Méndez-Díaz, J.D. Removal of diethyl phthalate from water solution by adsorption, photo-oxidation, ozonation and advanced oxidation process (UV/H2O2, O3/H2O2 and O3/activated carbon). Sci. Total Environ. 2013, 442, 26–35. [Google Scholar] [CrossRef]
- Velo-Gala, I.; López-Peñalver, J.J.; Sánchez-Polo, M.; Rivera-Utrilla, J. Activated carbon as photocatalyst of reactions in aqueous phase. Appl. Catal. B Environ. 2013, 142–143, 694–704. [Google Scholar] [CrossRef]
- Velo-Gala, I.; López-Peñalver, J.J.; Sánchez-Polo, M.; Rivera-Utrilla, J. Role of activated carbon on micropollutants degradation by different radiation processes. Mediterr. J. Chem. 2015, 4, 68–80. [Google Scholar] [CrossRef]
- Ocampo-Perez, R.; Rivera-Utrilla, J.; Abdel Daiem, M.M.; Sánchez-Polo, M. Integrated technologies based on the use of activated carbon and radiation to remove contaminants present in landfill leachates. In Landfills and Recycling Centers. Processing Systems, Impact on the Environment and Adverse Health Effects; Jackson, C.H., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2015; pp. 69–109. [Google Scholar]
- Orellana-García, F.; Álvarez, M.A.; López-Ramón, M.V.; Rivera-Utrilla, J.; Sánchez-Polo, M. Photoactivity of organic xerogels and aerogels in the photodegradation of herbicides from waters. Appl. Catal. B Environ. 2016, 181, 94–102. [Google Scholar] [CrossRef]
- Salazar-Rábago, J.J.; Sánchez-Polo, M.; Rivera-Utrilla, J.; Leyva-Ramos, R.; Ocampo-Pérez, R. Role of 1[O2]* in chlortetracycline degradation by solar radiation assisted by ruthenium metal complexes. Chem. Eng. J. 2016, 284, 896–904. [Google Scholar] [CrossRef]
- Salazar-Rábago, J.J.; Sánchez-Polo, M.; Rivera-Utrilla, J.; Leyva-Ramos, R.; Ocampo-Pérez, R.; Carrasco-Marin, F. Organic xerogels doped with Tris(2,2′-bipyridine) ruthenium (II) as hydroxyl radical promoters: Synthesis, characterization, and photoactivity. Chem. Eng. J. 2016, 306, 289–297. [Google Scholar] [CrossRef]
- Velo-Gala, I.; López-Peñalver, J.J.; Sánchez-Polo, M.; Rivera-Utrilla, J. Role of activated carbon surface chemistry in its photocatalytic activity and the generation of oxidant radicals under UV or solar radiation. Appl. Catal. B Environ. 2017, 207, 412–423. [Google Scholar] [CrossRef]
- Acosta-Rangel, A.; Sánchez-Polo, M.; Polo, A.M.S.; Rivera-Utrilla, J.; Berber-Mendoza, M.S. Tinidazole degradation assisted by solar radiation and iron-doped silica xerogels. Chem. Eng. J. 2018, 344, 21–33. [Google Scholar] [CrossRef]
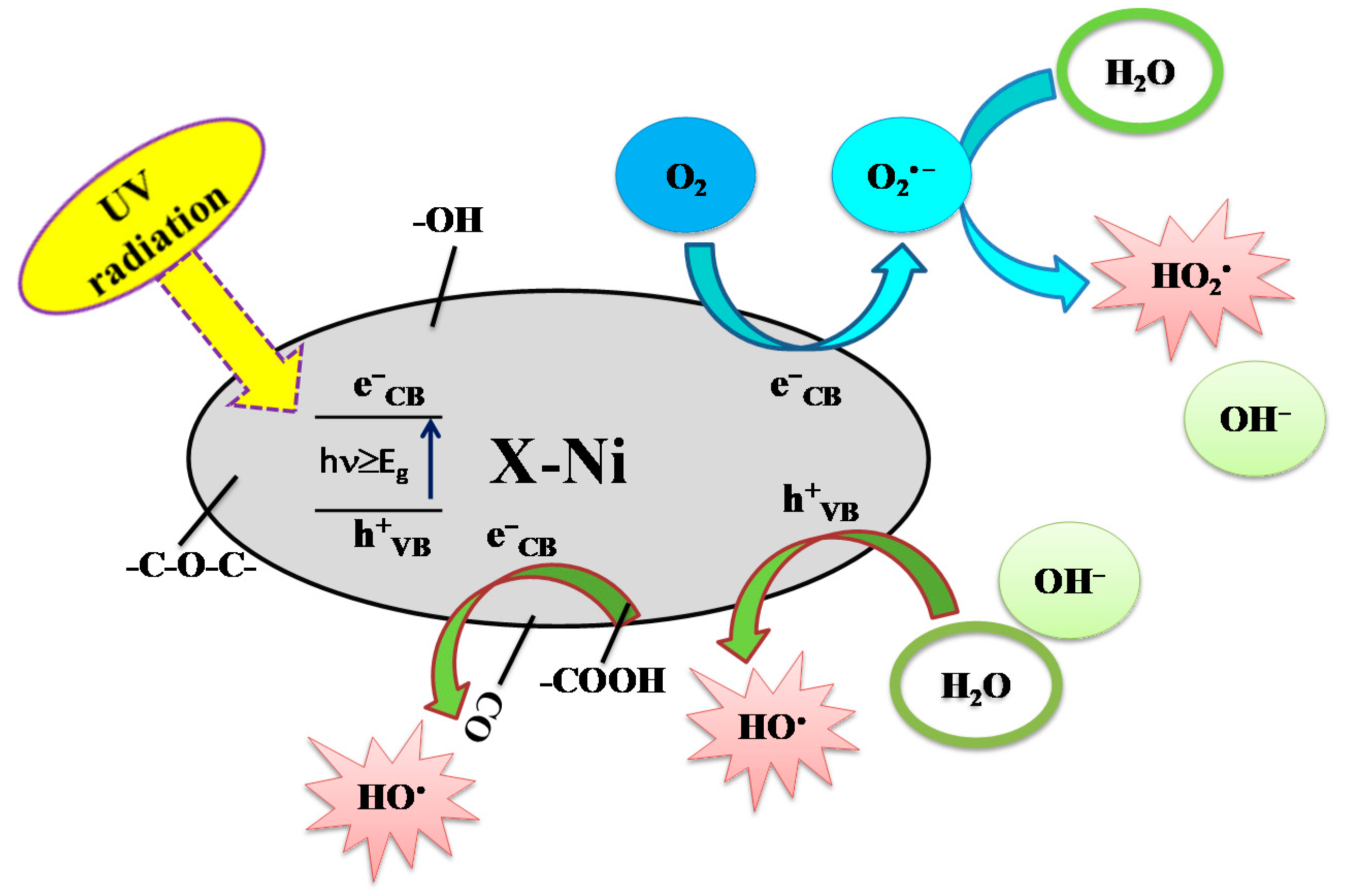
- Álvarez, M.A.; Orellana-García, F.; López-Ramón, M.V.; Rivera-Utrilla, J.; Sánchez-Polo, M. Influence of operational parameters on photocatalytic amitrole degradation using nickel organic xerogel under UV irradiation. Arab. J. Chem. 2018, 11, 564–572. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Costa, J.I.; Rivera-Utrilla, J.; Leyva-Ramos, R.; Sánchez-Polo, M.; Velo-Gala, I. Individual and simultaneous degradation of antibiotics sulfamethoxazole and trimethoprim by UV and solar radiation in aqueous solution using bentonite and vermiculite as photocatalysts. Appl. Clay Sci. 2018, 160, 217–225. [Google Scholar] [CrossRef]
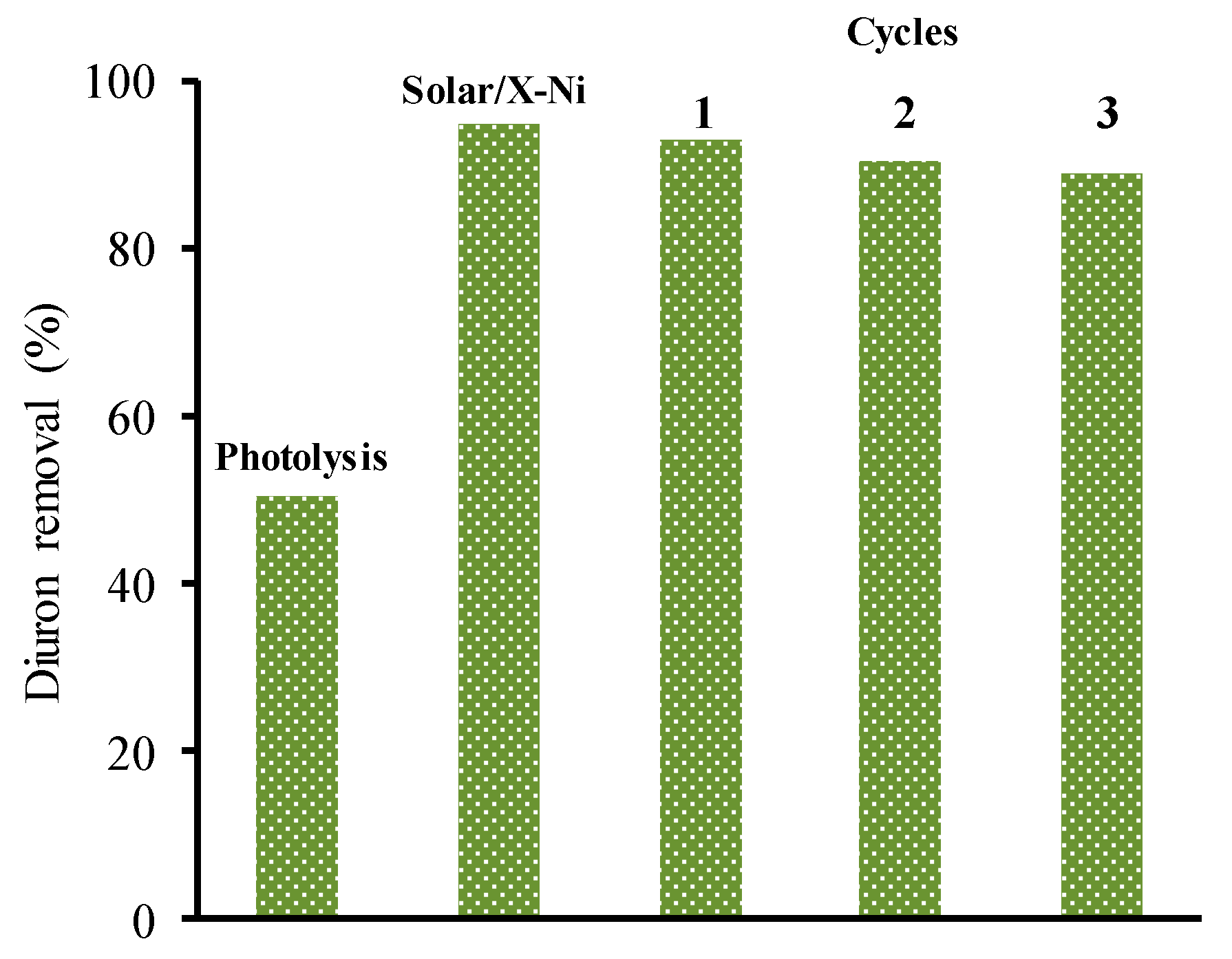
- López-Ramón, M.V.; Rivera-Utrilla, J.; Sánchez-Polo, M.; Polo, A.M.S.; Mota, A.J.; Orellana-García, F.; Álvarez, M.A. Photocatalytic oxidation of diuron using nickel organic xerogel under simulated solar irradiation. Sci. Total Environ. 2019, 650, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Perales, M.; Rozalen, M.; Sánchez-Polo, M.; Rivera-Utrilla, J.; López-Ramón, M.V.; Álvarez, M.A. Solar degradation of sulfamethazine using rGO/Bi composite photocatalysts. Catalysts 2020, 10, 573. [Google Scholar] [CrossRef]
- Ruidíaz-Martínez, M.; Álvarez, M.A.; López-Ramón, M.V.; Cruz-Quesada, G.; Rivera-Utrilla, J.; Sánchez-Polo, M. Hydrothermal synthesis of rGO-TiO2 composites as high-performance UV photocatalysts for ethylparaben degradation. Catalysts 2020, 10, 520. [Google Scholar] [CrossRef]
- Ahmed, S.; Rasul, M.G.; Brown, R.; Hashib, M.A. Influence of parameters on the heterogeneous photocatalytic degradation of pesticides and phenolic contaminants in wastewater: A short review. J. Environ. Manag. 2011, 92, 311–330. [Google Scholar] [CrossRef] [Green Version]
- Ledezma-Estrada, A.; Li, Y.Y.; Wang, A. Biodegradability enhancement of wastewater containing cefalexin by means of the electro-Fenton oxidation process. J. Hazard. Mater. 2012, 227–228, 41–48. [Google Scholar] [CrossRef]
- Sacco, O.; Vaiano, V.; Rizzo, L.; Sannino, D. Photocatalytic activity of a visible light active structured photocatalyst developed for municipal wastewater treatment. J. Clean. Prod. 2018, 175, 38–49. [Google Scholar] [CrossRef]
- Velo-Gala, I.; López-Peñalver, J.J.; Sánchez-Polo, M.; Rivera-Utrilla, J. Surface modifications of activated carbon by gamma radiation. Carbon 2014, 67, 236–249. [Google Scholar] [CrossRef]
- Kralchevska, K.; Milanova, M.; Tsvetkov, M.; Dimitrov, D.; Todorovsky, D. Influence of gamma-irradiation on the photocatalytic activity of Degussa P25 TiO2. J. Mater. Sci. 2012, 47, 4936–4945. [Google Scholar] [CrossRef]
- Tolosana-Moranchel, A.; Casas, J.A.; Carbajo, J.; Faraldos, M.; Bahamonde, A. Influence of TiO2 optical parameters in a slurry photocatalytic reactor: Kinetic modelling. Appl. Catal. B Environ. 2017, 200, 164–173. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/O-) in aqueous solutions. J. Phys. Chem. Ref. Data 1988, 17, 2593–2600. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Qiu, R.; Song, L.; Eric, B.; Mo, Y.; Huang, X. Role of oxygen active species in the photocatalytic degradation of phenol using polymer sintetized TiO2 under visible light irradiation. J. Hazard. Mater. 2009, 163, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Castilla, C.; Maldonado-Hódar, F.J. Carbon aerogels for catalysis applications: An overview. Carbon 2005, 43, 455–465. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Maldonado-Hódar, F.J.; Rivera-Utrilla, J.; Rodríguez-Castellón, E. Group 6 metal oxide-carbon aerogels. Their synthesis, characterization and catalytic activity in the skeletal isomerization of 1-butene. Appl. Catal. A Gen. 1999, 183, 345–356. [Google Scholar] [CrossRef]
- López, R.; Gómez, R. Band-gap energy estimation from diffuse reflectance measurements on sol–gel and commercial TiO2: A comparative study. J. Sol-Gel Sci. Technol. 2012, 61, 1–7. [Google Scholar] [CrossRef]
- Rodríguez-Reinoso, F.; Linares-Solano, A. Microporous structure of activated carbons as revealed by adsorption methods. In Chemistry and Physics of Carbon; Walker, P.L., Ed.; Marcel Dekker: New York, NY, USA, 1989; Volume 21, pp. 146–195. [Google Scholar]
- Zielke, U.; Huttinger, J.; Hoffman, W.P. Surface-oxidized carbon fibers: I. Surface structure and chemistry. Carbon 1994, 34, 983–998. [Google Scholar] [CrossRef]
- Tratnyek, P.G.; Hoigné, J. Photo-oxidation of 2,4,6-trimethylphenol in aqueous laboratory solutions and natural waters: Kinetics of reaction with singlet oxygen. J. Photochem. Photobiol. A Chem. 1994, 84, 153–160. [Google Scholar] [CrossRef]
- Stefan, M.I.; Bolton, J.R. Reinvestigation of the acetone degradation mechanism in dilute aqueous solution by the UV/H2O2 Process. Environ. Sci. Technol. 1999, 33, 870–873. [Google Scholar] [CrossRef]
- Hoigné, J. Inter-calibration of OH radical sources and water quality parameters. Water Sci. Technol. 1997, 35, 1–8. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Maldonado-Hódar, F.J.; Pérez-Cadenas, A. Physicochemical surface properties of Fe, Co, Ni, and Cu-doped monolithic organic aerogels. Langmuir 2003, 19, 5650–5655. [Google Scholar] [CrossRef]
- Czakkel, O.; Marthi, K.; Geissler, E.; Lásló, K. Influence of drying on the morphology of resorcinol-formaldehyde-based carbon gels. Micropor. Mesopor. Mater. 2005, 86, 124–133. [Google Scholar] [CrossRef]
- Milowska, K.Z.; Majewski, J.A. Functionalization of carbon nanotubes with –CHn, –NHn fragments, –COOH and –OH groups. J. Chem. Phys. 2013, 138, 194704–194714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, S.; Stanca, S.E.; Guasaquillo, I.; Thampi, K.R. Photocatalytic degradation of atrazine using suspended and supported TiO2. Appl. Catal. B Environ. 2004, 51, 107–116. [Google Scholar] [CrossRef]
- Fenoll, J.; Hellín, P.; Martínez, C.M.; Flores, P.; Navarro, S. Semiconductor-sensitized photodegradation of s-triazine and chloroacetanilide herbicides in leaching water using TiO2 and ZnO as catalyst under natural sunlight. J. Photochem. Photobiol. A 2012, 238, 81–87. [Google Scholar] [CrossRef]
- Wei, L.; Shifu, C.; Wei, Z.; Sujuan, Z. Titanium dioxide mediated photocatalytic degradation of methamidophos in aqueous phase. J. Hazard. Mater. 2009, 164, 154–160. [Google Scholar] [CrossRef]
- Wu, R.; Chen, C.; Chen, M.; Lu, C. Titanium dioxide-mediated heterogeneous photocatalytic degradation of terbufos: Parameter study and reaction pathways. J. Hazard. Mater. 2009, 162, 945–953. [Google Scholar] [CrossRef]
- Lin, C.; Lin, K. Photocatalytic oxidation of toxic organohalides with TiO2/UV: The effects of humic substances and organic mixtures. Chemosphere 2007, 66, 1872–1877. [Google Scholar] [CrossRef]
- Chen, J.; Wang, D.; Zhu, M.; Gao, C. Photocatalytic degradation of dimethoate using nanosized TiO2 powder. Desalination 2007, 207, 87–94. [Google Scholar] [CrossRef]
- Orellana-García, F.; Álvarez, M.A.; López-Ramón, M.V.; Rivera-Utrilla, J.; Sánchez-Polo, M. Effect of HO•, SO4•− and CO3•−/HCO3• radicals on the photodegradation of the herbicide amitrole by UV radiation in aqueous solution. Chem. Eng. J. 2015, 267, 182–190. [Google Scholar] [CrossRef]
