
Analyses of Genes Critical to Tumor Survival Reveal Potential ‘Supertargets’: Focus on Transcription
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Approach
3.2. Blood Malignancies
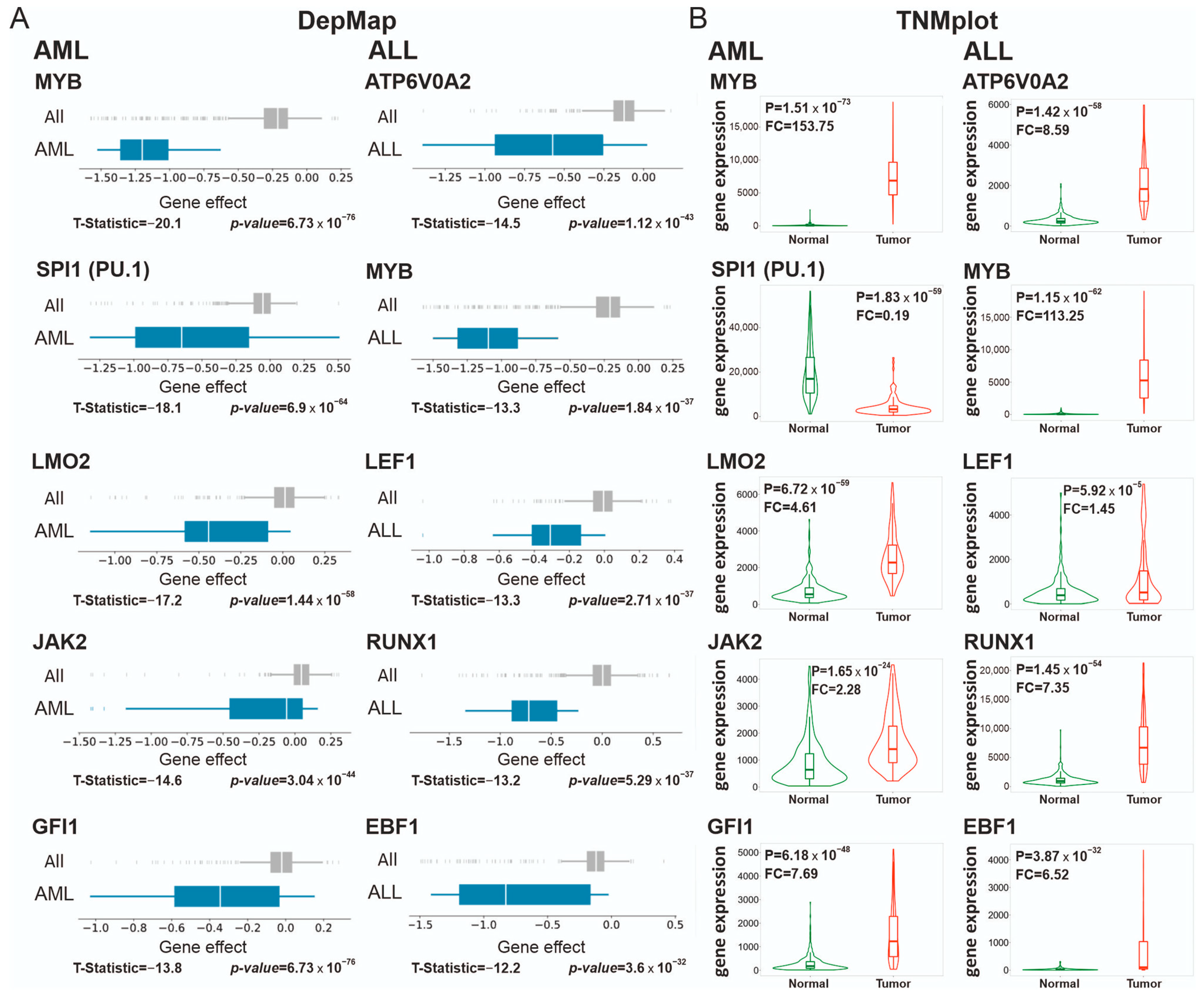
3.2.1. Supertargets in Acute Myeloid Leukemia (AML) and Acute Lymphocytic Leukemia (ALL)
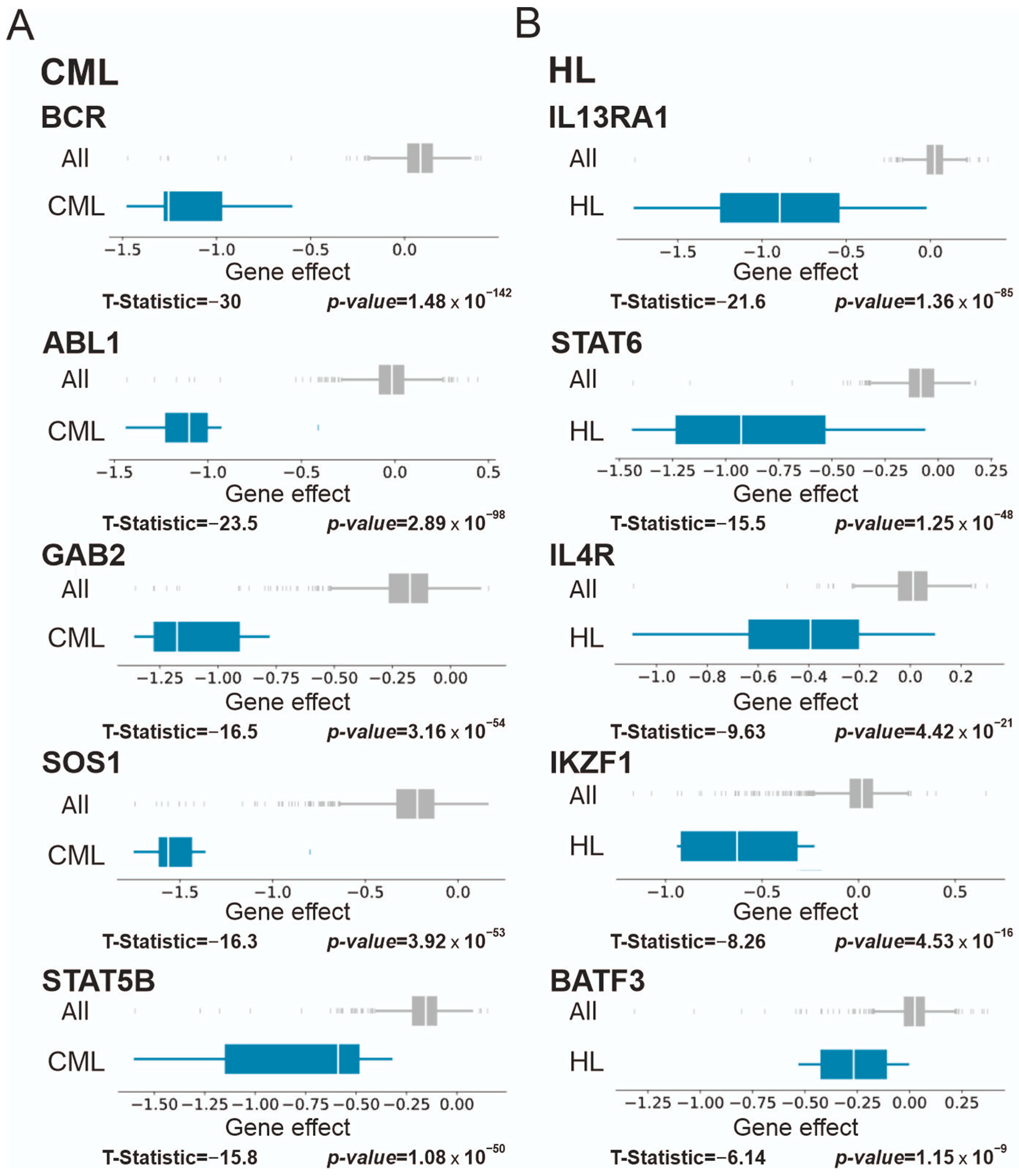
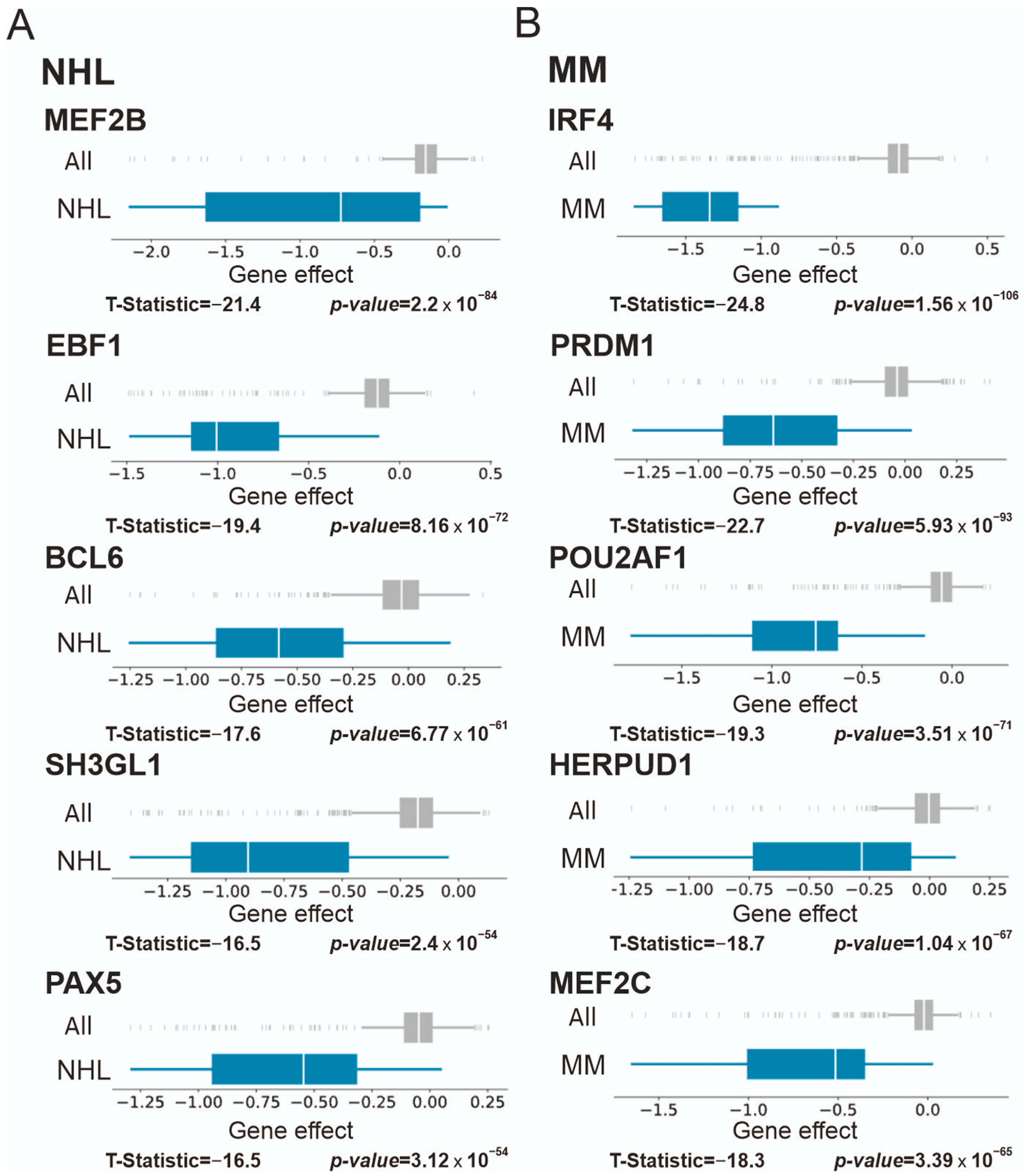
3.2.2. Chronic Myelogenous Leukemia (CML), Lymphoma, and Multiple Myeloma
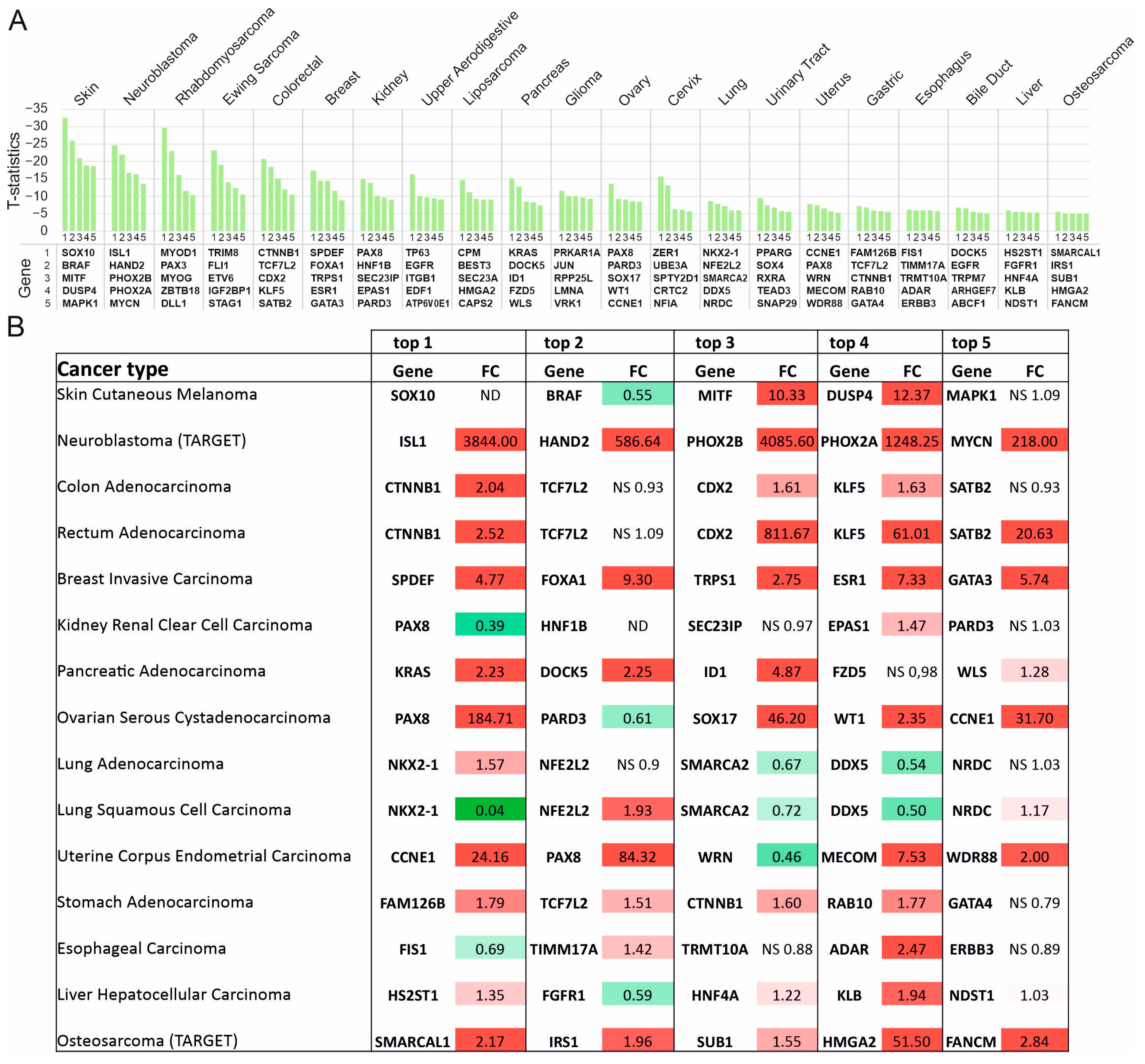
3.3. Supertargets in Solid Tumors
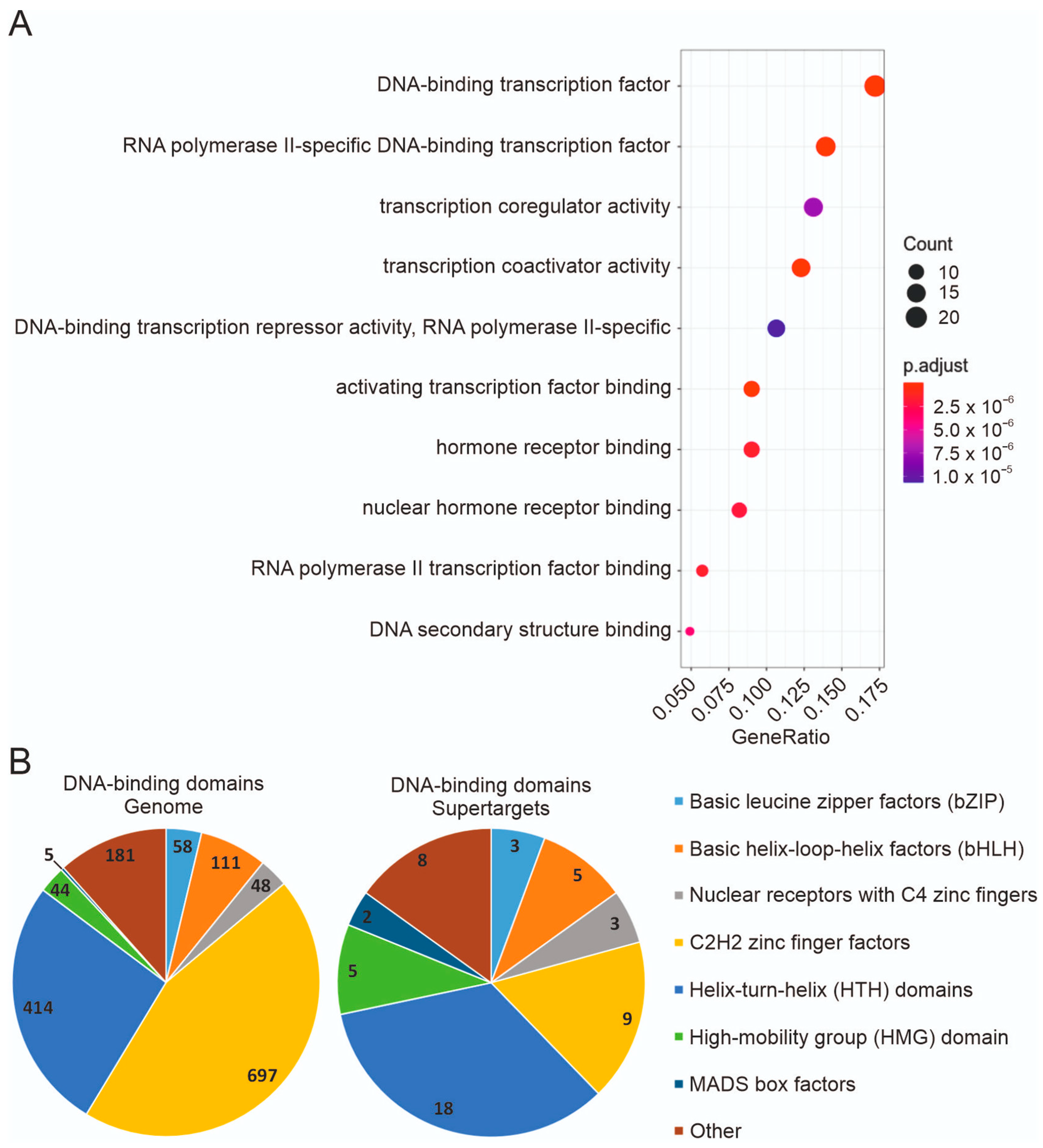
3.4. Expression in Tumor Cell Lines and Functional Types of Supertargets
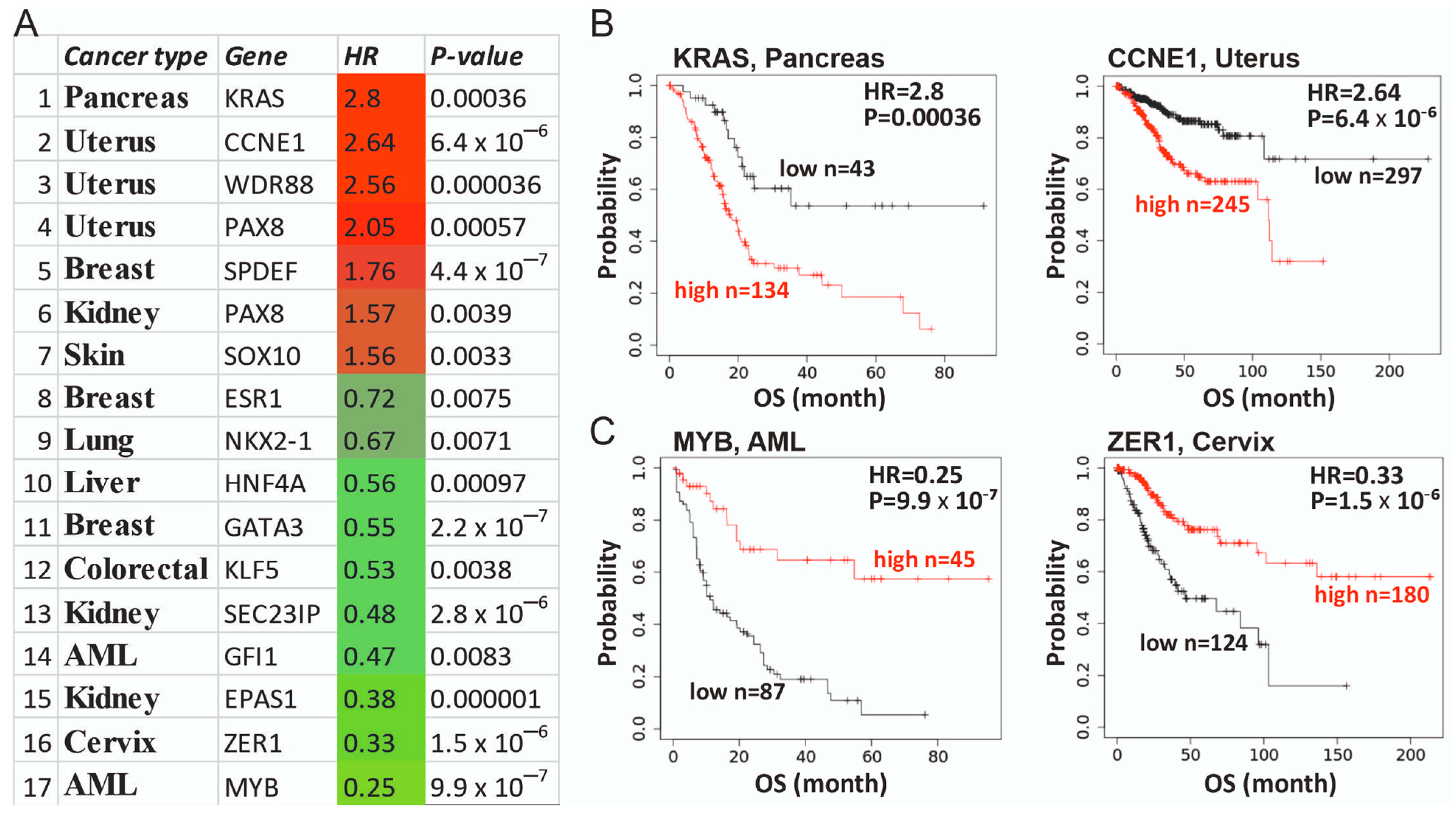
3.5. Clinical Utility of Supertargets
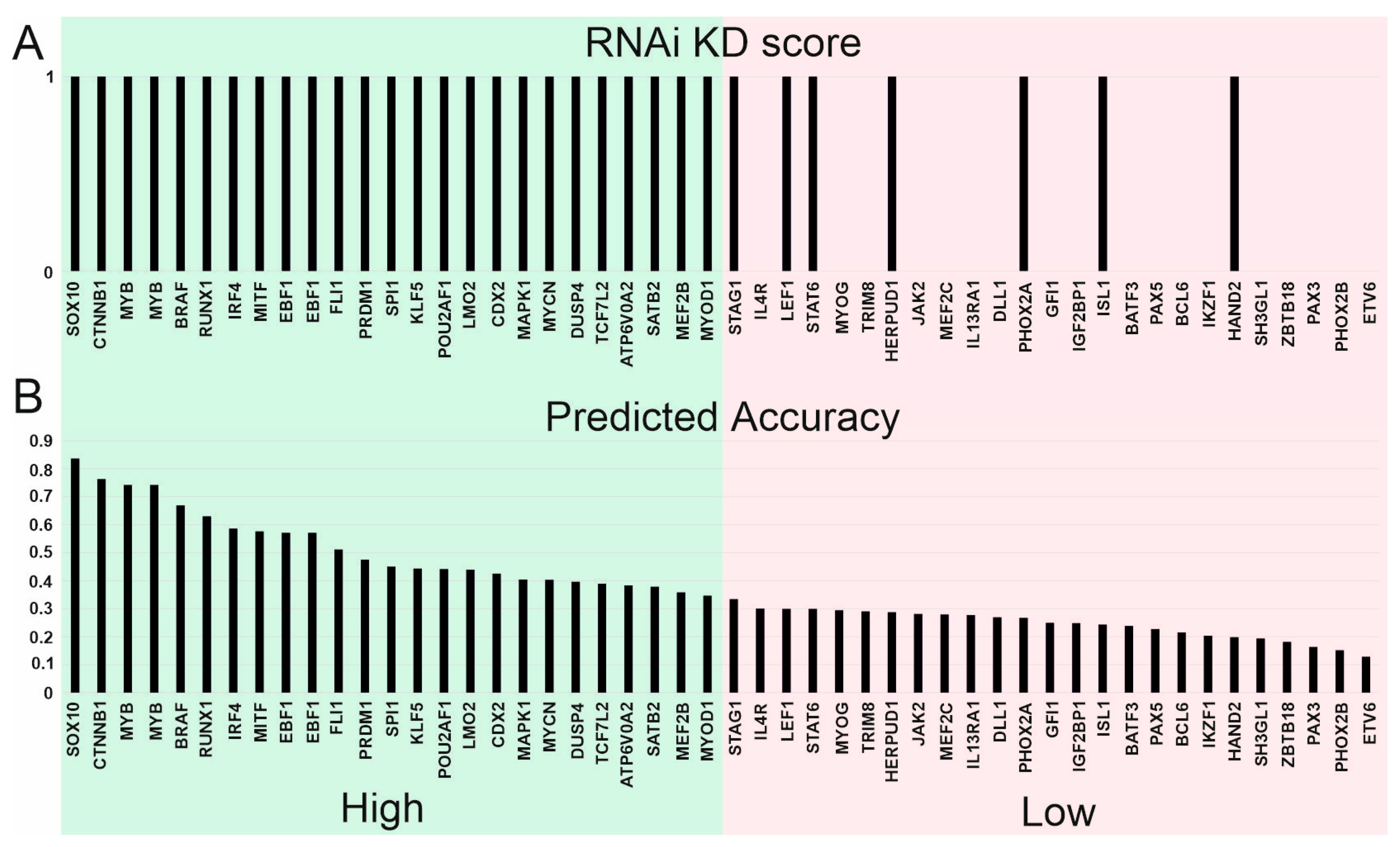
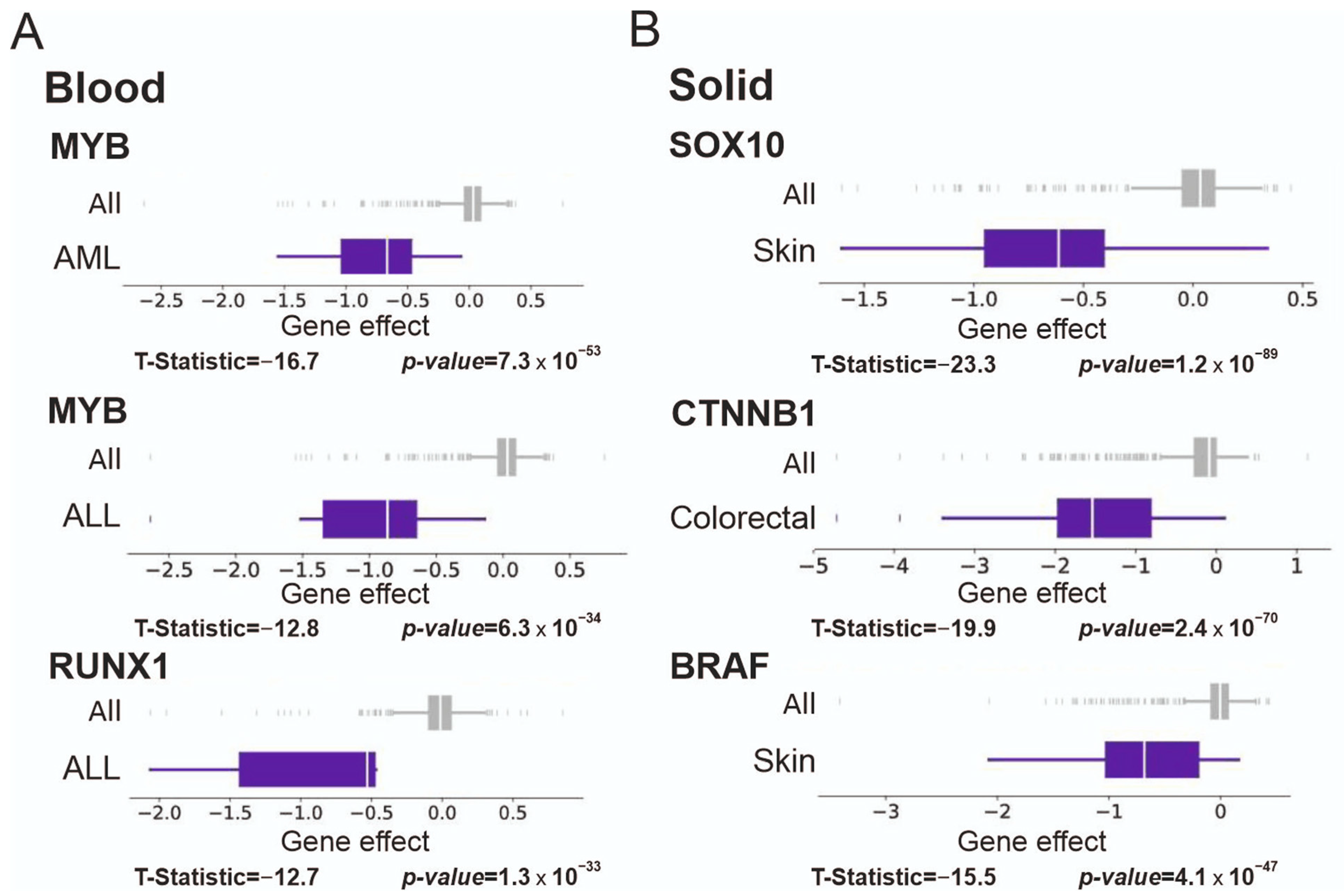
3.6. Significance of Supertargets Is Supported by RNAi DepMap Data
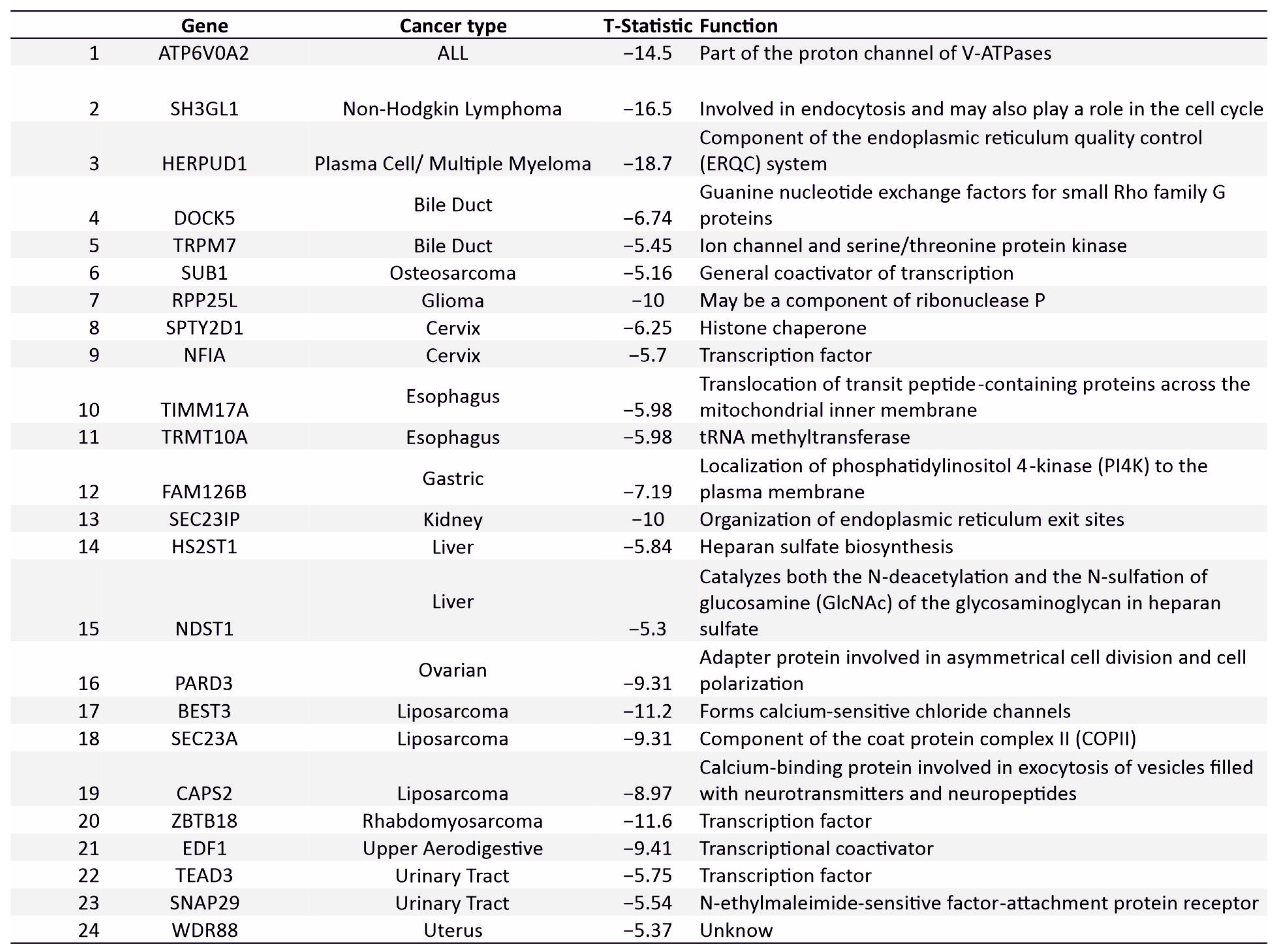
3.7. Novel Supertargets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Pfohl, U.; Pflaume, A.; Regenbrecht, M.; Finkler, S.; Graf Adelmann, Q.; Reinhard, C.; Regenbrecht, C.R.A.; Wedeken, L. Precision Oncology Beyond Genomics: The Future Is Here-It Is Just Not Evenly Distributed. Cells 2021, 10, 928. [Google Scholar] [CrossRef]
- Verma, M. Personalized Medicine and Cancer. J. Pers. Med. 2012, 2, 1–14. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational Correction of Copy Number Effect Improves Specificity of CRISPR-Cas9 Essentiality Screens in Cancer Cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Marcotte, R.; Brown, K.R.; Suarez, F.; Sayad, A.; Karamboulas, K.; Krzyzanowski, P.M.; Sircoulomb, F.; Medrano, M.; Fedyshyn, Y.; Koh, J.L.Y.; et al. Essential Gene Profiles in Breast, Pancreatic, and Ovarian Cancer Cells. Cancer Discov. 2012, 2, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Cowley, G.S.; Weir, B.A.; Vazquez, F.; Tamayo, P.; Scott, J.A.; Rusin, S.; East-Seletsky, A.; Ali, L.D.; Gerath, W.F.; Pantel, S.E.; et al. Parallel Genome-Scale Loss of Function Screens in 216 Cancer Cell Lines for the Identification of Context-Specific Genetic Dependencies. Sci. Data 2014, 1, 140035. