Hydroalkoxylation of Terminal and Internal Alkynes Catalyzed by Dinuclear Gold(I) Complexes with Bridging Di(N-Heterocyclic Carbene) Ligands




Abstract
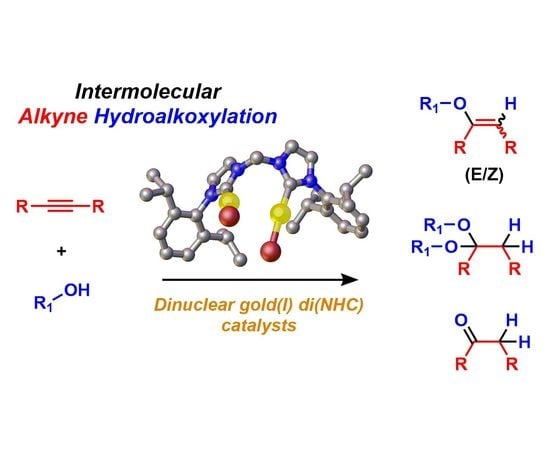
:
1. Introduction
2. Results and Discussion
2.1. Screening of the Reaction Conditions
2.2. Catalyst Screening
2.3. X-ray Structure Analysis
2.4. Substrates Scope and Analysis of the Selectivity in the Hydroalkoxylation of Alkynes
2.5. Comparison of The Catalytic Performances of Au2Br2L4 with Mononuclear Catalysts
3. Materials and Methods
3.1. Synthesis of the Mononuclear Gold(I) Complex {1-(2,6-Diisopropylphenyl)-3-Methyl Imidazol-2-Ylidene}Iodogold(I) AuIL7
3.2. X-ray Crystal Structure Determination of Au2Br2L4
3.3. Catalytic Tests on the Alkyne Hydroalkoxylation Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Teles, J.H.; Brode, S.; Chabanas, M. Cationic Gold(I) Complexes: Highly Efficient Catalysts for the Addition of Alcohols to Alkynes. Angew. Chem. Int. Ed. 1998, 37, 1415–1418. [Google Scholar] [CrossRef]
- Huguet, N.; Echavarren, A.M. Gold-Catalyzed O–H Bond Addition to Unsaturated Organic Molecules. In Hydrofunctionalization; Topics in Organometallic Chemistry; Ananikov, V.P., Tanaka, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 291–324. ISBN 978-3-642-33735-2. [Google Scholar]
- Zhdanko, A.; Maier, M.E. Explanation of “Silver Effects” in Gold(I)-Catalyzed Hydroalkoxylation of Alkynes. ACS Catal. 2015, 5, 5994–6004. [Google Scholar] [CrossRef]
- Corma, A.; Ruiz, V.R.; Leyva-Pérez, A.; Sabater, M.J. Regio- and Stereoselective Intermolecular Hydroalkoxylation of Alkynes Catalysed by Cationic Gold(I) Complexes. Adv. Synth. Catal. 2010, 352, 1701–1710. [Google Scholar] [CrossRef]
- Veenboer, R.M.P.; Dupuy, S.; Nolan, S.P. Stereoselective Gold(I)-Catalyzed Intermolecular Hydroalkoxlation of Alkynes. ACS Catal. 2015, 5, 1330–1334. [Google Scholar] [CrossRef] [Green Version]
- Ramón, R.S.; Pottier, C.; Gómez-Suárez, A.; Nolan, S.P. Gold(I)-Catalyzed Tandem Alkoxylation/Lactonization of γ-Hydroxy-α,β-Acetylenic Esters. Adv. Synth. Catal. 2011, 353, 1575–1583. [Google Scholar] [CrossRef]
- Zhdanko, A.; Maier, M.E. The Mechanism of Gold(I)-Catalyzed Hydroalkoxylation of Alkynes: An Extensive Experimental Study. Chem. Eur. J. 2014, 20, 1918–1930. [Google Scholar] [CrossRef]
- Larsen, M.H.; Houk, K.N.; Hashmi, A.S.K. Dual Gold Catalysis: Stepwise Catalyst Transfer via Dinuclear Clusters. J. Am. Chem. Soc. 2015, 137, 10668–10676. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Dual Gold Catalysis. Acc. Chem. Res. 2014, 47, 864–876. [Google Scholar] [CrossRef]
