
High Throughput Fluorescence-Based In Vitro Experimental Platform for the Identification of Effective Therapies to Overcome Tumour Microenvironment-Mediated Drug Resistance in AML
 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Determination of Cell Proliferation in eGFP-AML Cell Lines in Co-Culture with mCherry-HS5 Cells
2.3. Determination of AML Cell Viability by Flow Cytometry
2.4. Cell Cycle Analysis
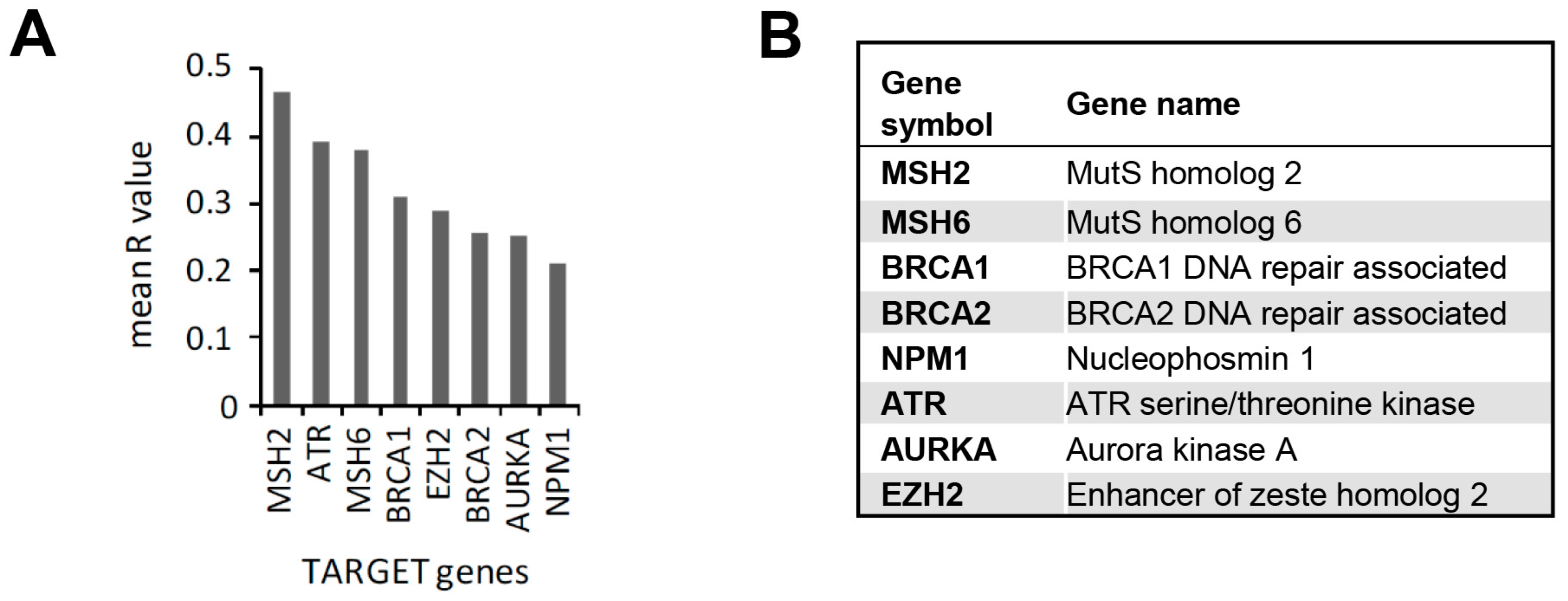
2.5. In Silico Analysis of the Correlation between the Expression of CRM1-Encoding XPO1 Gene and Genes in the TARGET Database
2.6. Statistics
3. Results
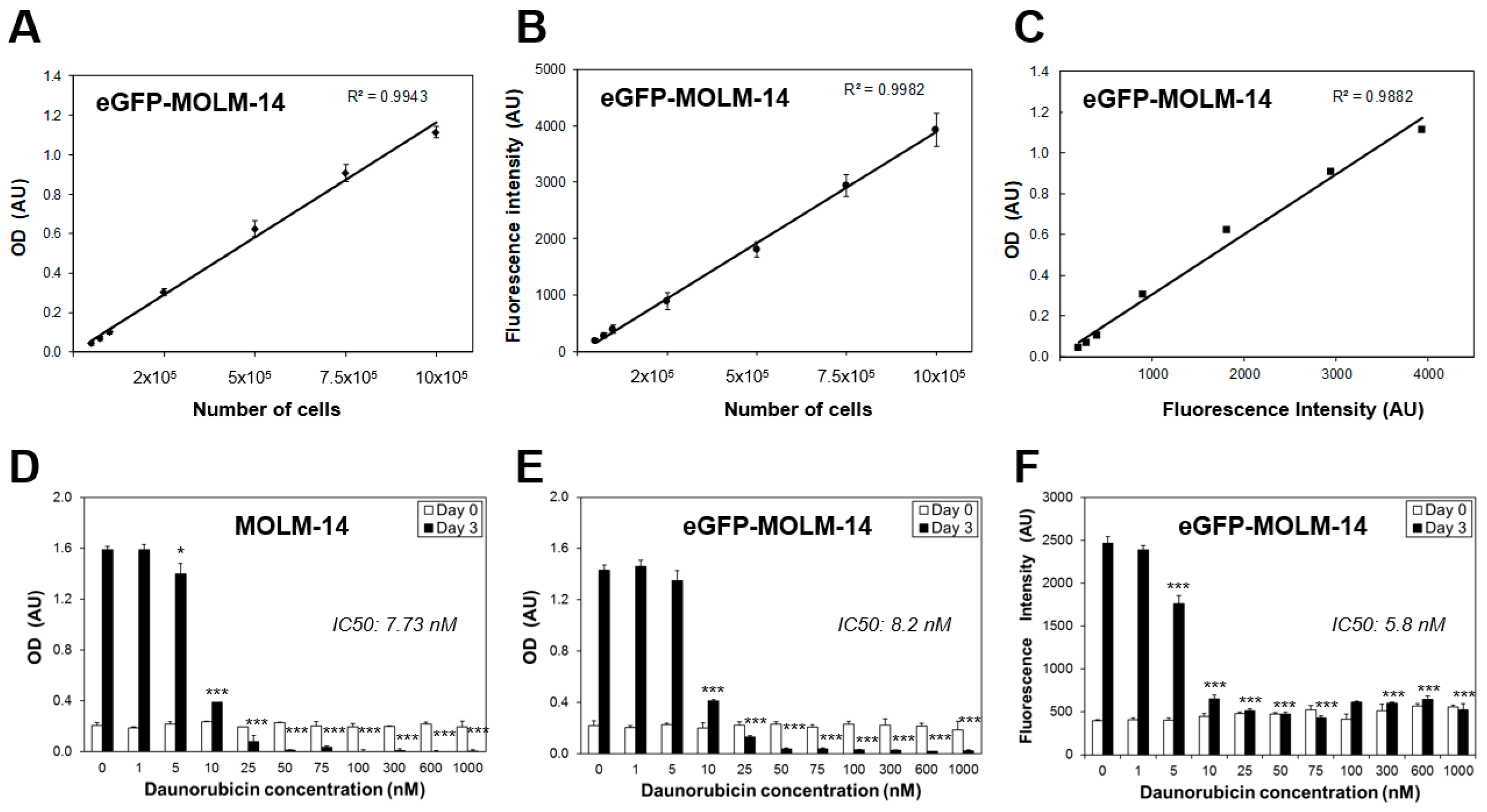
3.1. Characterisation of a High Throughput Experimental Platform to Distinguish Changes in the Proliferation of AML and BM Mesenchymal Stromal Cells in Co-Culture
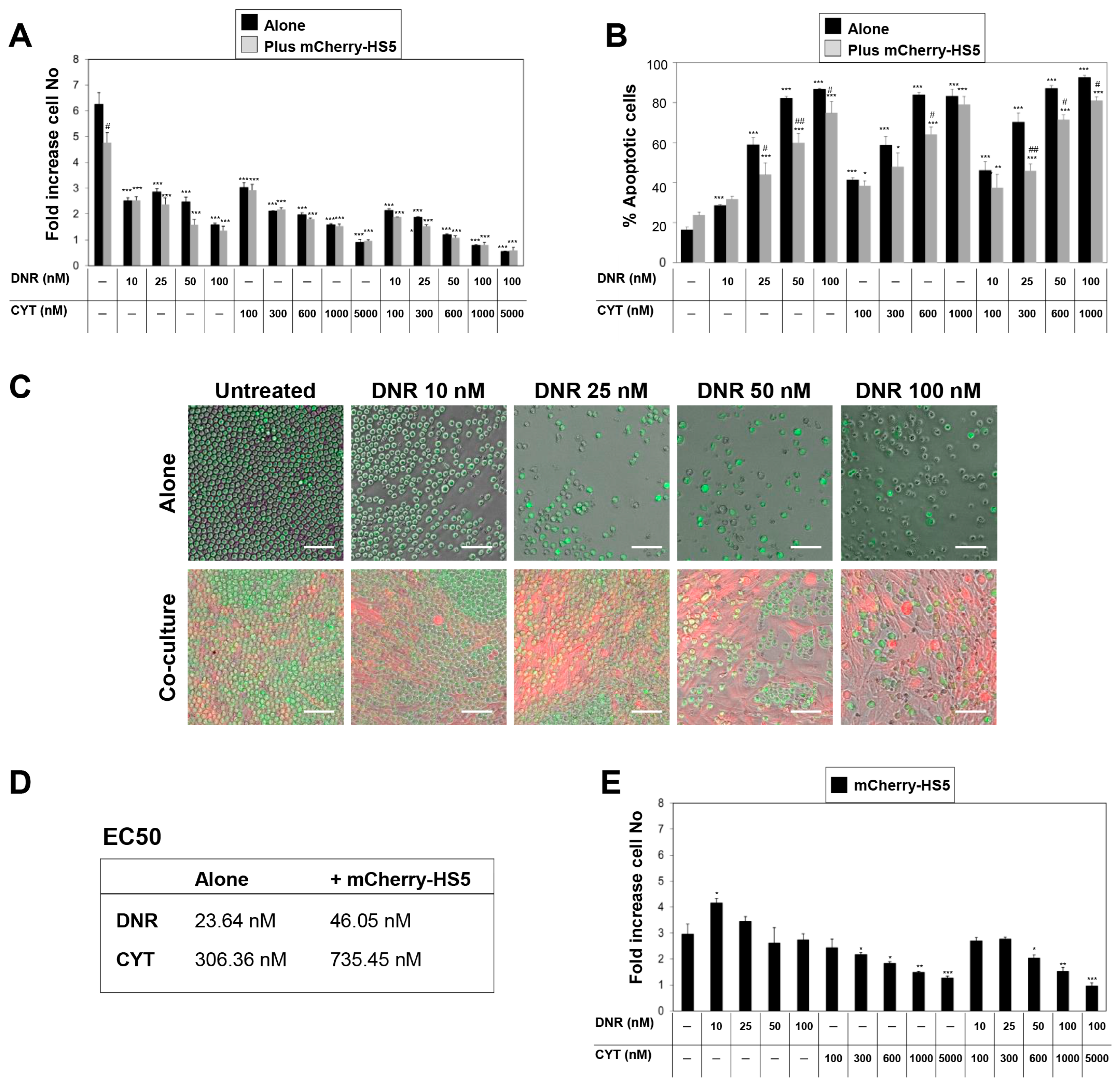
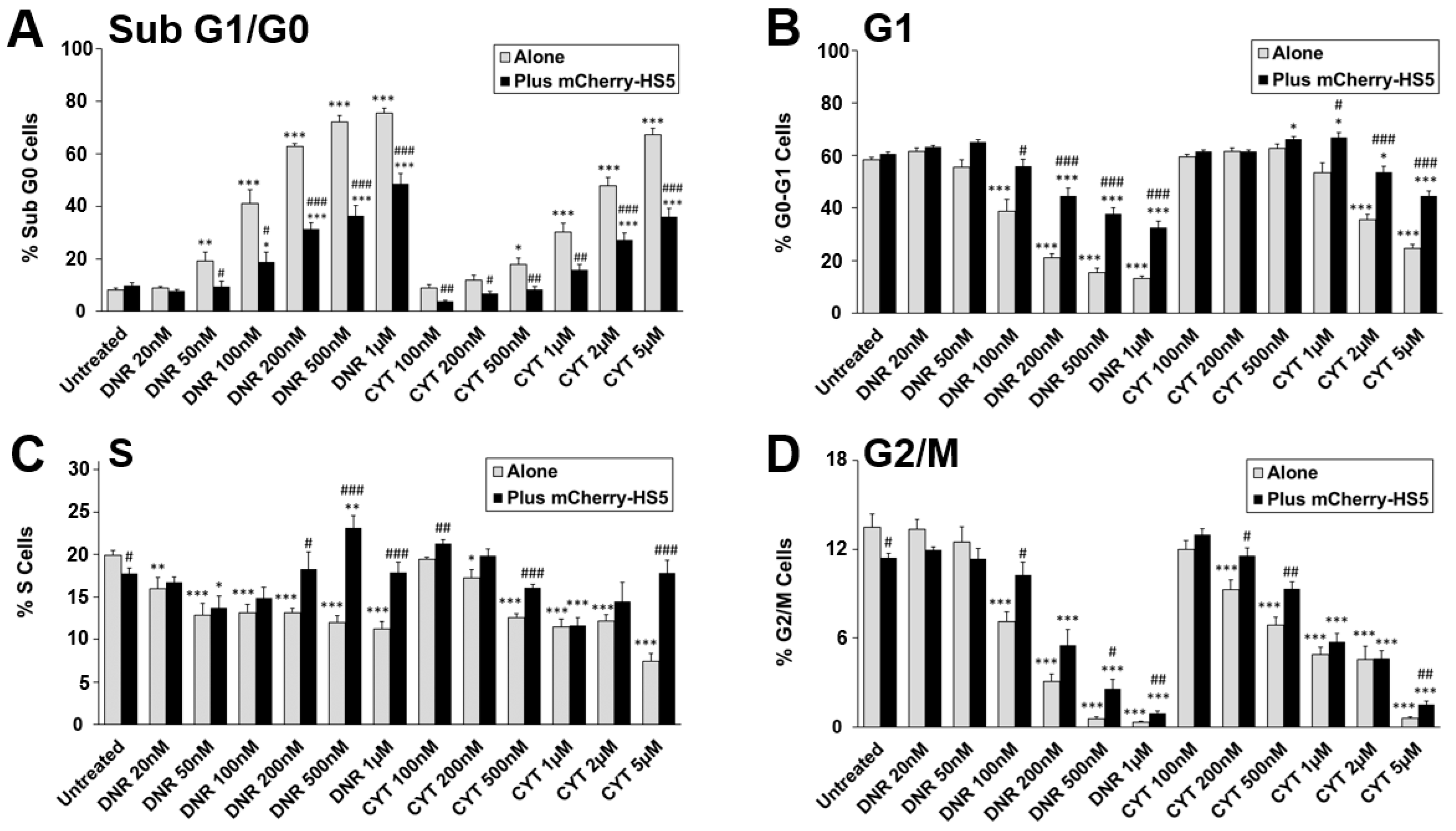
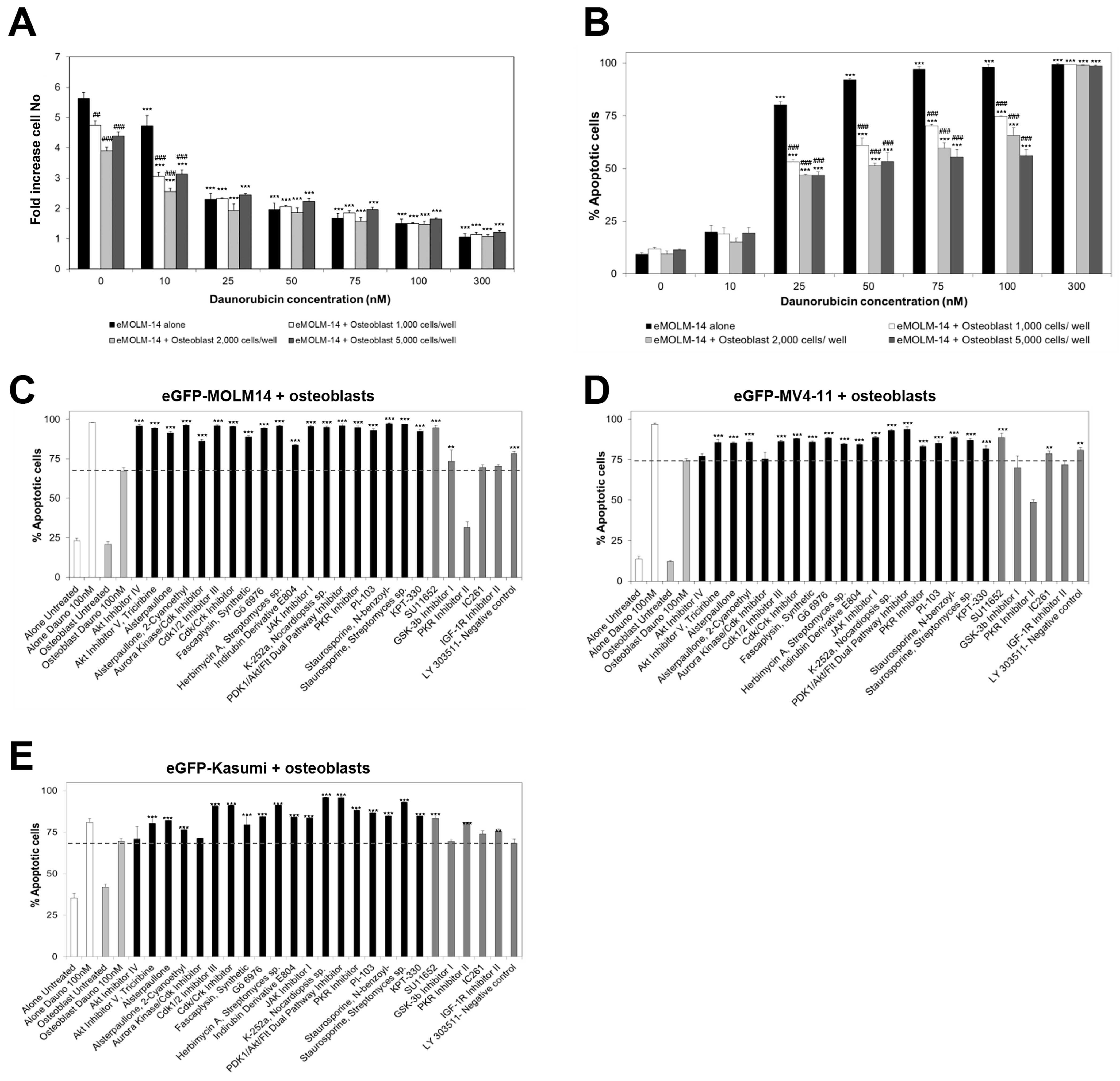
3.2. High Throughput Screening to Identify Drug Candidates and Signalling Pathways Involved in BM Stromal-Mediated Drug Resistance in AML
3.3. In Silico Identification of Potential New KPT-330-Based Combination Therapies against AML
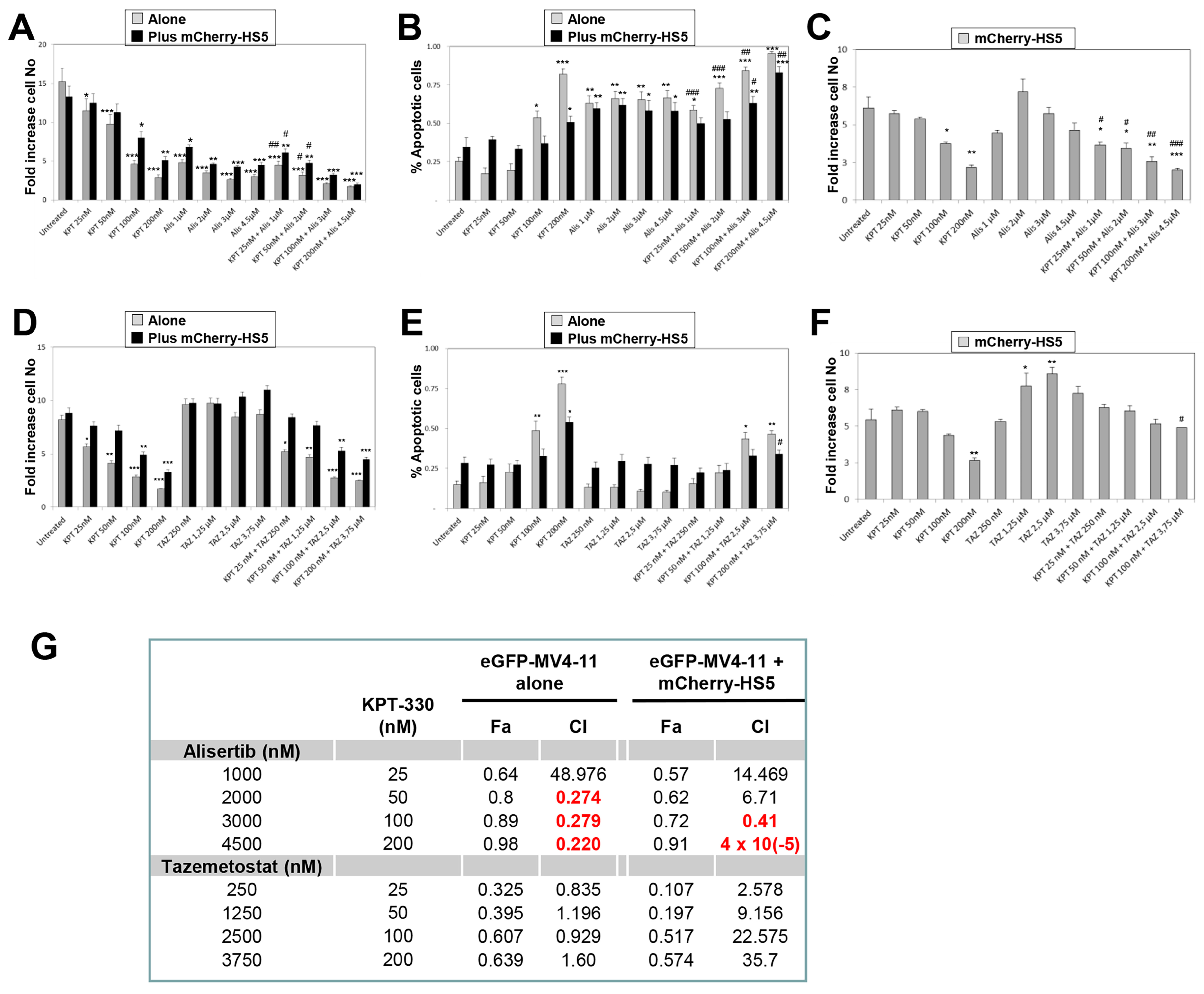
3.4. In Vitro Evaluation of Efficacy of Alisertib and Tazemetostat in Combination with KPT-330-to Overcome BM-Mediated Drug Resistance of AML Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ohno, R.; Kobayashi, T.; Tanimoto, M.; Hiraoka, A.; Imai, K.; Asou, N.; Tomonaga, M.; Tsubaki, K.; Takahashi, I.; Kodera, Y.; et al. Randomized study of individualized induction therapy with or without vincristine, and of maintenance-intensification therapy between 4 or 12 courses in adult acute myeloid leukemia. AML-87 Study of the Japan Adult Leukemia Study Group. Cancer 1993, 71, 3888–3895. [Google Scholar] [CrossRef] [PubMed]
- Hann, I.M.; Stevens, R.F.; Goldstone, A.H.; Rees, J.K.; Wheatley, K.; Gray, R.G.; Burnett, A.K. Randomized comparison of DAT versus ADE as induction chemotherapy in children and younger adults with acute myeloid leukemia. Results of the Medical Research Council’s 10th AML trial (MRC AML10). Adult and Childhood Leukaemia Working Parties of the Medical Research Council. Blood 1997, 89, 2311–2318. [Google Scholar]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M.; et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef]
- Dombret, H.; Gardin, C. An update of current treatments for adult acute myeloid leukemia. Blood 2016, 127, 53–61. [Google Scholar] [CrossRef]
- Luskin, M.R.; Lee, J.W.; Fernandez, H.F.; Abdel-Wahab, O.; Bennett, J.M.; Ketterling, R.P.; Lazarus, H.M.; Levine, R.L.; Litzow, M.R.; Paietta, E.M.; et al. Benefit of high-dose daunorubicin in AML induction extends across cytogenetic and molecular groups. Blood 2016, 127, 1551–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohty, R.; El Hamed, R.; Brissot, E.; Bazarbachi, A.; Mohty, M. New drugs before, during, and after hematopoietic stem cell transplantation for patients with acute myeloid leukemia. Haematologica 2023, 108, 321–341. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.J.; Blaise, D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood 2016, 127, 62–70. [Google Scholar] [CrossRef]