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, R.; Sayad, A.; Brown, K.R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner, J.E.; Bader, G.D.; Mills, G.B.; et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.W.; Cowley, G.S.; Weir, B.A.; Boehm, J.S.; Rusin, S.; Scott, J.A.; East, A.; Ali, L.D.; Lizotte, P.H.; Wong, T.C.; et al. Systematic Investigation of Genetic Vulnerabilities across Cancer Cell Lines Reveals Lineage-Specific Dependencies in Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 12372–12377. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. TNMplot.Com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER Version 14: More Genomes, a New PANTHER GO-Slim and Improvements in Enrichment Analysis Tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a Complete Collection of Transcription Factor Binding Models for Human and Mouse via Large-Scale ChIP-Seq Analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef]
- Ishii, H.; Yano, S. New Therapeutic Strategies for Adult Acute Myeloid Leukemia. Cancers 2022, 14, 2806. [Google Scholar] [CrossRef]
- Yu, J.; Jiang, P.Y.Z.; Sun, H.; Zhang, X.; Jiang, Z.; Li, Y.; Song, Y. Advances in Targeted Therapy for Acute Myeloid Leukemia. Biomark. Res. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Takao, S.; Forbes, L.; Uni, M.; Cheng, S.; Pineda, J.M.B.; Tarumoto, Y.; Cifani, P.; Minuesa, G.; Chen, C.; Kharas, M.G.; et al. Convergent Organization of Aberrant MYB Complex Controls Oncogenic Gene Expression in Acute Myeloid Leukemia. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, V.; Arniani, S.; Pierini, V.; di Giacomo, D.; Pierini, T.; Gorello, P.; Mecucci, C.; la Starza, R. T-Cell Acute Lymphoblastic Leukemia: Biomarkers and Their Clinical Usefulness. Genes 2021, 12, 1118. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, V.; Arniani, S.; Pierini, V.; Pierini, T.; di Giacomo, D.; Gorello, P.; Moretti, M.; Pellanera, F.; Elia, L.; Vitale, A.; et al. MYB Rearrangements and Over-Expression in T-Cell Acute Lymphoblastic Leukemia. Genes Chromosom. Cancer 2021, 60, 482–488. [Google Scholar] [CrossRef]
- Klempnauer, K.-H. C/EBPβ Sustains the Oncogenic Program of AML Cells by Cooperating with MYB and Co-Activator P300 in a Transcriptional Module. Exp. Hematol. 2022, 108, 8–15. [Google Scholar] [CrossRef]
