
The Tumor Microenvironment in Tumorigenesis and Therapy Resistance Revisited


Abstract
:Simple Summary
Abstract
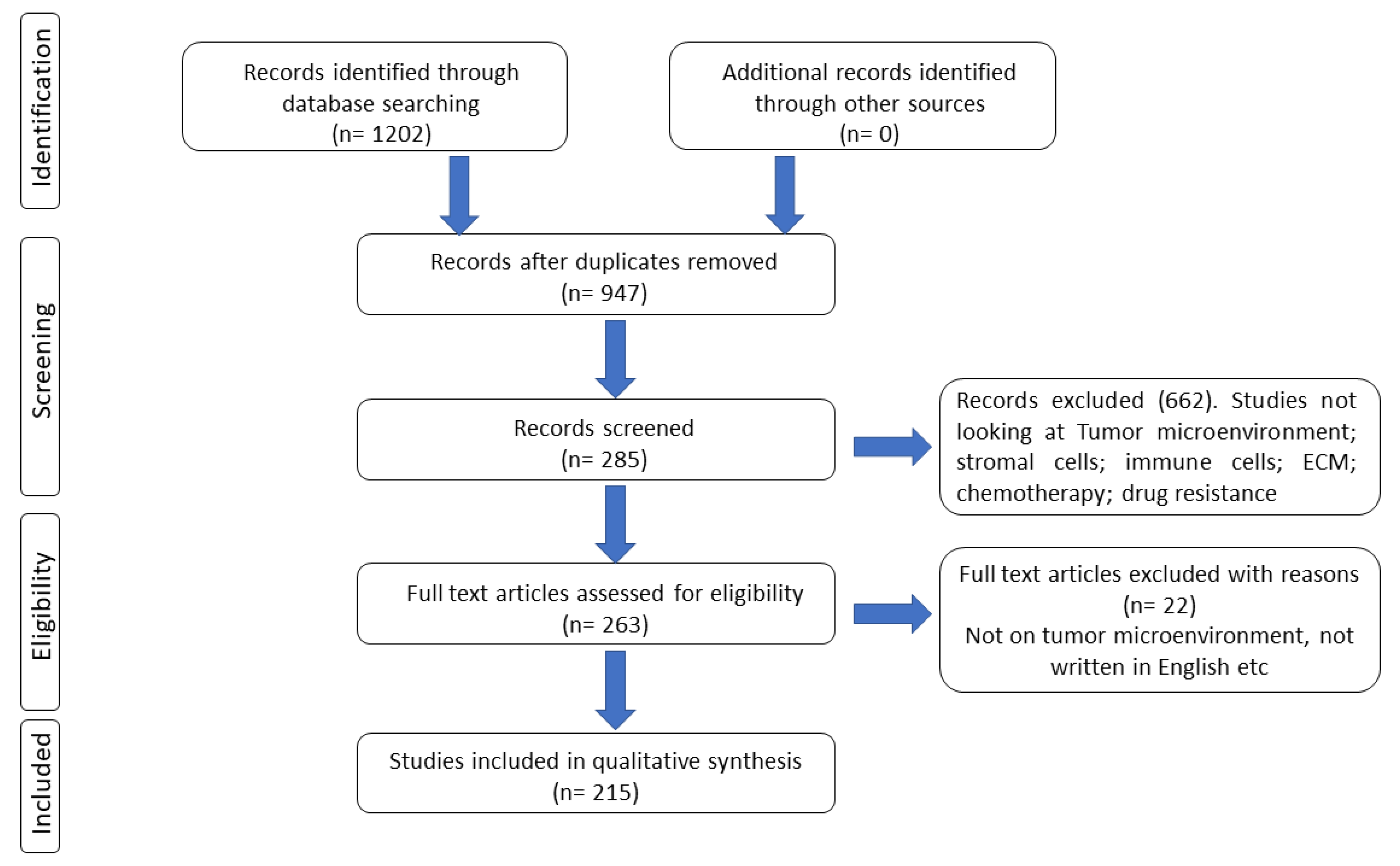
1. Methodology
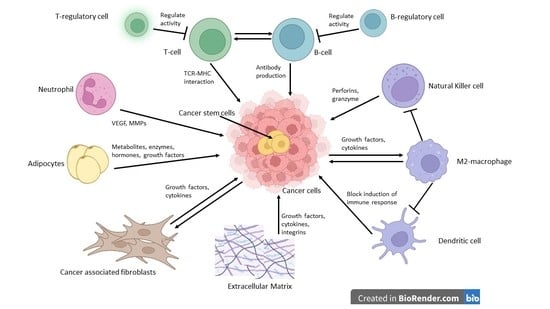
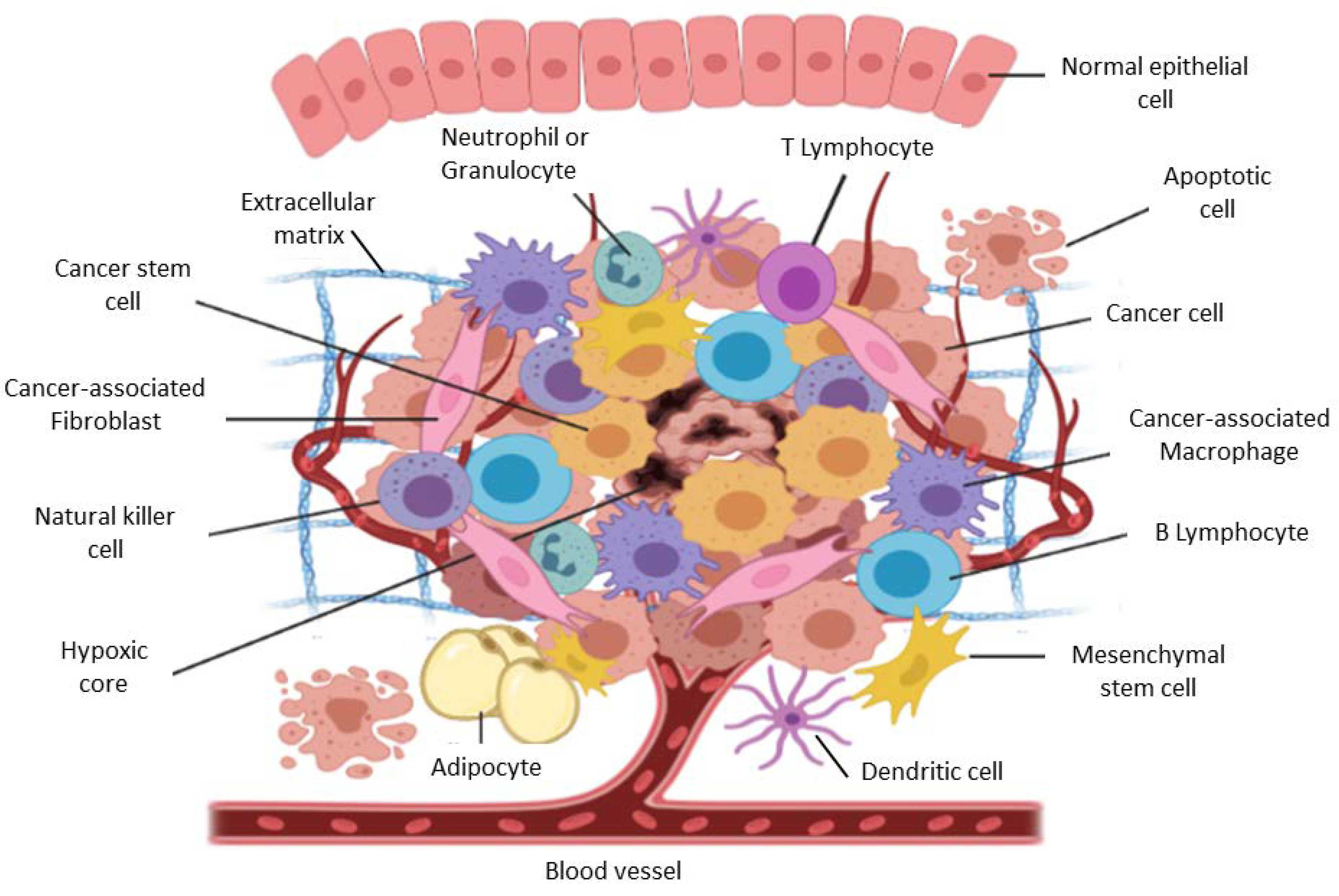
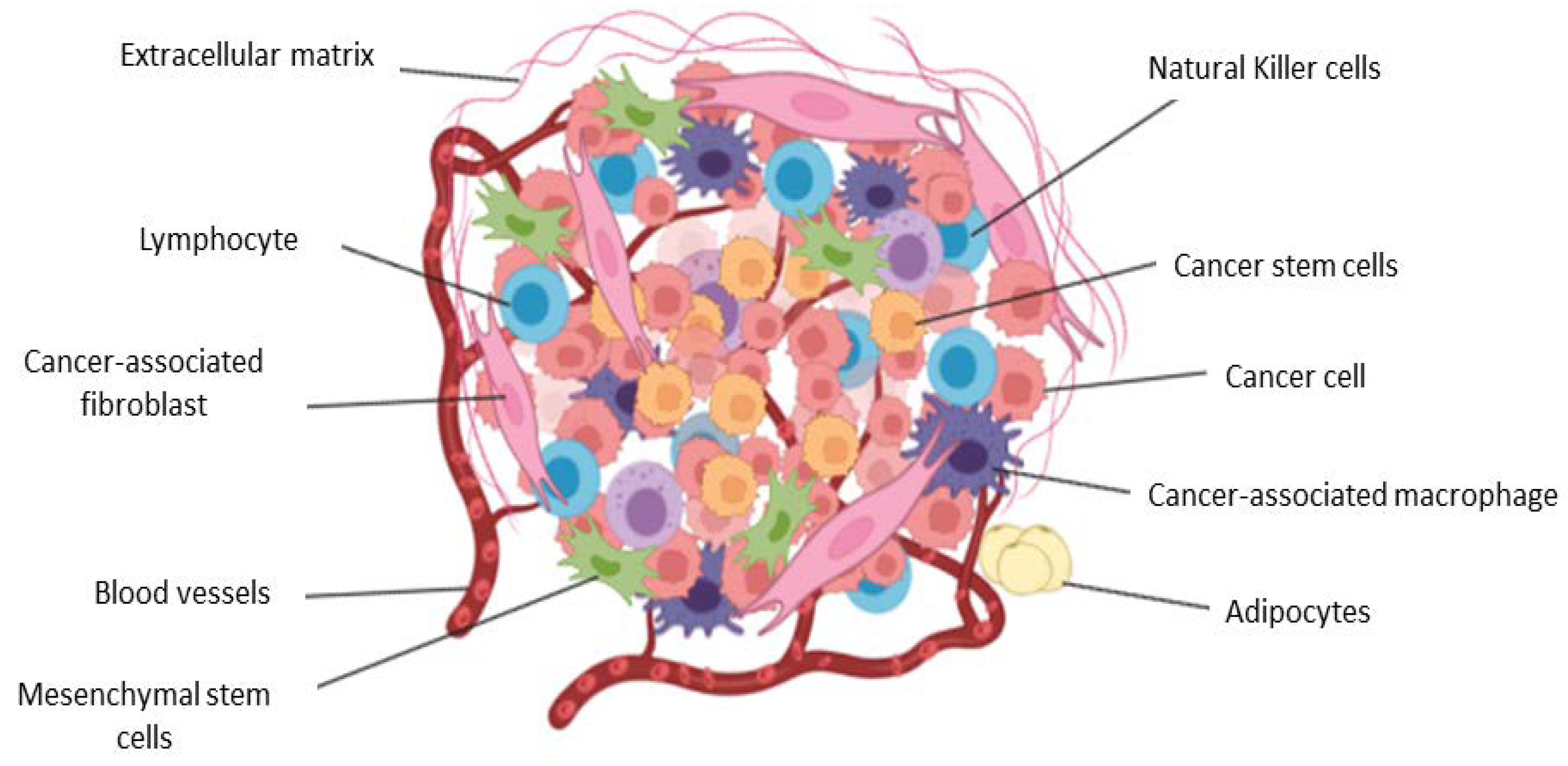
2. The Tumor Microenvironment in Brief
3. Biological Functions of Stromal and Immune Cells within the Tumor Microenvironment
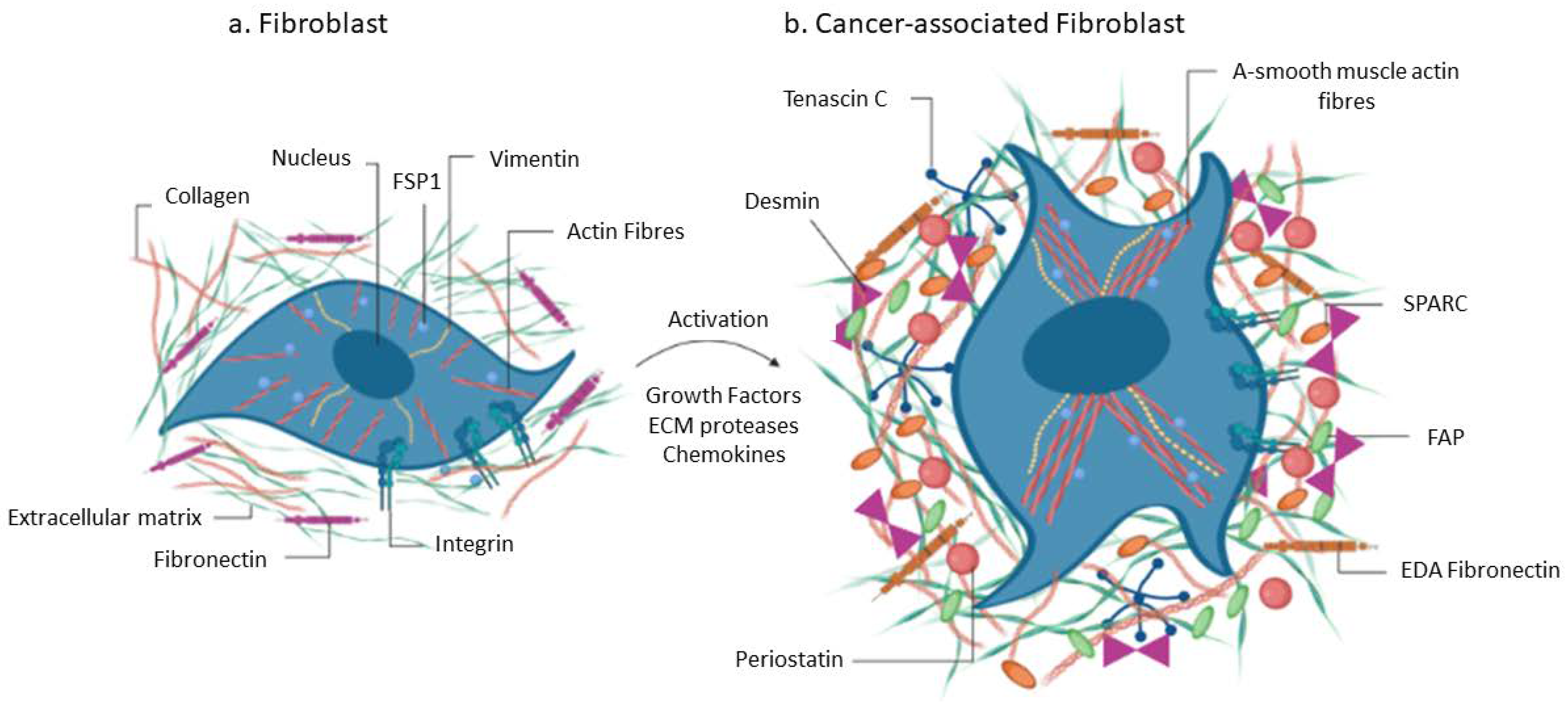
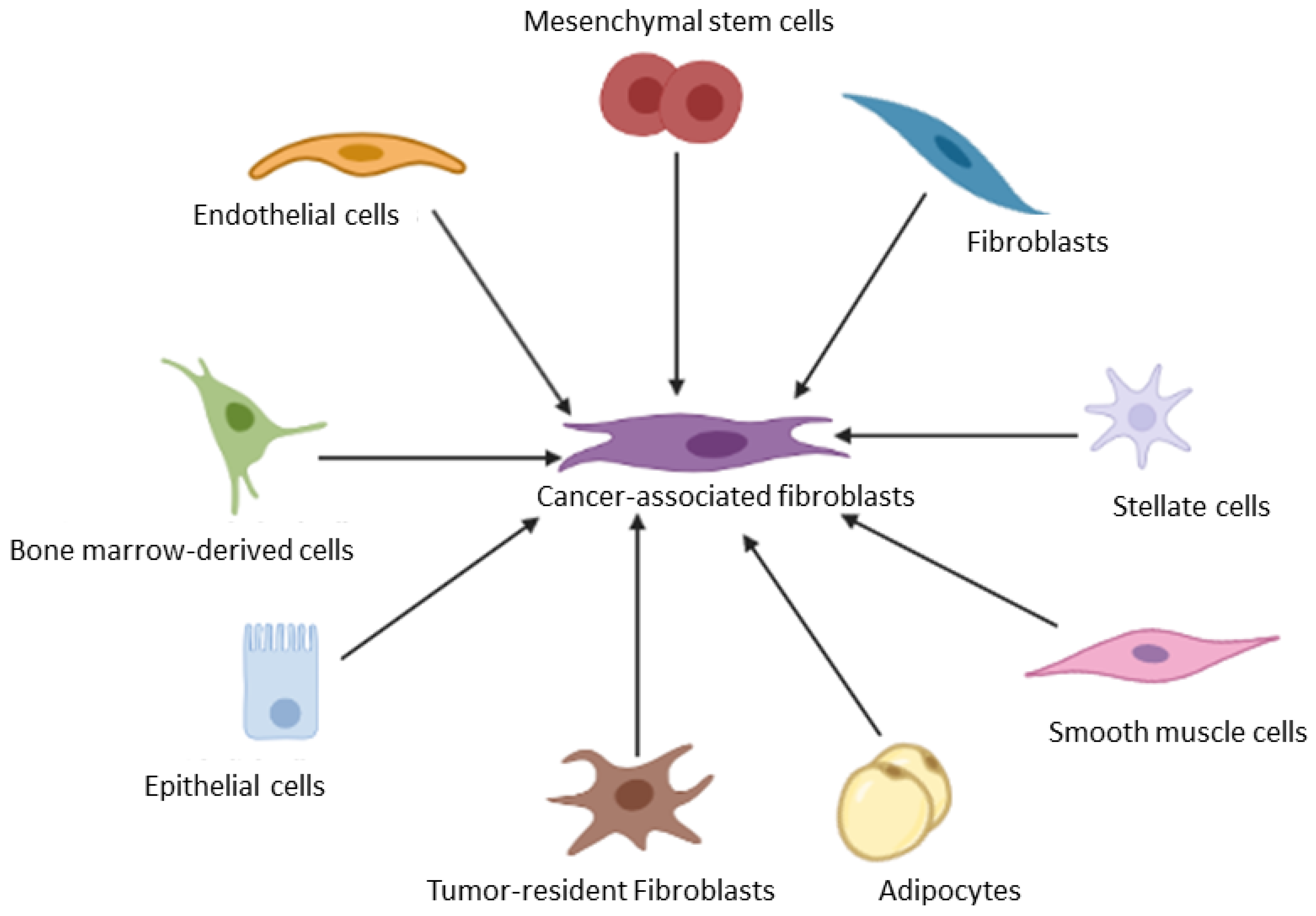
3.1. Cancer Associated Fibroblasts
3.2. Cancer Associated Endothelial Cells
3.3. Cancer-Associated Macrophages
3.4. Cancer-Associated Neutrophils
3.5. T Cells
3.6. B Cells
3.7. Natural Killer Cells
3.8. Dendritic Cells
3.9. Stellate Cells
3.10. Adipocytes
3.11. Mesenchymal Stem Cells
3.12. Pericytes
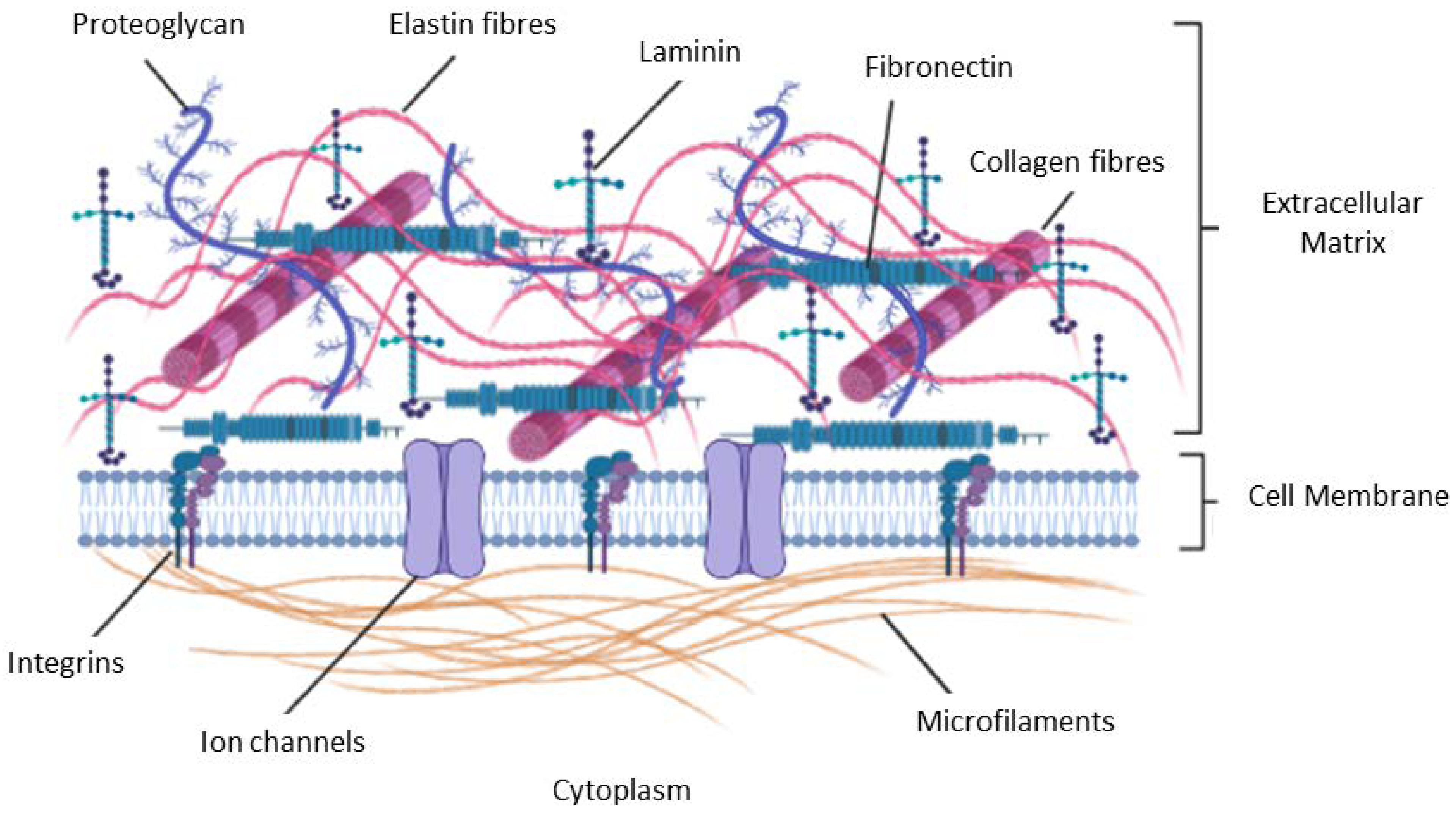
4. The Extracellular Matrix
5. Vascular Networks
6. Hypoxia within the TME
7. Exosomes and Exosomal miRNAs in Tumor Microenvironment
8. Advances in Therapeutic Targeting of TME
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016. [Google Scholar] [PubMed]
- Dzobo, K. Taking a Full Snapshot of Cancer Biology: Deciphering the Tumor Microenvironment for Effective Cancer Therapy in the Oncology Clinic. Omics 2020, 24, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erin, N.; Grahovac, J.; Brozovic, A.; Efferth, T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist. Updates 2020, 53, 100715. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Jonker, T.; Rowe, A.; Thomford, N.E.; Munro, D.; Dandara, C.; Wonkam, A.; Govender, D.; Calder, B.; Soares, N.C.; et al. The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices. Int. J. Mol. Sci. 2018, 19, 2861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [Green Version]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichoń, T.; Kułach, N. Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef]
- Kim, J.; Bae, J.S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediat. Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [Green Version]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K. Integrins Within the Tumor Microenvironment: Biological Functions, Importance for Molecular Targeting, and Cancer Therapeutics Innovation. Omics 2021, 25, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Dandara, C. Broadening Drug Design and Targets to Tumor Microenvironment? Cancer-Associated Fibroblast Marker Expression in Cancers and Relevance for Survival Outcomes. Omics A J. Integr. Biol. 2020, 24, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Dandara, C. Architecture of Cancer-Associated Fibroblasts in Tumor Microenvironment: Mapping Their Origins, Heterogeneity, and Role in Cancer Therapy Resistance. Omics 2020, 24, 314–339. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Senthebane, D.A.; Ganz, C.; Thomford, N.E.; Wonkam, A.; Dandara, C. Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review. Cells 2020, 9, 1896. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, A.E.; Roberts, E.W.; Fearon, D.T. Stromal Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2018, 1060, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, W.; Xiao, J.; Cao, B. The role of exosomes and “exosomal shuttle microRNA” in tumorigenesis and drug resistance. Cancer Lett. 2015, 356, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Hanley, C.J.; Mellone, M.; Ford, K.; Thirdborough, S.M.; Mellows, T.; Frampton, S.J.; Smith, D.M.; Harden, E.; Szyndralewiez, C.; Bullock, M.; et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype Through Inhibition of NOX4. J. Natl. Cancer Inst. 2018, 110, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar] [CrossRef]
- Busch, S.; Acar, A.; Magnusson, Y.; Gregersson, P.; Rydén, L.; Landberg, G. TGF-beta receptor type-2 expression in cancer-associated fibroblasts regulates breast cancer cell growth and survival and is a prognostic marker in pre-menopausal breast cancer. Oncogene 2015, 34, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Miyata, H.; Sugimura, K.; Fukuda, S.; Kanemura, T.; Yamashita, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. miR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis 2015, 36, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, A.; Zhu, C.; Peng, S.; Shuai, C.; Sun, L.; Han, Y.; Qian, Y.; Gao, S.; Su, T. Downregulation of Microrna-148a in Cancer-Associated Fibroblasts from Oral Cancer Promotes Cancer Cell Migration and Invasion by Targeting Wnt10b. J. Biochem. Mol. Toxicol. 2016, 30, 186–191. [Google Scholar] [CrossRef]
- Wiseman, B.S.; Werb, Z. Stromal effects on mammary gland development and breast cancer. Science 2002, 296, 1046–1049. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Lu, Z.; Tang, D.; Yao, J.; An, Y.; Wu, J.; Li, Q.; Gao, W.; Xu, Z.; Qian, Z.; et al. Galectin-1 secreted by activated stellate cells in pancreatic ductal adenocarcinoma stroma promotes proliferation and invasion of pancreatic cancer cells: An in vitro study on the microenvironment of pancreatic ductal adenocarcinoma. Pancreas 2011, 40, 832–839. [Google Scholar] [CrossRef]
- Sun, D.-Y.; Wu, J.-Q.; He, Z.-H.; He, M.-F.; Sun, H.-B. Cancer-associated fibroblast regulate proliferation and migration of prostate cancer cells through TGF-β signaling pathway. Life Sci. 2019, 235, 116791. [Google Scholar] [CrossRef]
- Maluccio, M.; Sharma, V.; Lagman, M.; Vyas, S.; Yang, H.; Li, B.; Suthanthiran, M. Tacrolimus enhances transforming growth factor-β1 expression and promotes tumor progression. Transplantation 2003, 76, 597–602. [Google Scholar] [CrossRef]
- Moses, H.; Barcellos-Hoff, M.H. TGF-β biology in mammary development and breast cancer. Cold Spring Harb. Perspect. Biol. 2011, 3, a003277. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cao, L.; Wang, H.; Liu, B.; Zhang, Q.; Meng, Z.; Wu, X.; Zhou, Q.; Xu, K. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget 2017, 8, 76116. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xiao, C.; Tan, L.; Wang, Q.; Li, X.; Feng, Y. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, S.T.D.; Pontes-Júnior, J.; Antunes, A.A.; Sousa-Canavez, J.M.D.; Abe, D.K.; Cruz, J.A.S.D.; Dall’Oglio, M.F.; Crippa, A.; Passerotti, C.C.; Ribeiro-Filho, L.A. Tgf-β1 expression as a biomarker of poor prognosis in prostate cancer. Clinics 2011, 66, 1143–1147. [Google Scholar] [PubMed]
- Robson, H.; Anderson, E.; James, R.D.; Schofield, P.F. Transforming growth factor β 1 expression in human colorectal tumours: An independent prognostic marker in a subgroup of poor prognosis patients. Br. J. Cancer 1996, 74, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Wang, H.; Liao, W.; Han, F.; Li, Y.Q.; Chen, S.W.; Lao, X.M. Transforming growth factor β is a poor prognostic factor and inhibits the favorable prognostic value of CD8+ CTL in human hepatocellular carcinoma. J. Immunother. 2017, 40, 175–186. [Google Scholar] [CrossRef]