- Wang, H.L.; Liang, W.Z.; Jiang, W.F. Solar photocatalytic degradation of 2-sec-butyl-4,6-dinitrophenol using TiO2/SiO2 aerogel composite photocatalysts. Mater. Chem. Phys. 2011, 130, 1372–1379. [Google Scholar] [CrossRef]
- Shi, L.; Yang, L.; Zhang, H.; Chang, K.; Zhao, G.; Kako, T. Implantation of iron(III) in porphyrinic metal organic frameworks for highly improved photocatalytic performance. Appl. Catal. B Environ. 2018, 224, 60–68. [Google Scholar] [CrossRef]
- de Escobar, C.C.; dos Santos, F.P.; dos Santos, J.H.Z. Effect of the amount and time of addition of a dye template on the adsorption and photocatalytic performance of molecularly imprinted silica. J. Environ. Chem. Eng. 2018, 6, 190–196. [Google Scholar] [CrossRef]
- Rebbouh, L.; Rosso, V.; Renotte, Y.; Lion, Y.; Grandjean, F.; Heinrichs, B.; Pirard, J.-P.; Delwiche, J.; Hubin-Franskin, M.-J.; Long, G.J. The nonlinear optical, magnetic, and Mösbauer spectral properties of some iron (III) doped silica xerogels. J. Mater. Sci. 2006, 41, 2839–2849. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, R.; Li, J.; Li, L.; Lin, S. First-principles study of transition metal doped anatase TiO2. Nanoscale Res. Lett. 2014, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Moradi, V.; Jun, M.B.; Blackburn, A.; Herring, R.A. Significant improvement in visible light photocatalytic activity of Fe doped TiO2 using an acid treatment process. Appl. Surf. Sci. 2018, 427, 791–799. [Google Scholar] [CrossRef]
- Rahmani, H.; Gholami, M.; Mahvi, A.; Alimohammadi, M.; Azarian, G.; Esrafili, A.; Rahmani, K.; Farzadkia, M. Tinidazole removal from aqueous solution by sonolysis in the presence of hydrogen peroxide. Bull. Environ. Contam. Toxicol. 2014, 92, 341–346. [Google Scholar] [CrossRef]
- Mangayayam, M.; Kiwi, J.; Giannakis, S.; Pulgarin, C.; Zivkovic, I.; Magrez, A.; Rtimi, S. FeOx magnetization enhancing E. coli inactivation by orders of magnitude on Ag-TiO2 nanotubes under sunlight. Appl. Catal. B Environ. 2017, 202, 438–445. [Google Scholar] [CrossRef] [Green Version]
- El mehdi Benacherine, M.; Debbache, N.; Ghoul, I.; Mameri, Y. Heterogeneous photoinduced degradation of amoxicillin by Goethite under artificial and natural irradiation. J. Photochem. Photobiol. A 2017, 335, 70–77. [Google Scholar] [CrossRef]
- Ocampo-Gaspar, M.; Cano-Guzmán, C.F.; Payan-Martínez, L.F.; González-Reyes, L.; Hernández-Pérez, I.; Garibay-Febles, V.; Pérez-Orozco, J.P.; Cabrera-Lara, L.I.; Ramón-García, M.I.; Galicia-Luis, L.; et al. Sizing the Fenton’s catalyst. J. Photochem. Photobiol. A 2018, 353, 527–535. [Google Scholar] [CrossRef]
- Schultz, D.M.; Yoon, T.P. Solar synthesis: Prospects in visible light photocatalysis. Science 2014, 343, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyanasundaram, K. Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Coord. Chem. Rev. 1982, 46, 159–244. [Google Scholar] [CrossRef]
- Miskoski, S.; Sanchez, E.; Garavano, M.; Lopez, M.; Soltermann, A.T.; Garcia, N.A. Singlet molecular oxygen-mediated photo-oxidation of tetracyclines: Kinetics, mechanism and microbiological implications. J. Photochem. Photobiol. B Biol. 1998, 43, 164–171. [Google Scholar] [CrossRef]
- Klavarioti, M.; Mantzavinos, D.; Kassinos, D. Removal of residual pharmaceuticals from aqueous systems by advanced oxidation processes. Environ. Int. 2009, 35, 402–417. [Google Scholar] [CrossRef]
- Raizada, P.; Sudhaik, A.; Singh, P. Photocatalytic water decontamination using Graphene and ZnO coupled: A review. Mater. Sci. Technol. 2019, 2, 509–525. [Google Scholar] [CrossRef]
- Upadhyay, R.K.; Soin, N.; Roy, S.S. Role of graphene metal oxide composites as photocatalysts, adsorbents and disinfectants in water treatment: In review. RSC Adv. 2014, 4, 3823–3848. [Google Scholar] [CrossRef]
- Pastrana-Martínez, L.M.; Morales-Torres, S.; Likodimos, V.; Figueiredo, J.L.; Faria, J.L.; Falaras, P.; Silva, A.M.T. Advanced nanostructured photocatalysts based on reduced graphene oxide-TiO2 composites for degradation of diphenhydramine pharmaceutical and methyl orange dye. Appl. Catal. B Environ. 2012, 123–124, 241–256. [Google Scholar] [CrossRef]
- Alamelu, K.; Raja, V.; Shiamala, L.; Jaffar Ali, B.M. Biphasic TiO2 nanoparticles decorated graphene nanosheets for visible light driven photocatalytic degradation of organic dyes. Appl. Surf. Sci. 2018, 430, 145–154. [Google Scholar] [CrossRef]
- Petala, A.; Noe, A.; Frontitis, Z.; Drivas, C.; Kennou, S.; Mantzavinos, D.; Kondarides, D.I. Synthesis and characterization of CoOx/BiVO4 photocatalysts for the degradation of propyl paraben. J. Hazard. Mater. 2019, 372, 198–206. [Google Scholar] [CrossRef]
- Bruny, R.; Bourbigot, M.M.; Doré, M. Oxidation of organic compounds through the combination ozone-hydrogen peroxide. Ozone Sci. Eng. 1985, 7, 241–257. [Google Scholar]
- Beltrán, F.J.; García Araya, J.F.; Acedo, B. Advanced oxidation of atrazine in water. II Ozonation combined with ultraviolet radiation. Water Res. 1994, 28, 2165–2174. [Google Scholar] [CrossRef]
- Logemann, F.P.; Anne, J.H.J. Water treatment with a fixed bed catalytic ozonation process. Water Sci. Technol. 1997, 35, 353–360. [Google Scholar] [CrossRef]
- Jans, U.; Hoigné, J. Activated carbon and carbon black catalyzed transformation of aqueous ozone into oh-radicals. Ozone Sci. Eng. 1998, 20, 67–90. [Google Scholar] [CrossRef]
- Álvarez, P.M.; García-Araya, J.F.; Beltrán, F.J.; Giráldez, I.; Jaramillo, J.; Gómez-Serrano, V. The influence of various factors on aqueous ozone decomposition by granular activated carbons and the development of a mechanistic approach. Carbon 2006, 44, 3102–3112. [Google Scholar] [CrossRef]
- Yeber, M.C.; Rodríguez, J.; Freer, J.; Baeza, J.; Durán, N.; Mansilla, H.D. Toxicity abatement and biodegradability by advanced chemical oxidation. Water. Sci. Technol. 1999, 40, 337–342. [Google Scholar] [CrossRef]
- Staehelin, J.; Hoigne, J. Decomposition of ozone in water in the presence of organic solutes acting as promoters and inhibitors of radical chain reactions. Environ. Sci. Technol. 1985, 19, 1206–1213. [Google Scholar] [CrossRef]
- Morgan, M.E.; Jenkins, R.G.; Walter, P.L. Inorganic constituents in American lignites. Fuel 1981, 60, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Castilla, C.; Carrasco-Marín, F.; Maldonado-Hódar, F.J.; Rivera-Utrilla, J. Effects of non-oxidant and oxidant acid treatments on the surface properties of an activated carbon with very low ash content. Carbon 1998, 36, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Elovitz, M.S.; von Gunten, U. Hydroxyl radical/ozone ratios during ozonation processes. I. The Rct concept. Ozone Sci. Eng. 1999, 21, 239–260. [Google Scholar] [CrossRef]
- von Gunten, U. Ozonation of drinking water: Part II. Disinfection and by-product formation in presence of bromide, iodide or chlorine. Water Res. 2003, 37, 1469–1487. [Google Scholar] [CrossRef]
- Li, W.; Reichenauer, G.; Fricke, J. Carbon aerogels derived from cresol-resorcinol-formaldehyde for supercapacitors. Carbon 2002, 40, 2955–2959. [Google Scholar] [CrossRef]
- Rotter, H.; Landau, M.V.; Carrera, M.; Goldfarb, D.; Herskowitz, M. High surface area chromia aerogel efficient catalyst and catalyst support for ethylacetate combustion. Appl. Catal. B Environ. 2004, 47, 111–126. [Google Scholar] [CrossRef]
- Yao, C.C.D.; Haag, W.R. Rate constants for direct reactions of ozone with several drinking water contaminants. Water Res. 1991, 27, 761–773. [Google Scholar]
- Andreozzi, R.; Insola, A.; Caprio, V.; D’Amore, G. The kinetics of Mn(II)-catalysed ozonation of oxalic acid in aqueous solution. Water Res. 1992, 26, 917–921. [Google Scholar] [CrossRef]
- Ma, J.; Graham, N.J.D. Degradation of atrazine by manganese-catalysed ozonation-influence of radical scavengers. Water Res. 2000, 34, 3822–3828. [Google Scholar] [CrossRef]
- Smit, B.; Maesen, T.L.M. Towards a molecular understanding of shape selectivity. Nature 2008, 451, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Valdés, H.; Farfán, V.J.; Manoli, J.A.; Zaror, C.A. Catalytic ozone aqueous decomposition promoted by natural zeolite and volcanic sand. J. Hazard. Mater. 2009, 165, 915–922. [Google Scholar] [CrossRef]
- Sagehashi, M.; Shiraishi, K.; Fujita, H.; Fujii, T.; Sakoda, A. Adsorptive ozonation of 2-methylisoborneol in natural water with preventing bromate formation. Water Res. 2005, 39, 3900–3908. [Google Scholar] [CrossRef]
- Sano, N.; Yamamoto, T.; Yamamoto, D.; Kim, S.-I.; Eiad-Ua, A.; Shinomiya, H.; Nakaiwa, M. Degradation of aqueous phenol by simultaneous use of ozone with silica-gel and zeolite. Chem. Eng. Process. 2007, 46, 513–519. [Google Scholar] [CrossRef]
- Beltrán, F.J.; García-Araya, J.F.; Álvarez, P.M. Sodium dodecylbenzenesulfonate removal from water and wastewater. 1. Kinetics of decomposition by ozonation. Ind. Eng. Chem. Res. 2000, 39, 2214–2220. [Google Scholar] [CrossRef]
- Von Gunten, U. Ozonation of drinking water: Part I. Oxidation kinetics and product formation. Water Res. 2003, 37, 1443–1467. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, C.; Zeng, G.; Tan, X.; Wang, H.; Huang, D.; Yang, K.; Wei, J.; Ma, C.; Nie, K. Design and engineering of layered double hydroxide based catalysts for water depollution by advanced oxidation processes: A review. J. Mater. Chem. A 2020, 8, 4141–4173. [Google Scholar] [CrossRef]
- Rekhate, C.V.; Srivastava, J.K. Recent advances in ozone-based advanced oxidation processes for treatment of wastewater—A review. Chem. Eng. J. Adv. 2020, 3, 100031. [Google Scholar] [CrossRef]