- Zuber, J.; Rappaport, A.R.; Luo, W.; Wang, E.; Chen, C.; Vaseva, A.V.; Shi, J.; Weissmueller, S.; Fellmann, C.; Taylor, M.J.; et al. An Integrated Approach to Dissecting Oncogene Addiction Implicates a Myb-Coordinated Self-Renewal Program as Essential for Leukemia Maintenance. Genes Dev. 2011, 25, 1628–1640. [Google Scholar] [CrossRef]
- Uttarkar, S.; Frampton, J.; Klempnauer, K.-H. Targeting the Transcription Factor Myb by Small-Molecule Inhibitors. Exp. Hematol. 2017, 47, 31–35. [Google Scholar] [CrossRef]
- Khan, I.; Eklund, E.E.; Gartel, A.L. Therapeutic Vulnerabilities of Transcription Factors in AML. Mol. Cancer Ther. 2021, 20, 229–237. [Google Scholar] [CrossRef]
- Antony-Debré, I.; Paul, A.; Leite, J.; Mitchell, K.; Kim, H.M.; Carvajal, L.A.; Todorova, T.I.; Huang, K.; Kumar, A.; Farahat, A.A.; et al. Pharmacological Inhibition of the Transcription Factor PU.1 in Leukemia. J. Clin. Investig. 2017, 127, 4297–4313. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.; Rabbitts, T.H. LMO2 at 25 Years: A Paradigm of Chromosomal Translocation Proteins. Open Biol. 2015, 5, 150062. [Google Scholar] [CrossRef]
- Wadman, I.A.; Osada, H.; Grütz, G.G.; Agulnick, A.D.; Westphal, H.; Forster, A.; Rabbitts, T.H. The LIM-Only Protein Lmo2 Is a Bridging Molecule Assembling an Erythroid, DNA-Binding Complex Which Includes the TAL1, E47, GATA-1 and Ldb1/NLI Proteins. EMBO J. 1997, 16, 3145–3157. [Google Scholar] [CrossRef] [PubMed]
- Nam, C.-H.; Rabbitts, T.H. The Role of LMO2 in Development and in T Cell Leukemia after Chromosomal Translocation or Retroviral Insertion. Mol. Ther. 2006, 13, 15–25. [Google Scholar] [CrossRef]
- Meyer, S.C.; Levine, R.L. Molecular Pathways: Molecular Basis for Sensitivity and Resistance to JAK Kinase Inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Senkevitch, E.; Durum, S. The Promise of Janus Kinase Inhibitors in the Treatment of Hematological Malignancies. Cytokine 2017, 98, 33–41. [Google Scholar] [CrossRef]
- Möröy, T.; Khandanpour, C. Role of GFI1 in Epigenetic Regulation of MDS and AML Pathogenesis: Mechanisms and Therapeutic Implications. Front. Oncol. 2019, 9, 824. [Google Scholar] [CrossRef]
- Tijchon, E.; Havinga, J.; van Leeuwen, F.N.; Scheijen, B. B-Lineage Transcription Factors and Cooperating Gene Lesions Required for Leukemia Development. Leukemia 2013, 27, 541–552. [Google Scholar] [CrossRef]