- Desmouliere, A.; Guyot, C.; Gabbiani, G. The stroma reaction myofibroblast: A key player in the control of tumor cell behavior. Int. J. Dev. Biol. 2004, 48, 509–517. [Google Scholar] [CrossRef]
- Huang, J.; Li, Z.; Ding, Z.; Luo, Q.; Lu, S. Different roles of myofibroblasts in the tumorigenesis of nonsmall cell lung cancer. Tumor Biol. 2016, 37, 15525–15534. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Feng, J.; Xie, J.; Mei, Z.; Shi, T.; Wang, S.; Du, Y.; Yang, G.; Wu, Y.; Cheng, X.; et al. Targeted blockade of TGF-β and IL-6/JAK2/STAT3 pathways inhibits lung cancer growth promoted by bone marrow-derived myofibroblasts. Sci. Rep. 2017, 7, 8660. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Matsuda, T.; Muraguchi, A.; Miyazono, K.; Kawabata, M. Cross-talk between IL-6 and TGF-β signaling in hepatoma cells. FEBS Lett. 2001, 492, 247–253. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; van Laar, J.M. Interleukin-6 (IL-6) Trans Signaling Drives a STAT3-dependent Pathway That Leads to Hyperactive Transforming Growth Factor-β (TGF-β) Signaling Promoting SMAD3 Activation and Fibrosis via Gremlin Protein. J. Biol. Chem. 2014, 289, 9952–9960. [Google Scholar] [CrossRef] [Green Version]
- Verbeke, H.; Struyf, S.; Laureys, G.; Van Damme, J. The expression and role of CXC chemokines in colorectal cancer. Cytokine Growth Factor Rev. 2011, 22, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, S.; Meng, Q.; Zhou, F.; Pan, B.; Liu, F.; Yu, Y. CXCR1 correlates to poor outcomes of EGFR-TKI against advanced non-small cell lung cancer by activating chemokine and JAK/STAT pathway. Pulm. Pharmacol. Ther. 2021, 67, 102001. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Liu, M. Chemokines and their receptors as biomarkers in esophageal cancer. Esophagus 2020, 17, 113–121. [Google Scholar] [CrossRef]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their receptors: Multifaceted roles in cancer progression and potential value as cancer prognostic markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [Green Version]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef]
- Karamanou, K.; Franchi, M.; Vynios, D.; Brézillon, S. Epithelial-to-mesenchymal transition and invadopodia markers in breast cancer: Lumican a key regulator. Semin. Cancer Biol. 2020, 62, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell. Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef]
- Pittayapruek, P.; Meephansan, J.; Prapapan, O.; Komine, M.; Ohtsuki, M. Role of Matrix Metalloproteinases in Photoaging and Photocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Investig. 2013, 123, 1902–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomperts, B.N.; Strieter, R.M. Fibrocytes in lung disease. J. Leukoc. Biol. 2007, 82, 449–456. [Google Scholar] [CrossRef]
- Barth, P.J.; Ebrahimsade, S.; Ramaswamy, A.; Moll, R. CD34+ fibrocytes in invasive ductal carcinoma, ductal carcinoma in situ, and benign breast lesions. Virchows Arch. 2002, 440, 298–303. [Google Scholar] [CrossRef]
- Barth, P.J.; Ebrahimsade, S.; Hellinger, A.; Moll, R.; Ramaswamy, A. CD34+ fibrocytes in neoplastic and inflammatory pancreatic lesions. Virchows Arch. 2002, 440, 128–133. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.-H. Adipose tissue derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor derived factors. Cell. Oncol. 2011, 34, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Fernández Moro, C.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsken, J.; Hanahan, D. A subset of cancer-associated fibroblasts determines therapy resistance. Cell 2018, 172, 643–644. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.; Qiu, W.; Haviv, I. Genetic changes in tumour microenvironments. J. Pathol. 2011, 223, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.P.; Cirillo, N.; Hassona, Y.; Wei, W.; Thurlow, J.K.; Cheong, S.C.; Pitiyage, G.; Parkinson, E.K.; Prime, S.S. Fibroblast gene expression profile reflects the stage of tumour progression in oral squamous cell carcinoma. J. Pathol. 2011, 223, 459–469. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Strell, C.; Paulsson, J.; Jin, S.B.; Tobin, N.P.; Mezheyeuski, A.; Roswall, P.; Mutgan, C.; Mitsios, N.; Johansson, H.; Wickberg, S.M.; et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J. Natl. Cancer Inst. 2019, 111, 983–995. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.; Tuxhorn, J.A.; Wheeler, T.M.; Frolov, A.; Scardino, P.T.; Ohori, M.; Wheeler, M.; Spitler, J.; Rowley, D.R. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin. Cancer Res. 2003, 9, 4792–4801. [Google Scholar]
- Muchlińska, A.; Nagel, A.; Popęda, M.; Szade, J.; Niemira, M.; Zieliński, J.; Skokowski, J.; Bednarz-Knoll, N.; Żaczek, A.J. Alpha-smooth muscle actin-positive cancer-associated fibroblasts secreting osteopontin promote growth of luminal breast cancer. Cell. Mol. Biol. Lett. 2022, 27, 1–14. [Google Scholar] [CrossRef]
- Sha, M.; Jeong, S.; Qiu, B.j.; Tong, Y.; Xia, L.; Xu, N.; Zhang, J.j.; Xia, Q. Isolation of cancer-associated fibroblasts and its promotion to the progression of intrahepatic cholangiocarcinoma. Cancer Med. 2018, 7, 4665–4677. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Sakakura, K.; Kudo, T.; Toyoda, M.; Kaira, K.; Oyama, T.; Chikamatsu, K. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 2017, 8, 8633. [Google Scholar] [CrossRef] [Green Version]
- Ni, W.-D.; Yang, Z.-T.; Cui, C.-A.; Cui, Y.; Fang, L.-Y.; Xuan, Y.-H. Tenascin-C is a potential cancer-associated fibroblasts marker and predicts poor prognosis in prostate cancer. Biochem. Biophys. Res. Commun. 2017, 486, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ni, W.; Cui, C.; Fang, L.; Xuan, Y. Tenascin C is a prognostic determinant and potential cancer-associated fibroblasts marker for breast ductal carcinoma. Exp. Mol. Pathol. 2017, 102, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Nishishita, R.; Morohashi, S.; Seino, H.; Wu, Y.; Yoshizawa, T.; Haga, T.; Saito, K.; Hakamada, K.; Fukuda, S.; Kijima, H. Expression of cancer-associated fibroblast markers in advanced colorectal cancer. Oncol. Lett. 2018, 15, 6195–6202. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Otero, N.; Clinch, A.B.; Hope, J.; Wang, W.; Reinhart-King, C.A.; King, M.R. Cancer associated fibroblasts confer shear resistance to circulating tumor cells during prostate cancer metastatic progression. Oncotarget 2020, 11, 1037. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Zhao, X.; He, Y.; Gao, J.; Fan, L.; Li, Z.; Yang, G.; Chen, H. Caveolin-1 expression level in cancer associated fibroblasts predicts outcome in gastric cancer. PLoS ONE 2013, 8, e59102. [Google Scholar] [CrossRef] [Green Version]