- Giwa, A.; Yusuf, A.; Balogun, H.A.; Sambudi, N.S.; Bilad, M.R.; Adeyemi, I.; Chakraborty, S.; Curcio, S. Recent advances in advanced oxidation processes for removal of contaminants from water: A comprehensive review. Process. Saf. Environ. 2020, 146, 220–256. [Google Scholar] [CrossRef]
- Tiwaria, S.K.; Sahoo, S.; Wang, N.; Huczko, A. Graphene research and their outputs: Status and prospect. J. Sci. Adv. Mater. 2020, 5, 10–29. [Google Scholar] [CrossRef]
- Karim, A.V.; Selvaraj, A. Graphene composites in photocatalytic oxidation of aqueous organic contaminants—A state of art. Process Saf. Environ. 2020, 146, 136–160. [Google Scholar] [CrossRef]
- Rodrigues, A.F.; Newman, L.; Jasim, D.; Mukherjee, S.P.; Wang, J.; Vacchi, I.A.; Ménard-Moyon, C.; Bianco, A.; Fadeel, B.; Kostarelos, K.; et al. Size-Dependent Pulmonary Impact of Thin Graphene Oxide Sheets in Mice: Toward Safe-by-Design. Adv. Sci. 2020, 7, 1903200. [Google Scholar] [CrossRef]
- Liao, K.H.; Lin, Y.S.; MacOsko, C.W.; Haynes, C.L. Cytotoxicity of graphene oxide and graphene in human erythrocytes and skin fibroblasts. ACS Appl. Mater. Interfaces 2011, 3, 2607–2615. [Google Scholar] [CrossRef]
- Ahmed, F.; Rodrigues, D.F. Investigation of acute effects of graphene oxide on wastewater microbial community: A case study. J. Hazard. Mater. 2013, 256–257, 33–39. [Google Scholar] [CrossRef]
- Kryuchkova, M.; Danilushkina, A.; Lvov, Y.; Fakhrullin, R. Evaluation of toxicity of nanoclays and graphene oxide in vivo: A Paramecium caudatum study. Environ. Sci. Nano 2016, 3, 442–452. [Google Scholar] [CrossRef] [Green Version]
- Bano, Z.; Mazari, S.A.; Saeed, R.M.Y.; Majeed, M.A.; Xia, M.; Memon, A.Q.; Abro, R.; Wang, F. Water decontamination by 3D graphene based materials: A review. J. Water Process. Eng. 2020, 36, 101404. [Google Scholar] [CrossRef]
- Yousefi, N.; Lu, X.; Elimelech, M.; Tufenkji, N. Environmental performance of graphene-based 3D macrostructures. Nat. Nanotechnol. 2019, 14, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Saufi, S.; Ismail, A. Fabrication of carbon membranes for gas separation-a review. Carbon 2004, 42, 241–259. [Google Scholar] [CrossRef]
- Liang, P.; Wei, A.; Zhang, Y.; Wu, J.; Zhang, X.; Li, S. Immobilisation of TiO2 films on activated carbon fibres by a hydrothermal method for photocatalytic degradation of toluene. Micro Nano Lett. 2016, 11, 539–544. [Google Scholar] [CrossRef]
- Stolz, A.; Floch, S.L.; Reinert, L.; Ramos, S.M.M.; Tuaillon-Combes, J.; Soneda, Y.; Chaudet, P.; Baillis, D.; Blanchard, N.; Duclaux, L.; et al. Melamine-derived carbon sponges for oil-water separation. Carbon 2016, 107, 198–208. [Google Scholar] [CrossRef]
- Yang, Y.; Chiang, K.; Burke, N. Porous carbon-supported catalysts for energy and environmental applications: A short review. Catal. Today 2011, 178, 197–205. [Google Scholar] [CrossRef]
- Vottero, E.; Carosso, M.; Jiménez-Ruiz, M.; Pellegrini, R.; Groppo, E.; Piovano, A. How do the graphenic domains terminate in activated carbons and carbon-supported metal catalysts? Carbon 2020, 169, 357–369. [Google Scholar] [CrossRef]
- Raji, M.; Mirbagheri, S.A.; Ye, F.; Dutta, J. Nano zero-valent iron on activated carbon cloth support as Fenton-like catalyst for efficient color and COD removal from melanoidin wastewater. Chemosphere 2020, 263, 127945. [Google Scholar] [CrossRef]
- Malik, S.N.; Ghosh, P.C.; Vaidya, A.N.; Mudliar, S.N. Hybrid ozonation process for industrial wastewater treatment: Principles and applications: A review. J. Water Process Eng. 2020, 35, 101193. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, H.; Wang, F.; Xiong, X.; Tian, K.; Sun, Y.; Yu, T. Application of heterogeneous catalytic ozonation for Refractory Organics in Wastewater. Catalysts 2019, 9, 241. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Park, S.J. Recent advances in preparations and applications of carbon aerogels: A review. Carbon 2020, 163, 1–18. [Google Scholar] [CrossRef]
- Maleki, H.; Hüsing, N. Current status, opportunities and challenges in catalytic and photocatalytic applications of aerogels: Environmental protection aspects. Appl. Catal. B Environ. 2018, 221, 530–555. [Google Scholar] [CrossRef]
- Suh, D.J. Catalytic applications of composite aerogels. J. Non-Cryst. Solids 2004, 350, 314–319. [Google Scholar] [CrossRef]
- Hasanpour, M.; Hatami, M. Photocatalytic performance of aerogels for organic dyes removal from wastewaters: Review study. J. Mol. Liq. 2020, 309, 113094. [Google Scholar] [CrossRef]
- Hu, E.; Shang, S.M.; Tao, X.; Jiang, S.; Chiu, K.L. Regeneration and reuse of highly polluting textile dyeing effluents through catalytic ozonation with carbon aerogel catalysts. J. Clean. Prod. 2016, 137, 1055–1065. [Google Scholar] [CrossRef]
- Xinbo, E.H.; Shang, W.S.; Tao, X.; Jiang, S.; Gan, L. Catalytic ozonation of simulated textile dyeing wastewater using mesoporous carbon aerogel supported copper oxide catalyst. J. Clean. Prod. 2016, 112, 4710–4718. [Google Scholar]
- Jiang, L.; Wang, Q.; Zhou, M.; Liang, L.; Li, K.; Yang, W.; Lu, X.; Zhang, Y. Role of adsorption and oxidation in porous carbon aerogel/persulfate system for non-radical degradation of organic contaminant. Chemosphere 2020, 241, 125066. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, Y.; Zhou, M.; Liang, L.; Li, K. Oxidation of Rhodamine B by persulfate activated with porous carbon aerogel through a non-radical mechanism. J. Hazard. Mater. 2018, 358, 53–61. [Google Scholar] [CrossRef]
- Rashid, T.; Iqbal, D.; Hazafa, A.; Hussain, S.; Sher, F.; Sher, F. Formulation of zeolite supported nano-metallic catalyst and applications in textile effluent treatment. J. Environ. Chem. Eng. 2020, 8, 104023. [Google Scholar] [CrossRef]
- Gonzalez-Olmos, R.; Holzer, F.; Kopinke, F.D.; Georgi, A. Indications of the reactive species in a heterogeneous Fenton-like reaction using Fe-containing zeolites. Appl. Catal. A 2011, 398, 44–53. [Google Scholar] [CrossRef]
- Bandala, E.R.; Sadek, R.; Gurgul, J.; Łątka, K.; Zimowska, M.; Valentin, L.; Rodriguez-Narvaez, O.M.; Dzwigaj, S. Assessment of the capability of Fe and Al modified BEA zeolites to promote advanced oxidation processes in aqueous phase. Chem. Eng. J. 2020. [Google Scholar] [CrossRef]
- Ismail, A.A.; Bahnemann, D.W. Photochemical splitting of water for hydrogen production by photocatalysis: A review. Sol. Energy Mat. Sol. C 2014, 128, 85–101. [Google Scholar] [CrossRef]
- Koyyada, G.; Pilli, N.S.; Jung, J.H.; Mandari, K.K.; Shanigaram, B.; Chandrasekharam, M. Shining light on panchromatic ruthenium sensitizers towards dye-sensitized photocatalytic hydrogen evolution. Int. J. Hydrog. Energy 2018, 43, 6963–6976. [Google Scholar] [CrossRef]
- Luis, E.T.; Iranmanesh, H.; Beves, J.E. Photosubstitution reactions in ruthenium(II) trisdiimine complexes: Implications for photoredox catalysis. Polyhedron 2019, 160, 1–9. [Google Scholar] [CrossRef]
- Bolobajev, J.; Kask, M.; Kreek, K.; Kulp, M.; Koel, M.; Goi, A. Metal-doped organic aerogels for photocatalytic degradation of trimethoprim. Chem. Eng. J. 2019, 357, 120–128. [Google Scholar] [CrossRef]
- Justh, N.; Mikula, G.J.; Bakos, L.P.; Nagy, B.; László, K.; Parditka, B.; Erdélyi, Z.; Takáts, V.; Mizsei, J.; Szilágyi, I.M. Photocatalytic properties of TiO2@polymer and TiO2@carbon aerogel composites prepared by atomic layer deposition. Carbon 2019, 147, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Bakos, L.P.; Mensah, J.; Lászlo, K.; Parditka, B.; Erdélyi, Z.; Székely, E.; Lukács, I.; Kónya, Z.; Cserháti, C.; Zhou, C.; et al. Nitrogen doped carbon aerogel composites with TiO2 and ZnO prepared by atomic layer deposition. J. Mater. Chem. C 2020, 8, 6891–6899. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Hu, H.; Yang, S.; Shanmugam, P.; Wei, W.; Selvaraj, M.; Xie, J. ZnS@carbonaceous aerogel composites fabricated in production of hydrogen and for removal of organic pollutants. J. Mater. Sci. Mater. Electron. 2018, 29, 8523–8534. [Google Scholar] [CrossRef]
- Maicaneanu, S.A.; McGhee, B.; Stefan, R.; Barbu-Tudoran, L.; Sedwick, C.; Lake, C.H. Investigations on Cationic Dye Degradation using iron-doped carbon xerogel. Chem. Eng. 2019, 3, 61. [Google Scholar] [CrossRef] [Green Version]
- De Moraes, N.P.; Valim, R.B.; da Silva Rocha, R.; da Silva, M.L.C.P.; Campos, T.M.B.; Thim, G.P.; Rodrigues, L.A. Effect of synthesis medium on structural and photocatalytic properties of ZnO/carbon xerogel composites for solar and visible light degradation of 4-chlorophenol and bisphenol A. Colloids Surf. A Physicochem. Eng. 2020, 584, 124034. [Google Scholar] [CrossRef]
- Metheniti, M.E.; Frontistis, Z.; Ribeiro, R.S.; Silva, A.M.T.; Faria, J.L.; Gomes, H.T.; Mantzavinos, D. Degradation of propyl paraben by activated persulfate using iron-containing magnetic carbon xerogels: Investigation of water matrix and process synergy effects. Environ. Sci. Pollut. Res. 2018, 25, 34801–34810. [Google Scholar] [CrossRef] [PubMed]
- Bailón-García, E.; Elmouwahidi, A.; Carrasco-Marín, F.; Pérez-Cadenas, A.F.; Maldonado-Hódar, F.J. Development of Carbon-ZrO2 composites with high performance as visible-light photocatalysts. Appl. Catal. B Environ. 2017, 217, 540–550. [Google Scholar] [CrossRef]
- Fathy, N.A.; El-Khouly, S.M.; Hassan, N.A.; Awad, M.S. Free- and Ni-doped carbon xerogels catalysts for wet peroxide oxidation of methyl orange. J. Water Process Eng. 2017, 16, 21–27. [Google Scholar] [CrossRef]
- Lima, L.F.d.S.; Coelho, C.R.; Gomes, G.H.M.; Mohallem, N.D.S. Nb2O5/SiO2 mesoporous monoliths synthetized by sol-gel process using ammonium niobate oxalate hydrate as porogenic agent. J. Sol-Gel Sci. Technol. 2020, 93, 168–174. [Google Scholar] [CrossRef]