- Eaton, A.F.; Merkulova, M.; Brown, D. The H+-ATPase (V-ATPase): From Proton Pump to Signaling Complex in Health and Disease. Am. J. Physiol. Physiol. 2021, 320, C392–C414. [Google Scholar] [CrossRef]
- Steidl, U.; Rosenbauer, F.; Verhaak, R.G.W.; Gu, X.; Ebralidze, A.; Otu, H.H.; Klippel, S.; Steidl, C.; Bruns, I.; Costa, D.B.; et al. Essential Role of Jun Family Transcription Factors in PU.1 Knockdown-Induced Leukemic Stem Cells. Nat. Genet. 2006, 38, 1269–1277. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, C.; Su, R.; Yin, Z.; Huang, G.; Yang, J.; Li, Z.; Zhang, K.; Fei, J. Targeting SOS1 Overcomes Imatinib Resistance with BCR-ABL Independence through Uptake Transporter SLC22A4 in CML. Mol. Ther. Oncolytics 2021, 23, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Sayad, A.; Chan, G.; Yang, W.; Lu, Z.; Virtanen, C.; van Etten, R.A.; Neel, B.G. SHP2 Is Required for BCR-ABL1-Induced Hematologic Neoplasia. Leukemia 2018, 32, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Able, A.A.; Burrell, J.A.; Stephens, J.M. STAT5-Interacting Proteins: A Synopsis of Proteins That Regulate STAT5 Activity. Biology 2017, 6, 20. [Google Scholar] [CrossRef]
- Suzuki, A.; Leland, P.; Joshi, B.H.; Puri, R.K. Targeting of IL-4 and IL-13 Receptors for Cancer Therapy. Cytokine 2015, 75, 79–88. [Google Scholar] [CrossRef]
- Xia, R.; Cheng, Y.; Han, X.; Wei, Y.; Wei, X. Ikaros Proteins in Tumor: Current Perspectives and New Developments. Front. Mol. Biosci. 2021, 8, 788440. [Google Scholar] [CrossRef]
- Kasprzyk, M.E.; Sura, W.; Dzikiewicz-Krawczyk, A. Enhancing B-Cell Malignancies-On Repurposing Enhancer Activity towards Cancer. Cancers 2021, 13, 3270. [Google Scholar] [CrossRef]
- Hammood, M.; Craig, A.W.; Leyton, J.V. Impact of Endocytosis Mechanisms for the Receptors Targeted by the Currently Approved Antibody-Drug Conjugates (ADCs)-A Necessity for Future ADC Research and Development. Pharmaceuticals 2021, 14, 674. [Google Scholar] [CrossRef]
- Song, S.; Matthias, P.D. The Transcriptional Regulation of Germinal Center Formation. Front. Immunol. 2018, 9, 2026. [Google Scholar] [CrossRef]
- Nie, X.; Liu, D.; Zheng, M.; Li, X.; Liu, O.; Guo, Q.; Zhu, L.; Lin, B. HERPUD1 Promotes Ovarian Cancer Cell Survival by Sustaining Autophagy and Inhibit Apoptosis via PI3K/AKT/MTOR and P38 MAPK Signaling Pathways. BMC Cancer 2022, 22, 1338. [Google Scholar] [CrossRef]