- Gómez-Suárez, A.; Nolan, S.P. Dinuclear Gold Catalysis: Are Two Gold Centers Better than One? Angew. Chem. Int. Ed. 2012, 51, 8156–8159. [Google Scholar] [CrossRef]
- Tkatchouk, E.; Mankad, N.P.; Benitez, D.; Goddard, W.A.; Toste, F.D. Two Metals Are Better Than One in the Gold Catalyzed Oxidative Heteroarylation of Alkenes. J. Am. Chem. Soc. 2011, 133, 14293–14300. [Google Scholar] [CrossRef] [Green Version]
- Hashmi, A.S.K.; Braun, I.; Nösel, P.; Schädlich, J.; Wieteck, M.; Rudolph, M.; Rominger, F. Simple Gold-Catalyzed Synthesis of Benzofulvenes—Gem-Diaurated Species as “Instant Dual-Activation” Precatalysts. Angew. Chem. Int. Ed. 2012, 51, 4456–4460. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Lauterbach, T.; Nösel, P.; Vilhelmsen, M.H.; Rudolph, M.; Rominger, F. Dual Gold Catalysis: σ,π-Propyne Acetylide and Hydroxyl-Bridged Digold Complexes as Easy-To-Prepare and Easy-To-Handle Precatalysts. Chem. Eur. J. 2013, 19, 1058–1065. [Google Scholar] [CrossRef]
- Oonishi, Y.; Gómez-Suárez, A.; Martin, A.R.; Nolan, S.P. Hydrophenoxylation of Alkynes by Cooperative Gold Catalysis. Angew. Chem. 2013, 125, 9949–9953. [Google Scholar] [CrossRef]
- Gómez-Suárez, A.; Oonishi, Y.; Martin, A.R.; Vummaleti, S.V.C.; Nelson, D.J.; Cordes, D.B.; Slawin, A.M.Z.; Cavallo, L.; Nolan, S.P.; Poater, A. On the Mechanism of the Digold(I)-Hydroxide-Catalysed Hydrophenoxylation of Alkynes. Chem. Eur. J. 2016, 22, 1125–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casals-Cruañas, È.; González-Belman, O.F.; Besalú-Sala, P.; Nelson, D.J.; Poater, A. The preference for dual-gold(I) catalysis in the hydro (alkoxylation vs. phenoxylation) of alkynes. Org. Biomol. Chem. 2017, 15, 6416–6425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccaccia, D.; Del Zotto, A.; Baratta, W. The pivotal role of the counterion in gold catalyzed hydration and alkoxylation of alkynes. Coord. Chem. Rev. 2019, 396, 103–116. [Google Scholar] [CrossRef]
- Gatto, M.; Baratta, W.; Belanzoni, P.; Belpassi, L.; Zotto, A.D.; Tarantelli, F.; Zuccaccia, D. Hydration and alkoxylation of alkynes catalyzed by NHC–Au–OTf. Green Chem. 2018, 20, 2125–2134. [Google Scholar] [CrossRef]
- Biasiolo, L.; Trinchillo, M.; Belanzoni, P.; Belpassi, L.; Busico, V.; Ciancaleoni, G.; D’Amora, A.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D. Unexpected Anion Effect in the Alkoxylation of Alkynes Catalyzed by N-Heterocyclic Carbene (NHC) Cationic Gold Complexes. Chem. Eur. J. 2014, 20, 14594–14598. [Google Scholar] [CrossRef]
- Trinchillo, M.; Belanzoni, P.; Belpassi, L.; Biasiolo, L.; Busico, V.; D’Amora, A.; D’Amore, L.; Del Zotto, A.; Tarantelli, F.; Tuzi, A.; et al. Extensive Experimental and Computational Study of Counterion Effect in the Reaction Mechanism of NHC-Gold(I)-Catalyzed Alkoxylation of Alkynes. Organometallics 2016, 35, 641–654. [Google Scholar] [CrossRef]