- Peter, A.; Mihaly-Cozmuta, A.; Nicula, C.; Mihaly-Cozmuta, L.; Jastrzębska, A.; Olszyna, A.; Baia, L. UV light-assisted degradation of methyl orange, methylene blue, phenol, salicylic acid, and rhodamine B: Photolysis versus Photocatalyis. Water Air Soil Pollut. 2017, 228, 41–53. [Google Scholar] [CrossRef]
- Mahy, J.G.; Hermans, S.; Tilkin, R.G.; Lamber, S.D. Influence of nucleating agent addition on the textural and photo-Fenton properties of Fe(III)/SiO2 catalysts. J. Phys. Chem. Solids 2020, 144, 109502. [Google Scholar] [CrossRef]
- De Oliveira Pereira, L.; Marques Sales, I.; Pereira Zampiere, L.; Vieira, S.S.; Guimarães, I.R.; Magalhães, F. Preparation of magnetic photocatalysts from TiO2, activated carbon and iron nitrate for environmental remediation. J. Photochem. Photobiol. A 2019, 382, 111907. [Google Scholar] [CrossRef]
- Baeza, P.; Aballay, P.; Matus, C.; Camú, E.; Ramirez, M.F.; Eyzaguirre, J.; Ojeda, J. Degradation of Paracetamol Adsorbed on Inorganic Supports Under UV Irradiation. Water Air Soil Pollut. 2019, 230, 34. [Google Scholar] [CrossRef]
- Matos, J.; Poon, P.S.; Montaña, R.; Romero, R.; Gonçalves, G.R.; Schettino, M.A., Jr.; Passamani, E.C.; Freitas, J.C.C. Photocatalytic activity of P-Fe/activated carbon nanocomposites under artificial solar irradiation. Catal. Today 2020, 356, 226–240. [Google Scholar] [CrossRef]
- Osawa, R.A.; Barrocas, B.T.; Monteiro, O.C.; Oliveira, M.C.; Florêncio, M.H. Photocatalytic degradation of cyclophosphamide and ifosfamide: Effects of wastewater matrix, transformation products and in silico toxicity prediction. Sci. Total Environ. 2019, 692, 503–510. [Google Scholar] [CrossRef]
- Akshatha, S.; Sreenivasa, S.; Kumar, K.Y.; Archana, S.; Prashanth, M.K.; Prasanna, B.P.; Chakraborty, P.; Krishnaiah, P.; Raghu, M.S.; Alrobei, H. Rutile, mesoporous ruthenium oxide decorated graphene oxide as an efficient visible light driven photocatalyst for hydrogen evolution reaction and organic pollutant degradation. Mat. Sci. Semicon. Proc. 2020, 116, 105156. [Google Scholar] [CrossRef]
- Pérez-Molina, A.; Morales-Torres, S.; Maldonado-Hódar, F.J.; Pastrana-Martínez, M.L. Functionalized graphene derivatives and TiO2 for high visible light photodegradation of azo dyes. Nanomaterials 2020, 10, 1106. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.; Gomez, C.; Duran-Valle, C.J.; Pastrana-Martínez, L.M.; Faria, J.L.; Silva, A.M.T.; Faraldos, M.; Bahamonde, A. bare TiO2 and graphene oxide TiO2 photocatalysts on the degradation of selected pesticides and influence of the water matrix. Appl. Surf. Sci. 2017, 416, 1013–1021. [Google Scholar] [CrossRef]
- Zouzelka, R.; Remzova, M.; Plsek, J.; Brabec, L.; Rathousky, J. Immobilized rGO/TiO2 photocatalyst for decontamination of water. Catalysts 2019, 9, 708. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Ma, X.; Ma, J.; Zhou, Y.; Liu, G.; Ma, D.; Deng, Z.; Luo, M.; Xin, Y. Fabrication of rGO and g-C3N4 co-modified TiO2 nanotube arrays photoelectrodes with enhanced photocatalytic performance. J. Colloid Interface Sci. 2020, 577, 75–85. [Google Scholar] [CrossRef]
- Shen, H.; Wang, J.; Jiang, J.; Luo, B.; Mao, B.; Shi, W. All-solid-state Z-scheme system of RGO-Cu2O/Bi2O3 for tetracycline degradation under visible-light irradiation. Chem. Eng. J. 2017, 313, 508–517. [Google Scholar] [CrossRef]
- Suresh, M.; Sivasamy, A. Bismuth oxide nanoparticles decorated graphene layers for the degradation of Methylene blue dye under visible light irradiations and antimicrobial activities. J. Environ. Chem. Eng. 2018, 6, 3745–3756. [Google Scholar] [CrossRef]
- Yang, J.; Xie, T.P.; Liu, C.L.; Xu, L.J. Facile fabrication of dumbbell-like beta-Bi2O3/graphene nanocomposites and their highly efficient photocatalytic activity. Materials 2018, 11, 1359. [Google Scholar] [CrossRef] [Green Version]
- Kakavandi, B.; Bahari, N.; Kalantary, R.R.; Fard, E.D. Enhanced sono-photocatalysis of tetracycline antibiotic using TiO2 decorated on magnetic activated carbon, MAC@T, coupled with US and UV: A new hybrid system. Ultrason. Sonochem. 2019, 55, 75–85. [Google Scholar] [CrossRef]
- Hamad, H.; Bailón-García, E.; Morales-Torres, S.; Carrasco-Marín, F.; Pérez-Cadenas, A.; Maldonado-Hódar, F.J. Functionalized cellulose for the controlled synthesis of novel carbon–Ti nanocomposites: Physicochemical and photocatalytic properties. Nanomaterials 2020, 10, 729. [Google Scholar] [CrossRef] [Green Version]
- El Mouchtari, E.M.; Daou, C.; Rafqah, S.; Najjar, F.; Anane, H.; Piram, A.; Hamade, A.; Briche, S.; Wong-Wah-Chung, P. TiO2 and activated carbon of Argania Spinosa tree nutshells composites for the adsorption photocatalysis removal of pharmaceuticals from aqueous solution. J. Photochem. Photobiol. A Chem. 2020, 388, 112183. [Google Scholar] [CrossRef]
- Ali, S.; Li, Z.; Chen, S.; Zada, A.; Khan, I.; Khan, I.; Ali, W.; Shaheen, S.; Qu, Y.; Jing, L. Synthesis of activated carbon-supported TiO2-based nano-photocatalysts with well recycling for efficiently degrading high-concentration pollutants. Catal. Today 2019, 335, 557–564. [Google Scholar] [CrossRef]
- Shokouhi, S.B.; Dehghanzadeh, R.; Aslani, H.; Shahmahdi, N. Activated carbon catalyzed ozonation (ACCO) of Reactive Blue 194 azo dye in aqueous saline solution: Experimental parameters, kinetic and analysis of activated carbon properties. J. Water Process Eng. 2020, 35, 101188. [Google Scholar] [CrossRef]
- Rajah, Z.; Guiza, M.; Solís, R.R.; Rivas, F.J.; Ouederni, A. Catalytic and photocatalytic ozonation with activated carbon as technologies in the removal of aqueous micropollutants. J. Photochem. Photobiol. A Chem. 2019, 382, 111961. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, G.Q.; Liu, C.; Zhang, R.N.; Chen, X.X.; Zhang, L. Synergistic effect of microbubbles and activated carbon on the ozonation treatment of synthetic dyeing wastewater. Sep. Purif. Technol. 2018, 201, 10–18. [Google Scholar] [CrossRef]
- Vatankhah, H.; Riley, M.S.; Murray, C.; Quiñones, O.; Steirer, K.X.; Dickenson, E.R.V.; Bellona, C. Simultaneous ozone and granular activated carbon for advanced treatment of micropollutants in municipal wastewater effluent. Chemosphere 2019, 234, 845–854. [Google Scholar] [CrossRef]
- Rozas, O.; Baeza, C.; Núñez, K.; Rossner, A.; Urrutia, R.; Mansilla, H.D. Organic micropollutants (OMPs) oxidation by ozone: Effect of activated carbon on toxicity abatement. Sci. Total Environ. 2017, 590–591, 430–439. [Google Scholar] [CrossRef]
- Hu, E.; Shang, S.M.; Tao, X.M.; Jiang, S.X.; Chiu, K.L. Minimizing freshwater consumption in the wash-off step in textile reactive dyeing by catalytic ozonation with carbon aerogel hosted bimetallic catalyst. Polymers 2018, 10, 193. [Google Scholar] [CrossRef] [Green Version]
- Hu, E.; Shang, S.; Chiu, K.L. Removal of reactive dyes in textile effluents by catalytic ozonation pursuing on-site effluent recycling. Molecules 2019, 24, 2755. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xiong, Z.; Wei, J.; Song, Y.; Ren, Y.; Xu, D.; Lai, B. Catalytic ozonation of penicillin G using cerium-loaded natural zeolite (CZ): Efficacy, mechanisms, pathways and toxicity assessment. Chem. Eng. J. 2020, 383, 123144. [Google Scholar] [CrossRef]
- Chen, C.; Yan, X.; Yoza, B.A.; Zhou, T.; Li, Y.; Zhan, Y.; Wang, Q.; Li, Q.X. Efficiencies and mechanisms of ZSM5 zeolites loaded with cerium, iron, or manganese oxides for catalytic ozonation of nitrobenzene in water. Sci. Total Environ. 2018, 612, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Ikhlaq, A.; Waheed, S.; Joya, K.S.; Kazmi, M. Catalytic ozonation of paracetamol on zeolite A: Non-radical mechanism. Catal. Commun. 2018, 112, 15–20. [Google Scholar] [CrossRef]
- Saeid, S.; Tolvanen, P.; Kumar, N.; Eränen, K.; Peltonen, J.; Peurla, M.; Mikkola, J.P.; Franz, A.; Salmi, T. Advanced oxidation process for the removal of ibuprofen from aqueous solution: A non-catalytic and catalytic ozonation study in a semi-batch reactor. Appl. Catal. B Environ. 2018, 230, 77–90. [Google Scholar] [CrossRef]
- Derco, J.; Dudáš, J.; Valičková, M.; Šimovičová, K.; Kecskés, J. Removal of micropollutants by ozone based processes. Chem. Eng. Process. 2015, 94, 78–84. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
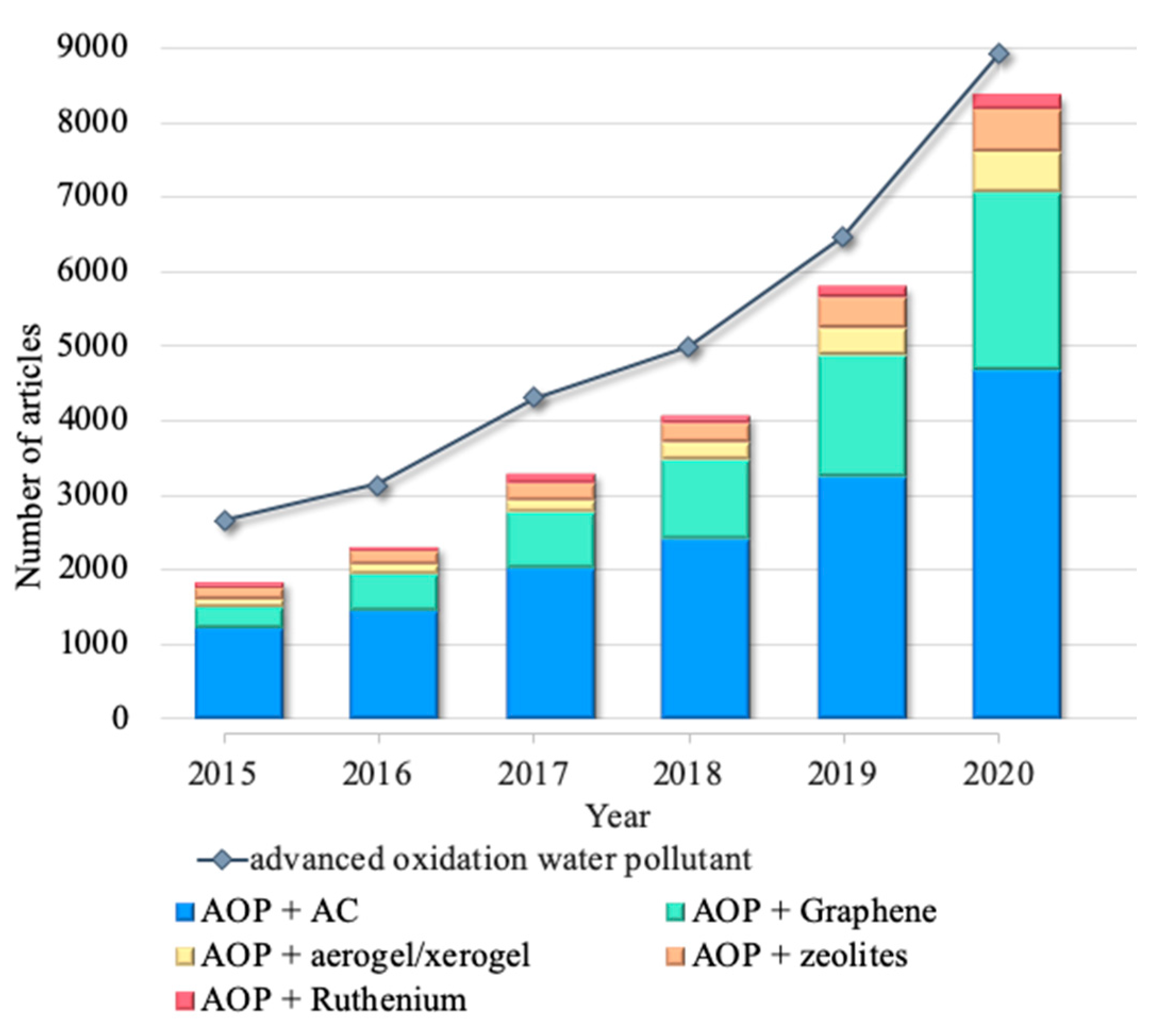{kind=link}
| Non-Photochemical Processes | Photochemical Processes |
|---|---|
|
|
| |
|
|
|
|
| |
| |
| |
|
| Ceca series | C | Original Ceca carbon |
| C-H• | Ceca carbon irradiated in the presence of H• radical | |
| C-e−aq | Ceca carbon irradiated in the presence of | |
| C-HO• | Ceca carbon irradiated in the presence of HO• radical | |
| C-0 | Ceca carbon irradiated in the presence of all three radicals | |
| C-a | Ceca carbon irradiated in the air (without water) | |
| Merck series | M | Original Merck carbon |
| M-H• | Merck carbon irradiated in the presence of H• radical | |
| M-e−aq | Merck carbon irradiated in the presence of | |
| M-HO• | Merck carbon irradiated in the presence of HO• radical | |
| M-0 | Merck carbon irradiated in the presence of all three radicals | |
| M-a | Merck carbon irradiated in the air (without water) | |
| Sorbo series | S | Original Sorbo carbon |
| S-H• | Sorbo carbon irradiated in the presence of H• radical | |
| S-e−aq | Sorbo carbon irradiated in the presence of | |
| S-HO• | Sorbo carbon irradiated in the presence of HO• radical | |
| S-0 | Sorbo carbon irradiated in the presence of all three radicals | |
| S-a | Sorbo carbon irradiated in the air (without water) | |
| Witco series | W | Original Witco carbon |
| W-H• | Witco carbon irradiated in the presence of H• radical | |
| W-e−aq | Witco carbon irradiated in the presence of | |