- Peng, Y.; Li, N.; Tang, F.; Qian, C.; Jia, T.; Liu, J.; Xu, Y. Corosolic Acid Sensitizes Ferroptosis by Upregulating HERPUD1 in Liver Cancer Cells. Cell Death Discov. 2022, 8, 376. [Google Scholar] [CrossRef]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective Gene Dependencies in MYCN-Amplified Neuroblastoma Include the Core Transcriptional Regulatory Circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef]
- Xu, M.; Sun, M.; Zhang, X.; Nguyen, R.; Lei, H.; Shern, J.F.; Thiele, C.J.; Liu, Z. HAND2 Assists MYCN Enhancer Invasion to Regulate a Noradrenergic Neuroblastoma Phenotype. Cancer Res. 2023, 83, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, C.; Bu, X.; Que, Y.; Zhang, L.; Zhang, Y.; Zhang, L.; Lu, S.; Huang, J.; Zhu, J.; et al. ISL1 Promoted Tumorigenesis and EMT via Aurora Kinase A-Induced Activation of PI3K/AKT Signaling Pathway in Neuroblastoma. Cell Death Dis. 2021, 12, 620. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.K.; Geradts, J.; Young, J. Prostate-Derived Ets Factor, an Oncogenic Driver in Breast Cancer. Tumor Biol. 2017, 39, 1010428317691688. [Google Scholar] [CrossRef] [PubMed]
- Porras, L.; Ismail, H.; Mader, S. Positive Regulation of Estrogen Receptor Alpha in Breast Tumorigenesis. Cells 2021, 10, 2966. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Huang, Y.; He, L.; Zhang, W.; Ren, J.; Wang, Y.; Wu, J.; Wu, X.; Shan, L.; et al. TRPS1 Drives Heterochromatic Origin Refiring and Cancer Genome Evolution. Cell Rep. 2021, 34, 108814. [Google Scholar] [CrossRef]
- Kondratick, C.M.; Washington, M.T.; Spies, M. Making Choices: DNA Replication Fork Recovery Mechanisms. Semin. Cell Dev. Biol. 2021, 113, 27–37. [Google Scholar] [CrossRef]
- Bansal, R.; Hussain, S.; Chanana, U.B.; Bisht, D.; Goel, I.; Muthuswami, R. SMARCAL1, the Annealing Helicase and the Transcriptional Co-Regulator. IUBMB Life 2020, 72, 2080–2096. [Google Scholar] [CrossRef]
- O’Rourke, J.J.; Bythell-Douglas, R.; Dunn, E.A.; Deans, A.J. ALT Control, Delete: FANCM as an Anti-Cancer Target in Alternative Lengthening of Telomeres. Nucleus 2019, 10, 221–230. [Google Scholar] [CrossRef]
