
Comprehensive Profiling and Therapeutic Insights into Differentially Expressed Genes in Hepatocellular Carcinoma

 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Mining and Processing
2.2. Functional Enrichment and Pathway Analysis
2.3. Identification of Hub Genes and Transcription Factors and Protein–Protein Interaction Network Elaboration
2.4. Drug Screening and the Association of Genes with Cancer Cell Events
3. Results
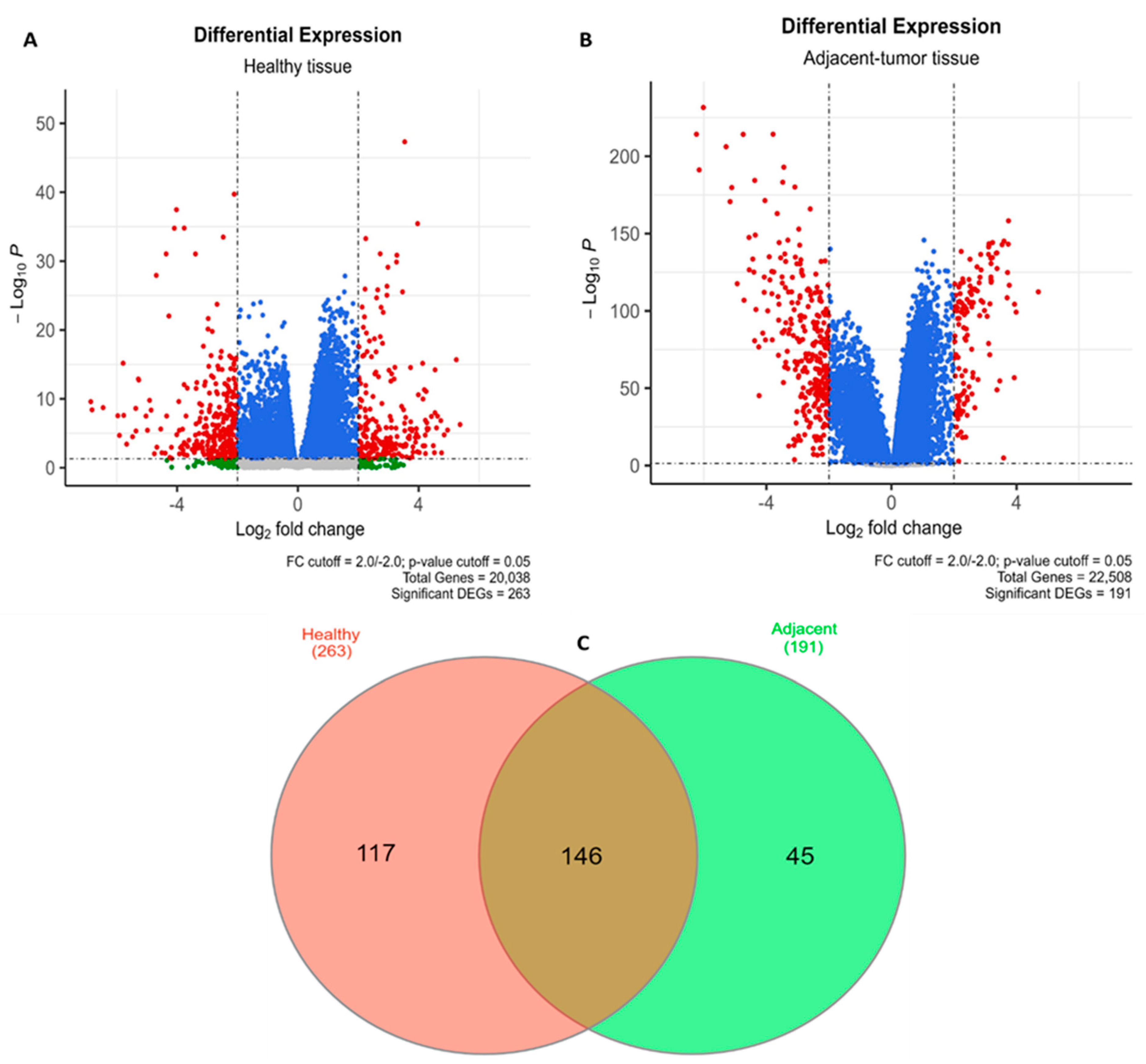
3.1. Data Mining, Filtering, and HCC—Associated Gene Identification
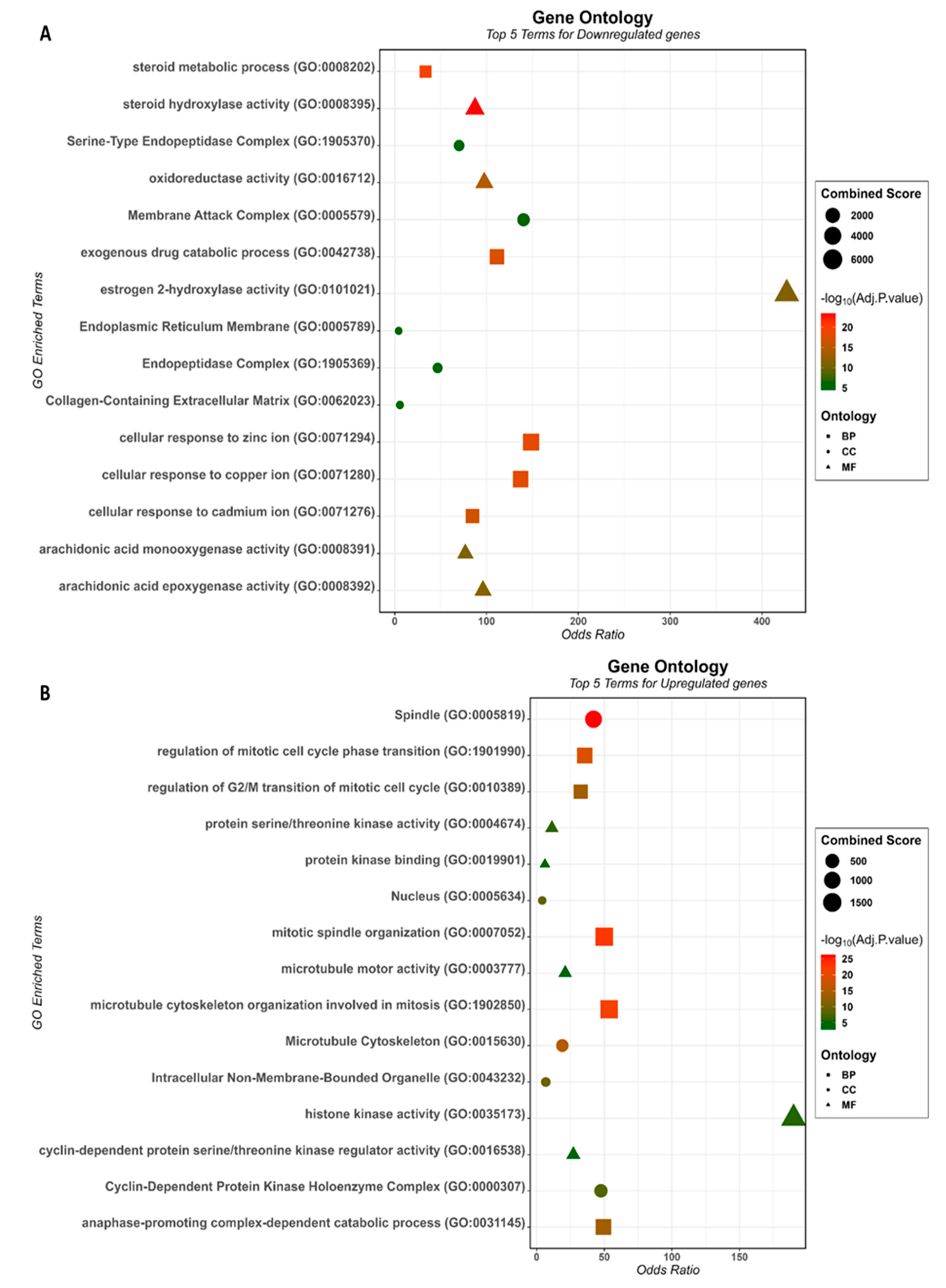
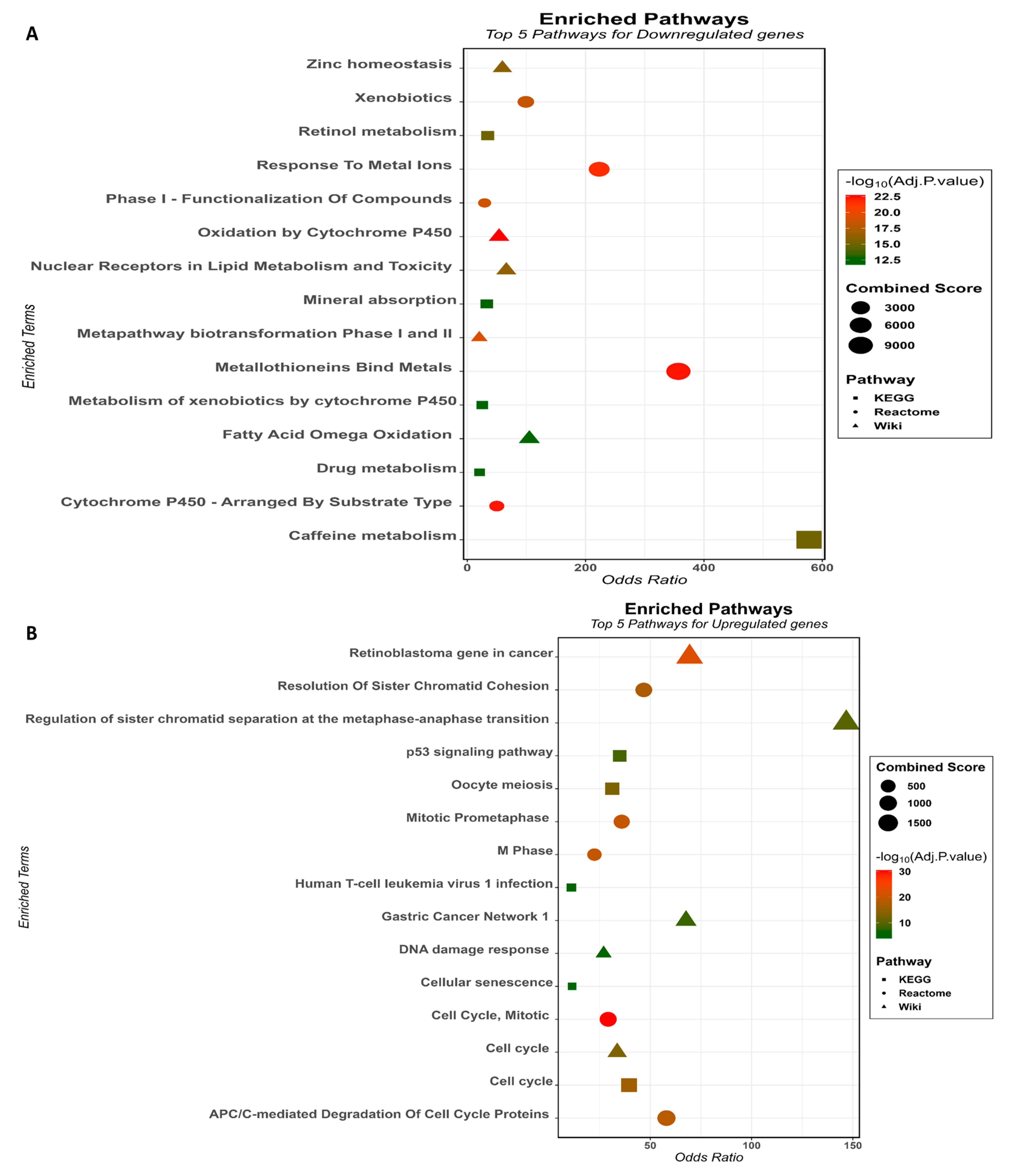
3.2. Gene Ontology and Pathway Enrichment Analysis of the HCC-Associated Genes
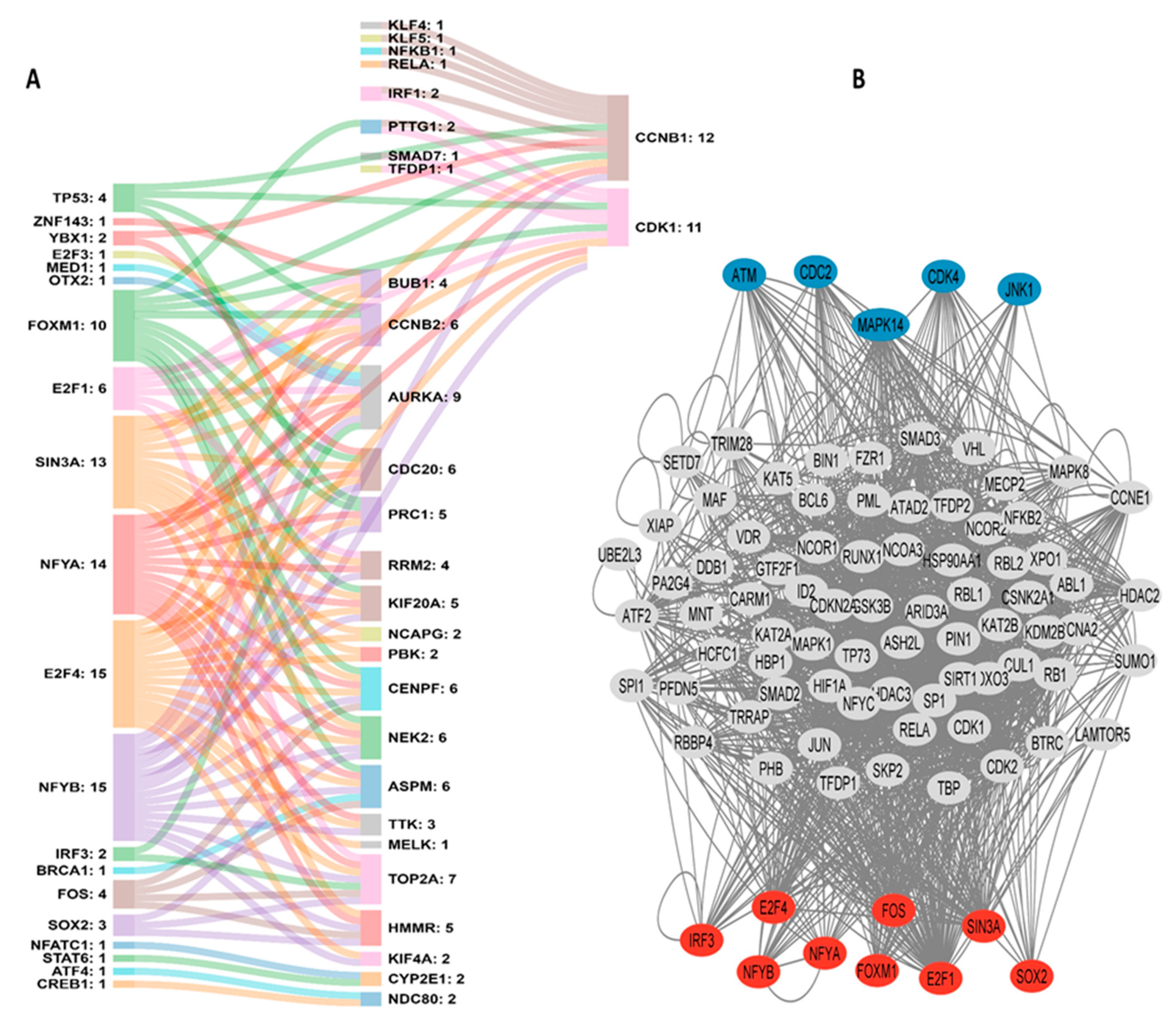
3.3. Hub Genes, Transcription Factors, and the PPI Network
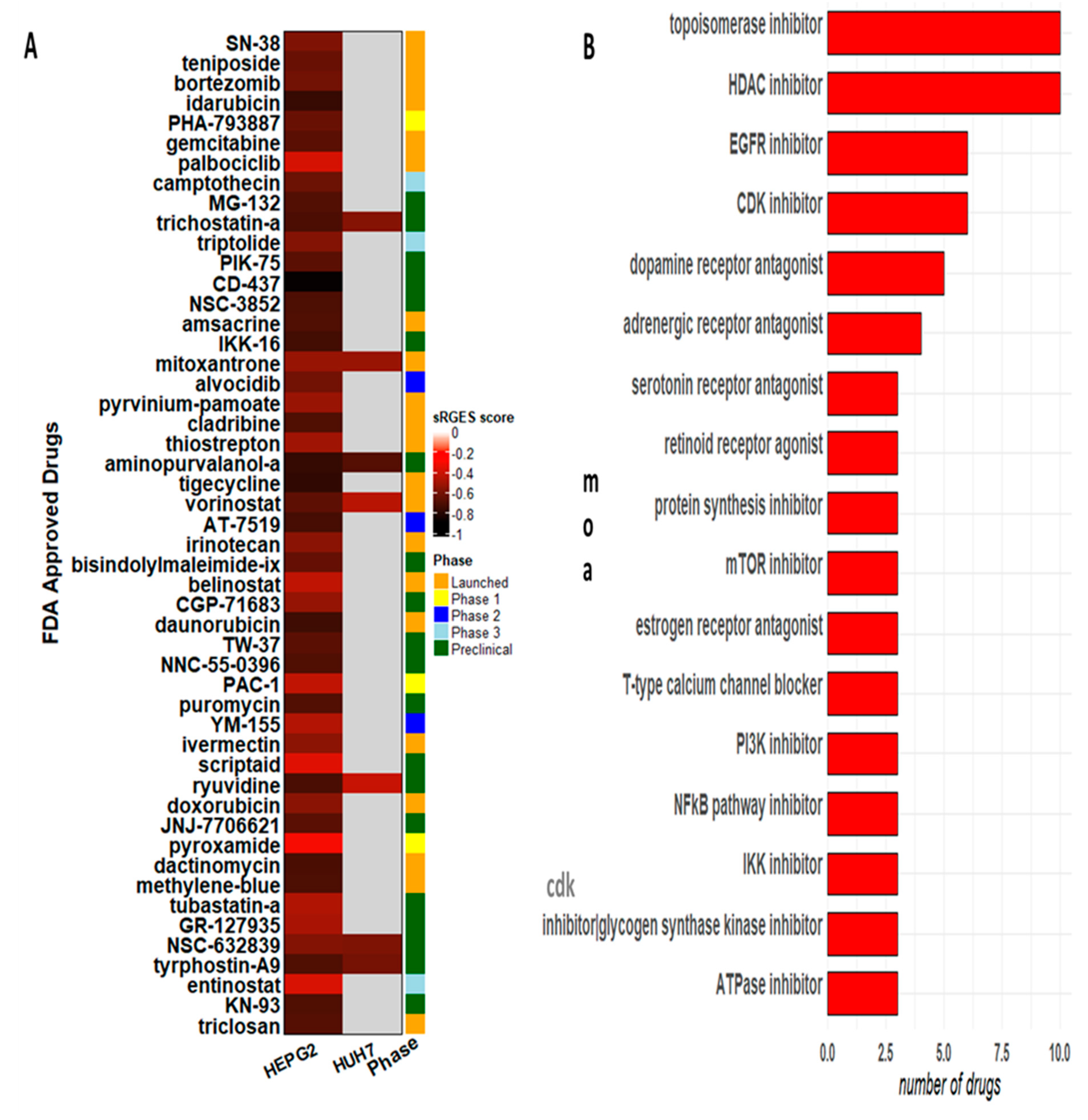
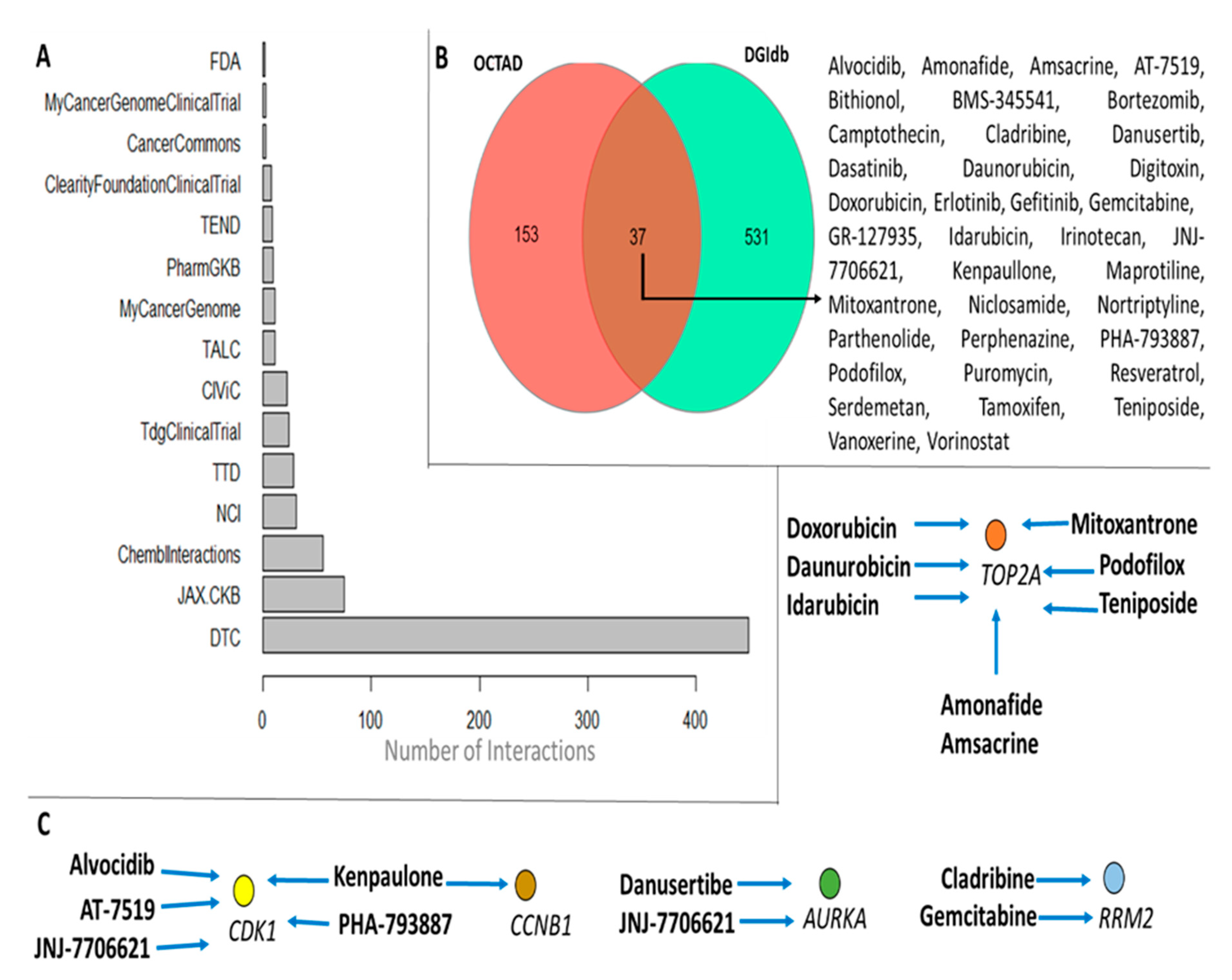
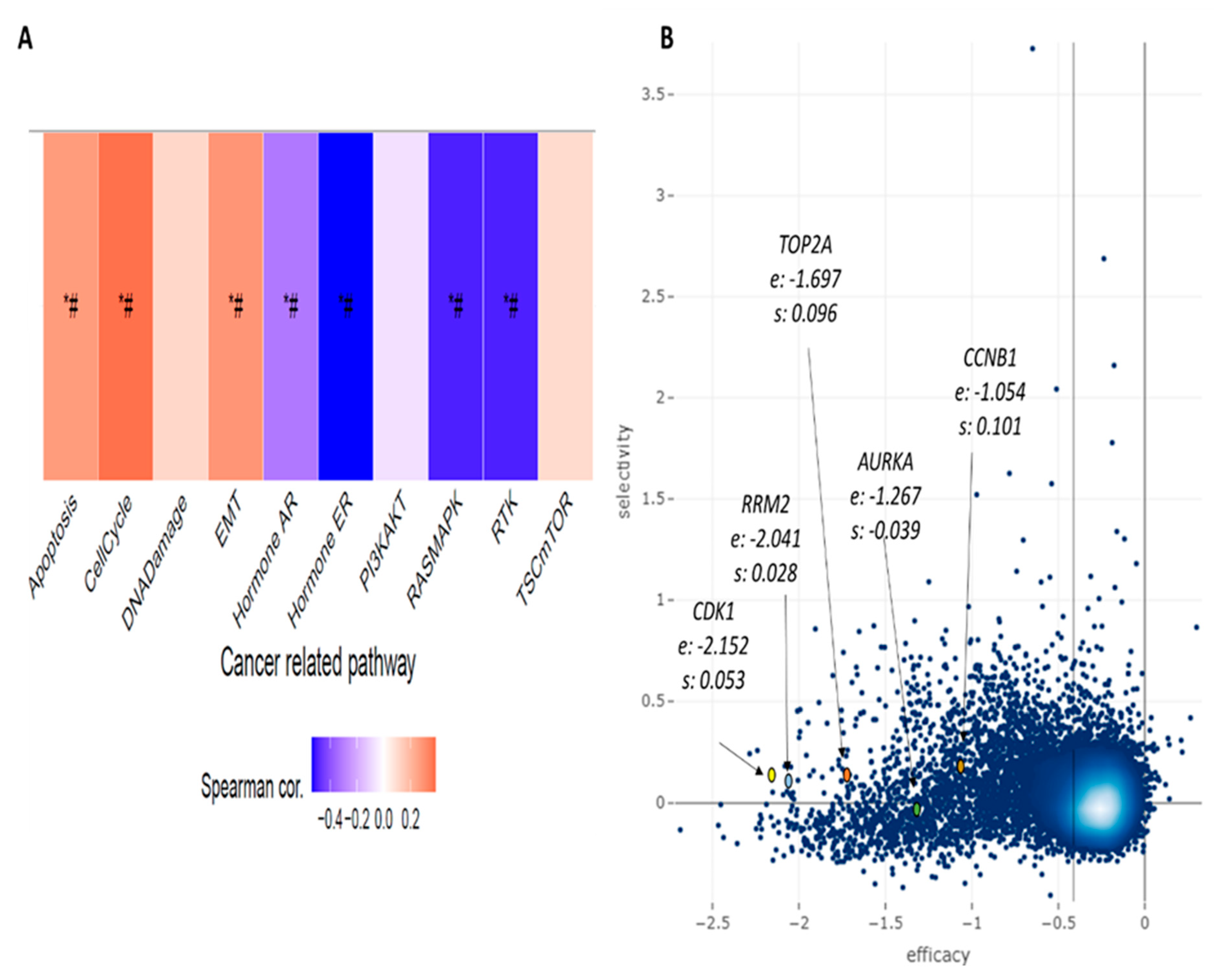
3.4. Novel Candidate Drugs and Their Gene Expression Reversal Potential
4. Discussion
5. Perspectives and Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, K.; Du, Y.; Li, L.; Wei, D.Q. Bioinformatics Approaches for Anti-cancer Drug Discovery. Curr. Drug Targets 2020, 21, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.S.Q.; Sloan, E.K.; Lambert, P.; Kirkpatrick, C.M.J.; Ilomaki, J. Drug repurposing using real-world data. Drug Discov. Today 2023, 28, 103422. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kaur, G.; Bansal, P.; Chawla, V.; Gupta, V. Bioinformatics Paradigms in Drug Discovery and Drug Development. Curr. Top. Med. Chem. 2023, 23, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Heimbach, J.K. New advances in the diagnosis and management of hepatocellular carcinoma. BMJ 2020, 371, m3544. [Google Scholar] [CrossRef] [PubMed]
- Parra, N.S.; Ross, H.M.; Khan, A.; Wu, M.; Goldberg, R.; Shah, L.; Mukhtar, S.; Beiriger, J.; Gerber, A.; Halegoua-DeMarzio, D. Advancements in the Diagnosis of Hepatocellular Carcinoma. Int. J. Transl. Med. 2023, 3, 51–65. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Tan, J.X.; Wang, F.; Dao, F.Y.; Zhang, Z.Y.; Lin, H. Early Diagnosis of Hepatocellular Carcinoma Using Machine Learning Method. Front. Bioeng. Biotechnol. 2020, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, X.; Zhang, S. Bioinformatics identification of crucial genes and pathways associated with hepatocellular carcinoma. Biosci. Rep. 2018, 38, BSR20181441. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Z.; Chen, L.; Li, Y.; Zhou, Z.; Chu, X. Identification of key genes and biological pathways in lung adenocarcinoma via bioinformatics analysis. Mol. Cell. Biochem. 2021, 476, 931–939. [Google Scholar] [CrossRef]
- Li, L.; Lei, Q.; Zhang, S.; Kong, L.; Qin, B. Screening and identification of key biomarkers in hepatocellular carcinoma: Evidence from bioinformatic analysis. Oncol. Rep. 2017, 38, 2607–2618. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, Y.; Hao, H.; Wang, Y.; Zhou, Z.; Wang, Z.; Chu, X. Screening Hub Genes as Prognostic Biomarkers of Hepatocellular Carcinoma by Bioinformatics Analysis. Cell Transpl. 2019, 28, 76S–86S. [Google Scholar] [CrossRef]
