
Identification of a Novel Eight-Gene Risk Model for Predicting Survival in Glioblastoma: A Comprehensive Bioinformatic Analysis


 , , , and
, , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Preprocessing
2.2. Analysis of Differentially Expressed Genes (DEGs)
2.3. Functional Enrichment Analysis of the DEGs
2.4. Risk Score Construction and Validation
2.5. Characterization of the Risk Model-Based Subgroups
2.6. Relationship between the Prognostic Genes and Immune Infiltration in GBM
3. Results
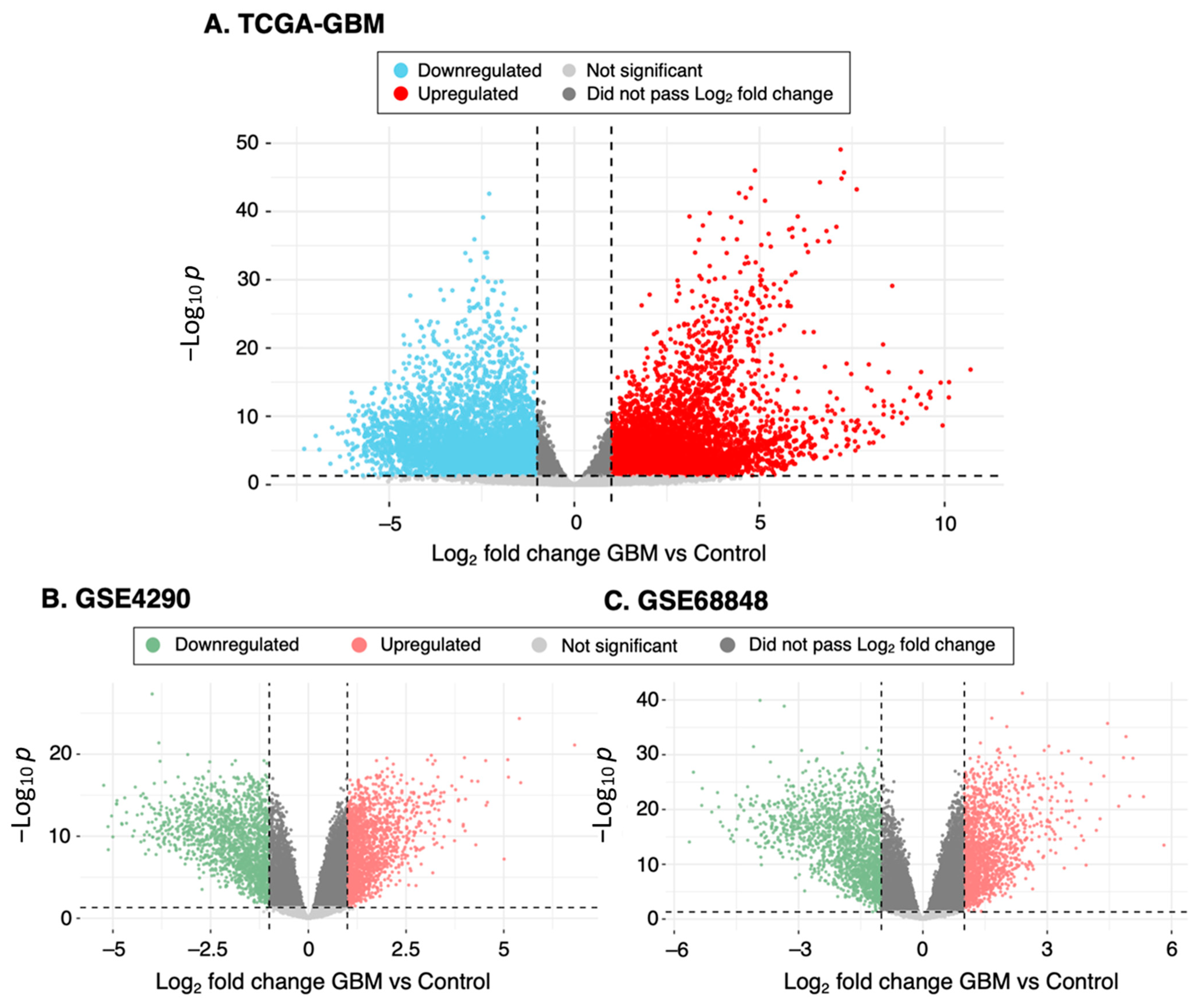
3.1. Identification of DEGs from GBM Datasets
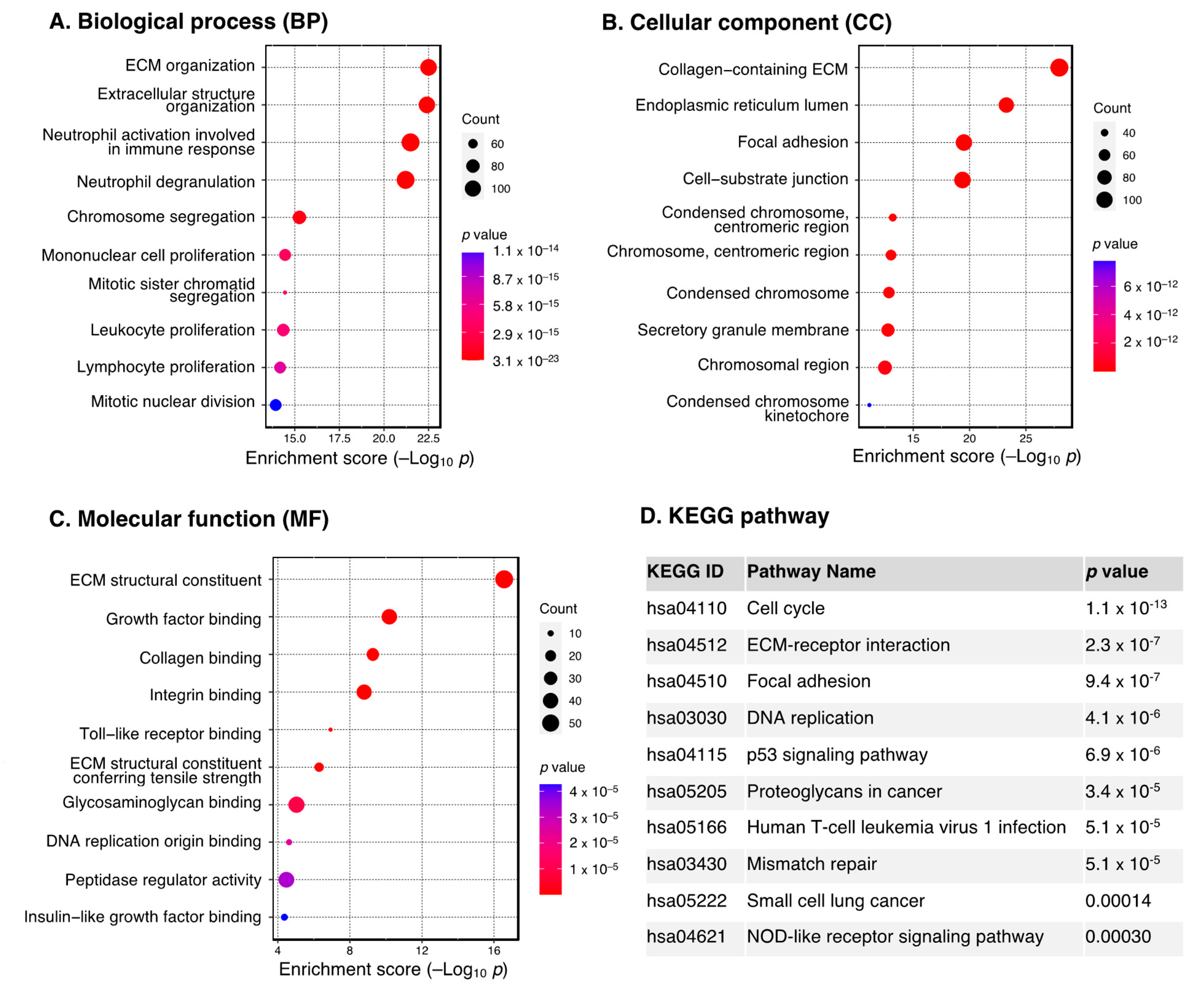
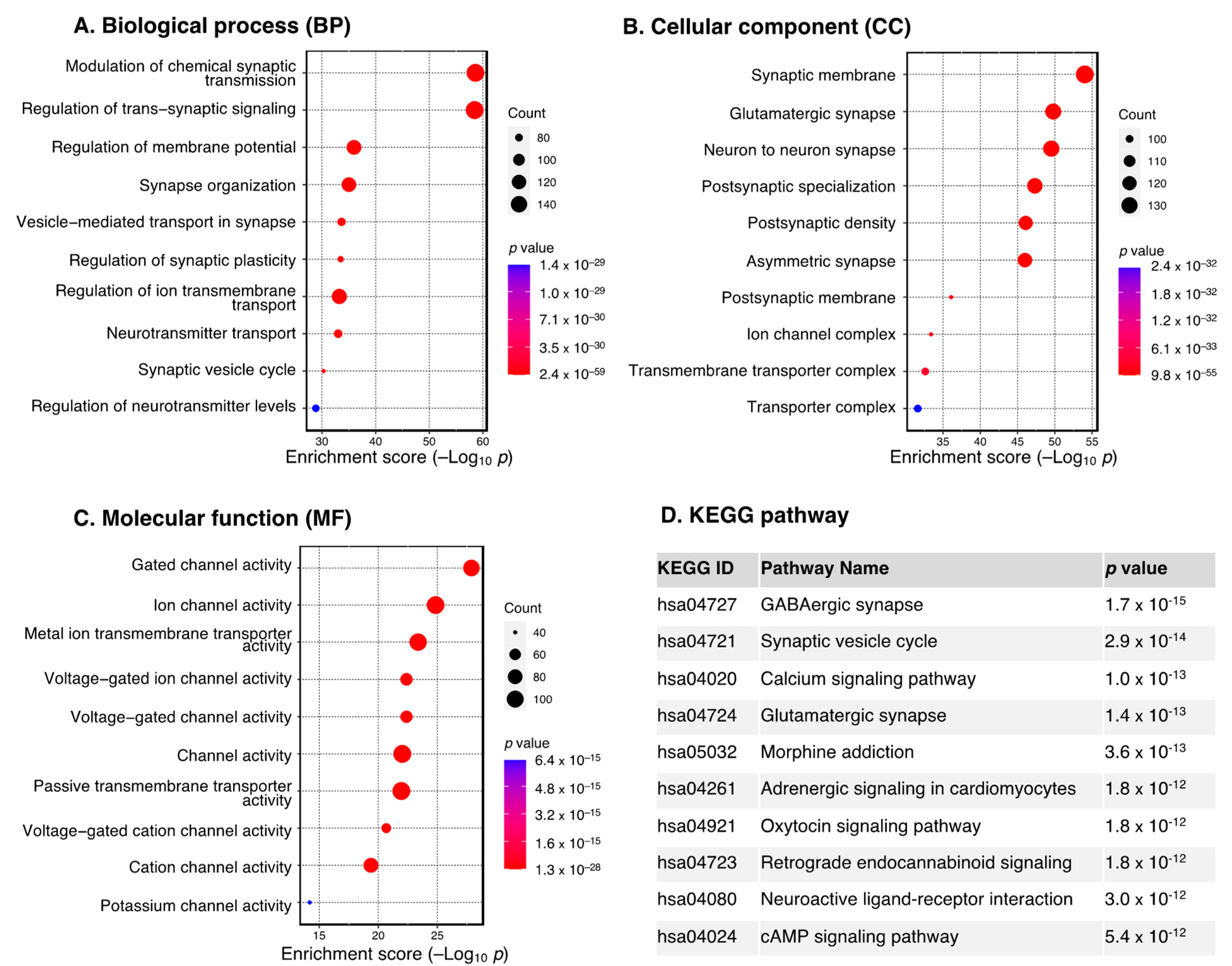
3.2. Functional Enrichment Analysis of DEGs
3.3. Identification of Prognosis-Related Genes and Construction of Risk Model
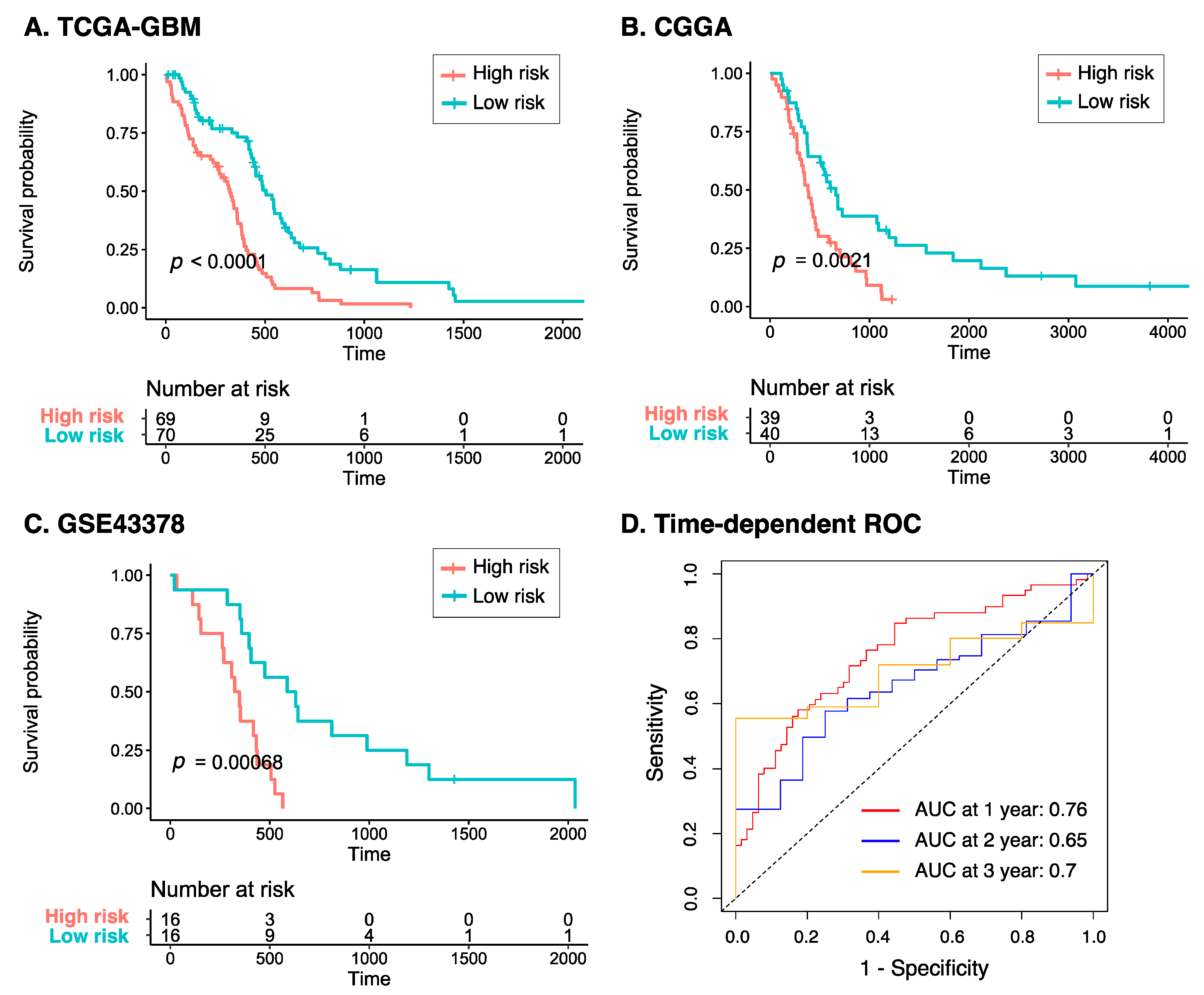
3.4. Characteristics of the Eight-Gene Risk Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncol. 2021, 23, iii1–iii105. [Google Scholar] [CrossRef]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From Molecular Pathology to Targeted Treatment. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, M.; Cai, J.; Yuan, Y.; Shi, Y.; Wu, H.; Liu, Q.; Yao, Y.; Chen, L.; Dang, W.; Zhang, X.; et al. A Four-Gene Signature-Derived Risk Score for Glioblastoma: Prospects for Prognostic and Response Predictive Analyses. Cancer Biol. Med. 2019, 16, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Du, M.; Lu, L. A Novel 16-Genes Signature Scoring System as Prognostic Model to Evaluate Survival Risk in Patients with Glioblastoma. Biomedicines 2022, 10, 317. [Google Scholar] [CrossRef]