- Boissel, N.; Renneville, A.; Leguay, T.; Lefebvre, P.C.; Recher, C.; Lecerf, T.; Delabesse, E.; Berthon, C.; Blanchet, O.; Prebet, T.; et al. Dasatinib in high-risk core binding factor acute myeloid leukemia in first complete remission: A French Acute Myeloid Leukemia Intergroup trial. Haematologica 2015, 100, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Man, C.H.; Fung, T.K.; Ho, C.; Han, H.H.; Chow, H.C.; Ma, A.C.; Choi, W.W.; Lok, S.; Cheung, A.M.; Eaves, C.; et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: Favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood 2012, 119, 5133–5143. [Google Scholar] [CrossRef]
- Feller, N.; van der Pol, M.A.; Van Stijn, A.; Weijers, G.W.; Westra, A.H.; Evertse, B.W.; Ossenkoppele, G.J.; Schuurhuis, G.J. MRD parameters using immunophenotypic detection methods are highly reliable in predicting survival in acute myeloid leukaemia. Leukemia 2004, 18, 1380–1390. [Google Scholar] [CrossRef]
- Kern, W.; Voskova, D.; Schoch, C.; Hiddemann, W.; Schnittger, S.; Haferlach, T. Determination of relapse risk based on assessment of minimal residual disease during complete remission by multiparameter flow cytometry in unselected patients with acute myeloid leukemia. Blood 2004, 104, 3078–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsunaga, T.; Fukai, F.; Miura, S.; Nakane, Y.; Owaki, T.; Kodama, H.; Tanaka, M.; Nagaya, T.; Takimoto, R.; Takayama, T.; et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia 2008, 22, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, S.W.; Scadden, D.T.; Gilliland, D.G. The leukemic stem cell niche: Current concepts and therapeutic opportunities. Blood 2009, 114, 1150–1157. [Google Scholar] [CrossRef] [Green Version]
- Lane, S.W.; Wang, Y.J.; Lo, C.C.; Ragu, C.; Bullinger, L.; Sykes, S.M.; Ferraro, F.; Shterental, S.; Lin, C.P.; Gilliland, D.G.; et al. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood 2011, 118, 2849–2856. [Google Scholar] [CrossRef] [Green Version]
- Schuringa, J.J.; Schepers, H. Ex vivo assays to study self-renewal and long-term expansion of genetically modified primary human acute myeloid leukemia stem cells. Methods Mol. Biol. 2009, 538, 287–300. [Google Scholar]
- Weisberg, E.; Liu, Q.; Zhang, X.; Nelson, E.; Sattler, M.; Liu, F.; Nicolais, M.; Zhang, J.; Mitsiades, C.; Smith, R.W.; et al. Selective Akt Inhibitors Synergize with Tyrosine Kinase Inhibitors and Effectively Override Stroma-Associated Cytoprotection of Mutant FLT3-Positive AML Cells. PLoS ONE 2013, 8, e56473. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Hemann, M.T. DNA damage-mediated induction of a chemoresistant niche. Cell 2010, 143, 355–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 Paracrine Network Links Cancer Chemoresistance and Metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, K.; Khatun, H.; Macpherson, L.; Caley, M.P.; Sturge, J.; Mufti, G.J.; Schey, S.A.; Calle, Y. Fluorescence-based experimental model to evaluate the concomitant effect of drugs on the tumour microenvironment and cancer cells. Br. J. Haematol. 2012, 157, 564–579. [Google Scholar] [CrossRef]
- McMillin, D.W.; Delmore, J.; Weisberg, E.; Negri, J.M.; Geer, D.C.; Klippel, S.; Mitsiades, N.; Schlossman, R.L.; Munshi, N.C.; Kung, A.L.; et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat. Med. 2010, 16, 483–489. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef]