- D’Amore, L.; Ciancaleoni, G.; Belpassi, L.; Tarantelli, F.; Zuccaccia, D.; Belanzoni, P. Unraveling the Anion/Ligand Interplay in the Reaction Mechanism of Gold(I)-Catalyzed Alkoxylation of Alkynes. Organometallics 2017, 36, 2364–2376. [Google Scholar] [CrossRef]
- Biffis, A.; Baron, M.; Tubaro, C. Chapter Five—Poly-NHC Complexes of Transition Metals: Recent Applications and New Trends. Adv. Organomet. Chem. 2015, 63, 203–288. [Google Scholar]
- Schmidbaur, H.; Schier, A. Aurophilic interactions as a subject of current research: An up-date. Chem. Soc. Rev. 2011, 41, 370–412. [Google Scholar] [CrossRef] [PubMed]
- Schmidbaur, H.; Schier, A. A briefing on aurophilicity. Chem. Soc. Rev. 2008, 37, 1931–1951. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Tubaro, C.; Biffis, A.; Basato, M.; Graiff, C.; Poater, A.; Cavallo, L.; Armaroli, N.; Accorsi, G. Blue-Emitting Dinuclear N-heterocyclic Dicarbene Gold(I) Complex Featuring a Nearly Unit Quantum Yield. Inorg. Chem. 2012, 51, 1778–1784. [Google Scholar] [CrossRef]
- Monticelli, M.; Tubaro, C.; Baron, M.; Basato, M.; Sgarbossa, P.; Graiff, C.; Accorsi, G.; Pell, T.P.; Wilson, D.J.D.; Barnard, P.J. Metal complexes with di(N-heterocyclic carbene) ligands bearing a rigid ortho-, meta or para-phenylene bridge. Dalton Trans. 2016, 45, 9540–9552. [Google Scholar] [CrossRef]
- Tubaro, C.; Baron, M.; Costante, M.; Basato, M.; Biffis, A.; Gennaro, A.; Isse, A.A.; Graiff, C.; Accorsi, G. Dinuclear gold(I) complexes with propylene bridged N-heterocyclic dicarbene ligands: Synthesis, structures, and trends in reactivities and properties. Dalton Trans. 2013, 42, 10952–10963. [Google Scholar] [CrossRef]
- Longhi, A.; Baron, M.; Rancan, M.; Bottaro, G.; Armelao, L.; Sgarbossa, P.; Tubaro, C. Possible Synthetic Approaches for Heterobimetallic Complexes by Using nNHC/tzNHC Heteroditopic Carbene Ligands. Molecules 2019, 24, 2305. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, M.; Baron, M.; Tubaro, C.; Bellemin-Laponnaz, S.; Graiff, C.; Bottaro, G.; Armelao, L.; Orian, L. Structural and Luminescent Properties of Homoleptic Silver(I), Gold(I), and Palladium(II) Complexes with nNHC-tzNHC Heteroditopic Carbene Ligands. ACS Omega 2019, 4, 4192–4205. [Google Scholar] [CrossRef]
- Baron, M.; Tubaro, C.; Basato, M.; Isse, A.A.; Gennaro, A.; Cavallo, L.; Graiff, C.; Dolmella, A.; Falivene, L.; Caporaso, L. Insights into the Halogen Oxidative Addition Reaction to Dinuclear Gold(I) Di(NHC) Complexes. Chem. Eur. J. 2016, 22, 10211–10224. [Google Scholar] [CrossRef]