- Kawaguchi, A.; Yajima, N.; Tsuchiya, N.; Homma, J.; Sano, M.; Natsumeda, M.; Takahashi, H.; Fujii, Y.; Kakuma, T.; Yamanaka, R. Gene Expression Signature-Based Prognostic Risk Score in Patients with Glioblastoma. Cancer Sci. 2013, 104, 1205–1210. [Google Scholar] [CrossRef]
- Wen, Y.-D.; Zhu, X.-S.; Li, D.-J.; Zhao, Q.; Cheng, Q.; Peng, Y. Proteomics-Based Prognostic Signature and Nomogram Construction of Hypoxia Microenvironment on Deteriorating Glioblastoma (GBM) Pathogenesis. Sci. Rep. 2021, 11, 17170. [Google Scholar] [CrossRef]
- Lei, C.; Chen, W.; Wang, Y.; Zhao, B.; Liu, P.; Kong, Z.; Liu, D.; Dai, C.; Wang, Y.; Wang, Y.; et al. Prognostic Prediction Model for Glioblastoma: A Metabolic Gene Signature and Independent External Validation. J. Cancer 2021, 12, 3796–3808. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Madhavan, S.; Zenklusen, J.-C.; Kotliarov, Y.; Sahni, H.; Fine, H.A.; Buetow, K. Rembrandt: Helping Personalized Medicine Become a Reality through Integrative Translational Research. Mol. Cancer Res. 2009, 7, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Sun, L.; Hui, A.-M.; Su, Q.; Vortmeyer, A.; Kotliarov, Y.; Pastorino, S.; Passaniti, A.; Menon, J.; Walling, J.; Bailey, R.; et al. Neuronal and Glioma-Derived Stem Cell Factor Induces Angiogenesis within the Brain. Cancer Cell 2006, 9, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Ringnér, M. What Is Principal Component Analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A Web Server for Functional Enrichment Analysis and Conversions of Gene Lists (2019 Update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.P.; Smith, B.; McAndrews-Hill, M.S.; Blake, J.A. Gene Ontology Annotations: What They Mean and Where They Come From. BMC Bioinform. 2008, 9, S2. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, K.-N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Blanche, P.; Dartigues, J.-F.; Jacqmin-Gadda, H. Estimating and Comparing Time-Dependent Areas under Receiver Operating Characteristic Curves for Censored Event Times with Competing Risks. Stat. Med. 2013, 32, 5381–5397. [Google Scholar] [CrossRef] [PubMed]
- Kamarudin, A.N.; Cox, T.; Kolamunnage-Dona, R. Time-Dependent ROC Curve Analysis in Medical Research: Current Methods and Applications. BMC Med. Res. Methodol. 2017, 17, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.; Pongor, L.; Su, Y.-T.; Xi, L.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Madurga, R.; García-Romero, N.; Jiménez, B.; Collazo, A.; Pérez-Rodríguez, F.; Hernández-Laín, A.; Fernández-Carballal, C.; Prat-Acín, R.; Zanin, M.; Menasalvas, E.; et al. Normal Tissue Content Impact on the GBM Molecular Classification. Brief. Bioinform. 2021, 22, bbaa129. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A Visual Analytics Platform for Comprehensive Gene Expression Profiling and Meta-Analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription Factor Regulation Inferred from Integrating