| W-HO• | Witco carbon irradiated in the presence of HO• radical | |
| W-0 | Witco carbon irradiated in the presence of all three radicals | |
| W-a | Witco carbon irradiated in the air (without water) |
| Activated | SBET a (N2) | VT b (N2) | DP c (N2) | V0 d (N2) | SExt e (N2) | V0 f (CO2) |
|---|---|---|---|---|---|---|
| Carbon | m2/g | cm3/g | nm | cm3/g | m2/g | cm3/g |
| C | 1294 | 0.65 | 2.02 | 0.55 | 82.68 | 0.34 |
| C-A | 1256 | 0.64 | 2.03 | 0.54 | 71.97 | 0.33 |
| C-0 | 1248 | 0.63 | 2.04 | 0.53 | 73.78 | 0.33 |
| M | 1302 | 0.66 | 2.02 | 0.55 | 84.00 | 0.39 |
| M-A | 1286 | 0.64 | 1.99 | 0.54 | 75.54 | 0.38 |
| M-0 | 1278 | 0.63 | 1.97 | 0.54 | 71.16 | 0.38 |
| S | 1143 | 0.57 | 2.02 | 0.49 | 53.84 | 0.29 |
| S-A | 1049 | 0.52 | 2.00 | 0.45 | 47.48 | 0.27 |
| S-0 | 1031 | 0.52 | 2.03 | 0.44 | 53.22 | 0.28 |
| W | 815 | 0.40 | 1.97 | 0.35 | 20.39 | 0.26 |
| W-A | 798 | 0.39 | 1.97 | 0.35 | 23.06 | 0.25 |
| W-0 | 794 | 0.39 | 1.97 | 0.35 | 20.14 | 0.24 |
| ACs | pHPZC | Acidic Groups a | Basic Groups b | Eg c |
|---|---|---|---|---|
| μeq/g | μeq/g | eV | ||
| C | 7.5 ± 0.2 | 240 | 409 | 3.65 ± 0.03 |
| C-A | 8.7 ± 0.2 | 107 | 657 | 3.25 ± 0.04 |
| C-0 | 7.7 ± 0.2 | 246 | 462 | 3.04 ± 0.02 |
| C-H• | 3.1 ± 0.2 | 488 | 158 | 3.36 ± 0.02 |
| C-e−aq | 7.2 ± 0.2 | 166 | 467 | 3.14 ± 0.02 |
| C-HO• | 7.7 ± 0.2 | 226 | 500 | 3.00 ± 0.02 |
| M | 10.0 ± 0.2 | 72 | 650 | 3.50 ± 0.02 |
| M-A | 10.7 ± 0.2 | 90 | 736 | 3.15 ± 0.04 |
| M-0 | 9.3 ± 0.1 | 105 | 468 | 3.33 ± 0.02 |
| M-H• | 4.2 ± 0.2 | 453 | 150 | 3.13 ± 0.02 |
| M-e−aq | 8.4 ± 0.2 | 68 | 389 | 3.20 ± 0.02 |
| M-HO• | 9.0 ± 0.2 | 48 | 431 | 3.23 ± 0.02 |
| S | 10.7 ± 0.1 | 108 | 1857 | 3.58 ± 0.02 |
| S-A | 11.9 ± 0.2 | 48 | 2376 | 3.28 ± 0.02 |
| S-0 | 10.0 ± 0.2 | 87 | 1052 | 2.98 ± 0.02 |
| S-H• | 4.4 ± 0.2 | 333 | 433 | 3.63 ± 0.02 |
| S-e−aq | 9.9 ± 0.2 | 68 | 968 | 3.16 ± 0.02 |
| S-HO• | 10.1 ± 0.2 | 88 | 1331 | 2.92 ± 0.02 |
| W | 8.4 ± 0.2 | 188 | 403 | 3.68 ± 0.02 |
| W-A | 9.2 ± 0.2 | 88 | 622 | 3.50 ± 0.04 |
| W-0 | 7.1 ± 0.1 | 128 | 340 | 3.23 ± 0.02 |
| W-H• | 4.7 ± 0.1 | 448 | 186 | 3.35 ± 0.02 |
| W-e−aq | 9.1 ± 0.2 | 100 | 558 | 3.15 ± 0.02 |
| W-HO• | 9.6 ± 0.1 | 50 | 688 | 3.10 ± 0.02 |
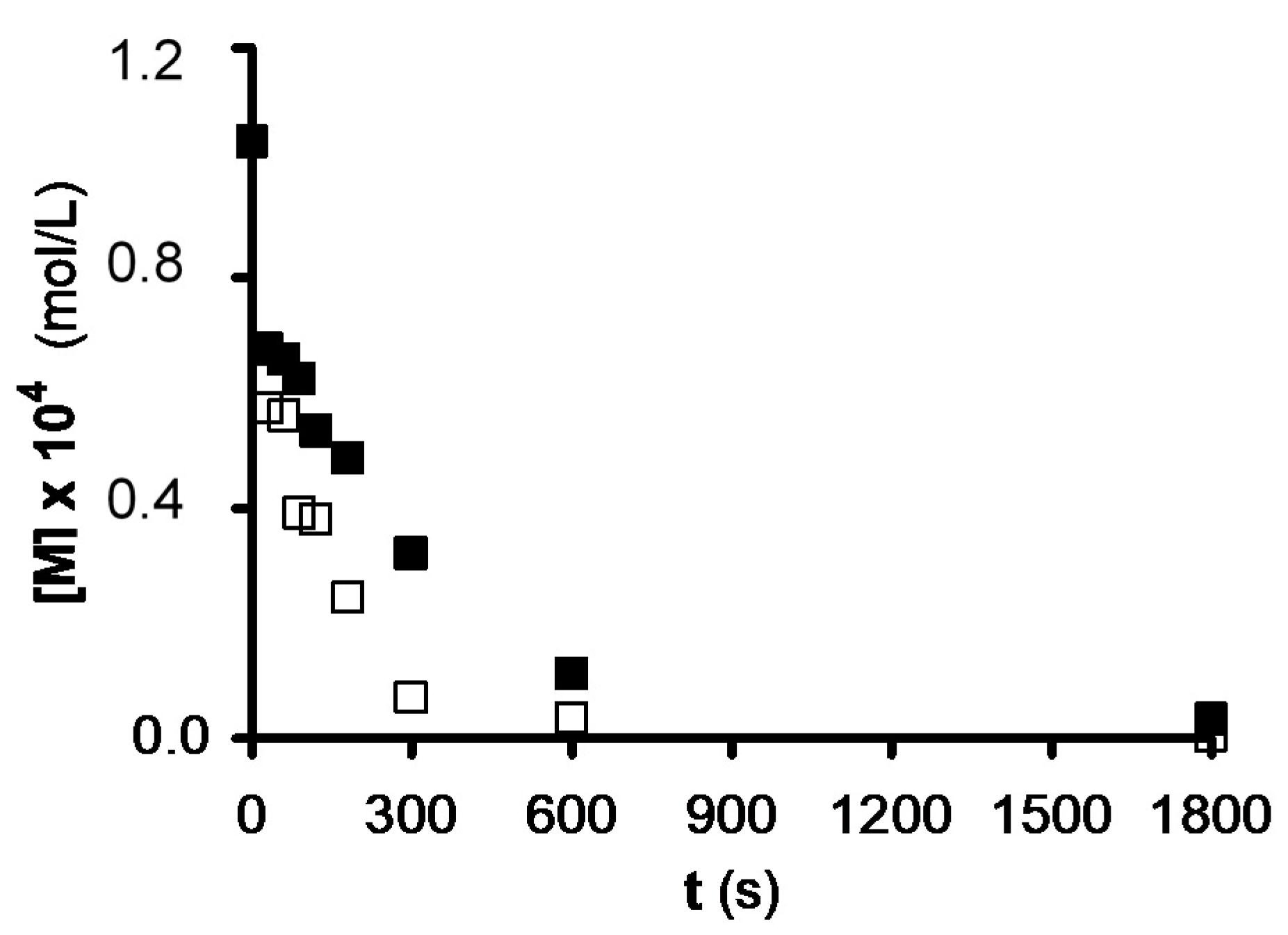
| Activated Carbon | kOB (15 min) | %UV Degradation | %Removal by Adsorption | %Removal by UV/AC | %Synergic Removal |
|---|---|---|---|---|---|
| min−1 | (1 min) | (1 min) | (1 min) | (1 min) | |
| C | 0.47 ± 0.01 | 9.29 | 14.20 | 51.50 | 28.01 |
| C-H• | 0.75 ± 0.04 | 9.29 | 11.70 | 49.79 | 28.80 |
| C-e−aq | 0.69 ± 0.03 | 9.29 | 11.60 | 44.61 | 23.72 |
| C-HO• | 1.05 ± 0.05 | 9.29 | 17.00 | 53.99 | 27.70 |
| C-0 | 2.05 ± 0.00 | 9.29 | 12.90 | 87.09 | 64.90 |
| M | 0.53 ± 0.04 | 9.29 | 18.70 | 66.60 | 38.61 |
| M-H• | 1.06 ± 0.02 | 9.29 | 30.40 | 65.38 | 25.69 |
| M-e−aq | 1.05 ± 0.04 | 9.29 | 0.86 | 65.04 | 54.89 |
| M-HO• | 0.93 ± 0.05 | 9.29 | 17.00 | 62.36 | 36.07 |
| M-0 | 0.69 ± 0.04 | 9.29 | 12.40 | 53.05 | 31.36 |
| S | 0.59 ± 0.03 | 9.29 | 28.80 | 71.00 | 32.91 |
| S-H• | 0.42 ± 0.05 | 9.29 | 8.49 | 45.00 | 27.22 |
| S-e−aq | 0.28 ± 0.02 | 9.29 | 9.07 | 32.49 | 14.13 |
| S-HO• | 1.03 ± 0.04 | 9.29 | 14.80 | 64.10 | 39.91 |
| S-0 | 1.07 ± 0.02 | 9.29 | 7.67 | 47.00 | 30.04 |
| W | 1.42 ± 0.04 | 9.29 | 12.80 | 75.20 | 53.11 |
| W-H• | 1.02 ± 0.04 | 9.29 | 1.04 | 69.47 | 59.14 |
| W-e−aq | 5.06 ± 0.06 | 9.29 | 1.02 | 100 | 89.69 |
| W-HO• | 5.26 ± 0.06 | 9.29 | 1.67 | 92.81 | 81.85 |
| W-0 | 0.99 ± 0.01 | 9.29 | 8.23 | 62.87 | 45.35 |
| Sample | SBET a | W0 b | Vm c | SCO2 d | Vmic e | V2 f | V3 g | pHPZC | Eg |
|---|---|---|---|---|---|---|---|---|---|
| m2/g | cm3/g | m2/g | cm3/g | eV | |||||
| A | 500 | - | - | 200 | 0.071 | 0.36 | 0.68 | 3.5 | - |
| A-Co(II)-15 | 562 | - | - | 206 | 0.073 | 0.43 | 0.97 | 3.8 | - |
| A-Ti(IV)-15 | 550 | - | - | 200 | 0.071 | 0.40 | 0.92 | 4.3 | - |
| A-Mn(II)-15 | 554 | - | - | 210 | 0.075 | 0.41 | 0.95 | 4.2 | - |
| A-Mn(II)-50 | 593 | - | - | 285 | 0.101 | 0.35 | 0.90 | 3.7 | - |
| A-Mn(11)-200 | 646 | - | - | 321 | 0.114 | 0.28 | 0.86 | 3.2 | - |
| A-Mn(II)-15C | 880 | - | - | 701 | 0.250 | 0.43 | 0.86 | 7.5 | - |
| X-Na | 2 | 0.002 | 0.030 | - | - | - | - | 3.6 | 3.81 |
| X-Co | 68 | 0.031 | 0.194 | - | - | - | - | 3.6 | 3.78 |
| X-Fe | 27 | 0.012 | 0.089 | - | - | - | - | 3.5 | 3.74 |
| X-Ni | 103 | 0.040 | 0.295 | - | - | - | - | 3.4 | 3.66 |
| A-Fe | 388 | 0.173 | 0.678 | - | - | - | - | 3.1 | 3.75 |
| A-Ni | 407 | 0.185 | 0.717 | - | - | - | - | 3.6 | 3.69 |
| XC-Fe | 338 | 0.134 | 0.225 | - | - | - | - | 7.3 | 4.35 |
| XC-Ni | 382 | 0.144 | 0.230 | - | - | - | - | 7.0 | 4.16 |
| AC-Fe | 568 | 0.221 | 0.396 | - | - | - | - | 7.1 | 4.47 |
| AC-Ni | 637 | 0.249 | 0.371 | - | - | - | - | 6.9 | 4.44 |
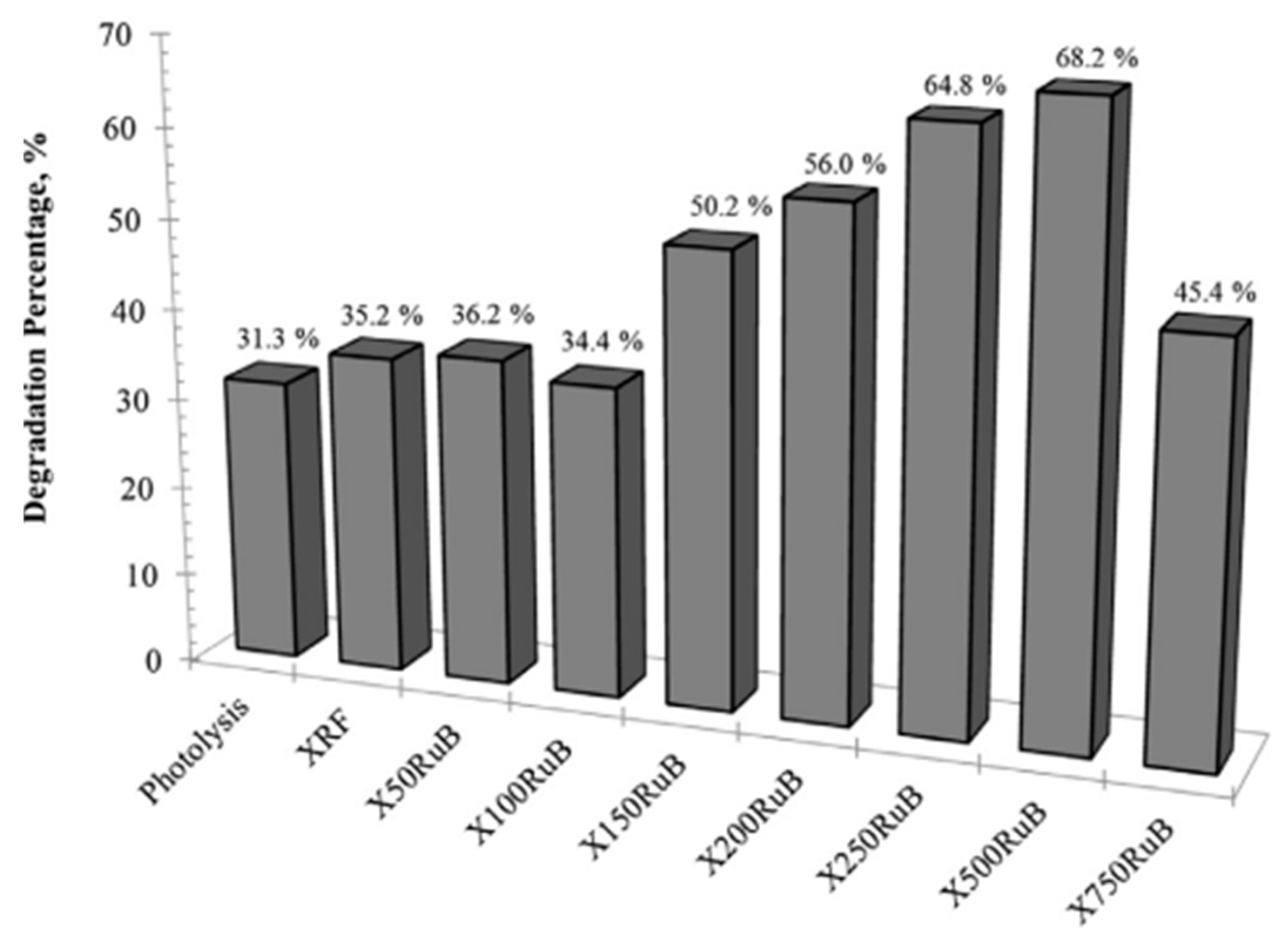
| XRF | <5 | - | - | 264 | 0.106 | - | - | - | 5.68 |
| X50RuB | <5 | - | - | 204 | 0.082 | - | - | - | - |
| X100RuB | <5 | - | - | 241 | 0.097 | - | - | - | - |
| X150RuB | <5 | - | - | 253 | 0.102 | - | - | - | 0.98 |
| X750RuB | <5 | - | - | 245 | 0.103 | - | - | - | - |
| Sample | O | –C=O/–OH | –C–O–C– | H2O |
|---|---|---|---|---|
| % | 532.2 ± 0.2 eV | 533.1 ± 0.2 eV | 535.1 ± 0.4 eV | |
| X-Na | 37.4 | 16.6 | 65.3 | 18.1 |
| X-Co | 37.6 | 18.0 | 66.2 | 15.8 |
| X-Fe | 38.1 | 20.4 | 60.1 | 19.5 |
| X-Ni | 32.0 | 24.2(28.4 a, 29.6 b) | 51.2(49.2 a, 51.9 b) | 24.6(22.4 a, 18.5 b) |
| XC-Fe | 3.9 | 7.9 | 38.0 | 54.1 |
| XC-Ni | 7.9 | 9.6 | 36.6 | 53.8 |
| A-Fe | 36.4 | 19.9 | 51.1 | 29.0 |
| A-Ni | 33.9 | 22.6 | 46.4 | 31.0 |
| AC-Fe | 9.7 | 10.3 | 30.8 | 58.9 |
| AC-Ni | 8.5 | 11.7 | 33.7 | 54.6 |
| Sample | Surface Area | Pore Volume | Average Pore |
|---|---|---|---|
| SBET (m2/g) | Vp (cm3/g) | Diameter, Dp (nm) | |
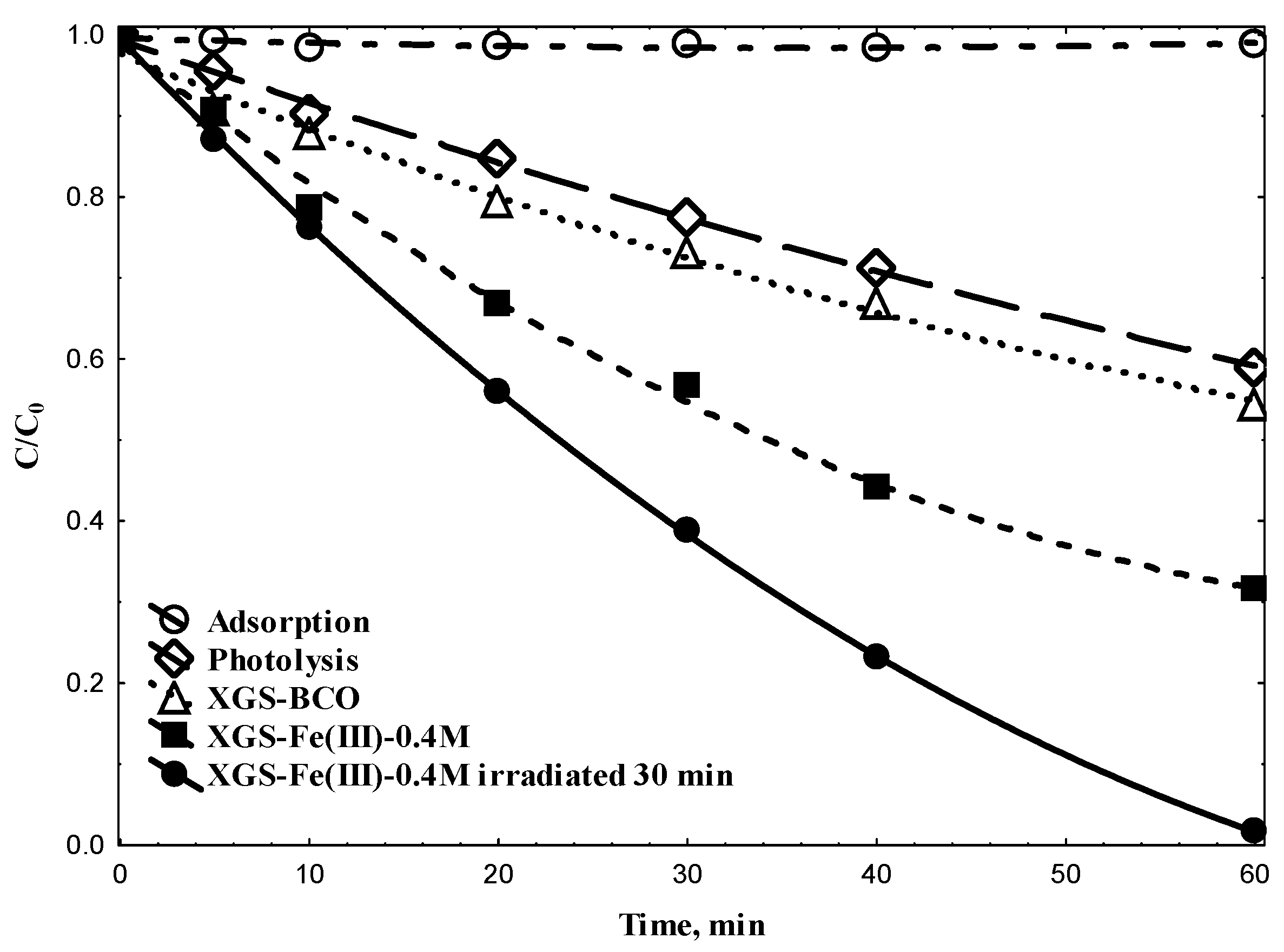
| XGS-BCO | 645.92 | 0.668 | 4.140 |
| XGS-Fe(III)-0.4 M-0.015 mm | 630.28 | 0.350 | 2.588 |
| XGS-Fe(III)-0.4 M-0.025 mm | 587.69 | 0.337 | 2.633 |
| XGS-Fe(III)-0.4 M-0.05 mm | 587.37 | 0.331 | 2.256 |
| [TNZ]0 | XGS | [Fe(III)] b | Particle Size | pH | k × 103 | Degradation |
|---|---|---|---|---|---|---|
| mg/L | g/L | M | nm | min−1 | % | |
| 25 | 0 | 0 | 0 | 7 | 8.63 | 41.03 |
| 25 | 1 a | 0 | 0.025 | 7 | 9.66 | 45.62 |
| 25 | 1 | 9 × 10−5 | 0.025 | 7 | 9.83 | 45.91 |
| 25 | 1 | 0.2 | 0.025 | 7 | 12.27 | 51.37 |
| 25 | 1 | 0.4 | 0.025 | 7 | 18.85 | 68.46 |
| 25 | 1 | 0.4 | 0.015 | 7 | 18.91 | 69.01 |
| 25 | 1 | 0.4 | 0.050 | 7 | 17.73 | 68.25 |
| 15 | 1 | 0.4 | 0.025 | 7 | 25.37 | 78.81 |
| 15 | 0 | 0 | 0 | 7 | 14.24 | 56.07 |
| 40 | 1 | 0.4 | 0.025 | 7 | 7.00 | 35.75 |
| 40 | 0 | 0 | 0 | 7 | 7.90 | 30.89 |
| 25 | 0.5 | 0.4 | 0.025 | 7 | 9.30 | 43.46 |
| 25 | 5 | 0.4 | 0.025 | 7 | 28.74 | 83.54 |
| 25 | 1 | 0.4 | 0.025 | 2 | 9.40 | 42.88 |
| 25 | 1 | 0.4 | 0.025 | 10 | 12.75 | 53.54 |
| 25 | 0 | 0 | 0 | 2 | 6.37 | 32.46 |
| 25 | 0 | 0 | 0 | 10 | 8.65 | 41.26 |
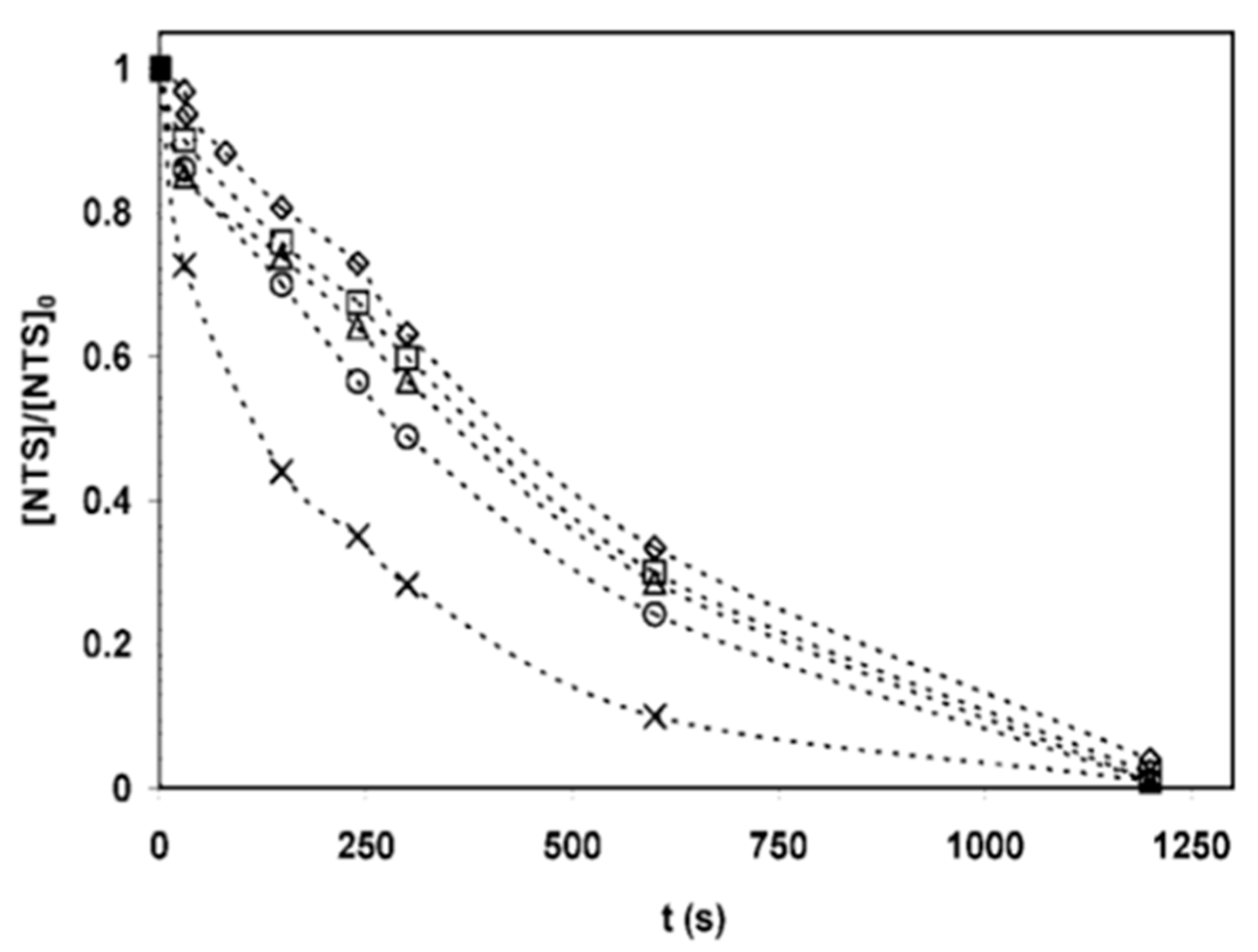
| 25 | 1 c | 0.4 | 0.025 | 7 | 62.94 | 98.38 |
| 25 | 1 d | 0.4 | 0.025 | 7 | 40.19 | 92.47 |
| 25 | 1 e | 0.4 | 0.025 | 7 | 28.72 | 83.53 |
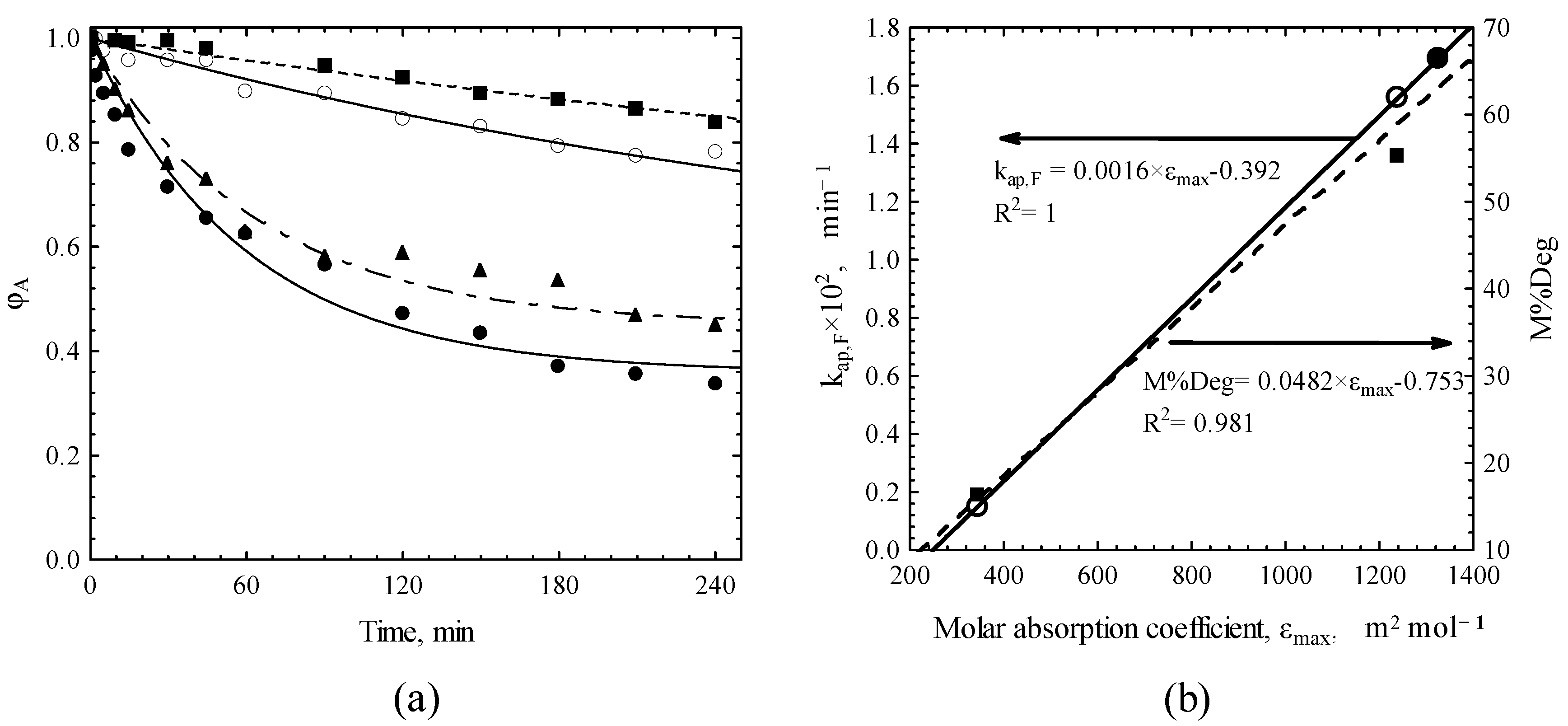
| CCTC0 | pHin | M%Deg | Kap,F × 102 |
|---|---|---|---|
| mg/L | min−1 | ||
| 15 | 7 | 25.29 | 0.36 |
| 25 | 7 | 21.74 | 0.29 |
| 35 | 7 | 16.58 | 0.16 |
| 25 | 2 | 9.00 | 0.27 |
| 25 | 11 | 44.39 | 5.44 |
| MOC | CMOC | pHin | M%Deg | Kap,S × 102 |
|---|---|---|---|---|
| mM | min−1 | |||
| Ru2B | 0.0134 | 7 | 16.19 | 0.15 |
| Ru3B | 0.0134 | 7 | 66.53 | 1.69 |
| Ru3P | 0.0134 | 7 | 55.21 | 1.56 |
| Ru3B | 0.0067 | 7 | 59.31 | 1.12 |
| Ru3B | 0.0268 | 7 | 72.82 | 3.49 |
| Ru3B | 0.0134 | 2 | 49.99 | 1.59 |
| Ru3B | 0.0134 | 11 | 51.50 | 10.02 |
| Sample | SBET a | V0 b | V0.95 c | Eg |
|---|---|---|---|---|
| m2/g | cm3/g | cm3/g | eV | |
| TiO2 | 81.5 | 0.030 | 0.375 | 3.20 |
| 4% rGO-TiO2 | 89.1 | 0.032 | 0.289 | 3.09 |
| 7% rGO-TiO2 | 97.7 | 0.036 | 0.242 | 2.75 |
| 10% rGO-TiO2 | 106.3 | 0.039 | 0.282 | 2.63 |
| 30% rGO-TiO2 | 141.1 | 0.051 | 0.273 | 2.55 |
| P25 | 57.0 | 0.020 | 0.138 | - |
| 4% rGO-P25 | 62.0 | 0.023 | 0.157 | - |
| 7% rGO-P25 | 66.8 | 0.024 | 0.171 | - |
| 10% rGO-P25 | 71.4 | 0.026 | 0.190 | - |
| 30% rGO-P25 | 115.9 | 0.043 | 0.236 | - |
| Bismuth subnitrate | 0.27 | - | - | - |
| 5% rGO/Bi | 26 | - | - | 3.51 |
| 10% rGO/Bi | 17 | - | - | 3.57 |
| 20% rGO/Bi | 20 | - | - | 3.73 |
| 30% rGO/Bi | 21 | - | - | 3.76 |
| 35% rGO/Bi | 21 | - | - | 3.76 |
| 40% rGO/Bi | 20 | - | - | 3.33 |
| 50% rGO/Bi | 21 | - | - | 3.04 |
| Sample | t1/2 a | t90% b | K c | EtP40 min d | TOC40 min e |
|---|---|---|---|---|---|
| min | min | min−1 | % | % | |
| UV | 30.2 | 100.4 | 0.023 | 61.5 | 14.0 |
| TiO2 | 22.4 | 75.2 | 0.031 | 72.5 | 21.8 |
| 4% rGO-TiO2 | 10.4 | 34.4 | 0.067 | 95.4 | 44.7 |
| 7% rGO-TiO2 | 7.2 | 23.9 | 0.096 | 98.6 | 56.6 |
| 10% rGO-TiO2 | 17.2 | 57.4 | 0.040 | 82.4 | 34.5 |
| 30% rGO-TiO2 | 28.9 | 96.1 | 0.024 | 60.7 | 24.9 |
| P25 | 28.1 | 93.3 | 0.025 | 64.5 | - |
| 4% rGO-P25 | 21.9 | 72.9 | 0.032 | 74.4 | - |
| 10% rGO-P25 | 34.5 | 114.6 | 0.020 | 53.3 | - |
| % rGO | Degradation Rate | % Degradation |
|---|---|---|
| min−1 | (2 h) | |
| 5 | 0.030 | 100 |
| 10 | 0.018 | 88 |
| 20 | 0.006 | 67 |
| 30 | 0.006 | 63 |
| 35 | 0.006 | 51 |
| 40 | 0.006 | 62 |
| 50 | 0.004 | 44 |
| Activated Carbon | SN2 a | V2 b | V3 c | pHPZC | Acid Groups d | Basic Groups e | Ash |
|---|---|---|---|---|---|---|---|
| m2/g | cm3/g | cm3/g | μeq/g | μeq/g | % | ||
| F400 | 1075 | 0.11 | 0.26 | 7.91 | 234 | 570 | 6.6 |
| Sorbo | 1295 | 0.06 | 0.37 | 9.42 | 88 | 1713 | 5.9 |
| Merck | 1301 | 0.09 | 0.26 | 7.89 | 114 | 582 | 5.2 |
| Ceca GAC | 966 | 0.13 | 0.16 | 6.83 | 323 | 99 | 12.0 |
| Ceca AC40 | 1201 | 0.07 | 0.32 | 5.29 | 438 | 102 | 8.3 |