- Yang, W.X.; Pan, Y.Y.; You, C.G. CDK1, CCNB1, CDC20, BUB1, MAD2L1, MCM3, BUB1B, MCM2, and RFC4 May Be Potential Therapeutic Targets for Hepatocellular Carcinoma Using Integrated Bioinformatic Analysis. BioMed Res. Int. 2019, 2019, 1245072. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tian, Y.; Chen, H.; Li, H.; Zheng, S. Key signaling pathways, genes and transcription factors associated with hepatocellular carcinoma. Mol. Med. Rep. 2018, 17, 8153–8160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, R.; Sun, L.; Guo, W.; Ji, X.; Hu, X. Comprehensive Analysis of Gene Expression Changes and Validation in Hepatocellular Carcinoma. Onco Targets Ther. 2021, 14, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, Z.; Tan, B.; Zhang, J.; Zhang, J.; Liu, S. Identification and Validation of Hub Genes Associated With Hepatocellular Carcinoma Via Integrated Bioinformatics Analysis. Front. Oncol. 2021, 11, 614531. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Camarda, R.; Odegaard, J.; Paik, H.; Oskotsky, B.; Krings, G.; Goga, A.; Sirota, M.; Butte, A.J. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 2017, 8, 1077. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Ma, L.; Paik, H.; Sirota, M.; Wei, W.; Chua, M.S.; So, S.; Butte, A.J. Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat. Commun. 2017, 8, 16022. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2023. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- HGNC Database, HUGO Gene Nomenclature Committee (HGNC), European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, United Kingdom. Available online: www.genenames.org (accessed on 10 May 2023).
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Carlson, M. Org.Hs.eg.db: Genome Wide Annotation for Human, version 3.8.2. 2019. Available online: https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html (accessed on 20 May 2023).
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling, Version 1.20.0. 2023. Available online: https://doi.org/10.18129/B9.bioc.EnhancedVolcano (accessed on 20 May 2023).
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Chen, H.M.; MacDonald, J.A. Network analysis of TCGA and GTEx gene expression datasets for identification of trait-associated biomarkers in human cancer. STAR Protoc. 2022, 3, 101168. [Google Scholar] [CrossRef]
- Wang, S.; Liu, X. The UCSCXenaTools R package: A toolkit for accessing genomics data from UCSC Xena platform, from cancer multi-omics to single-cell RNA-seq. J. Open Source Softw. 2019, 4, 1627. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Hadley, W. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 2015, 127, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- Clarke, D.J.B.; Kuleshov, M.V.; Schilder, B.M.; Torre, D.; Duffy, M.E.; Keenan, A.B.; Lachmann, A.; Feldmann, A.S.; Gundersen, G.W.; Silverstein, M.C.; et al. eXpression2Kinases (X2K) Web: Linking expression signatures to upstream cell signaling networks. Nucleic Acids Res. 2018, 46, W171–W179. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Liu, C.J.; Hu, F.F.; Xie, G.Y.; Miao, Y.R.; Li, X.W.; Zeng, Y.; Guo, A.Y. GSCA: An integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief. Bioinform. 2023, 24, bbac558. [Google Scholar] [CrossRef]