- Baron, M.; Tubaro, C.; Basato, M.; Biffis, A.; Graiff, C. Synthesis of dinuclear N-heterocyclic dicarbene Au(III)/Au(III) and Au(II)/Au(II) complexes via oxidative addition of chlorine or bromine to Au(I)/Au(I) species. J. Organomet. Chem. 2012, 714, 41–46. [Google Scholar] [CrossRef]
- Baron, M.; Tubaro, C.; Basato, M.; Natile, M.M.; Graiff, C. Oxidative halogenation of dinuclear N-heterocyclic dicarbene gold(I) complexes. J. Organomet. Chem. 2013, 723, 108–114. [Google Scholar] [CrossRef]
- Mageed, A.H.; Skelton, B.W.; Sobolev, A.N.; Baker, M.V. Formation of Dinuclear AuII and AuI/AuIII Mixed-Valence Complexes is Directed by Structural Constraints Imposed by Cyclophane-NHC Ligands. Eur. J. Inorg. Chem. 2018, 1, 109–120. [Google Scholar] [CrossRef]
- Baron, M.; Battistel, E.; Tubaro, C.; Biffis, A.; Armelao, L.; Rancan, M.; Graiff, C. Single-Step Synthesis of Dinuclear Neutral Gold(I) Complexes with Bridging Di(N-heterocyclic carbene) Ligands and Their Catalytic Performance in Cross Coupling Reactions and Alkyne Hydroamination. Organometallics 2018, 37, 4213–4223. [Google Scholar] [CrossRef]
- Goodwin, J.A.; Aponick, A. Regioselectivity in the Au-catalyzed hydration and hydroalkoxylation of alkynes. Chem. Commun. 2015, 51, 8730–8741. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, M.; Bergmann, M.; Müller-Borges, D.; Plenio, H. Bispentiptycenyl-N-Heterocyclic Carbene (NHC) Gold Complexes: Highly Active Catalysts for the Room Temperature Hydration of Alkynes. Adv. Synth. Catal. 2018, 360, 3572–3578. [Google Scholar] [CrossRef]
- Sirindil, F.; Nolan, S.P.; Dagorne, S.; Pale, P.; Blanc, A.; de Frémont, P. Synthesis, Characterization and Catalytic Activity of NHC Gold(I) Polyoxometalate Complexes. Chem. Eur. J. 2018, 24, 12630–12637. [Google Scholar] [CrossRef]
- Flahaut, A.; Roland, S.; Mangeney, P. Allylic alkylation and amination using mixed (NHC) (phosphine) palladium complexes under biphasic conditions. J. Organomet. Chem. 2007, 692, 5754–5762. [Google Scholar] [CrossRef]
- Brown, J.R.; Schwerdtfeger, P.; Schröder, D.; Schwarz, H. Experimental and theoretical studies of diatomic gold halides. J. Am. Soc. Mass Spectrom. 2002, 13, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Waghorne, W.E. Solubilities of the Silver Halides in Aqueous Mixtures of Methanol, Acetonitrile, and Dimethylsulfoxide. Mon. Chem. 2003, 134, 655–667. [Google Scholar] [CrossRef]
- Bruker. APEX3 and SAINT; Bruker AXS Inc.: Madison, WI, USA, 2015. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Cat | Co-Cat | T (°C) | t (h) | Alkyne Conversion (%) a |
|---|---|---|---|---|---|---|
| 1 | neat | Au2Br2L5 | AgSbF6 | rt | 22 | nr |
| 2 | neat | Au2Br2L5 | AgSbF6 | 40 | 1 | 100 |
