
Longitudinal Plasma Proteomics-Derived Biomarkers Predict Response to MET Inhibitors for MET-Dysregulated NSCLC

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
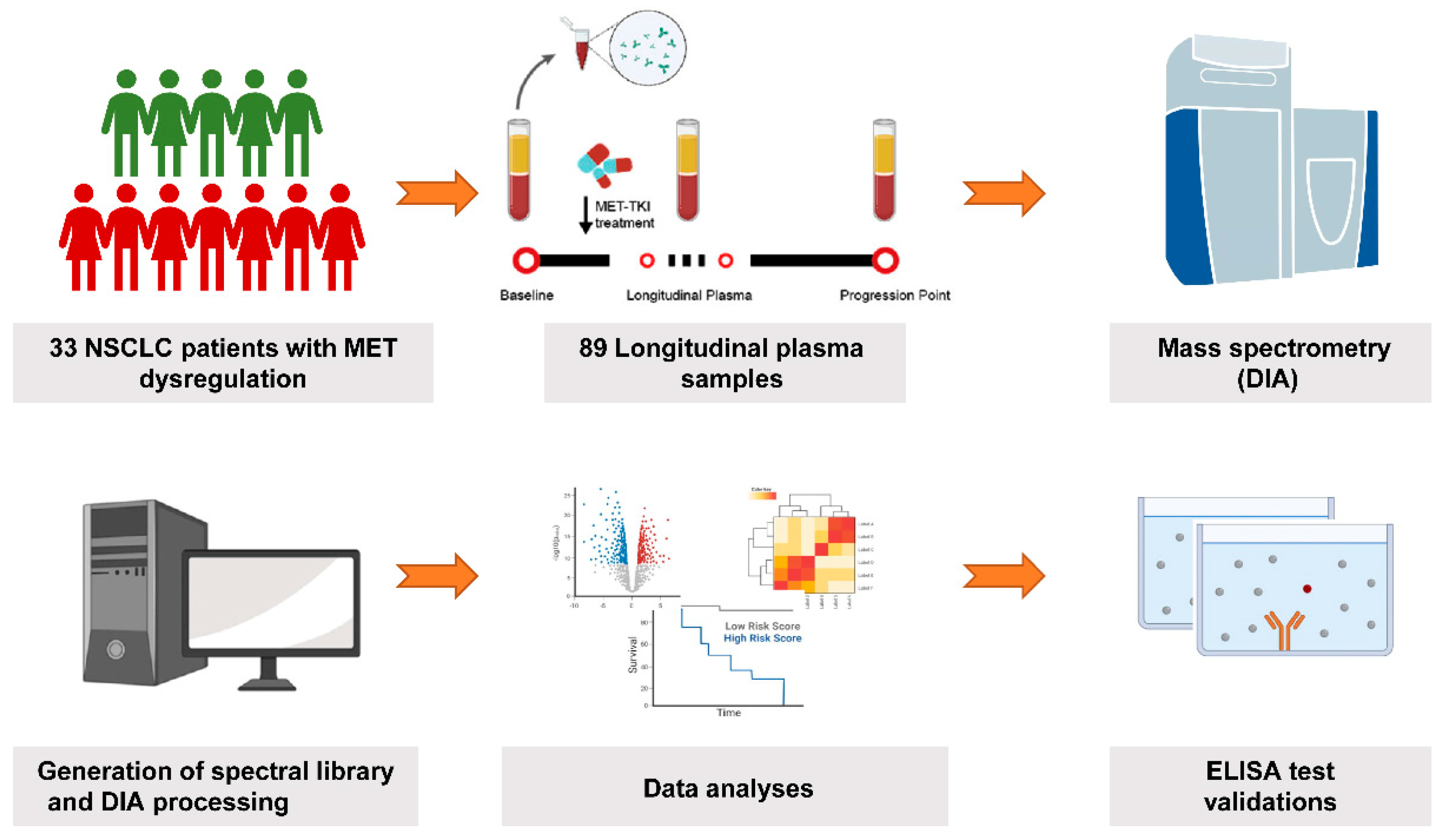
2. Materials and Methods
2.1. Patient Enrollment and Sample Collection
2.2. Plasma Sample Preparation for Spectral Library Generation
2.3. High-pH Reversed-Phase Fractionation
2.4. Liquid Chromatography
2.5. Mass Spectrometry
2.6. Generation of Spectral Libraries and DIA Data Analysis
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Statistical Analysis
3. Results
3.1. Patient Characteristics and Samples Collection
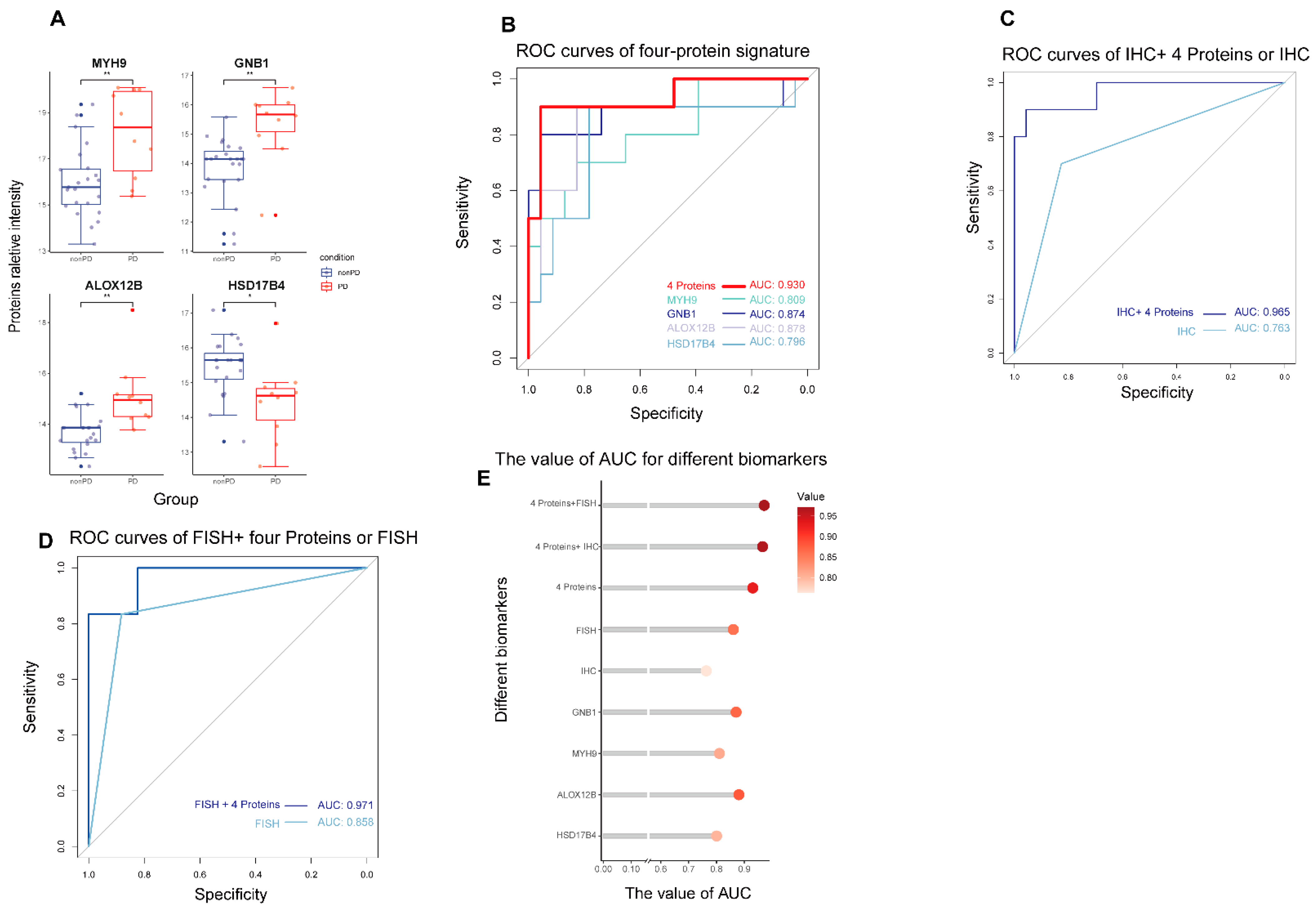
3.2. Global Proteomic Analysis of Peripheral Plasma and Predictive Biomarkers Selection for Patients Received MET Inhibitors
3.3. The Predictive Performance of Biomarkers for Response to MET Inhibitors in MET-Dysregulated NSCLC Patients
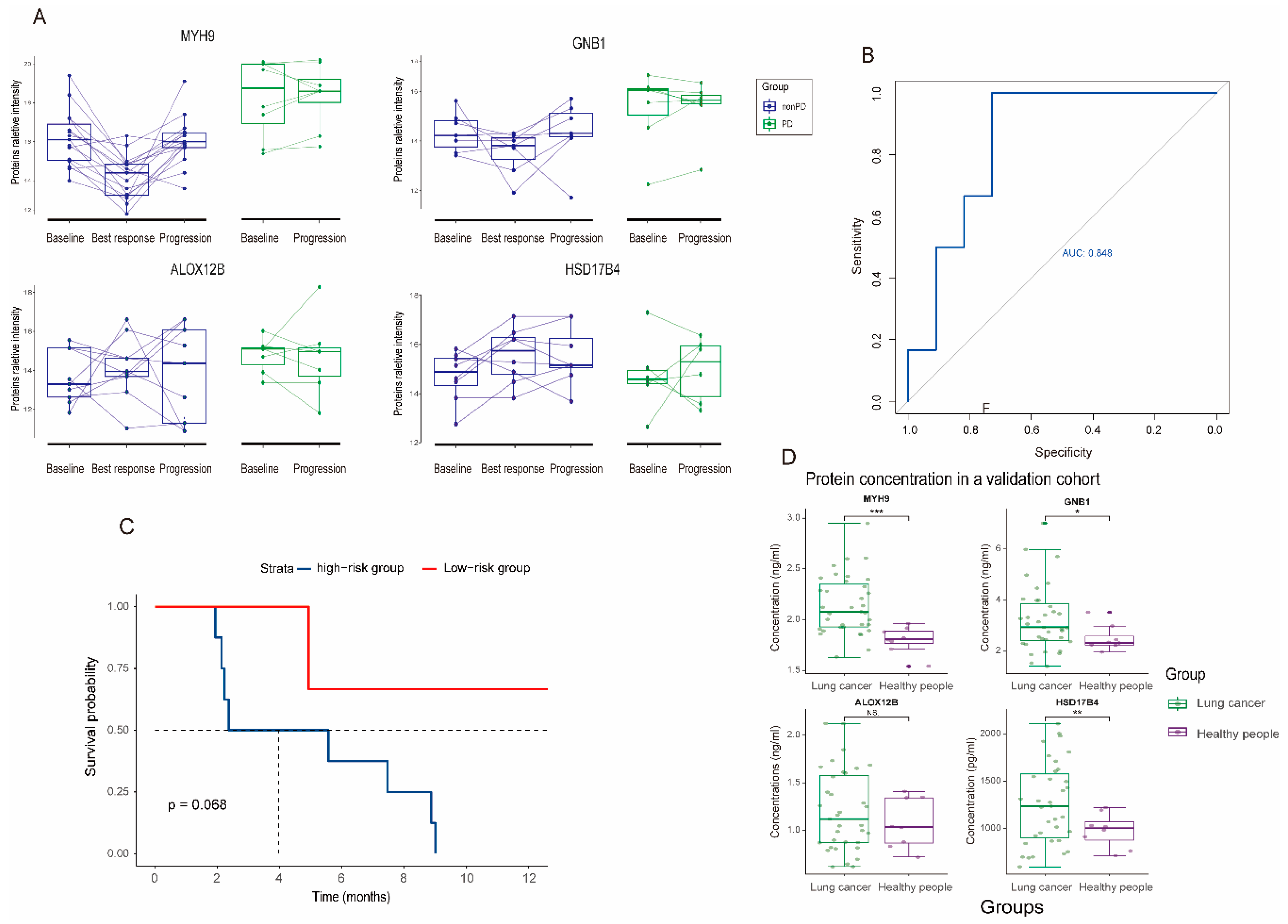
3.4. Dynamic Change and Validation of the Four Biomarker Candidates in Plasma following MET Inhibitors Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.W.; Seo, J.W.; Chung, D.H. High MET Gene Copy Number Leads to Shorter Survival in Patients with Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-Dependent Solid Tumours—Molecular Diagnosis and Targeted Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef]