Genome-Wide ChIP-X Experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA Interference Reveals That Oncogenic KRAS-Driven Cancers Require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the Population Abundance of Tissue-Infiltrating Immune and Stromal Cell Populations Using Gene Expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- McDermaid, A.; Monier, B.; Zhao, J.; Liu, B.; Ma, Q. Interpretation of Differential Gene Expression Results of RNA-Seq Data: Review and Integration. Brief. Bioinform. 2019, 20, 2044–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcu, E.; Sadler, M.C.; Lepik, K.; Auwerx, C.; Wood, A.R.; Weihs, A.; Sleiman, M.S.B.; Ribeiro, D.M.; Bandinelli, S.; Tanaka, T.; et al. Differentially Expressed Genes Reflect Disease-Induced Rather than Disease-Causing Changes in the Transcriptome. Nat. Commun. 2021, 12, 5647. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.R.K.; Mias, G.I. Data-Driven Analysis of Age, Sex, and Tissue Effects on Gene Expression Variability in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 392. [Google Scholar] [CrossRef] [Green Version]
- Dang, H.H.; Ta, H.D.K.; Nguyen, T.T.T.; Anuraga, G.; Wang, C.-Y.; Lee, K.-H.; Le, N.Q.K. Prospective Role and Immunotherapeutic Targets of Sideroflexin Protein Family in Lung Adenocarcinoma: Evidence from Bioinformatics Validation. Funct. Integr. Genom. 2022, 22, 1057–1072. [Google Scholar] [CrossRef]