| 3 | neat | Au2Br2L3 | AgOTf | rt | 22 | nr |
| 4 | neat | Au2Br2L5 | AgOTf | 40 | 1.5 18.5 | 86 89 |
| 5 | neat | Au2Br2L3 | AgOTf | 40 | 1 6.5 22 | 62 86 87 |
| 6 | CHCl3 | Au2Br2L5 | AgOTf | 40 | 1 | 100 |
| 7 | CH3CN b | Au2Br2L3 | AgOTf | 40 | 1 22 | <1 12 |
| 8 | neat | - | AgOTf | 40 | 22 | nr |
| Entry | Cat (mol%) | t (h) | Alkyne Conversion (%) a |
|---|---|---|---|
| 1 | Au2Br2L1 | 1.5 6.5 22 | 67 91 92 |
| 2 | Au2Br2L2 | 1.5 6.5 | 78 90 |
| 3 | Au2Br2L3 | 1.5 6.5 22 | 62 86 87 |
| 4 | Au2Br2L4 | 1 18.5 | 100 100 |
| 5 | Au2Br2L5 | 1 18.5 | 86 89 |
| 6 | Au2Br2L6 | 1 23 | 72 100 |
| Entry | Alkyne | Alcohol | t (h) | Alkyne Conversion (%) a | Yield (%) a |
|---|---|---|---|---|---|
| 1 | PhC≡CCO2Et | MeOH | 1 18.5 | 100 100 | (E)-3aa (33); 5a (67) 5a (100) |
| 2 | PhC≡CCO2Et | n-BuOH | 1 18.5 | 92 100 | (Z)-3ab (21); (E)-3ab (16); 5a (55) 5a (100) |
| 3 b | PhC≡CCO2Et | PhOH | 1 18.5 | 66 100 | (Z)-3ac (37); 5a (29) (Z)-3ac (31); 5a (69) |
| 4 | PhC≡CH | MeOH | 1 18.5 | 66 99 | 5b (66) 5b (99) |
| 5 | HC≡CCO2Et | MeOH | 1 18.5 | 50 100 | (Z)-3ca (15); (E)-3ca (19); 3ca’ (10); 4ca (6) (E)-3ca (26); 4ca (50); 5c (14) |
| 6 | EtC≡CEt | MeOH | 1 18.5 | 58 89 | 5d (58) 5d (89) |
| 7 | PhC≡CPh | MeOH | 1 18.5 | 11 63 | 5e (11) 5e (63) |
| 8 | PhC≡CCO2H | MeOH | 1 18.5 | 25 50 | 5b (14); 5g (6); 1g (5) 5b (37); 5g (3); 1g (10) |
| 9 c | PhC≡CPh | MeOH | 1 18.5 | 25 83 | (Z)-3ea (21); 5e (4) 5e (83) |
| Entry | Alkyne | Alcohol | Cat (mol%) | Co-Cat (mol%) | T (°C) | T (h) | Alkyne Conversion (%) a |
|---|---|---|---|---|---|---|---|
| 1 | EtC≡CEt | MeOH | IPrAuCl (2) | AgOTf (2) | 40 | 1 | 100 |
| 2 | EtC≡CEt | MeOH | AuIL7 (2) | AgOTf (2) | 40 | 1 18.5 | nr nr |
| 3 | PhC≡CCO2Et | MeOH | AuIL7 (2) | AgOTf (2) | 40 | 1 18.5 | nr nr |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcheggiani, E.; Tubaro, C.; Biffis, A.; Graiff, C.; Baron, M. Hydroalkoxylation of Terminal and Internal Alkynes Catalyzed by Dinuclear Gold(I) Complexes with Bridging Di(N-Heterocyclic Carbene) Ligands. Catalysts 2020, 10, 1. https://doi.org/10.3390/catal10010001
Marcheggiani E, Tubaro C, Biffis A, Graiff C, Baron M. Hydroalkoxylation of Terminal and Internal Alkynes Catalyzed by Dinuclear Gold(I) Complexes with Bridging Di(N-Heterocyclic Carbene) Ligands. Catalysts. 2020; 10(1):1. https://doi.org/10.3390/catal10010001
Chicago/Turabian StyleMarcheggiani, Elena, Cristina Tubaro, Andrea Biffis, Claudia Graiff, and Marco Baron. 2020. "Hydroalkoxylation of Terminal and Internal Alkynes Catalyzed by Dinuclear Gold(I) Complexes with Bridging Di(N-Heterocyclic Carbene) Ligands" Catalysts 10, no. 1: 1. https://doi.org/10.3390/catal10010001