- Lu, R.; O’Rourke, J.J.; Sobinoff, A.P.; Allen, J.A.M.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI Complex Suppresses Alternative Lengthening of Telomeres (ALT). Nat. Commun. 2019, 10, 2252. [Google Scholar] [CrossRef]
- Caldwell, R.B.; Braselmann, H.; Schoetz, U.; Heuer, S.; Scherthan, H.; Zitzelsberger, H. Positive Cofactor 4 (PC4) Is Critical for DNA Repair Pathway Re-Routing in DT40 Cells. Sci. Rep. 2016, 6, 28890. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Evers, B.; Helleday, T. PC4 Promotes Genome Stability and DNA Repair through Binding of SsDNA at DNA Damage Sites. Oncogene 2016, 35, 761–770. [Google Scholar] [CrossRef]
- Reiss, K.; del Valle, L.; Lassak, A.; Trojanek, J. Nuclear IRS-1 and Cancer. J. Cell. Physiol. 2012, 227, 2992–3000. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Ditzel, H.J.; Duijf, P.H.G.; Khaze, V.; Gjerstorff, M.F.; Baradaran, B. HMGA2 as a Critical Regulator in Cancer Development. Genes 2021, 12, 269. [Google Scholar] [CrossRef] [PubMed]
- Fedele, V.; Dai, F.; Masilamani, A.P.; Heiland, D.H.; Kling, E.; Gätjens-Sanchez, A.M.; Ferrarese, R.; Platania, L.; Soroush, D.; Kim, H.; et al. Epigenetic Regulation of ZBTB18 Promotes Glioblastoma Progression. Mol. Cancer Res. 2017, 15, 998–1011. [Google Scholar] [CrossRef] [PubMed]
- Bazzocco, S.; Dopeso, H.; Martínez-Barriocanal, Á.; Anguita, E.; Nieto, R.; Li, J.; García-Vidal, E.; Maggio, V.; Rodrigues, P.; de Marcondes, P.G.; et al. Identification of ZBTB18 as a Novel Colorectal Tumor Suppressor Gene through Genome-Wide Promoter Hypermethylation Analysis. Clin. Epigenet. 2021, 13, 88. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Pan, D. The Hippo Signaling Pathway in Development and Disease. Dev. Cell 2019, 50, 264–282. [Google Scholar] [CrossRef]
- Cunningham, R.; Hansen, C.G. The Hippo Pathway in Cancer: YAP/TAZ and TEAD as Therapeutic Targets in Cancer. Clin. Sci. 2022, 136, 197–222. [Google Scholar] [CrossRef]
- Zenker, M.; Bunt, J.; Schanze, I.; Schanze, D.; Piper, M.; Priolo, M.; Gerkes, E.H.; Gronostajski, R.M.; Richards, L.J.; Vogt, J.; et al. Variants in Nuclear Factor I Genes Influence Growth and Development. Am. J. Med Genet. Part C Semin. Med Genet. 2019, 181, 611–626. [Google Scholar] [CrossRef]