- De Toni, F.; Racaud-Sultan, C.; Chicanne, G.; Mas, V.M.; Cariven, C.; Mesange, F.; Salles, J.P.; Demur, C.; Allouche, M.; Payrastre, B.; et al. A crosstalk between the Wnt and the adhesion-dependent signaling pathways governs the chemosensitivity of acute myeloid leukemia. Oncogene 2006, 25, 3113–3122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobrial, I.M.; Detappe, A.; Anderson, K.C.; Steensma, D.P. The bone-marrow niche in MDS and MGUS: Implications for AML and MM. Nat. Rev. Clin. Oncol. 2018, 15, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, A.R.; Torrano, V.; Martin-Martin, N.; Caro-Maldonado, A.; Camacho, L.; Hermanova, I.; Guruceaga, E.; Lorenzo-Martin, L.F.; Caloto, R.; Gomis, R.R.; et al. CANCERTOOL: A Visualization and Representation Interface to Exploit Cancer Datasets. Cancer Res. 2018, 78, 6320–6328. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relashionships: The combined effects of multiple drugs or enzume inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Zwaan, C.M.; Meshinchi, S.; Radich, J.P.; Veerman, A.J.; Huismans, D.R.; Munske, L.; Podleschny, M.; Hahlen, K.; Pieters, R.; Zimmermann, M.; et al. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: Prognostic significance and relation to cellular drug resistance. Blood 2003, 102, 2387–2394. [Google Scholar] [CrossRef] [Green Version]
- Steudel, C.; Wermke, M.; Schaich, M.; Schakel, U.; Illmer, T.; Ehninger, G.; Thiede, C. Comparative analysis of MLL partial tandem duplication and FLT3 internal tandem duplication mutations in 956 adult patients with acute myeloid leukemia. Genes Chromosom. Cancer 2003, 37, 237–251. [Google Scholar] [CrossRef]
- Bogason, A.; Quartino, A.L.; Lafolie, P.; Masquelier, M.; Karlsson, M.O.; Paul, C.; Gruber, A.; Vitols, S. Inverse relationship between leukaemic cell burden and plasma concentrations of daunorubicin in patients with acute myeloid leukaemia. Br. J. Clin. Pharmacol. 2011, 71, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Krogh-Madsen, M.; Bender, B.; Jensen, M.K.; Nielsen, O.J.; Friberg, L.E.; Honore, P.H. Population pharmacokinetics of cytarabine, etoposide, and daunorubicin in the treatment for acute myeloid leukemia. Cancer Chemother. Pharm. 2012, 69, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Kashyap, T.; Yu, X.; Meng, X.; Lai, T.H.; McNeil, B.; Bhatnagar, B.; Shacham, S.; Kauffman, M.; Dorrance, A.M.; et al. XPO1 Inhibition using Selinexor Synergizes with Chemotherapy in Acute Myeloid Leukemia by Targeting DNA Repair and Restoring Topoisomerase IIalpha to the Nucleus. Clin. Cancer Res. 2016, 22, 6142–6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweet, K.; Komrokji, R.; Padron, E.; Cubitt, C.L.; Turner, J.G.; Zhou, J.; List, A.F.; Sallman, D.A.; Dawson, J.L.; Sullivan, D.M.; et al. Phase I Clinical Trial of Selinexor in Combination with Daunorubicin and Cytarabine in Previously Untreated Poor-Risk Acute Myeloid Leukemia. Clin. Cancer Res. 2020, 26, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashmi, M.S.; Sharif, M.A.; Jaan, A.; Salman, U.; Aziz, S.; Shahab, A.; Ahmed, Z.; Tariq, M.J.; Haider, M.Z.; Anwer, F. Efficacy and Safety of Selinexor for Acute Myeloid Leukemia: A Systematic Review. Blood 2020, 136, 15–16. [Google Scholar] [CrossRef]
- Janssen, J.; Lowenberg, B.; Manz, M.; Biemond, B.J.; Westerweel, P.E.; Klein, S.K.; Fehr, M.; Sinnige, H.A.M.; Efthymiou, A.; Legdeur, M.; et al. Addition of the nuclear export inhibitor selinexor to standard intensive treatment for elderly patients with acute myeloid leukemia and high risk myelodysplastic syndrome. Leukemia 2022, 36, 2189–2195. [Google Scholar] [CrossRef]