- Schildhaus, H.U.; Schultheis, A.M.; Ruschoff, J.; Binot, E.; Merkelbach-Bruse, S.; Fassunke, J.; Schulte, W.; Ko, Y.D.; Schlesinger, A.; Bos, M.; et al. MET Amplification Status in Therapy-Naive Adeno- and Squamous Cell Carcinomas of the Lung. Clin. Cancer Res. 2015, 21, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Niu, F.Y.; Han, J.F.; Lou, N.N.; Yang, J.J.; Zhang, X.C.; Zhou, Q.; Xie, Z.; Su, J.; Zhao, N.; et al. Predictive and Prognostic Value of De Novo MET Expression in Patients with Advanced Non-Small-Cell Lung Cancer. Lung Cancer 2015, 90, 375–380. [Google Scholar] [CrossRef]
- Reis, H.; Metzenmacher, M.; Goetz, M.; Savvidou, N.; Darwiche, K.; Aigner, C.; Herold, T.; Eberhardt, W.E.; Skiba, C.; Hense, J.; et al. MET Expression in Advanced Non-Small-Cell Lung Cancer: Effect on Clinical Outcomes of Chemotherapy, Targeted Therapy, and Immunotherapy. Clin. Lung Cancer 2018, 19, e441–e463. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib after Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients with EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Cheng, Y.; Zhou, J.; Lu, S.; Zhang, Y.; Zhao, J.; Kim, D.-W.; Soo, R.A.; Kim, S.-W.; Pan, H.; et al. Tepotinib Plus Gefitinib in Patients with EGFR-Mutant Non-Small-Cell Lung Cancer with MET Overexpression or MET Amplification and Acquired Resistance to Previous EGFR Inhibitor (INSIGHT Study): An Open-Label, Phase 1b/2, Multicentre, Randomised Trial. Lancet Respir. Med. 2020, 8, 1132–1143. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.-Y.; Ahn, M.-J.; Cho, B.C.; Yu, H.; Kim, S.-W.; Yang, J.C.-H.; Lee, J.S.; Su, W.-C.; Kowalski, D.; et al. Osimertinib Plus Savolitinib in Patients with EGFR Mutation-Positive, MET-Amplified, Non-Small-Cell Lung Cancer after Progression on EGFR Tyrosine Kinase Inhibitors: Interim Results from a Multicentre, Open-Label, Phase 1b Study. The Lancet Oncology 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; Del Grammastro, M.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET Gene Copy Number Negatively Affects Survival of Surgically Resected Non-Small-Cell Lung Cancer Patients. J. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noonan, S.A.; Berry, L.; Lu, X.; Gao, D.; Baron, A.E.; Chesnut, P.; Sheren, J.; Aisner, D.L.; Merrick, D.; Doebele, R.C.; et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number-Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J. Thorac. Oncol. 2016, 11, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, G.G.Y.; Guo, R.; Drilon, A.; Shao Weng Tan, D. Refining Patient Selection of MET-Activated Non-Small Cell Lung Cancer through Biomarker Precision. Cancer Treat. Rev. 2022, 110, 102444. [Google Scholar] [CrossRef]
- Guo, R.; Berry, L.D.; Aisner, D.L.; Sheren, J.; Boyle, T.; Bunn, P.A., Jr.; Johnson, B.E.; Kwiatkowski, D.J.; Drilon, A.; Sholl, L.M.; et al. Met Ihc Is a Poor Screen for MET Amplification or MET Exon 14 Mutations in Lung Adenocarcinomas: Data from a Tri-Institutional Cohort of the Lung Cancer Mutation Consortium. J. Thorac. Oncol. 2019, 14, 1666–1671. [Google Scholar] [CrossRef]
- Yin, W.; Guo, M.; Tang, Z.; Toruner, G.A.; Cheng, J.; Medeiros, L.J.; Tang, G. MET Expression Level in Lung Adenocarcinoma Loosely Correlates with MET Copy Number Gain/Amplification and Is a Poor Predictor of Patient Outcome. Cancers 2022, 14, 2433. [Google Scholar] [CrossRef]
- Zhang, B.; Whiteaker, J.R.; Hoofnagle, A.N.; Baird, G.S.; Rodland, K.D.; Paulovich, A.G. Clinical Potential of Mass Spectrometry-Based Proteogenomics. Nat. Rev. Clin. Oncol. 2019, 16, 256–268. [Google Scholar] [CrossRef]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting Biomarker Discovery by Plasma Proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef] [PubMed]
- Gillette, M.A.; Satpathy, S.; Cao, S.; Dhanasekaran, S.M.; Vasaikar, S.V.; Krug, K.; Petralia, F.; Li, Y.; Liang, W.W.; Reva, B.; et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 2020, 182, 200–225.e235. [Google Scholar] [CrossRef]
- Eltahir, M.; Isaksson, J.; Mattsson, J.S.M.; Karre, K.; Botling, J.; Lord, M.; Mangsbo, S.M.; Micke, P. Plasma Proteomic Analysis in Non-Small Cell Lung Cancer Patients Treated with PD-1/PD-L1 Blockade. Cancers 2021, 13, 3116. [Google Scholar] [CrossRef]