- Dang, H.-H.; Ta, H.D.K.; Nguyen, T.T.T.; Anuraga, G.; Wang, C.-Y.; Lee, K.-H.; Le, N.Q.K. Identifying GPSM Family Members as Potential Biomarkers in Breast Cancer: A Comprehensive Bioinformatics Analysis. Biomedicines 2021, 9, 1144. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Li, J.; Zhang, Y.; Yin, H.; Han, B. CRNDE, a Long-Noncoding RNA, Promotes Glioma Cell Growth and Invasion through MTOR Signaling. Cancer Lett. 2015, 367, 122–128. [Google Scholar] [CrossRef]
- Jing, S.-Y.; Lu, Y.-Y.; Yang, J.-K.; Deng, W.-Y.; Zhou, Q.; Jiao, B.-H. Expression of Long Non-Coding RNA CRNDE in Glioma and Its Correlation with Tumor Progression and Patient Survival. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3992–3996. [Google Scholar]
- Zhao, Z.; Liu, M.; Long, W.; Yuan, J.; Li, H.; Zhang, C.; Tang, G.; Jiang, W.; Yuan, X.; Wu, M.; et al. Knockdown LncRNA CRNDE Enhances Temozolomide Chemosensitivity by Regulating Autophagy in Glioblastoma. Cancer Cell Int. 2021, 21, 456. [Google Scholar] [CrossRef]
- Sun, H.-T.; Cheng, S.-X.; Tu, Y.; Li, X.-H.; Zhang, S. FoxQ1 Promotes Glioma Cells Proliferation and Migration by Regulating NRXN3 Expression. PLoS ONE 2013, 8, e55693. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, P.; Dong, X.; Li, H.; Li, S.; Cheng, S.; Yuan, J.; Yang, X.; Qian, Z.; Dong, J. Circ_0001367 Inhibits Glioma Proliferation, Migration and Invasion by Sponging MiR-431 and Thus Regulating NRXN3. Cell Death Dis. 2021, 12, 536. [Google Scholar] [CrossRef]
- Liu, L.; Yang, S.; Lin, K.; Yu, X.; Meng, J.; Ma, C.; Wu, Z.; Hao, Y.; Chen, N.; Ge, Q.; et al. Sp1 Induced Gene TIMP1 Is Related to Immune Cell Infiltration in Glioblastoma. Sci. Rep. 2022, 12, 11181. [Google Scholar] [CrossRef]