- Zeng, B.; Glicksberg, B.S.; Newbury, P.; Chekalin, E.; Xing, J.; Liu, K.; Wen, A.; Chow, C.; Chen, B. OCTAD: An open workspace for virtually screening therapeutics targeting precise cancer patient groups using gene expression features. Nat. Protoc. 2021, 16, 728–753. [Google Scholar] [CrossRef]
- Wagner, A.H.; Coffman, A.C.; Ainscough, B.J.; Spies, N.C.; Skidmore, Z.L.; Campbell, K.M.; Krysiak, K.; Pan, D.; McMichael, J.F.; Eldred, J.M.; et al. DGIdb 2.0: Mining clinically relevant drug-gene interactions. Nucleic Acids Res. 2016, 44, D1036–D1044. [Google Scholar] [CrossRef]
- Carvalho, R.F.; do Canto, L.M.; Cury, S.S.; Frøstrup Hansen, T.; Jensen, L.H.; Rogatto, S.R. Drug Repositioning Based on the Reversal of Gene Expression Signatures Identifies TOP2A as a Therapeutic Target for Rectal Cancer. Cancers 2021, 13, 5492. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Bhalla, S.; Kaur, D.; Raghava, G.P. CancerLivER: A database of liver cancer gene expression resources and biomarkers. Database 2020, 2020, baaa012. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed]
- Petrini, I.; Cecchini, R.L.; Mascaro, M.; Ponzoni, I.; Carballido, J.A. Papillary Thyroid Carcinoma: A thorough Bioinformatic Analysis of Gene Expression and Clinical Data. Genes 2023, 14, 1250. [Google Scholar] [CrossRef] [PubMed]
- Chuffa, L.G.d.A.; de Souza, M.C.; Cruz, E.M.S.; Ferreira, F.B.; de Morais, J.M.B.; Seiva, F.R.F. Hepatocellular carcinoma and miRNAs: An in silico approach revealing potential therapeutic targets for polyphenols. Phytomedicine Plus 2022, 2, 100259. [Google Scholar] [CrossRef]
- Wu, M.; Liu, Z.; Zhang, A.; Li, N. Identification of key genes and pathways in hepatocellular carcinoma: A preliminary bioinformatics analysis. Medicine 2019, 98, e14287. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhou, J.; Zhang, Y.; Zeng, Y.; Jie, G.; Wang, S.; Yang, A.; Zhang, M. Identifying potential prognosis markers in hepatocellular carcinoma via integrated bioinformatics analysis and biological experiments. Front. Genet. 2022, 13, 942454. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, H.; Safarpour, H.; Moossavi, M.; Khorashadizadeh, M. Identification of Potential Prognostic Markers and Key Therapeutic Targets in Hepatocellular Carcinoma Using Weighted Gene Co-Expression Network Analysis: A Systems Biology Approach. Iran. J. Biotechnol. 2022, 20, e2968. [Google Scholar] [CrossRef]
- Wang, Y.B.; Zhou, B.X.; Ling, Y.B.; Xiong, Z.Y.; Li, R.X.; Zhong, Y.S.; Xu, M.X.; Lu, Y.; Liang, H.; Chen, G.H.; et al. Decreased expression of ApoF associates with poor prognosis in human hepatocellular carcinoma. Gastroenterol. Rep. 2019, 7, 354–360. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, X.; Lei, Y.; Wang, G.; Liu, M. Glypican-3: A Novel and Promising Target for the Treatment of Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 824208. [Google Scholar] [CrossRef]
- Jing, Q.; Yuan, C.; Zhou, C.; Jin, W.; Wang, A.; Wu, Y.; Shang, W.; Zhang, G.; Ke, X.; Du, J.; et al. Comprehensive analysis identifies CLEC1B as a potential prognostic biomarker in hepatocellular carcinoma. Cancer Cell Int. 2023, 23, 113. [Google Scholar] [CrossRef]