| Norit | 968 | 0.10 | 0.42 | 9.18 | 139 | 2050 | 4.8 |
| Witco | 808 | 0.04 | 0.05 | 6.85 | 183 | 253 | 0.3 |
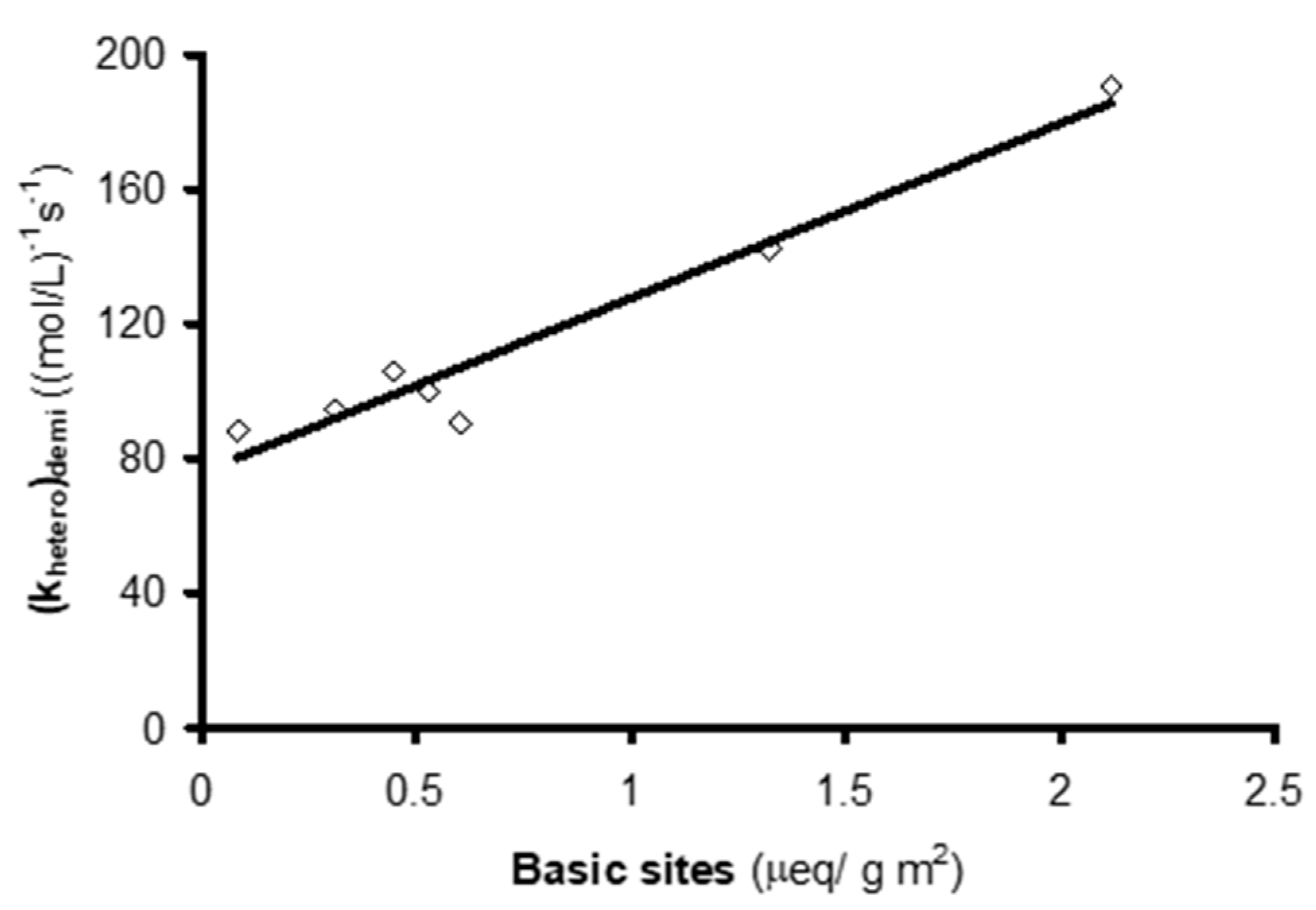
| Activated Carbon | kobs | khetero | (khetero)demi |
|---|---|---|---|
| s−1 | (mol/L)−1 s−1 | (mol/L)−1 s−1 | |
| F400 | 0.0115 | 134.6 | 106.1 |
| Sorbo | 0.0152 | 189.4 | 142.3 |
| Merck | 0.1005 | 114.4 | 105.7 |
| Ceca GAC | 0.0155 | 195.2 | 90.4 |
| Ceca AC40 | 0.0086 | 95.2 | 88.5 |
| Norit | 0.0653 | 210.5 | 190.4 |
| Witco | 0.0085 | 94.2 | 94.2 |
| Experiment | Sample | Carbon Dose | [O3] | kD | Rct |
|---|---|---|---|---|---|
| g/L | M | s−1 | |||
| 1 | Without carbon | 0.00 | 2 × 10−5 | 6.0 × 10−4 | 2.7 × 10−9 |
| 2 | F400 | 0.50 | 2 × 10−5 | 3.2 × 10−3 | 1.2 × 10−8 |
| 3 | F400 | 0.50 | 4 × 10−5 | 3.6 × 10−3 | 1.6 × 10−8 |
| 4 | F400 | 0.50 | 6 × 10−5 | 4.0 × 10−3 | 4.7 × 10−8 |
| 5 | F400 | 0.01 | 2 × 10−5 | 6.1 × 10−4 | 3.0 × 10−9 |
| 6 | F400 | 0.25 | 2 × 10−5 | 9.0 × 10−4 | 6.0 × 10−9 |
| 7 | F400 | 0.85 | 2 × 10−5 | 8.0 × 10−3 | 5.7 × 10−8 |
| 10 | F400-1 | 0.50 | 2 × 10−5 | 2.9 × 10−3 | 1.4 × 10−8 |
| 11 | F400-2 | 0.50 | 2 × 10−5 | 2.6 × 10−3 | 1.5 × 10−8 |
| 12 | F400-3 | 0.50 | 2 × 10−5 | 2.4 × 10−3 | 1.5 × 10−8 |
| 13 | F400-10 | 0.50 | 2 × 10−5 | 1.0 × 10−3 | 5.6 × 10−9 |
| 14 | F400-120 | 0.50 | 2 × 10−5 | 4.0 × 10−4 | 3.8 × 10−9 |
| Sample | KOH/Coke | SN2 | Vmic | V2 a | V3 b | pHPZC |
|---|---|---|---|---|---|---|
| m2/g | cm3/g | cm3/g | cm3/g | |||
| C | 0 | <30 | 0.02 | Nil | 0.011 | 6.5 |
| C-1 | 1 | 1619 | 0.55 | 0.063 | 0.132 | 8.4 |
| C-2 | 2 | 1261 | 0.41 | 0.061 | 0.154 | 8.8 |
| C-3 | 3 | 1021 | 0.25 | 0.058 | 0.176 | 9.3 |
| C-4 | 4 | 970 | 0.20 | 0.051 | 0.263 | 9.7 |
| Sample | C | C-1 | C-2 | C-3 | C-4 |
| khetero (mol/L)−1 s−1 | 57.7 | 105.8 | 86.5 | 76.9 | 67.3 |
| Sample | SN2 | Vmicro | V2 | V3 |
|---|---|---|---|---|
| m2/g | cm3/g | cm3/g | cm3/g | |
| W | 812 | 0.238 | 0.040 | 0.050 |
| W-A | 904 | 0.206 | 0.047 | 0.091 |
| W-C | 825 | 0.222 | 0.042 | 0.102 |
| W-U | 1057 | 0.289 | 0.064 | 0.122 |
| Sample | Pyridine | Pyridone | Pyrrole | N-Oxide |
|---|---|---|---|---|
| (398.5 ± 0.2 eV) | (399.5 ± 0.2 eV) | (400.5 ± 0.2 eV) | (402.5 ± 0.2 eV) | |
| W-U | 8 | 42 | 50 | - |
| W-U (ozonated) | 7 | 47 | 16 | 30 |
| Sample | SN2 | SCO2 | Vmicro | V2 | V3 |
|---|---|---|---|---|---|
| m2/g | m2/g | cm3/g | cm3/g | cm3/g | |
| A | 500 | 200 | 0.07 | 0.36 | 0.68 |
| A-Co(II)-15 | 562 | 206 | 0.07 | 0.43 | 0.97 |
| A-Ti(IV)-15 | 550 | 203 | 0.07 | 0.40 | 0.92 |
| A-Mn(II)-15 | 554 | 210 | 0.07 | 0.41 | 0.95 |
| A-Mn(II)-15-1 | 540 | 200 | 0.07 | 0.41 | 0.94 |
| A-Mn(II)-15-2 | 534 | 204 | 0.07 | 0.40 | 0.93 |
| A-Mn(II)-15-3 | 546 | 206 | 0.07 | 0.41 | 0.95 |
| A-Co(II)-15-1 | 560 | 210 | 0.07 | 0.41 | 0.92 |
| A-Ti(IV)-15-1 | 538 | 200 | 0.07 | 0.38 | 0.90 |
| Sample | pHPZC | C | O | Co(II) | Mn(II) | Mn(III) | Mn(IV) | Ti(IV) |
|---|---|---|---|---|---|---|---|---|
| % | % | % | % | % | % | % | ||
| A | 3.5 | 68 | 32 | - | - | - | - | - |
| A-Co(II)-15 | 3.8 | 64 | 22 | 14 | - | - | - | - |
| A-Ti(IV)-15 | 4.3 | 64 | 21 | - | - | - | - | 15 |
| A-Mn(II)-15 | 4.2 | 62 | 22 | - | 16 | - | - | - |
| A-Mn(II)-15-1 | 4.0 | 54 | 30 | - | 10 | 4 | 2 | - |
| A-Mn(II)-15-2 | 4.1 | 49 | 35 | - | 8 | 4 | 4 | - |
| A-Mn(II)-15-3 | 3.9 | 42 | 42 | - | 6 | 4 | 6 | - |
| A-Co(II)-15-1 | 3.9 | 55 | 31 | 14 | - | - | - | - |
| A-Ti(IV)-15-1 | 4.2 | 53 | 32 | - | - | - | - | 15 |
| Experiment | Sample | Carbon Dose | kD | Rct |
|---|---|---|---|---|
| mg | s−1 | |||
| 1 | Without aerogel | 0 | 6.0 × 10−4 | 2.68 × 10−9 |
| 2 | A | 2.5 | 6.2 × 10−4 | 2.74 × 10−9 |
| 3 | A-Co(II)-15 | 2.5 | 5.8 × 10−4 | 2.56 × 10−9 |
| 4 | A-Ti(IV)-15 | 2.5 | 6.1 × 10−4 | 2.73 × 10−9 |
| 5 | A-Mn(II)-15 | 2.5 | 4.2 × 10−3 | 5.36 × 10−8 |
| 10 | A-Mn(II)-15-1 | 2.5 | 2.6 × 10−3 | 3.35 × 10−8 |
| 11 | A-Mn(II)-15-2 | 2.5 | 2.1 × 10−3 | 2.68 × 10−8 |
| 12 | A-Mn(II)-15-3 | 2.5 | 1.4 × 10−3 | 1.78 × 10−8 |
| Sample | SN2 | W0 (N2) a | W0 (CO2) b | L0 (N2) c | L0 (CO2) d |
|---|---|---|---|---|---|
| m2/g | cm3/g | cm3/g | nm | nm | |
| Z-2 | 7.1 | 0.037 | 0.144 | 2.02 | 1.00 |
| Z-13X | 341.3 | 0.166 | 0.184 | 0.48 | 0.30 |
| ZSM-5 | 314.4 | 0.161 | 0.245 | 0.41 | 0.74 |
| ZSM-5-NaOH | 326.4 | 0.162 | 0.320 | 0.70 | 1.14 |
| Sample | Composition (%) | pHPZC | Si/Al | |||
|---|---|---|---|---|---|---|
| Zeolite-A | Sodalite | ZSM-5 | Faujasite | |||
| Z-2 | 97.3 | 2.7 | 0 | 0 | 11.4 | 0.89 |
| Z-13X | 0 | 0 | 0 | 100 | 11.1 | 1.73 |
| ZSM-5 | 0 | 0 | 100 | 0 | 2.8 | 598.50 |
| ZSM-5-NaOH | 0 | 0 | 100 | 0 | 7.3 | 394.44 |
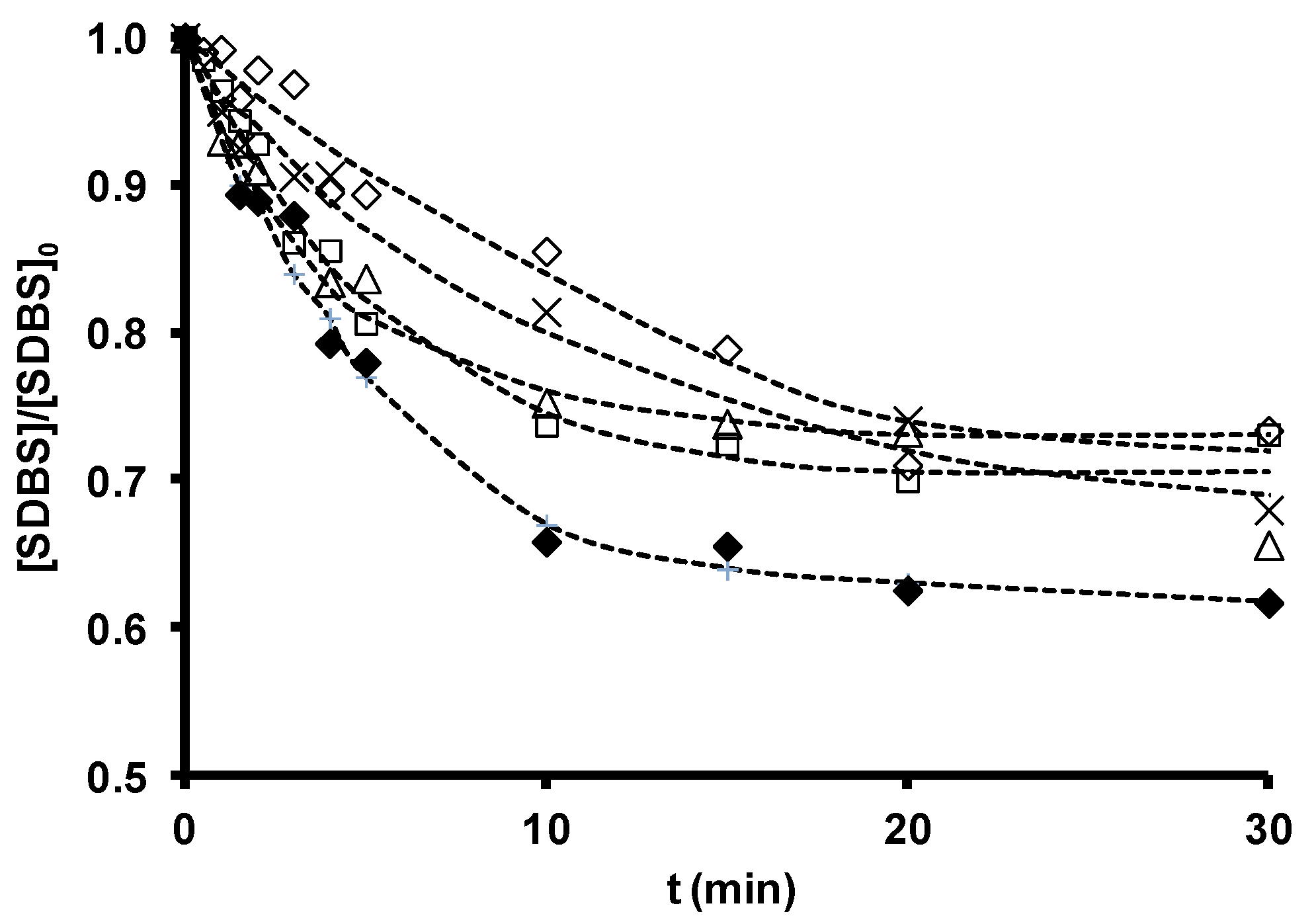
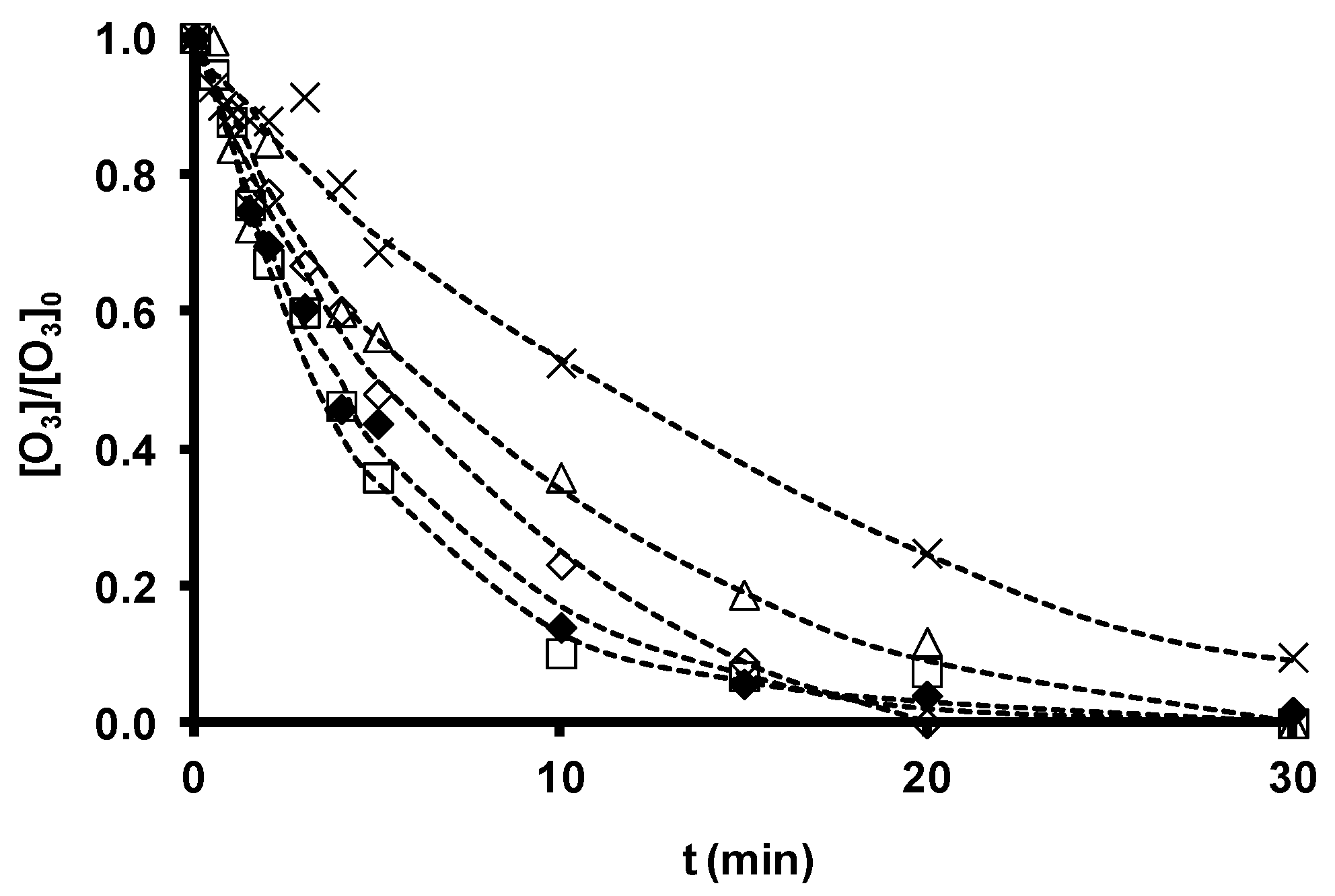
| Zeolite | Dose | Kobs (O3) | R2 | Kobs (SDBS) | R2 | (CHO•)het |
|---|---|---|---|---|---|---|
| mg/L | s−1 × 103 | s−1 × 104 | mol/L × 1015 | |||
| Without zeolite | 0 | 1.27 ± 0.04 | 0.997 | 2.75 ± 0.66 | 0.971 | |
| Z-2 | 100 | 1.73 ± 0.17 | 0.984 | 4.05 ± 1.16 | 0.959 | 1.36 |
| Z-13X | 100 | 3.83 ± 0.28 | 0.993 | 7.36 ± 1.16 | 0.975 | 4.61 |
| ZSM-5 | 100 | 2.50 ± 0.21 | 0.993 | 2.69 ± 0.37 | 0.983 | 0 |
| ZSM-5-NaOH | 100 | 3.24 ± 0.22 | 0.981 | 6.25 ± 1.66 | 0.965 | 3.55 |
| Catalyst | Contaminants | System | Experimental Conditions | Observations | Ref. |
|---|---|---|---|---|---|
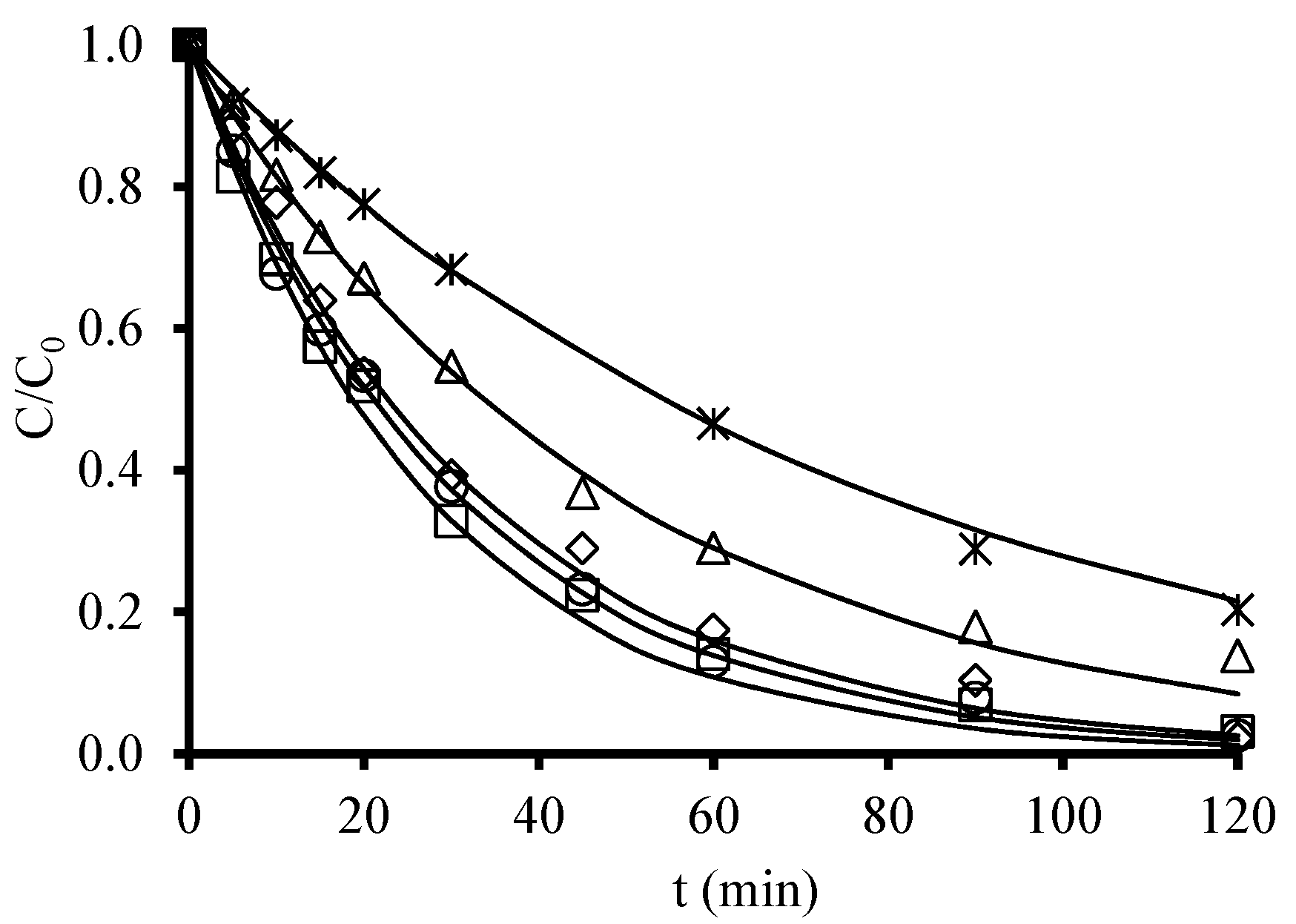
| Ni-, Co-, Fe-, Zn-doped organic aerogels | Trimethoprim [10 μM] | UV/metal–carbon aerogel | Photoreactor equipped with a low-pressure Hg UV lamp (11 W) with radiation intensity between 315 and 400 nm. Temperature = 22 °C [Catalyst]0 = 10 mg/L Time 120 min | Trimethoprim was resistant to photolytic degradation. Apparent rate constants did not significantly vary among Fe-, Zn-, Ni-, and Co-doped aerogels. Ni-doped aerogel had a slightly higher capacity to catalyze trimethoprim degradation. % degradation = 80% with Ni doped aerogel after 2 h of irradiation. | [182] |
| TiO2-carbon aerogel | Methyl orange [8 × 10−5 M] | UV/TiO2-carbon aerogel | Two parallel UV lamps (Osram 18 W UV-A blacklights) Temperature = 25 °C [Catalyst]0 = 330 mg/L Time 240 min | The carbon aerogel (RFCA) composites had higher photocatalytic activity versus polymer aerogel (RFA) due to the larger surface area and more numerous active reaction sites of the substrate. Unexpectedly, the bare RFCA substrate was a more active photocatalyst versus its composites. % degradation = 65% with RFCA after 4 h of irradiation. | [183] |
| ZnO or TiO2- Nitrogen-doped carbon aerogel composites | Methyl orange [4 × 10−5 M] | UV/carbon aerogel composites | Two parallel UV lamps (Osram 18 W UV-A blacklights) Temperature = 25 °C [Catalyst]0 = 330 mg/L Time 240 min | The bare carbon aerogel and TiO2-coated samples were the most active ones and were even more efficacious than P25. The highest photocatalytic efficacy was observed for the sample coated with thinner layers of TiO2, which decomposed around 40% of methyl orange dye after 240 min. | [184] |
| ZnS-carbonaceous aerogel composites | Methylene blue [40 mg/L] | Visible/carbon aerogel composites | 500 W xenon lamp with 420 nm cutoff filter. [Catalyst]0 = 1000 mg/L Time 120 min | The 55 wt % ZnS/CA sample exhibits excellent photocatalytic performances for methylene blue degradation: 91% degradation after 180 min of irradiation. | [185] |
| Iron-doped carbon xerogel | Brilliant green, crystal violet, methyl green [100 mg/L] | Heterogeneous Fenton | Temperature = 25 °C, 50 rpm [Catalyst]0 = 4 g/L pH = 5.5–6 Time 180 min | Dyes and TOC removal efficacies up to 99% and 65%, respectively, were obtained for all dyes. % efficiencies obtained for all dyes directly correlate with the iron content. | [186] |
| ZnO-carbon xerogel composites | Chlorophenol, bisphenol A [10 mg/L] | UV or solar/carbon xerogel composites | Source of solar radiation was an Osram Ultra Vitalux 300 W lamp; source of visible radiation source was an Osram Powerstar HQI-T W/D Pro 400 W. Temperature = 25 °C [Catalyst]0 = 500 mg/L Time 300 min | All composites showed photocatalytic activity to degrade 4-chlorophenol and bisphenol A under both solar and visible radiation. Materials with intermediate proportions between carbon xerogel and zinc oxide were optimal for the photodegradation. The maximum values for the degradation of 4-chlorophenol were 88% and 49% after 5 h under solar and visible radiation, respectively % bisphenol A degradation values were 78% and 42%, respectively. | [187] |
| Iron-doped carbon xerogel | Propyl paraben (PP) [420 μg/L] | Activated persulfate (PS) oxidation | [PS] = 25–750 mg/L pH = 3, 6, and 9 Temperature = 25 °C [Catalyst]0 = 63 mg/L Time 60 min | The pseudo-first-order degradation rate of PP increased with higher PS concentration and lower PP concentration and solution pH. The water matrix had a detrimental effect on kinetics depending on the complexity and concentration of matrix constituents. | [188] |
| ZrO2-carbon xerogel composites | Orange G [10 mg/L] | Visible/carbon xerogel composites | Glass photoreactor equipped with visible ReptoLux 2.0 14 W lamp. Temperature = 25 °C [Catalyst]0 = 1000 mg/L Time 400 min | The composites were more efficacious versus the pure samples, obtaining % degradation values of 47.7%, 63.0%, and 98.0% for catalysts with 20, 30, and 40% ZrO2 content, respectively. There was a very high synergic effect between carbon xerogel and ZrO2, with a higher catalytic efficacy versus P25, providing an effective and more rapid degradation of Orange G. | [189] |