- Song, G.; Xu, S.; Zhang, H.; Wang, Y.; Xiao, C.; Jiang, T.; Wu, L.; Zhang, T.; Sun, X.; Zhong, L.; et al. TIMP1 Is a Prognostic Marker for the Progression and Metastasis of Colon Cancer through FAK-PI3K/AKT and MAPK Pathway. J. Exp. Clin. Cancer Res. 2016, 35, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toker, A.; Ohashi, P.S. Chapter Five—Expression of Costimulatory and Inhibitory Receptors in FoxP3+ Regulatory T Cells within the Tumor Microenvironment: Implications for Combination Immunotherapy Approaches. In Advances in Cancer Research; Tew, K.D., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 144, pp. 193–261. [Google Scholar]
- Blank, A.-E.; Baumgarten, P.; Zeiner, P.; Zachskorn, C.; Löffler, C.; Schittenhelm, J.; Czupalla, C.J.; Capper, D.; Plate, K.H.; Harter, P.N.; et al. Tumour Necrosis Factor Receptor Superfamily Member 9 (TNFRSF9) Is up-Regulated in Reactive Astrocytes in Human Gliomas. Neuropathol. Appl. Neurobiol. 2015, 41, e56–e67. [Google Scholar] [CrossRef]
- Yu, Z.; Yang, H.; Song, K.; Fu, P.; Shen, J.; Xu, M.; Xu, H. Construction of an Immune-Related Gene Signature for the Prognosis and Diagnosis of Glioblastoma Multiforme. Front. Oncol. 2022, 12, 938679. [Google Scholar] [CrossRef]
- Rana, P.; Thai, P.; Dinh, T.; Ghosh, P. Relevant and Non-Redundant Feature Selection for Cancer Classification and Subtype Detection. Cancers 2021, 13, 4297. [Google Scholar] [CrossRef] [PubMed]