- Simpkins, S.A.; Hanby, A.M.; Holliday, D.L.; Speirs, V. Clinical and functional significance of loss of caveolin-1 expression in breast cancer-associated fibroblasts. J. Pathol. 2012, 227, 490–498. [Google Scholar] [CrossRef]
- Musumeci, M.; Coppola, V.; Addario, A.; Patrizii, M.; Maugeri-Saccà, M.; Memeo, L.; Colarossi, C.; Francescangeli, F.; Biffoni, M.; Collura, D.; et al. Control of tumor and microenvironment cross-talk by miR-15a and miR-16 in prostate cancer. Oncogene 2011, 30, 4231–4242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronisz, A.; Godlewski, J.; Wallace, J.A.; Merchant, A.S.; Nowicki, M.O.; Mathsyaraja, H.; Srinivasan, R.; Trimboli, A.J.; Martin, C.K.; Li, F.; et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat. Cell Biol. 2011, 14, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aprelikova, O.; Palla, J.; Hibler, B.; Yu, X.; Greer, Y.E.; Yi, M.; Stephens, R.; Maxwell, G.L.; Jazaeri, A.; Risinger, J.I.; et al. Silencing of miR-148a in cancer-associated fibroblasts results in WNT10B-mediated stimulation of tumor cell motility. Oncogene 2013, 32, 3246–3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Li, Y.; Zou, L.; Zhu, Z. Role of exosomes in crosstalk between cancer-associated fibroblasts and cancer cells. Front. Oncol. 2019, 9, 356. [Google Scholar] [CrossRef]
- Davies, G.; Cunnick, G.H.; Mansel, R.E.; Mason, M.D.; Jiang, W.G. Levels of expression of endothelial markers specific to tumour-associated endothelial cells and their correlation with prognosis in patients with breast cancer. Clin. Exp. Metastasis 2004, 21, 31–37. [Google Scholar] [CrossRef]
- De Sanctis, F.; Ugel, S.; Facciponte, J.; Facciabene, A. The dark side of tumor-associated endothelial cells. In Proceedings of the Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2018; pp. 35–47. [Google Scholar]
- Ronca, R.; Van Ginderachter, J.A.; Turtoi, A. Paracrine interactions of cancer-associated fibroblasts, macrophages and endothelial cells: Tumor allies and foes. Curr. Opin. Oncol. 2018, 30, 45–53. [Google Scholar] [CrossRef]
- Rankin, E.á.; Giaccia, A. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.L. Hypoxia in the tumorigenesis of gliomas and as a potential target for therapeutic measures. Neurosurg. Focus 2006, 20, E24. [Google Scholar] [CrossRef]
- Ye, J.; Koumenis, C. ATF4, an ER stress and hypoxia-inducible transcription factor and its potential role in hypoxia tolerance and tumorigenesis. Curr. Mol. Med. 2009, 9, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.L. Brain tumor hypoxia: Tumorigenesis, angiogenesis, imaging, pseudoprogression, and as a therapeutic target. J. Neuro-Oncol. 2009, 92, 317–335. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Yehya, A.H.S.; Asif, M.; Petersen, S.H.; Subramaniam, A.V.; Kono, K.; Majid, A.M.S.A.; Oon, C.E. Angiogenesis: Managing the culprits behind tumorigenesis and metastasis. Medicina 2018, 54, 8. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [Green Version]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [Green Version]
- Wesseling, M.; Sakkers, T.; De Jager, S.; Pasterkamp, G.; Goumans, M. The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis. Vasc. Pharmacol. 2018, 106, 1–8. [Google Scholar] [CrossRef]
- Motz, G.T.; Coukos, G. The parallel lives of angiogenesis and immunosuppression: Cancer and other tales. Nat. Rev. Immunol. 2011, 11, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Frumento, G.; Piazza, T.; Di Carlo, E.; Ferrini, S. Targeting tumor-related immunosuppression for cancer immunotherapy. Endocr. Metab. Immune Disord.-Drug Targets Former. Curr. Drug Targets-Immune Endocr. Metab. Disord. 2006, 6, 223–237. [Google Scholar] [CrossRef]
- Virrey, J.J.; Guan, S.; Li, W.; Schönthal, A.H.; Chen, T.C.; Hofman, F.M. Increased survivin expression confers chemoresistance to tumor-associated endothelial cells. Am. J. Pathol. 2008, 173, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.-Q.; Sun, H.-C.; Zhang, W.; Zhu, X.-D.; Zhuang, P.-Y.; Zhang, J.-B.; Wang, L.; Wu, W.-z.; Qin, L.-X.; Tang, Z.-Y. Human Hepatocellular Carcinoma Tumor–derived Endothelial Cells Manifest Increased Angiogenesis Capability and Drug Resistance Compared with Normal Endothelial CellsTEC Cells Increase Drug Resistance. Clin. Cancer Res. 2009, 15, 4838–4846. [Google Scholar] [CrossRef]
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.-C.; Totsuka, Y.; Klagsbrun, M. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.J.; Lin, Y.-C.; Yao, P.-L.; Yuan, A.; Chen, H.-Y.; Shun, C.-T.; Tsai, M.-F.; Chen, C.-H.; Yang, P.-C. Tumor-associated macrophages: The double-edged sword in cancer progression. J. Clin. Oncol. 2005, 23, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: Technicalities and challenges in routine clinical practice. Front. Oncol. 2020, 9, 1512. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Li, X.; Zhang, C.; Yang, Y.; Jiang, J.; Wu, C. Tumor-associated macrophages in cancers. Clin. Transl. Oncol. 2016, 18, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Almatroodi, S.A.; McDonald, C.F.; Darby, I.A.; Pouniotis, D.S. Characterization of M1/M2 tumour-associated macrophages (TAMs) and Th1/Th2 cytokine profiles in patients with NSCLC. Cancer Microenviron. 2016, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Heusinkveld, M.; van Der Burg, S.H. Identification and manipulation of tumor associated macrophages in human cancers. J. Transl. Med. 2011, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Paulus, P.; Stanley, E.R.; Schäfer, R.; Abraham, D.; Aharinejad, S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res. 2006, 66, 4349–4356. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, E.M.; Heusinkveld, M.; Tummers, B.; Vogelpoel, L.T.; Goedemans, R.; Jha, V.; Nortier, J.W.; Welters, M.J.; Kroep, J.R.; van der Burg, S.H. Chemotherapy Alters Monocyte Differentiation to Favor Generation of Cancer-Supporting M2 Macrophages in the Tumor MicroenvironmentEffect of Chemotherapy on Tumor Microenvironment. Cancer Res. 2013, 73, 2480–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinushi, M.; Komohara, Y. Tumor-associated macrophages as an emerging target against tumors: Creating a new path from bench to bedside. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2015, 1855, 123–130. [Google Scholar] [CrossRef]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-associated neutrophils in cancer: Going pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef] [Green Version]
- Gregory, A.D.; McGarry Houghton, A. Tumor-associated neutrophils: New targets for cancer therapy. Cancer Res. 2011, 71, 2411–2416. [Google Scholar] [CrossRef] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Tolle, F.; Umansky, V.; Utikal, J.; Kreis, S.; Bréchard, S. Neutrophils in Tumorigenesis: Missing Targets for Successful Next Generation Cancer Therapies? Int. J. Mol. Sci. 2021, 22, 6744. [Google Scholar] [CrossRef] [PubMed]