| Ni-doped carbon xerogels | Methyl orange [25, 50, and 100 mg/L] | Catalytic wet peroxide oxidation | [H2O2] = 0.026, 0.08, 0.4 M pH = 3, 5, and 8 Temperature = 50 and 70 °C [Catalyst]0 = 500 and 1000 mg/L Time 150 min | The dye decomposition rate was strongly dependent on the H2O2 dose, temperature, catalyst dose, and initial concentration of dye. The catalytic decomposition of dye was complete in 30 min using CX-Ni-500 at pH 3. Incorporation of metallic Ni by impregnation-reduction method onto the carbon xerogel surface was largely responsible for the dye decomposition efficacy. | [190] |
| Nb2O5-silica xerogels | Methylene blue (MB) [12 mg/L] | UVC/catalyst | Lamp with UVC light (Phillips/254 nm) [Catalyst]0 = 20,000 mg/L Time 500 min | The pure silica xerogel showed no photoactivity. Nanocomposites heated at 500 and 700 °C are formed by small crystallites with low photoactivity dispersed in the matrix, while samples heated at 900 °C showed good activity, reducing the concentration of MB by 80% after 500 min. | [191] |
| Ag/titania–silica xerogel | Methyl orange, methylene blue, phenol, salicylic acid, and rhodamine B [100 μM] | UV/catalyst | UV reactor system equipped with medium pressure 150 W Hg lamp, UV light range 350–400 nm. Temperature = 20 °C [Catalyst]0 = 375 mg/L Time 120 min | The efficacy to degrade methyl orange and phenol was higher by photolysis than by photocatalysis. The amount of rhodamine B degraded was higher by photocatalysis than by photolysis after the first 30 min. The methylene blue degradation rate was higher by photocatalysis than by photolysis because of the affinity of the positively charged molecule for the negative surface of the catalyst. The salicylic acid degradation rate was the same by photolysis and photocatalysis. | [192] |
| Fe-SiO2 xerogels | p-Nitrophenol (PNP) [10−4 M] | Photo-Fenton process | Halogen lamp (300 W, 220 V) with a continuous spectrum from 300 to 800 nm. [H2O2] = 10−3 M Temperature = 20 °C [Catalyst]0 = 550 mg/L Time 24 h | Photo-Fenton activities of the two iron-doped samples can be directly linked to their specific morphologies. The iron-doped sample without EDAS had the greatest PNP degradation efficacy (99%) due to the direct availability of iron. Nevertheless, the PNP degradation was higher for the sample with EDAS (76%) than for the blank experiment without catalyst (64%). The catalysts displayed stable catalytic activity after 72 h of operation. | [193] |
| Magnetic AC | Remazol black dye (RB) [40mg/L] | UV/AC-Fe | Radiation source: low-pressure mercury lamp; emission spectrum 254 nm (30W-Philips, I = 1.20mW cm−2). [Catalyst]0 = 300 mg/L Volume = 200 mL Time 240 min Temperature = 25 °C | Percentage discoloration of RB by photocatalysis was 16% for the AC-Fe catalyst and 17–67% for AC-Fe catalysts with different Ti concentrations. AC-Fe evidenced photoactive behavior. | [194] |
| Activated carbon codumil | Paracetamol [50 mg/L] | UV/AC | Radiation source: one or two 8 W Phillips mercury vapor lamps; emission spectrum 254 nm (I = 59.8 mW/cm2 and 119.6 mW/cm2). [Catalyst]0 = 250 mg in 50 mL Volume = 50 mL Time: 120 min | There was a higher degradation of paracetamol pre-adsorbed on AC. The percentage degradation of paracetamol adsorbed on AC with UV-C irradiance had a maximum value of 79%, while the maximum values for alumina and silica were 65% and 77%, respectively. | [195] |
| Commercial AC and endocarp of babassu coconut (EBC) chemically activated with H3PO4 (85%) | Methylene blue (MB) [20 mg/L] | AC Merck, ECB and EBC/Fe, simulated solar radiation | Radiation source: solar simulator box; Xe-lamp solar spectrum (I (UV region) = 105W/m2, corresponding to 9% of the total radiation I of 1162W/m2). [Catalyst]0 = 50 mg/L Volume = 125mL Time: 300 min | MB concentration was reduced by 22% with commercial AC, showing that the photochemical activity of this AC is low but not negligible. The most striking performance was achieved with the EBC/Fe-700° nanocomposite (ca. 58% MB converted after 5 h). Formation of the FePO4 phase at 700 °C is the main factor responsible for the increase in photoactivity. | [196] |
| Ruthenium doped titanate nanowires (Ru-TNW) | Cyclophosphamide (CP) and ifosfamide (IF) | UV–vis/Ru-TNW | Radiation source: 450 W medium-pressure mercury-vapor lamp (Hanovia, Slough, UK); total I energy = 40–48% in the UV range and 40–43% in the visible region. [Catalyst]0 = 130 mg/L Volume = 150 mL [CP]0 = [IF]0 = 10mg/L Time 120 min Water matrix: distilled water (DW) and wastewater (WW) from secondary wastewater treatment. | The removal rate of both pollutants with Ru-TNW as photocatalyst under UV–vis radiation was two-fold that obtained with photolysis in DW. The t1/2 was 10.6 min and 8.0 min for CP and IF, respectively. In WW, the increase in t1/2 was 4.5 min and 4.9 min, respectively. | [197] |
| RuO2-GO composite | Methylene blue (MB) | Sunlight/RuO2 Sunlight/GO Sunlight/RuO2-GO | Radiation source: solar light from 11.30 a.m. to 2.30 p.m. during the summer in Bengaluru, Karnataka (India). I = 68,000 flux. [Catalyst]0 = 500 mg/L Volume = 40 mL [MB]0 = 20mg/L Time: 60 min | After irradiation for 60 min, the MB removal rate was 95% in the presence of RuO2-GO composite versus 40% with pure RuO2 and 20% with GO. The enhanced photocatalytic activity of the RuO2-GO composite was due to the synergistic effect between mesoporous RuO2 anchored on GO via Ru–O–C bond. | [198] |
| rGO-TiO2 | Orange G [20 mg/L] | solar/rGO-TiO2 | SOLAR BOX 1500 e with a 1500 W Xenon lamp Temperature = 28 °C pH = 6 [Catalyst]0 = 1000 mg/L Time 60 min | Orange G photodegradation was always higher with graphene derivative-TiO2 composites than with TiO2 under simulated solar light. Orange G conversion rates of 99.8%, 90.0%, 98.2%, 96.5%, and 47.6% were obtained at 60 min with GO-T, rGO-T, rGONS-T, rGOB-T, and TiO2, respectively. The photocatalytic activity of the samples decreased with increased initial pH. | [199] |
| rGO-TiO2 | Diuron [7.5 mg/L], alachlor [12.5 mg/L], isoproturon [12.5 mg/L] and atrazine [6.25 mg/L] | UV, visible/rGO-TiO2 | Six UV black blue light lamps (Narva LT 15 W/073) and 4 Daylight lamps (Narva LT 15 W/865) for UV–vis experiments. Under near-visible, 10 daylight fluorescent lamps were employed [Catalyst]0 = 500 mg/L Time 300 min | Pesticide photodegradation and TOC abatement were greater with the GO-TiO2 composite than with bare TiO2. This increase in photoefficacy was even greater in natural water. | [200] |
| rGO-TiO2 | 4-chlorophenol [0.1 mmol/L] | UV/rGO-TiO2 | Sylvania Lynx-S 11WBLB lamp, UV light (365 nm), density of 1.0 mW/cm2 Temperature = 25 °C pH = 6 [Catalyst]0 = 1000 mg/L Time 300 min | The presence of rGO in the composite increased the formation of hydroxyl/HO• radicals to attack the 4-chlorophenol molecules, explaining the improvement in photocatalytic performance. In comparison to TiO2, the rGO/TiO2 photocatalyst increased the first-order reaction rate constant by around 70%. % degradation = 85% after 5 h of irradiation. | [201] |
| rGO@g-C3N4-TiO2 nanotube | Tetracycline hydrochloride [20 mg/L] | UV/catalyst | Xenon lamp (150 W) Time 120 min | rGO@g-C3N4/TNAs photoelectrodes exhibited higher photocatalytic activity. The removal rate of tetracycline by rGO@g-C3N4/TiO2 nanotube photoelectrodes reached 90% after 120 min, and the reaction kinetic constant was 2.38-fold higher than for TiO2 nanotube photoelectrodes. | [202] |
| rGO-Cu2O/Bi2O3 | Tetracycline [10 mg/L] | Visible/rGO-Bi2O3 | 250 W Xe lamp with UV cutoff filters [Catalyst]0 = 500 mg/L Time 180 min | TC photodegradation in the presence of rGO-Cu2O/Bi2O3 and rGO/Bi2O3 was 38% and 24.5%, respectively, after 3 h. The photocatalytic activity of the composites was greater, with an increase in GO from 20 to 50 mg. | [203] |
| rGO-Bi2O3 | Methylene blue [0–50 mg/L] | Visible/rGO-Bi2O3 | Visible photoreactor. 500 W tungsten halogen lamp (400–800 nm) [Catalyst]0 = 300–2000 mg/L Time 500 min | Maximum MB degradation of 98% was achieved at neutral pH with 1 g/L of catalyst. The rGO-Bi2O3 nanocomposite exhibited higher photocatalytic activity (98%) versus pristine Bi2O3 (55%) or rGO (20%) under visible light irradiation after 240 min. | [204] |
| rGO-Bi2O3 | Rhodamine B [10 mg/L] | Visible/rGO-Bi2O3 | 300-W xenon lamp Temperature = 25 °C [Catalyst]0 = 1000 mg/L Time 90 min | Composites with 1% rGO showed the highest photocatalytic activity (100% degradation of rhodamine B after 30 min), which was three-fold higher versus bare Bi2O3. This high photocatalytic activity was attributed to the presence of graphene, which served as an electron collector and transporter to increase the lifetime of photogenerated charge carriers from Bi2O3. | [205] |
| Activated carbon-TiO2 | Tetracycline [30 mg/L] | OV or US/catalyst | UV light from UV-C lamp (PHILIPS) with λ = 254 nm and intensity of 6 W. Temperature = 25 °C [Catalyst]0 = 300 mg/L Time 180 min | The degradation rate was higher when AC-TiO2 was coupled with US and UV irradiations. Removal rates of 93 and 50.4% were achieved for TC and TOC, respectively, after 180 min irradiation. The anions exerted an inhibitory effect. | [206] |
| Activated carbon-TiO2 | Orange G [10 mg/L] | UV/carbon-TiO2 | Low-pressure mercury vapor lamp (TNN 15/32, 15 W, λ = 254 nm [Catalyst]0 = 1000 mg/L Time 40 min | Carbon-Ti materials with a developed micro-mesoporosity reduced the bandgap, being more active in orange G photodegradation under UV irradiation versus TiO2, reaching 100% degradation after 30 min of irradiation. Catalytic activity decreased with higher carbonization temperature. | [207] |
| Activated carbon-TiO2 | Carbamazepine [50 mg/L] | solar/carbon-TiO2 | LOT Quantum Design xenon lamp system (300 W)Temperature = 20 °C [Catalyst]0 = 100 mg/L Time 300 min | The composite with 9% TiO2 was selected because it had the highest adsorption and degradation rate and lowest production cost. % degradation = 85% after 300 min of irradiation. | [208] |