- Zhang, W.; Ly, C.; Ishizawa, J.; Mu, H.; Ruvolo, V.; Shacham, S.; Daver, N.; Andreeff, M. Combinatorial targeting of XPO1 and FLT3 exerts synergistic anti-leukemia effects through induction of differentiation and apoptosis in FLT3-mutated acute myeloid leukemias: From concept to clinical trial. Haematologica 2018, 103, 1642–1653. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.S.; Garzon, R.; Lapalombella, R. Selinexor for advanced hematologic malignancies. Leuk. Lymphoma 2020, 61, 2335–2350. [Google Scholar] [CrossRef]
- Kojima, K.; Kornblau, S.M.; Ruvolo, V.; Dilip, A.; Duvvuri, S.; Davis, R.E.; Zhang, M.; Wang, Z.; Coombes, K.R.; Zhang, N.; et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood 2013, 121, 4166–4174. [Google Scholar] [CrossRef]
- Saulino, D.M.; Younes, P.S.; Bailey, J.M.; Younes, M. CRM1/XPO1 expression in pancreatic adenocarcinoma correlates with survivin expression and the proliferative activity. Oncotarget 2018, 9, 21289–21295. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Camacho, S.C.; Silvers, T.R.; Razak, A.R.; Gabrail, N.Y.; Gerecitano, J.F.; Kalir, E.; Pereira, E.; Evans, B.R.; Ramus, S.J.; et al. Inhibition of the Nuclear Export Receptor XPO1 as a Therapeutic Target for Platinum-Resistant Ovarian Cancer. Clin. Cancer Res. 2017, 23, 1552–1563. [Google Scholar] [CrossRef] [Green Version]
- Shen, A.; Wang, Y.; Zhao, Y.; Zou, L.; Sun, L.; Cheng, C. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery 2009, 65, 153–159; discussion 159–160. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Saltarski, J.M.; White, M.A.; Scaglioni, P.P.; Gerber, D.E. Therapeutic Targeting of Nuclear Export Inhibition in Lung Cancer. J. Thorac. Oncol. 2017, 12, 1446–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arango, N.P.; Yuca, E.; Zhao, M.; Evans, K.W.; Scott, S.; Kim, C.; Gonzalez-Angulo, A.M.; Janku, F.; Ueno, N.T.; Tripathy, D.; et al. Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res. 2017, 19, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subhash, V.V.; Yeo, M.S.; Wang, L.; Tan, S.H.; Wong, F.Y.; Thuya, W.L.; Tan, W.L.; Peethala, P.C.; Soe, M.Y.; Tan, D.S.P.; et al. Anti-tumor efficacy of Selinexor (KPT-330) in gastric cancer is dependent on nuclear accumulation of p53 tumor suppressor. Sci. Rep. 2018, 8, 12248. [Google Scholar] [CrossRef] [Green Version]
- Gravina, G.L.; Mancini, A.; Sanita, P.; Vitale, F.; Marampon, F.; Ventura, L.; Landesman, Y.; McCauley, D.; Kauffman, M.; Shacham, S.; et al. KPT-330, a potent and selective exportin-1 (XPO-1) inhibitor, shows antitumor effects modulating the expression of cyclin D1 and survivin [corrected] in prostate cancer models. BMC Cancer 2015, 15, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aladhraei, M.; Kassem Al-Thobhani, A.; Poungvarin, N.; Suwannalert, P. Association of XPO1 Overexpression with NF-kappaB and Ki67 in Colorectal Cancer. Asian Pac. J. Cancer Prev. 2019, 20, 3747–3754. [Google Scholar] [CrossRef] [Green Version]
- Azizian, N.G.; Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol. 2020, 13, 61. [Google Scholar] [CrossRef]
- Taylor, J.; Sendino, M.; Gorelick, A.N.; Pastore, A.; Chang, M.T.; Penson, A.V.; Gavrila, E.I.; Stewart, C.; Melnik, E.M.; Herrejon Chavez, F.; et al. Altered Nuclear Export Signal Recognition as a Driver of Oncogenesis. Cancer Discov. 2019, 9, 1452–1467. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Qiao, W.; Sun, Q.; Luo, Y. Chromosome Region Maintenance 1 (XPO1/CRM1) as an Anticancer Target and Discovery of Its Inhibitor. J. Med. Chem. 2021, 64, 15534–15548. [Google Scholar] [CrossRef]