- Houghton, A.M. The paradox of tumor-associated neutrophils: Fueling tumor growth with cytotoxic substances. Cell Cycle 2010, 9, 1732–1737. [Google Scholar] [CrossRef] [Green Version]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Bui, T.M.; Yalom, L.K.; Sumagin, R. Tumor-associated neutrophils: Orchestrating cancer pathobiology and therapeutic resistance. Expert Opin. Ther. Targets 2021, 25, 573–583. [Google Scholar] [CrossRef]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in tumor microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vlachonikola, E.; Stamatopoulos, K.; Chatzidimitriou, A. T cells in chronic lymphocytic leukemia: A two-edged sword. Front. Immunol. 2021, 11, 612244. [Google Scholar] [CrossRef]
- Andersen, M.H.; Schrama, D.; thor Straten, P.; Becker, J.C. Cytotoxic T cells. J. Investig. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Finlay, D.; Cantrell, D.A. Metabolism, migration and memory in cytotoxic T cells. Nat. Rev. Immunol. 2011, 11, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Navasardyan, I.; Bonavida, B. Regulation of T Cells in Cancer by Nitric Oxide. Cells 2021, 10, 2655. [Google Scholar] [CrossRef]
- Tang, X.X.; Shimada, H.; Ikegaki, N. Clinical Relevance of CD4 Cytotoxic T Cells in High-Risk Neuroblastoma. Front. Immunol. 2021, 12, 650427. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, Z.; Han, Y.; Han, L.; Zou, X.; Zhou, B.; Hu, R.; Hao, J.; Bai, S.; Xiao, H.; et al. Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment. Nat. Commun. 2020, 11, 6268. [Google Scholar] [CrossRef] [PubMed]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Regulatory T cell targeting in cancer: Emerging strategies in immunotherapy. Eur. J. Immunol. 2021, 51, 280–291. [Google Scholar] [CrossRef]
- Elkord, E.; Sasidharan Nair, V. T-Regulatory Cells in Health and Disease. J. Immunol. Res. 2018, 2018, 5025238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasidharan Nair, V.; Saleh, R.; Toor, S.M.; Cyprian, F.S.; Elkord, E. Metabolic reprogramming of T regulatory cells in the hypoxic tumor microenvironment. Cancer Immunol. Immunother. 2021, 70, 2103–2121. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Wang, Y.; Brown, Z.J.; Xia, Y.; Huang, Z.; Shen, C.; Hu, Z.; Beane, J.; Ansa-Addo, E.A.; et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 1271–1283. [Google Scholar] [CrossRef]
- Kordasti, S.Y.; Ingram, W.; Hayden, J.; Darling, D.; Barber, L.; Afzali, B.; Lombardi, G.; Wlodarski, M.W.; Maciejewski, J.P.; Farzaneh, F.; et al. CD4+CD25high Foxp3+ regulatory T cells in myelodysplastic syndrome (MDS). Blood 2007, 110, 847–850. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.P.; Chun, N.A.; Ismail, S.M.; Abraham, M.T.; Yusoff, M.N.; Zain, R.B.; Ngeow, W.C.; Ponniah, S.; Cheong, S.C. CD4+CD25hiCD127low regulatory T cells are increased in oral squamous cell carcinoma patients. PLoS ONE 2014, 9, e103975. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.A.; Cui, X.; Schmittling, R.J.; Sanchez-Perez, L.; Snyder, D.J.; Congdon, K.L.; Archer, G.E.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; et al. Monoclonal antibody blockade of IL-2 receptor α during lymphopenia selectively depletes regulatory T cells in mice and humans. Blood 2011, 118, 3003–3012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.E.; Greenberger, B.A.; Taylor, J.M.; Edelman, M.J.; Lu, B. The Underappreciated Role of the Humoral Immune System and B Cells in Tumorigenesis and Cancer Therapeutics: A Review. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 38–45. [Google Scholar] [CrossRef]
- Corsiero, E.; Delvecchio, F.R.; Bombardieri, M.; Pitzalis, C. B cells in the formation of tertiary lymphoid organs in autoimmunity, transplantation and tumorigenesis. Curr. Opin. Immunol. 2019, 57, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Mintz, M.A.; Cyster, J.G. T follicular helper cells in germinal center B cell selection and lymphomagenesis. Immunol. Rev. 2020, 296, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Roghanian, A.; Fraser, C.; Kleyman, M.; Chen, J. B Cells Promote Pancreatic Tumorigenesis. Cancer Discov. 2016, 6, 230–232. [Google Scholar] [CrossRef] [Green Version]
- Bruno, T.C.; Ebner, P.J.; Moore, B.L.; Squalls, O.G.; Waugh, K.A.; Eruslanov, E.B.; Singhal, S.; Mitchell, J.D.; Franklin, W.A.; Merrick, D.T.; et al. Antigen-Presenting Intratumoral B Cells Affect CD4(+) TIL Phenotypes in Non-Small Cell Lung Cancer Patients. Cancer Immunol. Res. 2017, 5, 898–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Jensen, P.E. The role of B lymphocytes as antigen-presenting cells. Arch. Immunol. Ther. Exp. 2008, 56, 77–83. [Google Scholar] [CrossRef]
- Ghosh, D.; Jiang, W.; Mukhopadhyay, D.; Mellins, E.D. New insights into B cells as antigen presenting cells. Curr. Opin. Immunol. 2021, 70, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, I.; Jeon, D.; Moseman, J.E.; Muralidhar, A.; Potluri, H.K.; McNeel, D.G. Role of B cells as antigen presenting cells. Front. Immunol. 2022, 13, 954936. [Google Scholar] [CrossRef]
- Edechi, C.A.; Ikeogu, N.; Uzonna, J.E.; Myal, Y. Regulation of Immunity in Breast Cancer. Cancers 2019, 11, 1080. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.L.; Khan, N.; Basu, S.; Soni, V. B-Cell-Based Immunotherapy: A Promising New Alternative. Vaccines 2022, 10, 879. [Google Scholar] [CrossRef]
- Kuroda, H.; Jamiyan, T.; Yamaguchi, R.; Kakumoto, A.; Abe, A.; Harada, O.; Masunaga, A. Tumor microenvironment in triple-negative breast cancer: The correlation of tumor-associated macrophages and tumor-infiltrating lymphocytes. Clin. Transl. Oncol. 2021, 23, 2513–2525. [Google Scholar] [CrossRef]
- Catalán, D.; Mansilla, M.A.; Ferrier, A.; Soto, L.; Oleinika, K.; Aguillón, J.C.; Aravena, O. Immunosuppressive Mechanisms of Regulatory B Cells. Front. Immunol. 2021, 12, 611795. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Fu, Y.; Chu, Y. Regulatory B Cells. Adv. Exp. Med. Biol. 2020, 1254, 87–103. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Terrén, I.; Orrantia, A.; Vitallé, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10, 2278. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.S.; Knútsdóttir, H.; Ramakrishnan, G.; Padmanaban, V.; Warrier, M.; Ramirez, J.C.; Dunworth, M.; Zhang, H.; Jaffee, E.M.; Bader, J.S.; et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J. Cell Biol. 2020, 219, e202001134. [Google Scholar] [CrossRef]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef]
- López-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar]