- Chen, K.-S.; Lim, J.W.C.; Richards, L.J.; Bunt, J. The Convergent Roles of the Nuclear Factor I Transcription Factors in Development and Cancer. Cancer Lett. 2017, 410, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lee, D.; Dhiman, V.; Jiang, P.; Xu, J.; McGillivray, P.; Yang, H.; Liu, J.; Meyerson, W.; Clarke, D.; et al. An Integrative ENCODE Resource for Cancer Genomics. Nat. Commun. 2020, 11, 3696. [Google Scholar] [CrossRef]
- Vishnoi, K.; Viswakarma, N.; Rana, A.; Rana, B. Transcription Factors in Cancer Development and Therapy. Cancers 2020, 12, 2296. [Google Scholar] [CrossRef]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.-H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef]
- Bushweller, J.H. Targeting Transcription Factors in Cancer—From Undruggable to Reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Illendula, A.; Pulikkan, J.A.; Zong, H.; Grembecka, J.; Xue, L.; Sen, S.; Zhou, Y.; Boulton, A.; Kuntimaddi, A.; Gao, Y.; et al. A Small-Molecule Inhibitor of the Aberrant Transcription Factor CBFβ-SMMHC Delays Leukemia in Mice. Science 2015, 347, 779–784. [Google Scholar] [CrossRef]
- Konopleva, M.Y.; Röllig, C.; Cavenagh, J.; Deeren, D.; Girshova, L.; Krauter, J.; Martinelli, G.; Montesinos, P.; Schäfer, J.A.; Ottmann, O.; et al. Idasanutlin plus Cytarabine in Relapsed or Refractory Acute Myeloid Leukemia: Results of the MIRROS Trial. Blood Adv. 2022, 6, 4147–4156. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DeAngelo, D.J.; Chromik, J.; Chatterjee, M.; Bauer, S.; Lin, C.-C.; Suarez, C.; de Vos, F.; Steeghs, N.; Cassier, P.A.; et al. Results from a First-in-Human Phase I Study of Siremadlin (HDM201) in Patients with Advanced Wild-Type TP53 Solid Tumors and Acute Leukemia. Clin. Cancer Res. 2022, 28, 870–881. [Google Scholar] [CrossRef]
- Karadkhelkar, N.M.; Lin, M.; Eubanks, L.M.; Janda, K.D. Demystifying the Druggability of the MYC Family of Oncogenes. J. Am. Chem. Soc. 2023, 145, 3259–3269. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Neklesa, T.K. Targeting Nuclear Receptors with PROTAC Degraders. Mol. Cell. Endocrinol. 2019, 493, 110452. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; Gordon, D.A.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110: An Oral Androgen Receptor PROTAC Degrader for Prostate Cancer. J. Clin. Oncol. 2019, 37, 259. [Google Scholar] [CrossRef]