- Lu, X.; Lu, X.; Wang, Z.C.; Iglehart, J.D.; Zhang, X.; Richardson, A.L. Predicting features of breast cancer with gene expression patterns. Breast Cancer Res. Treat. 2008, 108, 191–201. [Google Scholar] [CrossRef]
- Ivshina, A.V.; George, J.; Senko, O.; Mow, B.; Putti, T.C.; Smeds, J.; Lindahl, T.; Pawitan, Y.; Hall, P.; Nordgren, H.; et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006, 66, 10292–10301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawitan, Y.; Bjohle, J.; Amler, L.; Borg, A.L.; Egyhazi, S.; Hall, P.; Han, X.; Holmberg, L.; Huang, F.; Klaar, S.; et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: Derived and validated in two population-based cohorts. Breast Cancer Res. 2005, 7, R953–R964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Klijn, J.G.; Zhang, Y.; Sieuwerts, A.M.; Look, M.P.; Yang, F.; Talantov, D.; Timmermans, M.; Meijer-van Gelder, M.E.; Yu, J.; et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005, 365, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Chitale, D.; Gong, Y.; Taylor, B.S.; Broderick, S.; Brennan, C.; Somwar, R.; Golas, B.; Wang, L.; Motoi, N.; Szoke, J.; et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene 2009, 28, 2773–2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma; Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Yin, X.; Walter, V.; Zhao, N.; Cabanski, C.R.; Hayward, M.C.; Miller, C.R.; Socinski, M.A.; Parsons, A.M.; Thorne, L.B.; et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS ONE 2012, 7, e36530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinsky, G.V.; Glinskii, A.B.; Stephenson, A.J.; Hoffman, R.M.; Gerald, W.L. Gene expression profiling predicts clinical outcome of prostate cancer. J. Clin. Investig. 2004, 113, 913–923. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, J.; Li, C.; Higgins, J.P.; van de Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorissen, R.N.; Gibbs, P.; Christie, M.; Prakash, S.; Lipton, L.; Desai, J.; Kerr, D.; Aaltonen, L.A.; Arango, D.; Kruhoffer, M.; et al. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clin. Cancer Res. 2009, 15, 7642–7651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, K.; Versloot, M.; Cameron, K.; Colak, S.; de Sousa e Melo, F.; de Jong, J.H.; Bleackley, J.; Vermeulen, L.; Versteeg, R.; Koster, J.; et al. Mutations in the Ras-Raf Axis underlie the prognostic value of CD133 in colorectal cancer. Clin. Cancer Res. 2012, 18, 3132–3141. [Google Scholar] [CrossRef] [Green Version]
- Laibe, S.; Lagarde, A.; Ferrari, A.; Monges, G.; Birnbaum, D.; Olschwang, S.; Project, C.O.L. A seven-gene signature aggregates a subgroup of stage II colon cancers with stage III. OMICS 2012, 16, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Marisa, L.; de Reynies, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene expression classification of colon cancer into molecular subtypes: Characterization, validation, and prognostic value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [Green Version]
- Roepman, P.; Schlicker, A.; Tabernero, J.; Majewski, I.; Tian, S.; Moreno, V.; Snel, M.H.; Chresta, C.M.; Rosenberg, R.; Nitsche, U.; et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int. J. Cancer 2014, 134, 552–562. [Google Scholar] [CrossRef]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [Green Version]