| Activated carbon-TiO2 | 2,4-dichlorophenol [100 mg/L] | solar/carbon-TiO2 | 300-W xenon lamp [Catalyst]0 = 250 mg/L Time 300 min | AC can accept the photogenerated electrons, improving charge separation and considerably improving photocatalytic activities to degrade 2,4 dichlorophenol. The photogenerated hydroxyl/HO• radicals may predominate in the photocatalytic degradation of 2,4 dichlorophenol. | [209] |
| Granulated activated carbon (GAC) | Reactive Blue 194 (RB194) [10–200 mg/L] | Activated carbon catalyzed ozonation (ACCO) | Continuous 178.8 mg O3/L Gas flow 1.5 L/min Temperature = 24, 35 and 50 °C [Catalyst]0 = a column packed with 2.6 L GAC and 1.2 L dye solution Time: 40 min [NaCl] = 5−50 g/L | With ozonation alone, an increase in NaCl concentration had a negligible effect on RB194 removal, but enhanced color removal by ACCO, obtaining % color removal of 61 and 84% after 5 min of contact with NaCl concentrations of 5 and 50 g/L, respectively. 100% color removal was achieved by O3 after 20 min. Besides the synergistic effect of GAC in the ozone system, the higher dye removal rate was attributed to the production of reactive chlorine species. | [210] |
| Olive stone activated carbon (OSAC) | Clopyralid [1 mg/L] | O3/OSAC | Continuous 15 mg O3/L Gas flow 30 L/h Temperature = 25 °C [Catalyst]0 = 0.1–1 g/L Time: 60 min solar simulator (SSR): Xe arc lamp 500 W/m2 of radiation over 300 nm. | The use of OSAC increased the removal of clopyralid from 66% to 96% after 30 min versus ozone alone. The OSAC concentration of 0.75 g/L yielded the best removal of clopyralid with O3. Kinetic rate constant values were: O3/OSAC (0.106 min−1) > O3/OSAC/SSR (0.099 min−1) > O3/SSR (0.097 min−1) > O3 (0.063 min−1). OSAC can be considered not only as an adsorbent but also as a catalyst. | [211] |
| Commercial granular activated carbon (GAC) | Acid red 3R [100 mg/L] | O3 MB/GAC | Continuous 12.5 mg O3/L Gas flow 0.3 L/min [Catalyst]0 = 2.0 g/L Time 120 min Diameter of O3 microbubbles (MBs): 50 µm | Decolorization rate constants were 0.342 min−1 and 0.173 min−1 for MB ozonation with and without AC, respectively. O3 self-decomposition rate constants in water in MB aeration and coarse bubble aeration were 0.049 min−1 and 0.013 min−1, respectively. When AC was added to MB aeration, the O3 self-decomposition rate constant increased to 0.093 min−1 because O3 decomposition was enhanced by the catalytic effect of AC. The catalytic activity of this commercial AC appeared stable after five cycles. | [212] |
| Commercial granular activated carbon (GAC) | Drugs and additives mix as micropollutants (MP) | O3/GAC at pilot-scale in wastewater effluent, over a long-term experiment | Continuous 0.3 and 0.5 mg O3/mg DOC. Gas flow 5 L/min [Catalyst]0 = 0.5 and 2.0 g/L Time 20 h. Influent: tertiary-filtered wastewater effluent from a reclamation facility in Nevada, USA. MP mix (ng/L): triclosan (27.00), sulfamethoxazole (742.5), carbamazepine (142.5), naproxen (19.25), trimethoprim (15.25), gemfibrozil (1.37), atenolol (37.5), fluoxetine (26.5), DEET (135.25), primidone (145.0), sucralose (48500), meprobamate (242.5), NMOR (15.5), TCEP (240.0). | O3/GAC improved the abatement of MPs (TCEP, sucralose, and meprobamate) versus ozonation alone at 20 h of operation. However, the overall effectiveness of O3/GAC markedly decreased after longer O3 exposure, and the improvement ceased for most of the MPs (except sucralose and NMOR) after 20 h of operation due to O3-induced changes in GAC surface properties (structural damage, reduced surface area, and increase in surface acidic functional groups). | [213] |
| Powdered activated carbon (AC-1230) (75 µm particle size) | Drugs mix as micropollutants (MP) | O3/PAC | Continuous 0.5–2.0 mg O3/L Gas flow 24 L/h [Catalyst]0 = 20 mg/L Time 30 min Influent: ultrapure (UPW) and natural water (NW) from the Biobío river. Toxicity test with Daphnia magna. MP (2.30–2.90 mg/L): atrazine (ATZ), carbamazepine (CBZ), diclofenac (DCL), triclosan (TCS) | Treatment with O3 and O3/PAC showed the slowest transformation rate for ATZ (>90% at 30 min) but a very fast rate for CBZ, DCL and TCS (>90% at 5 min). ATZ oxidation was mainly driven by HO• radicals, while CBZ, DCL and TCS were directly removed by O3. Addition of PAC in the ozonation process markedly improved acute toxicity removal. For a similar TOC reduction, the reaction time was reduced from 60 to 10 min in O3/PAC. | [214] |
| Carbon aerogel | Reactive blue 19 | O3/carbon aerogel | Continuous Temperature = 24 °C 2.5 mg O3/min [Catalyst]0 = 4 g/L Time 20 min | The presence of carbon aerogel increased decolorization versus ozonation alone. The percentage of decolorization was almost 99% after 5 min in the presence of carbon aerogel. | [172] |
| Carbon aerogel | Reactive blue 19 | O3/carbon aerogel | Continuous 8 mg O3/L Gas flow 0.5 L/min Temperature = 24 °C [Catalyst]0 = 2.0 g/L or 2.4 g/L Time 30 min | The participation of carbon aerogel strikingly enhanced the removal of dye versus ozonation alone. Decolorization rates of 85%, 73% 55% were obtained after 5 min using Ag-Fe2O3/carbon aerogel, carbon aerogel, and ozone alone, respectively. | [215] |
| Carbon aerogel | Reactive blue 19 | O3/carbon aerogel | Continuous 8 mg O3/L Gas flow 0.5 L/min Temperature = 24 °C [Catalyst]0 = 1.0 g/L or 1.2 g/L Time 30 min | Ozonation alone was generally not able to completely degrade organic contaminants and needed the addition of a catalyst to promote degradation. After 10 min, the effluent was treated by catalytic ozonation was 100% decolorized, whereas ozonation-treated effluent remained bluish. | [216] |
| Cerium-loaded natural zeolite (CZ) | Penicillin G (PG) [50mg/L] | O3/CZ | Continuous 6 mg O3/min Gas flow 0.6 L/min Temperature = 25 °C [Catalyst]0 = 0–3 g/L Time 15 min Cerium = 0–8% | PG degradation efficacy of the O3/CZ system was higher than that of O3 alone. Optimal operating conditions were: Ce loading mass = 4%, CZ dosage = 2 g/L, O3 dosage = 6 mg/min, and initial solution Ph = 4.5. PG removal was 99.5% after 15 min. These optimal conditions produced the highest specific catalyst surface area of the catalyst with no agglomeration of Ce particles, and the appropriate ratio between O3 and catalyst was not limited by ozone dosage. | [217] |
| High-silica zeolites ZSM5, loaded with metallic (Ce, Fe, or Mn) oxides | Nitrobenzene [100 mg/L] | O3/ZSM5 | Continuous 5 mg O3/min Gas flow 3 L/min Temperature = 30 °C [Catalyst]0 = 2.5 g/L Time 60 minCe = Fe = Mn = 0.8 wt % Pristine ZSM5 zeolites: NaZ38 (Na type of counter ion, molar ratio SiO2/Al2O3 = 38, Na2O at 4.08 wt %); HZ38 (H type, SiO2/Al2O3 ratio = 38, Na2O at 0.13 wt %), and NaZ100 (Na type, SiO2/Al2O3 = 100, Na2O at 1.57 wt %). | Adsorption and direct ozonation contributed to early TOC removal in O3/ZSM5 with HZ38 and NaZ100 zeolites. HO• radicals dominate the removal process. Among the ZSM5 zeolites studied, Ce/NaZ38 achieved the highest TOC removal, attributable to the surface SiO bonds. | [218] |
| Zeolite 4A (Z4A) | Paracetamol [50mg/L] | O3/Z4A | Continuous 0.9 mg O3/min Temperature = 25 °C [Catalyst]0 = 11.11 g/L Time 60 min | Z4A did not promote the decomposition of ozone to produce HO• and O2•− radicals. Thus, paracetamol ozonation in the presence of Z4A followed a non-radical mechanism. Z4A removed paracetamol by direct reaction with the O3 molecule. The effect of Z4A was attributed to the approximation of both pollutant and O3 molecules by adsorption on its surface. | [219] |
| H- and Fe-modified Beta zeolite | Ibuprofen (IBU) [10–100 mg/L] | O3/Beta-Zeolite | Continuous 60 mg O3/L Gas flow 0.25–1.1 L/min Temperature = 5–30 °C [Catalyst]0 = 0.25–1.0 g/L Time 4 h Catalysts: H-Beta-25, Fe-H-Beta-EIM-25 and Fe- H-Beta-150-SSIE | The presence of zeolite catalysts improved IBU removal, especially at lower temperatures, because the O3 concentration was higher. Catalysts with a larger amount of Brønsted and Lewis acid sites and higher Fe concentration achieved greater degradation of IBU. | [220] |
| Natural zeolite | Standard mix of organochlorinated compounds (Supelco Co.) in high-quality (99%) | O3/Zeolite vs. O3/GAC | Continuous 5 g O3/h Gas flow 0.5 L/min [Catalyst]0 = not specified Time 30 min Organochlorinated compounds: heptachlor (HCH), hexachlorobutadiene (HCHBD), lindane (LIN), pentachlorobenzene (PCHB), hexachlorobenzene (HCHB). | Adsorptive O3/GAC and O3/Zeolite ozonation processes significantly reduced the reaction time versus O3 or O3/UV. O3/GAC showed greater potential for organochlorine compound removal versus O3/Zeolite. | [221] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera-Utrilla, J.; López-Ramón, M.V.; Sánchez-Polo, M.; Álvarez, M.Á.; Velo-Gala, I. Characteristics and Behavior of Different Catalysts Used for Water Decontamination in Photooxidation and Ozonation Processes. Catalysts 2020, 10, 1485. https://doi.org/10.3390/catal10121485
Rivera-Utrilla J, López-Ramón MV, Sánchez-Polo M, Álvarez MÁ, Velo-Gala I. Characteristics and Behavior of Different Catalysts Used for Water Decontamination in Photooxidation and Ozonation Processes. Catalysts. 2020; 10(12):1485. https://doi.org/10.3390/catal10121485
Chicago/Turabian StyleRivera-Utrilla, José, María Victoria López-Ramón, Manuel Sánchez-Polo, Miguel Ángel Álvarez, and Inmaculada Velo-Gala. 2020. "Characteristics and Behavior of Different Catalysts Used for Water Decontamination in Photooxidation and Ozonation Processes" Catalysts 10, no. 12: 1485. https://doi.org/10.3390/catal10121485