- Birbrair, A.; Zhang, T.; Wang, Z.-M.; Messi, M.L.; Olson, J.D.; Mintz, A.; Delbono, O. Type-2 pericytes participate in normal and tumoral angiogenesis. Am. J. Physiol.-Cell Physiol. 2014, 307, C25–C38. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Marcus, A.; Raulet, D.H. Immunosurveillance and immunotherapy of tumors by innate immune cells. Curr. Opin. Immunol. 2016, 38, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Chong, A.S.-F.; Markham, P.N.; Gebel, H.M.; Bines, S.D.; Coon, J.S. Diverse multidrug-resistance-modification agents inhibit cytolytic activity of natural killer cells. Cancer Immunol. Immunother. 1993, 36, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Savas, B.; Cole, S.; Akoglu, T.; Pross, H. P-glycoprotein-mediated multidrug resistance and cytotoxic effector cells. Nat Immun 1992, 11, 177–192. [Google Scholar] [PubMed]
- Takahashi, M.; Misawa, Y.; Watanabe, N.; Kawanishi, T.; Tanaka, H.; Shigenobu, K.; Kobayashi, Y. Role of P-glycoprotein in human natural killer-like cell line-mediated cytotoxicity. Exp. Cell Res. 1999, 253, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Radford, K.J. The role of dendritic cells in cancer. Int. Rev. Cell Mol. Biol. 2019, 348, 123–178. [Google Scholar] [CrossRef]
- Volovitz, I.; Melzer, S.; Amar, S.; Bocsi, J.; Bloch, M.; Efroni, S.; Ram, Z.; Tárnok, A. Dendritic Cells in the Context of Human Tumors: Biology and Experimental Tools. Int. Rev. Immunol. 2016, 35, 116–135. [Google Scholar] [CrossRef]
- Zhou, B.; Lawrence, T.; Liang, Y. The Role of Plasmacytoid Dendritic Cells in Cancers. Front. Immunol. 2021, 12, 749190. [Google Scholar] [CrossRef]
- Aspord, C.; Pedroza-Gonzalez, A.; Gallegos, M.; Tindle, S.; Burton, E.C.; Su, D.; Marches, F.; Banchereau, J.; Palucka, A.K. Breast cancer instructs dendritic cells to prime interleukin 13–secreting CD4+ T cells that facilitate tumor development. J. Exp. Med. 2007, 204, 1037–1047. [Google Scholar] [CrossRef]
- Vicari, A.P.; Caux, C.; Trinchieri, G. Tumour escape from immune surveillance through dendritic cell inactivation. In Proceedings of the Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2002; pp. 33–42. [Google Scholar]
- Barry, A.E.; Baldeosingh, R.; Lamm, R.; Patel, K.; Zhang, K.; Dominguez, D.A.; Kirton, K.J.; Shah, A.P.; Dang, H. Hepatic Stellate Cells and Hepatocarcinogenesis. Front. Cell Dev. Biol. 2020, 8, 709. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Jiang, K.; Werner, J.; Bazhin, A.V.; D’Haese, J.G. The Role of Stellate Cells in Pancreatic Ductal Adenocarcinoma: Targeting Perspectives. Front. Oncol. 2020, 10, 621937. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Mi, Y.; Zheng, B.; Wei, P.; Gu, Y.; Zhang, Z.; Xu, Y.; Cai, S.; Li, X.; Li, D. Highly-metastatic colorectal cancer cell released miR-181a-5p-rich extracellular vesicles promote liver metastasis by activating hepatic stellate cells and remodelling the tumour microenvironment. J. Extracell. Vesicles 2022, 11, e12186. [Google Scholar] [CrossRef]
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elechalawar, C.K.; Hossen, M.N.; Shankarappa, P.; Peer, C.J.; Figg, W.D.; Robertson, J.D.; Bhattacharya, R.; Mukherjee, P. Targeting Pancreatic Cancer Cells and Stellate Cells Using Designer Nanotherapeutics In Vitro. Int. J. Nanomed. 2020, 15, 991–1003. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.X.; Gong, W.Z.; Zhou, K.; Xiao, Z.G.; Hou, F.T.; Huang, T.; Zhang, L.; Dong, H.Y.; Zhang, W.L.; Liu, Y.; et al. CXCR4/TGF-β1 mediated hepatic stellate cells differentiation into carcinoma-associated fibroblasts and promoted liver metastasis of colon cancer. Cancer Biol. Ther. 2020, 21, 258–268. [Google Scholar] [CrossRef]
- Lua, I.; Li, Y.; Zagory, J.A.; Wang, K.S.; French, S.W.; Sévigny, J.; Asahina, K. Characterization of hepatic stellate cells, portal fibroblasts, and mesothelial cells in normal and fibrotic livers. J. Hepatol. 2016, 64, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senoo, H.; Mezaki, Y.; Fujiwara, M. The stellate cell system (vitamin A-storing cell system). Anat. Sci. Int. 2017, 92, 387–455. [Google Scholar] [CrossRef] [PubMed]
- Senoo, H.; Yoshikawa, K.; Morii, M.; Miura, M.; Imai, K.; Mezaki, Y. Hepatic stellate cell (vitamin A-storing cell) and its relative--past, present and future. Cell Biol. Int. 2010, 34, 1247–1272. [Google Scholar] [CrossRef] [PubMed]
- Ferdek, P.E.; Jakubowska, M.A. Biology of pancreatic stellate cells—More than just pancreatic cancer. Pflügers Arch.-Eur. J. Physiol. 2017, 469, 1039–1050. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, S.-K.; Khawar, I.A.; Jeong, S.-Y.; Chung, S.; Kuh, H.-J. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dalin, S.; Sullivan, M.R.; Lau, A.N.; Grauman-Boss, B.; Mueller, H.S.; Kreidl, E.; Fenoglio, S.; Luengo, A.; Lees, J.A.; Vander Heiden, M.G. Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine ResistanceDeoxycytidine from Stellate Cells Confers Drug Resistance. Cancer Res. 2019, 79, 5723–5733. [Google Scholar] [CrossRef] [Green Version]
- Habisch, H.; Zhou, S.; Siech, M.; Bachem, M.G. Interaction of stellate cells with pancreatic carcinoma cells. Cancers 2010, 2, 1661–1682. [Google Scholar] [CrossRef] [Green Version]
- Hessmann, E.; Patzak, M.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.; Frese, K.K.; Gopinathan, A.; Richards, F. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hefetz-Sela, S.; Scherer, P.E. Adipocytes: Impact on tumor growth and potential sites for therapeutic intervention. Pharmacol. Ther. 2013, 138, 197–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Chen, X.; He, Q.; Gimple, R.C.; Liao, Y.; Wang, L.; Wu, R.; Xie, Q.; Rich, J.N.; Shen, K. Adipocytes promote breast tumorigenesis through TAZ-dependent secretion of Resistin. Proc. Natl. Acad. Sci. USA 2020, 117, 33295–33304. [Google Scholar] [CrossRef]
- De Pergola, G.; Silvestris, F. Obesity as a major risk factor for cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef] [Green Version]
- Ntikoudi, E.; Kiagia, M.; Boura, P.; Syrigos, K. Hormones of adipose tissue and their biologic role in lung cancer. Cancer Treat. Rev. 2014, 40, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Y.; Lehuédé, C.; Laurent, V.; Dirat, B.; Dauvillier, S.; Bochet, L.; Le Gonidec, S.; Escourrou, G.; Valet, P.; Muller, C. Adipose tissue and breast epithelial cells: A dangerous dynamic duo in breast cancer. Cancer Lett. 2012, 324, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Camarda, R.; Williams, J.; Malkov, S.; Zimmerman, L.J.; Manning, S.; Aran, D.; Beardsley, A.; Van de Mark, D.; Chen, Y.; Berdan, C.A. Tumor cell-adipocyte gap junctions activate lipolysis and are essential for breast tumorigenesis. bioRxiv 2018, 277939. [Google Scholar] [CrossRef] [Green Version]
- Strong, A.L.; Strong, T.A.; Rhodes, L.V.; Semon, J.A.; Zhang, X.; Shi, Z.; Zhang, S.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Obesity associated alterations in the biology of adipose stem cells mediate enhanced tumorigenesis by estrogen dependent pathways. Breast Cancer Res. 2013, 15, 1–15. [Google Scholar] [CrossRef] [Green Version]