- Amunjela, J.N.; Swan, A.H.; Brand, T. The Role of the Popeye Domain Containing Gene Family in Organ Homeostasis. Cells 2019, 8, 1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and Microarray in Transcriptome Profiling of Activated T Cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Hentschel, B.; Simon, M.; Westphal, M.; Schackert, G.; Tonn, J.C.; Loeffler, M.; Reifenberger, G.; Pietsch, T.; von Deimling, A.; et al. Long-Term Survival in Primary Glioblastoma with versus without Isocitrate Dehydrogenase Mutations. Clin. Cancer Res. 2013, 19, 5146–5157. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
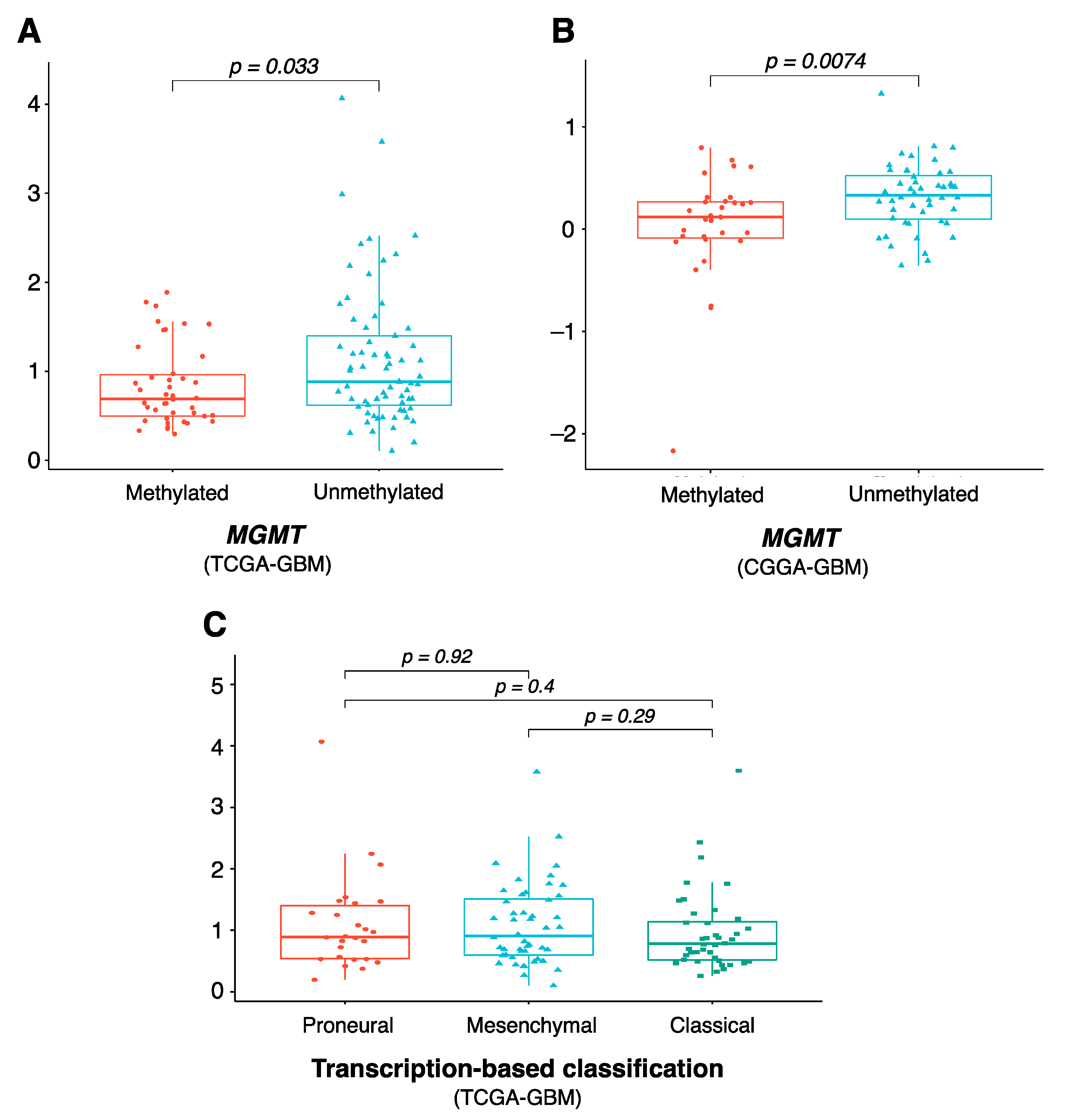{kind=link}
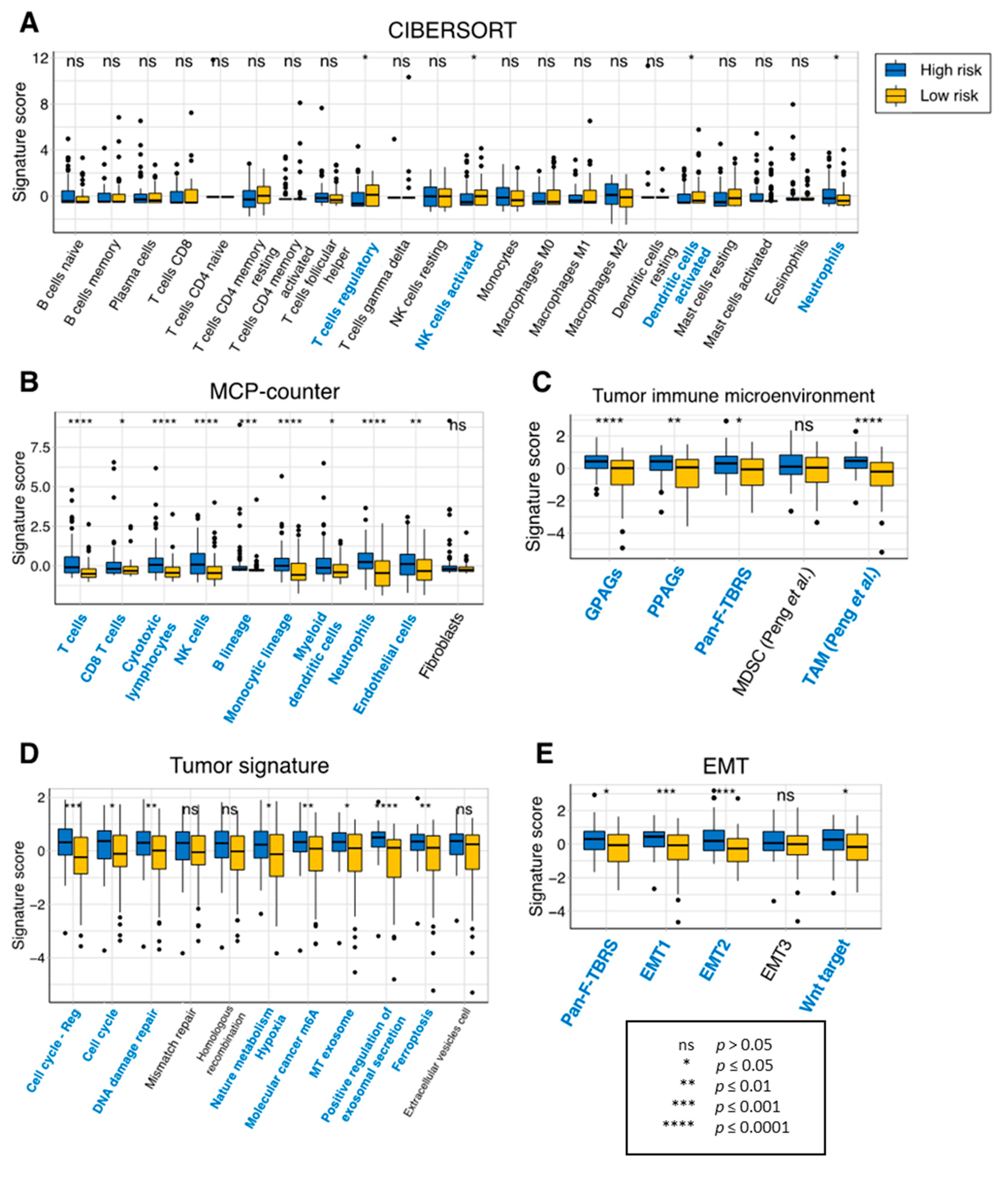{kind=link}
| Dataset | Sample Size | Clinical Information | Platform | Application |
|---|---|---|---|---|
| TCGA-GBM | 142 GBM | Female 50 (35.2%) Age 61.6 ± 11.9 (range 24–89) | Illumina | Differential expression analysis, risk score construction |
| GTEx (BA9) | 209 normal frontal cortex samples | Female 56 (26.8%) Age group 60–70 (57.4%), 50–59 (30.1%), 40–49 (8.1%), 30–39 (2%), 20–29 (2.4%) | Illumina TruSeq | Differential expression analysis |
| GSE4290 | 81 GBM vs. 23 non-tumor (epilepsy) | Not available | Affymetrix HG-U133Plus2 | Differential expression analysis |
| GSE68848 | 228 GBM vs. 28 non-tumor | Not available | Affymetrix HG-U133Plus2 | Differential expression analysis |
| CGGA | 79 GBM | Not applicable | Not applicable | Risk score validation |
| GSE43378 | 32 GBM | Not applicable | Not applicable | Risk score validation |