- Yu, J.; Xia, X.; Dong, Y.; Gong, Z.; Li, G.; Chen, G.G.; Lai, P.B.S. CYP1A2 suppresses hepatocellular carcinoma through antagonizing HGF/MET signaling. Theranostics 2021, 11, 2123–2136. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, P.; Wen, J.; Gu, Y.; Yang, Z.; Lan, J.; Fan, H.; Liu, Z.; Guo, D. FCN3 inhibits the progression of hepatocellular carcinoma by suppressing SBDS-mediated blockade of the p53 pathway. Int. J. Biol. Sci. 2023, 19, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Joachim, J.H.; Mehta, K.J. Hepcidin in hepatocellular carcinoma. Br. J. Cancer 2022, 127, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.L.; Pan, B.; Pan, J.H.; Gan, M.F. Metallothionein 1M suppresses tumorigenesis in hepatocellular carcinoma. Oncotarget 2017, 8, 33037–33046. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.; Gadi, S.; Lin, M.; Qi, Q.; Niture, S.S.; Moore, J.T.; Bodnar, W.; Fernando, R.A.; Levine, K.E.; Kumar, D. Cadmium modulates steatosis, fibrosis, and oncogenic signaling in liver cancer cells by activating notch and AKT/mTOR pathways. Environ. Toxicol. 2023, 38, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Men, H.; Young, J.L.; Zhou, W.; Zhang, H.; Wang, X.; Xu, J.; Lin, Q.; Tan, Y.; Zheng, Y.; Cai, L. Early-Life Exposure to Low-Dose Cadmium Accelerates Diethylnitrosamine and Diet-Induced Liver Cancer. Oxidative Med. Cell. Longev. 2021, 2021, 1427787. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yan, C.; Ma, J.; Peng, P.; Ren, X.; Cai, S.; Shen, X.; Wu, Y.; Zhang, S.; Wang, X.; et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat. Metab. 2023, 5, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.L.; Wang, K.H.; Hsieh, H.P.; Yen, W.C. Novel FLT3/AURK multikinase inhibitor is efficacious against sorafenib-refractory and sorafenib-resistant hepatocellular carcinoma. J. Biomed. Sci. 2022, 29, 5. [Google Scholar] [CrossRef]
- Lai, X.; Wu, Y.K.; Hong, G.Q.; Li, J.K.; Luo, Q.; Yuan, J.; Dai, G.H.; Liu, S.Q.; Feng, H.G. A Novel Gene Signature Based on CDC20 and FCN3 for Prediction of Prognosis and Immune Features in Patients with Hepatocellular Carcinoma. J. Immunol. Res. 2022, 2022, 9117205. [Google Scholar] [CrossRef]
- Chen, M.; Wu, G.B.; Xie, Z.W.; Shi, D.L.; Luo, M. A novel diagnostic four-gene signature for hepatocellular carcinoma based on artificial neural network: Development, validation, and drug screening. Front. Genet. 2022, 13, 942166. [Google Scholar] [CrossRef] [PubMed]
- Azuaje, F.J. Selecting biologically informative genes in co-expression networks with a centrality score. Biol. Direct 2014, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Ma, H.; Jin, B. Expression of FOXM1 and PLK1 predicts prognosis of patients with hepatocellular carcinoma. Oncol. Lett. 2022, 23, 146. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; You, C.; Huang, J.; Zhang, M. NFYAv1 is a Tumor-Promoting Transcript Associated with Poor Prognosis of Hepatocellular Carcinoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2023, 29, e938410. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Fu, Q.; Xu, J.; Gu, X.; Zhou, H.; Zhi, C. Transcription factor E2F4 is an indicator of poor prognosis and is related to immune infiltration in hepatocellular carcinoma. J. Cancer 2021, 12, 1792–1803. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Wigle, T.J.; Tsvetkova, E.V.; Welch, S.A.; Kim, R.B. DPYD and Fluorouracil-Based Chemotherapy: Mini Review and Case Report. Pharmaceutics 2019, 11, 199. [Google Scholar] [CrossRef]