- El Atat, O.; Antonios, D.; Hilal, G.; Hokayem, N.; Abou-Ghoch, J.; Hashim, H.; Serhal, R.; Hebbo, C.; Moussa, M.; Alaaeddine, N. An evaluation of the stemness, paracrine, and tumorigenic characteristics of highly expanded, minimally passaged adipose-derived stem cells. PLoS ONE 2016, 11, e0162332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacIsaac, Z.M.; Shang, H.; Agrawal, H.; Yang, N.; Parker, A.; Katz, A.J. Long-term in-vivo tumorigenic assessment of human culture-expanded adipose stromal/stem cells. Exp. Cell Res. 2012, 318, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzobo, K. Recent trends in multipotent human mesenchymal stem/stromal cells: Learning from history and advancing clinical applications. OMICS A J. Integr. Biol. 2021, 25, 342–357. [Google Scholar] [CrossRef]
- Dzobo, K.; Turnley, T.; Wishart, A.; Rowe, A.; Kallmeyer, K.; Van Vollenstee, F.A.; Thomford, N.E.; Dandara, C.; Chopera, D.; Pepper, M.S. Fibroblast-derived extracellular matrix induces chondrogenic differentiation in human adipose-derived mesenchymal stromal/stem cells in vitro. Int. J. Mol. Sci. 2016, 17, 1259. [Google Scholar] [CrossRef] [Green Version]
- Kass, L.; Erler, J.T.; Dembo, M.; Weaver, V.M. Mammary epithelial cell: Influence of extracellular matrix composition and organization during development and tumorigenesis. Int. J. Biochem. Cell Biol. 2007, 39, 1987–1994. [Google Scholar] [CrossRef] [Green Version]
- Vieweg, J.; Su, Z.; Dahm, P.; Kusmartsev, S. Reversal of tumor-mediated immunosuppression. Clin. Cancer Res. 2007, 13, 727s–732s. [Google Scholar] [CrossRef] [Green Version]
- Papait, A.; Stefani, F.R.; Cargnoni, A.; Magatti, M.; Parolini, O.; Silini, A.R. The multifaceted roles of MSCs in the tumor microenvironment: Interactions with immune cells and exploitation for therapy. Front. Cell Dev. Biol. 2020, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.G. The detached pericyte hypothesis: A novel explanation for many puzzling aspects of tumorigenesis. Org. J. Biol. Sci. 2018, 2, 25–42. [Google Scholar]
- Xian, X.; Håkansson, J.; Ståhlberg, A.; Lindblom, P.; Betsholtz, C.; Gerhardt, H.; Semb, H. Pericytes limit tumor cell metastasis. J. Clin. Investig. 2006, 116, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.I.; Shields, D.J.; Barillas, S.G.; Acevedo, L.M.; Murphy, E.; Huang, J.; Scheppke, L.; Stockmann, C.; Johnson, R.S.; Angle, N. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 2008, 456, 809–813. [Google Scholar] [CrossRef]
- Viski, C.; König, C.; Kijewska, M.; Mogler, C.; Isacke, C.M.; Augustin, H.G. Endosialin-expressing pericytes promote metastatic dissemination. Cancer Res. 2016, 76, 5313–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, A.; Franklin, M.J.; Dudek, A.Z. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am. J. Hematol. 2010, 85, 593–598. [Google Scholar] [CrossRef]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef] [Green Version]
- Pieterse, Z.; Sinha, D.; Kaur, P. Pericytes in metastasis. Adv. Exp. Med. Biol. 2019, 1147, 125–135. [Google Scholar]
- Gerhardt, H.; Semb, H. Pericytes: Gatekeepers in tumour cell metastasis? J. Mol. Med. 2008, 86, 135–144. [Google Scholar] [CrossRef]
- Prazeres, P.H.D.M.; Sena, I.F.G.; da Terra Borges, I.; de Azevedo, P.O.; Andreotti, J.P.; de Paiva, A.E.; de Almeida, V.M.; de Paula Guerra, D.A.; Dos Santos, G.S.P.; Mintz, A. Pericytes are heterogeneous in their origin within the same tissue. Dev. Biol. 2017, 427, 6–11. [Google Scholar] [CrossRef]
- Prete, A.; Lo, A.S.; Sadow, P.M.; Bhasin, S.S.; Antonello, Z.A.; Vodopivec, D.M.; Ullas, S.; Sims, J.N.; Clohessy, J.; Dvorak, A.M. Pericytes Elicit Resistance to Vemurafenib and Sorafenib Therapy in Thyroid Carcinoma via the TSP-1/TGFβ1 AxisTSP-1, TGFβ1, and Drug Resistance in Thyroid Cancer. Clin. Cancer Res. 2018, 24, 6078–6097. [Google Scholar] [CrossRef] [Green Version]
- Nilendu, P.; Sarode, S.C.; Jahagirdar, D.; Tandon, I.; Patil, S.; Sarode, G.S.; Pal, J.K.; Sharma, N.K. Mutual concessions and compromises between stromal cells and cancer cells: Driving tumor development and drug resistance. Cell. Oncol. 2018, 41, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Lin, Y.; Wang, C.; Deng, L.; Chen, M.; Assaraf, Y.G.; Chen, Z.-S.; Ye, W.; Zhang, D. New insights into antiangiogenic therapy resistance in cancer: Mechanisms and therapeutic aspects. Drug Resist. Updates 2022, 64, 100849. [Google Scholar] [CrossRef]
- Dzobo, K.; Leaner, V.D.; Parker, M.I. Feedback regulation of the alpha2(1) collagen gene via the Mek-Erk signaling pathway. IUBMB Life 2012, 64, 87–98. [Google Scholar] [CrossRef]
- Dzobo, K.; Leaner, V.D.; Parker, M.I. Absence of feedback regulation in the synthesis of COL1A1. Life Sci. 2014, 103, 25–33. [Google Scholar] [CrossRef]
- Stupack, D.G.; Cheresh, D.A. ECM remodeling regulates angiogenesis: Endothelial integrins look for new ligands. Sci. STKE 2002, 2002, pe7. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Sirica, A.E.; Gores, G.J. Desmoplastic stroma and cholangiocarcinoma: Clinical implications and therapeutic targeting. Hepatol. (Baltim. Md.) 2014, 59, 2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Holle, A.W.; Young, J.L.; Spatz, J.P. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv. Drug Deliv. Rev. 2016, 97, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Morin, P.J. Drug resistance and the microenvironment: Nature and nurture. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2003, 6, 169–172. [Google Scholar] [CrossRef]
- Campbell, N.E.; Kellenberger, L.; Greenaway, J.; Moorehead, R.A.; Linnerth-Petrik, N.M.; Petrik, J. Extracellular Matrix Proteins and Tumor Angiogenesis. J. Oncol. 2010, 2010, 586905. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.R. The matrix in cancer. Nat. Rev. Cancer 2021, 21, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2011, 14, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhao, S.; Han, J.; Zheng, L.; Yang, Z.; Zhao, L. Hypoxia-inducible factor-1α induces multidrug resistance protein in colon cancer. OncoTargets Ther. 2015, 8, 1941–1948. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761. [Google Scholar] [CrossRef] [Green Version]
- Franco, P.I.R.; Rodrigues, A.P.; de Menezes, L.B.; Miguel, M.P. Tumor microenvironment components: Allies of cancer progression. Pathol.-Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaahtomeri, K.; Alitalo, K. Lymphatic Vessels in Tumor Dissemination versus ImmunotherapyLymphatics in Metastasis vs. Immunotherapy. Cancer Res. 2020, 80, 3463–3465. [Google Scholar] [CrossRef]
- Nathanson, S.D. Insights into the mechanisms of lymph node metastasis. Cancer 2003, 98, 413–423. [Google Scholar] [CrossRef]