- Raaphorst, F.M.; Meijer, C.J.; Fieret, E.; Blokzijl, T.; Mommers, E.; Buerger, H.; Packeisen, J.; Sewalt, R.A.; Otte, A.P.; van Diest, P.J. Poorly differentiated breast carcinoma is associated with increased expression of the human polycomb group EZH2 gene. Neoplasia 2003, 5, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Sudo, T.; Utsunomiya, T.; Mimori, K.; Nagahara, H.; Ogawa, K.; Inoue, H.; Wakiyama, S.; Fujita, H.; Shirouzu, K.; Mori, M. Clinicopathological significance of EZH2 mRNA expression in patients with hepatocellular carcinoma. Br. J. Cancer 2005, 92, 1754–1758. [Google Scholar] [CrossRef]
- Damodaran, A.P.; Vaufrey, L.; Gavard, O.; Prigent, C. Aurora A Kinase Is a Priority Pharmaceutical Target for the Treatment of Cancers. Trends Pharm. Sci. 2017, 38, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Kim, C.G.; Lim, Y.; Shin, S.Y. Aurora kinase A inhibitor TCS7010 demonstrates pro-apoptotic effect through the unfolded protein response pathway in HCT116 colon cancer cells. Oncol. Lett. 2017, 14, 6571–6577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Hojfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Xu, Y. Effects of Enhancer of Zeste Homolog 2 (EZH2) Expression on Brain Glioma Cell Proliferation and Tumorigenesis. Med. Sci. Monit. 2018, 24, 7249–7255. [Google Scholar] [CrossRef] [PubMed]
- Dawei, H.; Honggang, D.; Qian, W. AURKA contributes to the progression of oral squamous cell carcinoma (OSCC) through modulating epithelial-to-mesenchymal transition (EMT) and apoptosis via the regulation of ROS. Biochem. Biophys. Res. Commun. 2018, 507, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, K.; Sogaard Helbo, A.; Fibiger Munch-Petersen, H.; Sjo, L.; Christensen, J.; Sommer Kristensen, L.; Asmar, F.; Hermansen, N.E.U.; O’Connel, C.; Gimsing, P.; et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol. Oncol. 2018, 12, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Felgenhauer, J.; Tomino, L.; Selich-Anderson, J.; Bopp, E.; Shah, N. Dual BRD4 and AURKA Inhibition Is Synergistic against MYCN-Amplified and Nonamplified Neuroblastoma. Neoplasia 2018, 20, 965–974. [Google Scholar] [CrossRef]
- Fioravanti, R.; Stazi, G.; Zwergel, C.; Valente, S.; Mai, A. Six Years (2012–2018) of Researches on Catalytic EZH2 Inhibitors: The Boom of the 2-Pyridone Compounds. Chem. Rec. 2018, 18, 1818–1832. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Marachelian, A.; Fox, E.; Kudgus, R.A.; Reid, J.M.; Groshen, S.; Malvar, J.; Bagatell, R.; Wagner, L.; Maris, J.M.; et al. Phase I Study of the Aurora A Kinase Inhibitor Alisertib in Combination with Irinotecan and Temozolomide for Patients With Relapsed or Refractory Neuroblastoma: A NANT (New Approaches to Neuroblastoma Therapy) Trial. J. Clin. Oncol. 2016, 34, 1368–1375. [Google Scholar] [CrossRef] [Green Version]
- Argon, E.K.; Nguyen, F.; Rajarethinam, A.; Kansra, V.; Pettitt, A. Abstract CT170: Phase 1 study to characterize the pharmacokinetics and safety of tazemetostat in b-cell lymphomas or advanced solid tumors. Cancer Res. 2020, 80, CT170. [Google Scholar] [CrossRef]
- Schepers, K.; Campbell, T.B.; Passegue, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem. Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Strowig, T.; Verma, R.; Koduru, S.; Hafemann, A.; Hopf, S.; Kocoglu, M.H.; Borsotti, C.; Zhang, L.; Branagan, A.; et al. Microenvironment-dependent growth of preneoplastic and malignant plasma cells in humanized mice. Nat. Med. 2016, 22, 1351–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarnerio, J.; Mendez, L.M.; Asada, N.; Menon, A.V.; Fung, J.; Berry, K.; Frenette, P.S.; Ito, K.; Pandolfi, P.P. A non-cell-autonomous role for Pml in the maintenance of leukemia from the niche. Nat. Commun. 2018, 9, 66. [Google Scholar] [CrossRef] [Green Version]