- da Silva, M.C.; Fabiano, L.C.; da Costa Salomao, K.C.; de Freitas, P.L.Z.; Neves, C.Q.; Borges, S.C.; de Souza Carvalho, M.D.G.; Breithaupt-Faloppa, A.C.; de Thomaz, A.A.; Dos Santos, A.M.; et al. A Rodent Model of Human-Dose-Equivalent 5-Fluorouracil: Toxicity in the Liver, Kidneys, and Lungs. Antioxidants 2023, 12, 1005. [Google Scholar] [CrossRef]
- Jaeger, S.; Aloy, P. From protein interaction networks to novel therapeutic strategies. IUBMB Life 2012, 64, 529–537. [Google Scholar] [CrossRef]
- Jonas, B.A.; Hou, J.Z.; Roboz, G.J.; Alvares, C.L.; Jeyakumar, D.; Edwards, J.R.; Erba, H.P.; Kelly, R.J.; Rollig, C.; Fiedler, W.; et al. A phase 1b study of venetoclax and alvocidib in patients with relapsed/refractory acute myeloid leukemia. Hematol. Oncol. 2023, 41, 743–752. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, L.; Zhang, Y.; Jiang, Z.; Lu, H.; Xie, Y.; Han, W.; Zhao, W.; He, J.; Shi, Z.; et al. The CDK inhibitor AT7519 inhibits human glioblastoma cell growth by inducing apoptosis, pyroptosis and cell cycle arrest. Cell Death Dis. 2023, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhu, F.; Zhu, T.; Zhao, Z.; Li, L.; Lin, S.; Zhao, H.; Yang, L.; Zhao, C.; Wang, L.; et al. AT7519 against lung cancer via the IL6/STAT3 signaling pathway. Biochem. Biophys. Res. Commun. 2022, 609, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Santo, L.; Vallet, S.; Hideshima, T.; Cirstea, D.; Ikeda, H.; Pozzi, S.; Patel, K.; Okawa, Y.; Gorgun, G.; Perrone, G.; et al. AT7519, A novel small molecule multi-cyclin-dependent kinase inhibitor, induces apoptosis in multiple myeloma via GSK-3beta activation and RNA polymerase II inhibition. Oncogene 2010, 29, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.S.; Cooke, L.; Lock, V.; Qi, W.; Lewis, E.J.; Thompson, N.T.; Lyons, J.F.; Mahadevan, D. AT7519, a cyclin-dependent kinase inhibitor, exerts its effects by transcriptional inhibition in leukemia cell lines and patient samples. Mol. Cancer Ther. 2010, 9, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Karas, K.; Karwaciak, I.; Chalaskiewicz, K.; Salkowska, A.; Pastwinska, J.; Bachorz, R.A.; Ratajewski, M. Anti-hepatocellular carcinoma activity of the cyclin-dependent kinase inhibitor AT7519. Biomed. Pharmacother. 2023, 164, 115002. [Google Scholar] [CrossRef] [PubMed]
- Camins, A.; Verdaguer, E.; Folch, J.; Canudas, A.M.; Pallas, M. The role of CDK5/P25 formation/inhibition in neurodegeneration. Drug News Perspect. 2006, 19, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Trzcinska-Daneluti, A.M.; Nguyen, L.; Jiang, C.; Fladd, C.; Uehling, D.; Prakesch, M.; Al-awar, R.; Rotin, D. Use of kinase inhibitors to correct DeltaF508-CFTR function. Mol. Cell. Proteom. 2012, 11, 745–757. [Google Scholar] [CrossRef]