- Cady, B. Lymph node metastases: Indicators, but not governors of survival. Arch. Surg. 1984, 119, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Som, P.M. Detection of metastasis in cervical lymph nodes: CT and MR criteria and differential diagnosis. AJR Am. J. Roentgenol. 1992, 158, 961–969. [Google Scholar] [CrossRef]
- Deng, J.-Y.; Liang, H. Clinical significance of lymph node metastasis in gastric cancer. World J. Gastroenterol. WJG 2014, 20, 3967. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Giaccia, A.J. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998, 58, 1408–1416. [Google Scholar] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [Green Version]
- Finger, E.C.; Giaccia, A.J. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010, 29, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 2007, 80, 51. [Google Scholar] [PubMed]
- Hung, J.-J.; Yang, M.-H.; Hsu, H.-S.; Hsu, W.-H.; Liu, J.; Wu, K. Prognostic significance of hypoxia-inducible factor-1α, TWIST1 and Snail expression in resectable non-small cell lung cancer. Thorax 2009, 64, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Sun, B.; Zhao, X.; Wang, X.; Li, Y.; Qiu, Z.; Liu, T.; Gu, Q.; Dong, X.; Zhang, Y. Hypoxia promotes vasculogenic mimicry formation by the Twist1-Bmi1 connection in hepatocellular carcinoma. Int. J. Mol. Med. 2015, 36, 783–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilkes, D.M.; Bajpai, S.; Chaturvedi, P.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J. Biol. Chem. 2013, 288, 10819–10829. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, A.C. Biomechanical forces shape the tumor microenvironment. Ann. Biomed. Eng. 2011, 39, 1379–1389. [Google Scholar] [CrossRef]
- Comito, G.; Giannoni, E.; Segura, C.; Barcellos-de-Souza, P.; Raspollini, M.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, C.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.B.; Bhadauria, S. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014, 5, 5350. [Google Scholar] [CrossRef] [Green Version]
- Chiu, D.K.-C.; Tse, A.P.-W.; Xu, I.M.-J.; Di Cui, J.; Lai, R.K.-H.; Li, L.L.; Koh, H.-Y.; Tsang, F.H.-C.; Wei, L.L.; Wong, C.-M. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Švastová, E.; Hulíková, A.; Rafajová, M.; Zat’ovičová, M.; Gibadulinová, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.r. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef] [Green Version]
- du Souich, P.; Fradette, C. The effect and clinical consequences of hypoxia on cytochrome P450, membrane carrier proteins activity and expression. Expert Opin. Drug Metab. Toxicol. 2011, 7, 1083–1100. [Google Scholar] [CrossRef]
- Park, T.-E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; FitzGerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M. Hypoxia-enhanced Blood-Brain Barrier Chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Brouqui, P.; Amrane, S.; Million, M.; Cortaredona, S.; Parola, P.; Lagier, J.-C.; Raoult, D. Asymptomatic hypoxia in COVID-19 is associated with poor outcome. Int. J. Infect. Dis. 2021, 102, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Kashani, K.B. Hypoxia in COVID-19: Sign of severity or cause for poor outcomes. In Proceedings of the Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1094–1096. [Google Scholar]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Milman, N.; Ginini, L.; Gil, Z. Exosomes and their role in tumorigenesis and anticancer drug resistance. Drug Resist. Updates 2019, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [Green Version]
- Su, L.-L.; Chang, X.-J.; Zhou, H.-D.; Hou, L.-B.; Xue, X.-Y. Exosomes in esophageal cancer: A review on tumorigenesis, diagnosis and therapeutic potential. World J. Clin. Cases 2019, 7, 908. [Google Scholar] [CrossRef]
- Yu, D.D.; Wu, Y.; Shen, H.Y.; Lv, M.M.; Chen, W.X.; Zhang, X.H.; Zhong, S.L.; Tang, J.H.; Zhao, J.H. Exosomes in development, metastasis and drug resistance of breast cancer. Cancer Sci. 2015, 106, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Najminejad, H.; Kalantar, S.M.; Abdollahpour-Alitappeh, M.; Karimi, M.H.; Seifalian, A.M.; Gholipourmalekabadi, M.; Sheikhha, M.H. Emerging roles of exosomal miRNAs in breast cancer drug resistance. IUBMB Life 2019, 71, 1672–1684. [Google Scholar] [CrossRef]
- Uen, Y.; Wang, J.-W.; Wang, C.; Jhang, Y.; Chung, J.-Y.; Tseng, T.; Sheu, M.; Lee, S. Mining of potential microRNAs with clinical correlation-regulation of syndecan-1 expression by miR-122-5p altered mobility of breast cancer cells and possible correlation with liver injury. Oncotarget 2018, 9, 28165. [Google Scholar] [CrossRef] [Green Version]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Gu, Y.; Du, Y.; Liu, J. Exosomes: Diagnostic biomarkers and therapeutic delivery vehicles for cancer. Mol. Pharm. 2019, 16, 3333–3349. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, E.; Lee, M.Y. Exosomes as diagnostic biomarkers in cancer. Mol. Cell. Toxicol. 2018, 14, 113–122. [Google Scholar] [CrossRef]
- An, T.; Qin, S.; Xu, Y.; Tang, Y.; Huang, Y.; Situ, B.; Inal, J.M.; Zheng, L. Exosomes serve as tumour markers for personalized diagnostics owing to their important role in cancer metastasis. J. Extracell. Vesicles 2015, 4, 27522. [Google Scholar] [CrossRef]
- Jafari, R.; Rahbarghazi, R.; Ahmadi, M.; Hassanpour, M.; Rezaie, J. Hypoxic exosomes orchestrate tumorigenesis: Molecular mechanisms and therapeutic implications. J. Transl. Med. 2020, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, Y.; Inoue, H.; Furukawa, M.; Kakudo, K.; Nozaki, M. Heparin-binding epidermal growth factor-like growth factor is a potent regulator of invasion activity in oral squamous cell carcinoma. Oncol. Rep. 2012, 27, 954–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zheng, Y.; Zhao, M. Exosome-based cancer therapy: Implication for targeting cancer stem cells. Front. Pharmacol. 2017, 7, 533. [Google Scholar] [CrossRef] [Green Version]
- Chitadze, G.; Bhat, J.; Lettau, M.; Janssen, O.; Kabelitz, D. Generation of Soluble NKG 2 D Ligands: Proteolytic Cleavage, Exosome Secretion and Functional Implications. Scand. J. Immunol. 2013, 78, 120–129. [Google Scholar] [CrossRef]
- Lundholm, M.; Schröder, M.; Nagaeva, O.; Baranov, V.; Widmark, A.; Mincheva-Nilsson, L.; Wikström, P. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLoS ONE 2014, 9, e108925. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, S.E.; Lakhal, S.; Mäger, I.; Wood, M.J. Exosomes for targeted siRNA delivery across biological barriers. Adv. Drug Deliv. Rev. 2013, 65, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, A.; Graziano, F.; Loupakis, F.; Santini, D.; Catalano, V.; Bisonni, R.; Ficarelli, R.; Fontana, A.; Andreoni, F.; Falcone, A. Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFIRI chemotherapy. Pharm. J. 2008, 8, 278–288. [Google Scholar] [CrossRef]