- Sendino, M.; Omaetxebarria, M.J.; Prieto, G.; Rodriguez, J.A. Using a Simple Cellular Assay to Map NES Motifs in Cancer-Related Proteins, Gain Insight into CRM1-Mediated NES Export, and Search for NES-Harboring Micropeptides. Int. J. Mol. Sci. 2020, 21, 6341. [Google Scholar] [CrossRef]
- Arnaoutov, A.; Azuma, Y.; Ribbeck, K.; Joseph, J.; Boyarchuk, Y.; Karpova, T.; McNally, J.; Dasso, M. Crm1 is a mitotic effector of Ran-GTP in somatic cells. Nat. Cell Biol. 2005, 7, 626–632. [Google Scholar] [CrossRef]
- Xu, D.; Grishin, N.V.; Chook, Y.M. NESdb: A database of NES-containing CRM1 cargoes. Mol. Biol. Cell 2012, 23, 3673–3676. [Google Scholar] [CrossRef] [PubMed]
- Garitano-Trojaola, A.; Sancho, A.; Gotz, R.; Eiring, P.; Walz, S.; Jetani, H.; Gil-Pulido, J.; Da Via, M.C.; Teufel, E.; Rhodes, N.; et al. Actin cytoskeleton deregulation confers midostaurin resistance in FLT3-mutant acute myeloid leukemia. Commun. Biol. 2021, 4, 799. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, R.; Wang, H.; Sun, M.; Lee, D.G.; Peng, J.; Thiele, C.J. Combining selinexor with alisertib to target the p53 pathway in neuroblastoma. Neoplasia 2022, 26, 100776. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % eGFP-MOLM-14 Apoptotic Cells | % Inhib Prolif mCh-HS5 | |
|---|---|---|
| KPT-330 | 92.7 | 57.3 |
| Staurosporine, Streptomyces sp. (PKC inhibitor) | 99.9 | 78.1 |
| Staurosporine, N-benzoyl- (PKC inhibitor) | 78.1 | 39.7 |
| PKR Inhibitor | 99.9 | 42.5 |
| PI-103 (PI3K/mTOR inhibitor) | 81.4 | 47.8 |
| PDK1/Akt/Flt Dual Pathway Inhibitor | 99.9 | 87.9 |
| PDGF RTK Inhibitor | 76.2 | 13.5 |
| K-252a, Nocardiopsis sp. (PKA, PKC, PKG, others) | 100.0 | 51.2 |
| JAK Inhibitor I | 98.1 | 11.5 |
| Indirubin Derivative E804 (Src-STA3 inhibitor) | 80.9 | 8.9 |
| IKK-2 Inhibitor IV * | 81.5 | 0.8 |
| Herbimycin A, Streptomyces sp. (Src inhibitor) | 99.2 | 36.7 |
| GSK-3 Inhibitor XIII * | 75.3 | 0.6 |
| Gö 6976 (PKC inhibitor) | 94.9 | 40.2 |
| Fascaplysin, (CDK4 inhibitor) | 82.1 | 60.1 |
| Cdk1/2 Inhibitor III | 99.8 | 45.2 |
| Cdk/Crk Inhibitor | 99.9 | 56.2 |
| Aurora Kinase/Cdk Inhibitor * | 81.9 | -15.2 |
| Alsterpaullone, 2-Cyanoethyl (GSK-3β, CDK5/p25, CDK1/cyclin B) | 99.7 | 55 |
| Alsterpaullone * (GSK-3β, CDK5/p25, CDK1/cyclin B) | 97.1 | 6.3 |
| Akt Inhibitor V, Triciribine | 81.1 | 24.7 |
| Akt Inhibitor IV | 89.4 | 56.2 |
| eMOLM-14 Alone | eMOLM-14 + mCherry-HS5 | ||||
|---|---|---|---|---|---|
| Dauno (nM) | Fa | CI | Fa | CI | |
| Cytarabine | |||||
| 100 nM | 10 | 0.709 | 0.94 | 0.415 | 3.083 |
| 300 nM | 25 | 0.855 | 1.446 | 0.573 | 2.271 |
| 600 nM | 50 | 0.988 | 0.754 | 0.685 | 1.79 |
| 1 µM | 100 | 0.995 | 1.005 | 0.799 | 1.178 |
| Cdk/Crk Inhibitor | |||||
| 10 nM | 10 | 0.397 | 1.175 | 0.452 | 1.133 |
| 20 nM | 25 | 0.877 | 1.052 | 0.653 | 1.104 |
| 30 nM | 50 | 0.992 | 0.644 | 0.828 | 0.789 |
| 40 nM | 100 | 0.994 | 1.082 | 0.916 | 0.601 |
| Fascaplysin | |||||
| 200 nM | 10 | 0.907 | 0.925 | 0.530 | 1.076 |
| 300 nM | 25 | 0.984 | 0.957 | 0.766 | 0.605 |
| 400 nM | 50 | 0.990 | 1.337 | 0.867 | 0.537 |
| 600 nM | 100 | 0.993 | 2.097 | 0.957 | 0.426 |
| PDK1/Akt/Flt Inhibitor | |||||
| 100 nM | 10 | 0.340 | 1.522 | 0.324 | 2.129 |
| 200 nM | 25 | 0.966 | 0.834 | 0.667 | 0.988 |