- Joo, H.C.; Choi, J.W.; Moon, H.; Lee, C.Y.; Yoo, K.J.; Kim, S.W.; Hwang, K.C. Protective effects of kenpaullone on cardiomyocytes following H(2)O(2)-induced oxidative stress are attributed to inhibition of connexin 43 degradation by SGSM3. Biochem. Biophys. Res. Commun. 2018, 499, 368–373. [Google Scholar] [CrossRef]
- Rybak, L.P.; Dhukhwa, A.; Mukherjea, D.; Ramkumar, V. Local Drug Delivery for Prevention of Hearing Loss. Front. Cell. Neurosci. 2019, 13, 300. [Google Scholar] [CrossRef]
- Zhuang, L.; Yang, Z.; Meng, Z. Upregulation of BUB1B, CCNB1, CDC7, CDC20, and MCM3 in Tumor Tissues Predicted Worse Overall Survival and Disease-Free Survival in Hepatocellular Carcinoma Patients. BioMed Res. Int. 2018, 2018, 7897346. [Google Scholar] [CrossRef]
- Wu, B.; Yang, W.; Fu, Z.; Xie, H.; Guo, Z.; Liu, D.; Ge, J.; Zhong, S.; Liu, L.; Liu, J.; et al. Selected using bioinformatics and molecular docking analyses, PHA-793887 is effective against osteosarcoma. Aging 2021, 13, 16425–16444. [Google Scholar] [CrossRef] [PubMed]
- Ly, C.Y.; Pfannenstiel, J.; Pant, A.; Yang, Z.; Fehr, A.R.; Rodzkin, M.S.; Davido, D.J. Inhibitors of One or More Cellular Aurora Kinases Impair the Replication of Herpes Simplex Virus 1 and Other DNA and RNA Viruses with Diverse Genomes and Life Cycles. Microbiol. Spectr. 2023, 11, e0194322. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Byrne, D.P.; Burgess, S.G.; Bormann, J.; Bakovic, J.; Huang, Y.; Zhyvoloup, A.; Yu, B.Y.K.; Peak-Chew, S.; Tran, T.; et al. Covalent Aurora A regulation by the metabolic integrator coenzyme A. Redox Biol. 2020, 28, 101318. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Wang, J.; Wang, X.; Huang, T.; Fang, Z.; Yan, L.; Fan, Y.; Xu, D. Widespread genomic/molecular alterations of DNA helicases and their clinical/therapeutic implications across human cancer. Biomed. Pharmacother. 2023, 158, 114193. [Google Scholar] [CrossRef] [PubMed]
- Benten, D.; Keller, G.; Quaas, A.; Schrader, J.; Gontarewicz, A.; Balabanov, S.; Braig, M.; Wege, H.; Moll, J.; Lohse, A.W.; et al. Aurora kinase inhibitor PHA-739358 suppresses growth of hepatocellular carcinoma in vitro and in a xenograft mouse model. Neoplasia 2009, 11, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, B. MicroRNA-582-3p targeting ribonucleotide reductase regulatory subunit M2 inhibits the tumorigenesis of hepatocellular carcinoma by regulating the Wnt/beta-catenin signaling pathway. Bioengineered 2022, 13, 12876–12887. [Google Scholar] [CrossRef] [PubMed]
- Molica, M.; Breccia, M.; Capria, S.; Trisolini, S.; Foa, R.; Jabbour, E.; Kadia, T.M. The role of cladribine in acute myeloid leukemia: An old drug up to new tricks. Leuk. Lymphoma 2020, 61, 536–545. [Google Scholar] [CrossRef]
- Goyal, G.; Ravindran, A.; Young, J.R.; Shah, M.V.; Bennani, N.N.; Patnaik, M.M.; Nowakowski, G.S.; Thanarajasingam, G.; Habermann, T.M.; Vassallo, R.; et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica 2020, 105, 348–357. [Google Scholar] [CrossRef]
- Stamatellos, V.P.; Papazisis, G. Safety and Monitoring of the Treatment with Disease-Modifying Therapies (DMTs) for Multiple Sclerosis (MS). Curr. Rev. Clin. Exp. Pharmacol. 2023, 18, 39–50. [Google Scholar] [CrossRef]