- Koroleva, O.A.; Dutikova, Y.V.; Trubnikov, A.V.; Zenov, F.A.; Manasova, E.V.; Shtil, A.A.; Kurkin, A.V. PROTAC: Targeted Drug Strategy. Principles and Limitations. Russ. Chem. Bull. 2022, 71, 2310–2334. [Google Scholar] [CrossRef]
- Chetverina, D.A.; Lomaev, D.V.; Erokhin, M.M. Polycomb and Trithorax Group Proteins: The Long Road from Mutations in Drosophila to Use in Medicine. Acta Naturae 2020, 12, 66–85. [Google Scholar] [CrossRef]
- Bracken, A.P.; Brien, G.L.; Verrijzer, C.P. Dangerous Liaisons: Interplay between SWI/SNF, NuRD, and Polycomb in Chromatin Regulation and Cancer. Genes Dev. 2019, 33, 936–959. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF Complex in Cancer—Biology, Biomarkers and Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Erokhin, M.; Chetverina, O.; Győrffy, B.; Tatarskiy, V.V.; Mogila, V.; Shtil, A.A.; Roninson, I.B.; Moreaux, J.; Georgiev, P.; Cavalli, G.; et al. Clinical Correlations of Polycomb Repressive Complex 2 in Different Tumor Types. Cancers 2021, 13, 3155. [Google Scholar] [CrossRef]
- Yee, N.S. Role of TRPM7 in Cancer: Potential as Molecular Biomarker and Therapeutic Target. Pharmaceuticals 2017, 10, 39. [Google Scholar] [CrossRef]
- Köferle, A.; Schlattl, A.; Hörmann, A.; Thatikonda, V.; Popa, A.; Spreitzer, F.; Ravichandran, M.C.; Supper, V.; Oberndorfer, S.; Puchner, T.; et al. Interrogation of Cancer Gene Dependencies Reveals Paralog Interactions of Autosome and Sex Chromosome-Encoded Genes. Cell Rep. 2022, 39, 110636. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chetverina, D.; Vorobyeva, N.E.; Gyorffy, B.; Shtil, A.A.; Erokhin, M. Analyses of Genes Critical to Tumor Survival Reveal Potential ‘Supertargets’: Focus on Transcription. Cancers 2023, 15, 3042. https://doi.org/10.3390/cancers15113042
Chetverina D, Vorobyeva NE, Gyorffy B, Shtil AA, Erokhin M. Analyses of Genes Critical to Tumor Survival Reveal Potential ‘Supertargets’: Focus on Transcription. Cancers. 2023; 15(11):3042. https://doi.org/10.3390/cancers15113042
Chicago/Turabian StyleChetverina, Darya, Nadezhda E. Vorobyeva, Balazs Gyorffy, Alexander A. Shtil, and Maksim Erokhin. 2023. "Analyses of Genes Critical to Tumor Survival Reveal Potential ‘Supertargets’: Focus on Transcription" Cancers 15, no. 11: 3042. https://doi.org/10.3390/cancers15113042