| 300 nM | 50 | 0.991 | 0.917 | 0.728 | 1.285 |
| 400 nM | 100 | 0.993 | 1.466 | 0.975 | 0.116 |
| Go6976 | |||||
| 500 nM | 10 | 0.883 | 0.605 | 0.498 | 0.753 |
| 600 nM | 25 | 0.989 | 0.396 | 0.795 | 0.372 |
| 700 nM | 50 | 0.993 | 0.644 | 0.837 | 0.484 |
| 800 nM | 100 | 0.996 | 1.031 | 0.919 | 0.39 |
| Staurosporine, N-benzoyl | |||||
| 200 nM | 10 | 0.975 | 0.46 | 0.471 | 0.909 |
| 400 nM | 25 | 0.993 | 0.441 | 0.629 | 0.841 |
| 600 nM | 50 | 0.993 | 0.791 | 0.765 | 0.6 |
| 800 nM | 100 | 0.992 | 1.501 | 0.916 | 0.192 |
| Staurosporine, Streptomyces sp. | |||||
| 5 nM | 10 | 0.992 | 0.247 | 0.454 | 1.216 |
| 10 nM | 25 | 0.990 | 0.84 | 0.763 | 0.529 |
| 20 nM | 50 | 0.991 | 1.357 | 0.901 | 0.425 |
| 30 nM | 100 | 0.994 | 1.279 | 0.949 | 0.363 |
| KPT-330 | |||||
| 100 nM | 10 | 0.756 | 1.118 | 0.485 | 1.097 |
| 130 nM | 25 | 0.959 | 1.024 | 0.693 | 0.748 |
| 160 nM | 50 | 0.992 | 0.957 | 0.824 | 0.549 |
| 200 nM | 100 | 0.994 | 1.404 | 0.929 | 0.34 |
| Cancer Type | Study/Reference | Cohort Size | ID |
|---|---|---|---|
| Breast cancer | Lu et al., 2008 [50] | 131 | GEO: GSE5460 |
| Ivshina et al., 2016 [51] | 249 | GEO: GSE4922 | |
| TCGA | 522 | ||
| Pawitan et al., 2005 [52] | 159 | GEO: GSE1456 | |
| Wang et al., 2005 [53] | 286 | GEO: GSE2034 | |
| Lung cancer | Chitale et al., 2009 [54] | 128 | |
| Sheden et al., 2008 [55] | 442 | GEO: GSE68465 | |
| TCGA | 514 | ||
| Wilkerson et al., 2012 [56] | 116 | GEO: GSE26939 | |
| Prostate cancer | Glinsky et al., 2004 [57] | 79 | |
| Grasso et al., 2012 [58] | 88 | GEO: GSE35988 | |
| Lapointe et al., 2004 [59] | 26 | GEO: GSE3933 | |
| Taylor et al., 2010 [60] | 179 | GEO: GSE21034 | |
| TCGA | 496 | ||
| Varambally et al., 2005 [61] | 19 | GEO: GSE3325 | |
| Colorectal cancer | Colonomics | 246 | GEO: GSE44076 |
| Jorissen et al., 2009 [62] | 290 | GEO: GSE14333 | |
| Kemper et al., 2012 [63] | 90 | GEO: GSE33113 | |
| Laibe et al., 2012 [64] | 130 | GEO: GSE37892 | |
| Marisa et al., 2013 [65] | 585 | GEO: GSE39582 | |
| Roepman et al., 2014 [66] | 188 | GEO: GSE42284 | |
| TCGA | 374 | ||
| Acute myeloid leukaemia (AML) | Tyner et al., 2018 [67] | 672 | dbGaP: 30641 |
| Liu et al., 2018 [68] | 200 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arroyo-Berdugo, Y.; Sendino, M.; Greaves, D.; Nojszewska, N.; Idilli, O.; So, C.W.; Di Silvio, L.; Quartey-Papafio, R.; Farzaneh, F.; Rodriguez, J.A.; et al. High Throughput Fluorescence-Based In Vitro Experimental Platform for the Identification of Effective Therapies to Overcome Tumour Microenvironment-Mediated Drug Resistance in AML. Cancers 2023, 15, 1988. https://doi.org/10.3390/cancers15071988
Arroyo-Berdugo Y, Sendino M, Greaves D, Nojszewska N, Idilli O, So CW, Di Silvio L, Quartey-Papafio R, Farzaneh F, Rodriguez JA, et al. High Throughput Fluorescence-Based In Vitro Experimental Platform for the Identification of Effective Therapies to Overcome Tumour Microenvironment-Mediated Drug Resistance in AML. Cancers. 2023; 15(7):1988. https://doi.org/10.3390/cancers15071988
Chicago/Turabian StyleArroyo-Berdugo, Yoana, Maria Sendino, David Greaves, Natalia Nojszewska, Orest Idilli, Chi Wai So, Lucy Di Silvio, Ruby Quartey-Papafio, Farzin Farzaneh, Jose Antonio Rodriguez, and et al. 2023. "High Throughput Fluorescence-Based In Vitro Experimental Platform for the Identification of Effective Therapies to Overcome Tumour Microenvironment-Mediated Drug Resistance in AML" Cancers 15, no. 7: 1988. https://doi.org/10.3390/cancers15071988