- Hoffmann, K.; Franz, C.; Xiao, Z.; Mohr, E.; Serba, S.; Buchler, M.W.; Schemmer, P. Sorafenib modulates the gene expression of multi-drug resistance mediating ATP-binding cassette proteins in experimental hepatocellular carcinoma. Anticancer Res. 2010, 30, 4503–4508. [Google Scholar]
- Gallois, L.; Fiallo, M.; Garnier-Suillerot, A. Comparison of the interaction of doxorubicin, daunorubicin, idarubicin and idarubicinol with large unilamellar vesicles. Circular dichroism study. Biochim. Et Biophys. Acta 1998, 1370, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, Q.; Li, X.; Ren, X.; Li, J.; Zhang, Y.; Zeng, S.; Xu, L.; Dong, X.; Zhai, B. TOP2A inhibition reverses drug resistance of hepatocellular carcinoma to regorafenib. Am. J. Cancer Res. 2022, 12, 4343–4360. [Google Scholar] [PubMed]
- Guiu, B.; Assenat, E. Doxorubicin for the treatment of hepatocellular carcinoma: GAME OVER! Ann. Transl. Med. 2020, 8, 1693. [Google Scholar] [CrossRef] [PubMed]
- Sanduzzi-Zamparelli, M.; Forner, A. Systemic doxorubicin and hepatocellular carcinoma: The end of an era never risen up. Lancet Gastroenterol. Hepatol. 2019, 4, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Guiu, B.; Chevallier, P.; Assenat, E.; Barbier, E.; Merle, P.; Bouvier, A.; Dumortier, J.; Nguyen-Khac, E.; Gugenheim, J.; Rode, A.; et al. Idarubicin-loaded Beads for Chemoembolization of Hepatocellular Carcinoma: The IDASPHERE II Single-Arm Phase II Trial. Radiology 2019, 291, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Padia, S.A. Is Idarubicin the Future of TACE? Radiology 2019, 291, 809–810. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Bramley, R.L.; Cowell, I.G.; Jackson, G.H.; Austin, C.A. Proteasomal inhibition potentiates drugs targeting DNA topoisomerase II. Biochem. Pharmacol. 2016, 103, 29–39. [Google Scholar] [CrossRef]
- Shah, Z.; Gohar, U.F.; Jamshed, I.; Mushtaq, A.; Mukhtar, H.; Zia-Ui-Haq, M.; Toma, S.I.; Manea, R.; Moga, M.; Popovici, B. Podophyllotoxin: History, Recent Advances and Future Prospects. Biomolecules 2021, 11, 603. [Google Scholar] [CrossRef]
- Yu, X.; Che, Z.; Xu, H. Recent Advances in the Chemistry and Biology of Podophyllotoxins. Chemistry 2017, 23, 4467–4526. [Google Scholar] [CrossRef]
- Aita, P.; Robieux, I.; Sorio, R.; Tumolo, S.; Corona, G.; Cannizzaro, R.; Colussi, A.M.; Boiocchi, M.; Toffoli, G. Pharmacokinetics of oral etoposide in patients with hepatocellular carcinoma. Cancer Chemother. Pharmacol. 1999, 43, 287–294. [Google Scholar] [CrossRef]
- Lu, M.; Xiao, L.; Xu, B.; Gao, Q. Identification of Novel Genes and Associated Drugs in Advanced Clear Cell Renal Cell Carcinoma by Bioinformatic Methods. Tohoku J. Exp. Med. 2022, 258, 79–90. [Google Scholar] [CrossRef]
- Ma, J.; Li, L.; Yue, K.; Zhang, Z.; Su, S.; Chen, Y.; Yu, L.; Zhang, P.; Ma, R.; Li, Y.; et al. A naphthalimide-polyamine conjugate preferentially accumulates in hepatic carcinoma metastases as a lysosome-targeted antimetastatic agent. European J. Med. Chem. 2021, 221, 113469. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chiou, J.T.; Wang, L.J.; Chen, Y.J.; Chang, L.S. Amsacrine downregulates BCL2L1 expression and triggers apoptosis in human chronic myeloid leukemia cells through the SIDT2/NOX4/ERK/HuR pathway. Toxicol. Appl. Pharmacol. 2023, 474, 116625. [Google Scholar] [CrossRef]