- Harries, M.; Gore, M. Part I: Chemotherapy for epithelial ovarian cancer–treatment at first diagnosis. Lancet Oncol. 2002, 3, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Flemming, A. PD1 makes waves in anticancer immunotherapy. Nat. Rev. Drug Discov. 2012, 11, 601. [Google Scholar] [CrossRef]
- Haanen, J.B.; Robert, C. Immune checkpoint inhibitors. Immuno-Oncol. 2015, 42, 55–66. [Google Scholar]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chafe, S.C.; McDonald, P.C.; Saberi, S.; Nemirovsky, O.; Venkateswaran, G.; Burugu, S.; Gao, D.; Delaidelli, A.; Kyle, A.H.; Baker, J.H. Targeting Hypoxia-Induced Carbonic Anhydrase IX Enhances Immune-Checkpoint Blockade Locally and SystemicallyCAIX Inhibition Enhances Immune-Checkpoint Blockade. Cancer Immunol. Res. 2019, 7, 1064–1078. [Google Scholar] [CrossRef] [Green Version]
- Elinav, E.; Garrett, W.S.; Trinchieri, G.; Wargo, J. The cancer microbiome. Nat. Rev. Cancer 2019, 19, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.V. Developing combination strategies using PD-1 checkpoint inhibitors to treat cancer. In Proceedings of the Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2019; pp. 21–30. [Google Scholar]
- Decazes, P.; Bohn, P. Immunotherapy by immune checkpoint inhibitors and nuclear medicine imaging: Current and future applications. Cancers 2020, 12, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grywalska, E.; Pasiarski, M.; Góźdź, S.; Roliński, J. Immune-checkpoint inhibitors for combating T-cell dysfunction in cancer. OncoTargets Ther. 2018, 11, 6505. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.J.; Mao, X.B.; Ren, L.L.; Chu, X.Y. Inhibition of CXCL12/CXCR4 axis as a potential targeted therapy of advanced gastric carcinoma. Cancer Med. 2017, 6, 1424–1436. [Google Scholar] [CrossRef]
- Bule, P.; Aguiar, S.I.; Aires-Da-Silva, F.; Dias, J.N.R. Chemokine-directed tumor microenvironment modulation in cancer immunotherapy. Int. J. Mol. Sci. 2021, 22, 9804. [Google Scholar] [CrossRef] [PubMed]
- Perdih, A.; Sollner Dolenc, M. Small molecule antagonists of integrin receptors. Curr. Med. Chem. 2010, 17, 2371–2392. [Google Scholar] [CrossRef] [PubMed]
- Keely, P.J. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J. Mammary Gland Biol. Neoplasia 2011, 16, 205–219. [Google Scholar] [CrossRef]
- Gehler, S.; Ponik, S.M.; Riching, K.M.; Keely, P.J. Bi-directional signaling: Extracellular matrix and integrin regulation of breast tumor progression. Crit. Rev. Eukaryot. Gene Expr. 2013, 23, 139–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tentori, L.; Dorio, A.S.; Muzi, A.; Lacal, P.M.; Ruffini, F.; Navarra, P.; Graziani, G. The integrin antagonist cilengitide increases the antitumor activity of temozolomide against malignant melanoma. Oncol. Rep. 2008, 19, 1039–1043. [Google Scholar] [CrossRef]
- Unruh, A.; Ressel, A.; Mohamed, H.G.; Johnson, R.S.; Nadrowitz, R.; Richter, E.; Katschinski, D.M.; Wenger, R.H. The hypoxia-inducible factor-1α is a negative factor for tumor therapy. Oncogene 2003, 22, 3213–3220. [Google Scholar] [CrossRef] [Green Version]
- Rohwer, N.; Dame, C.; Haugstetter, A.; Wiedenmann, B.; Detjen, K.; Schmitt, C.A.; Cramer, T. Hypoxia-inducible factor 1α determines gastric cancer chemosensitivity via modulation of p53 and NF-κB. PLoS ONE 2010, 5, e12038. [Google Scholar] [CrossRef] [Green Version]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F. Acidity Generated by the Tumor Microenvironment Drives Local InvasionAcid-Mediated Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Kraus, M.; Wolf, B. Implications of acidic tumor microenvironment for neoplastic growth and cancer treatment: A computer analysis. Tumor Biol. 1996, 17, 133–154. [Google Scholar] [CrossRef]
- Chen, Q.; Feng, L.; Liu, J.; Zhu, W.; Dong, Z.; Wu, Y.; Liu, Z. Intelligent albumin–MnO2 nanoparticles as pH-/H2O2-responsive dissociable nanocarriers to modulate tumor hypoxia for effective combination therapy. Adv. Mater. 2016, 28, 7129–7136. [Google Scholar] [CrossRef]
- Saggar, J.K.; Tannock, I.F. Activity of the hypoxia-activated pro-drug TH-302 in hypoxic and perivascular regions of solid tumors and its potential to enhance therapeutic effects of chemotherapy. Int. J. Cancer 2014, 134, 2726–2734. [Google Scholar] [CrossRef]
- Hajj, C.; Russell, J.; Hart, C.P.; Goodman, K.A.; Lowery, M.A.; Haimovitz-Friedman, A.; Deasy, J.O.; Humm, J.L. A combination of radiation and the hypoxia-activated prodrug evofosfamide (TH-302) is efficacious against a human orthotopic pancreatic tumor model. Transl. Oncol. 2017, 10, 760–765. [Google Scholar] [CrossRef]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef]
- Feldman, D.E.; Chauhan, V.; Koong, A.C. The unfolded protein response: A novel component of the hypoxic stress response in tumors. Mol. Cancer Res. 2005, 3, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 1–16. [Google Scholar] [CrossRef]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell. Physiol. 2019, 234, 5700–5721. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAFs Marker | Description and Function of Protein | Effect within TME |
|---|---|---|
| α-SMA | Actin isoform: cellular contraction and maintenance of structure | Promote tumor cell proliferation; involved in immunosuppression [73,74,75] |
| Tenascin-C | Extracellular matrix glycoprotein: cell migration; wound healing | Impeding drug delivery; protect tumor cells [76,77] |
| Vimentin | Type III intermediate filament protein: cell migration; cell structure maintenance | Tumor cell migration and invasion [25,34,74,75] |
| PDGFRα/β | Protein tyrosine kinase receptor: cellular signaling | Macrophage polarization; angiogenesis [16,78] |
| FAP | Membrane-bound gelatinase: protease activity; ECM remodeling | Angiogenesis, macrophage polarization; immunosuppression; metastasis [16,17,79] |
| GPR77 | Complement component 5a receptor 2: Activation of complement; promote inflammation | Maintains tumor cell stemness; Drug resistance [80] |
| Caveolin-1 | Scaffolding protein within caveolar membranes: maintains cellular structure and signaling | Low caveolin-1 linked to poor prognosis [81,82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dzobo, K.; Senthebane, D.A.; Dandara, C. The Tumor Microenvironment in Tumorigenesis and Therapy Resistance Revisited. Cancers 2023, 15, 376. https://doi.org/10.3390/cancers15020376
Dzobo K, Senthebane DA, Dandara C. The Tumor Microenvironment in Tumorigenesis and Therapy Resistance Revisited. Cancers. 2023; 15(2):376. https://doi.org/10.3390/cancers15020376
Chicago/Turabian StyleDzobo, Kevin, Dimakatso A. Senthebane, and Collet Dandara. 2023. "The Tumor Microenvironment in Tumorigenesis and Therapy Resistance Revisited" Cancers 15, no. 2: 376. https://doi.org/10.3390/cancers15020376