| Gene | Coefficient | Hazard Ratio | 95% Confidence Interval | p Value |
|---|---|---|---|---|
| CRNDE | 0.00047 | 1.00047 | 1.00021–1.000739 | 0.0004 |
| NRXN3 | 0.00067 | 1.00067 | 1.000294–1.001064 | 0.0005 |
| POPDC3 | 0.00202 | 1.00202 | 1.000876–1.003172 | 0.0005 |
| PTPRN | 0.00017 | 1.00017 | 1.000106–1.000246 | 8.8 × 10−7 |
| PTPRN2 | 0.00012 | 1.00012 | 1.000054–1.000205 | 0.0007 |
| SLC46A2 | 0.06594 | 1.06816 | 1.037777–1.098549 | 2.1 × 10−5 |
| TIMP1 | 9.03 × 10−6 | 1.000009 | 1.000004–1.000014 | 0.0004 |
| TNFSF9 | 0.00361 | 1.00362 | 1.001559–1.005691 | 0.0005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, H.-H.; Ta, H.D.K.; Nguyen, T.T.T.; Wang, C.-Y.; Lee, K.-H.; Le, N.Q.K. Identification of a Novel Eight-Gene Risk Model for Predicting Survival in Glioblastoma: A Comprehensive Bioinformatic Analysis. Cancers 2023, 15, 3899. https://doi.org/10.3390/cancers15153899
Dang H-H, Ta HDK, Nguyen TTT, Wang C-Y, Lee K-H, Le NQK. Identification of a Novel Eight-Gene Risk Model for Predicting Survival in Glioblastoma: A Comprehensive Bioinformatic Analysis. Cancers. 2023; 15(15):3899. https://doi.org/10.3390/cancers15153899
Chicago/Turabian StyleDang, Huy-Hoang, Hoang Dang Khoa Ta, Truc Tran Thanh Nguyen, Chih-Yang Wang, Kuen-Haur Lee, and Nguyen Quoc Khanh Le. 2023. "Identification of a Novel Eight-Gene Risk Model for Predicting Survival in Glioblastoma: A Comprehensive Bioinformatic Analysis" Cancers 15, no. 15: 3899. https://doi.org/10.3390/cancers15153899