- Goodwin, B.; Lou, J.; Butchy, M.; Wilson, T.; Atabek, U.; Spitz, F.; Hong, Y. Hepatocellular-Cholangiocarcinoma Collision Tumors: An Update of Current Management Practices. Am. Surg. 2023, 89, 2685–2692. [Google Scholar] [CrossRef]
- Shen, Z.; Yin, L.; Zhou, H.; Ji, X.; Jiang, C.; Zhu, X.; He, X. Combined inhibition of AURKA and HSF1 suppresses proliferation and promotes apoptosis in hepatocellular carcinoma by activating endoplasmic reticulum stress. Cell. Oncol. 2021, 44, 1035–1049. [Google Scholar] [CrossRef]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSE | Plataform | Type | Tumor Sample | Non-Tumor Sample |
|---|---|---|---|---|
| 102079 | GPL 570 | Adjacent-tissue | 152 | 91 |
| 121248 | GPL 570 | Adjacent-tissue | 70 | 37 |
| 12941 | GPL 5175 | Adjacent-tissue | 10 | 10 |
| 136247 | GPL 17586 | Adjacent-tissue | 39 | 30 |
| 14520 | GPL 571 | Adjacent-tissue | 225 | 220 |
| 22405 | GPL 10553 | Adjacent-tissue | 24 | 24 |
| 25097 | GPL 10687 | Adjacent-tissue | 268 | 243 |
| 36376 | GPL 10558 | Adjacent-tissue | 240 | 193 |
| 39791 | GPL 10558 | Adjacent-tissue | 72 | 72 |
| 41804 | GPL 570 | Adjacent-tissue | 20 | 20 |
| 45267 | GPL 570 | Adjacent-tissue | 46 | 41 |
| 57957 | GPL 10558 | Adjacent-tissue | 39 | 39 |
| 60502 | GPL 96 | Adjacent-tissue | 18 | 18 |
| 64041 | GPL 6244 | Adjacent-tissue | 60 | 60 |
| 76427 | GPL 10558 | Adjacent-tissue | 115 | 52 |
| 84005 | GPL 5175 | Adjacent-tissue | 38 | 38 |
| 84402 | GPL 570 | Adjacent-tissue | 14 | 14 |
| 102079 | GPL 570 | Healthy liver | 152 | 14 |
| 112790 | GPL 570 | Healthy liver | 183 | 15 |
| 62232 | GPL 570 | Healthy liver | 81 | 10 |
| 50579 | GPL 14550 | Healthy-liver | 67 | 10 |
| TCGA x GTEx | RNA-Seq | - | 359 | 110 |
| Term | Ratio | FDR |
|---|---|---|
| Liver carcinoma | 68/3593 | 4.63 × 10−21 |
| Carcinogenesis | 55/4065 | 3.02 × 10−9 |
| Liver diseases | 19/606 | 1.50 × 10−7 |
| Chronic liver disease | 10/129 | 3.18 × 10−7 |
| Hepatocarcinogenesis | 17/527 | 4.73 × 10−7 |
| Liver neoplasms | 26/1321 | 8.22 × 10−7 |
| Malignant neoplasm of liver | 20/805 | 1.26 × 10−6 |
| Liver and Intrahepatic Biliary Tract Carcinoma | 15/607 | 6.31 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caputo, W.L.; de Souza, M.C.; Basso, C.R.; Pedrosa, V.d.A.; Seiva, F.R.F. Comprehensive Profiling and Therapeutic Insights into Differentially Expressed Genes in Hepatocellular Carcinoma. Cancers 2023, 15, 5653. https://doi.org/10.3390/cancers15235653
Caputo WL, de Souza MC, Basso CR, Pedrosa VdA, Seiva FRF. Comprehensive Profiling and Therapeutic Insights into Differentially Expressed Genes in Hepatocellular Carcinoma. Cancers. 2023; 15(23):5653. https://doi.org/10.3390/cancers15235653
Chicago/Turabian StyleCaputo, Wesley Ladeira, Milena Cremer de Souza, Caroline Rodrigues Basso, Valber de Albuquerque Pedrosa, and Fábio Rodrigues Ferreira Seiva. 2023. "Comprehensive Profiling and Therapeutic Insights into Differentially Expressed Genes in Hepatocellular Carcinoma" Cancers 15, no. 23: 5653. https://doi.org/10.3390/cancers15235653