- Niu, L.; Thiele, M.; Geyer, P.E.; Rasmussen, D.N.; Webel, H.E.; Santos, A.; Gupta, R.; Meier, F.; Strauss, M.; Kjaergaard, M.; et al. Noninvasive Proteomic Biomarkers for Alcohol-Related Liver Disease. Nat. Med. 2022, 28, 1277–1287. [Google Scholar] [CrossRef]
- Zhu, Y.; Aebersold, R.; Mann, M.; Guo, T. Snapshot: Clinical Proteomics. Cell 2021, 184, 4840. [Google Scholar] [CrossRef] [PubMed]
- Mignard, X.; Ruppert, A.M.; Antoine, M.; Vasseur, J.; Girard, N.; Mazieres, J.; Moro-Sibilot, D.; Fallet, V.; Rabbe, N.; Thivolet-Bejui, F.; et al. C-MET Overexpression as a Poor Predictor of MET Amplifications or Exon 14 Mutations in Lung Sarcomatoid Carcinomas. J. Thorac. Oncol. 2018, 13, 1962–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.X.; Jie, G.L.; Li, A.N.; Liu, S.Y.; Sun, H.; Zheng, M.M.; Zhou, J.Y.; Zhang, J.T.; Zhang, X.C.; Zhou, Q.; et al. MET Amplification Identified by Next-Generation Sequencing and Its Clinical Relevance for Met Inhibitors. Exp. Hematol. Oncol. 2021, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.P.; Yang, S.R.; Choudhury, N.J.; Ptashkin, R.N.; Eslamdoost, N.; Falcon, C.J.; Martin, A.; Plodkowski, A.; Wilhelm, C.; Shen, R.; et al. Bioinformatically-Expanded Next-Generation Sequencing Analysis Optimizes Identification of Therapeutically Relevant MET Copy Number Alterations in >50,000 Tumors. Clin. Cancer Res. 2022, 28, 4649–4659. [Google Scholar] [CrossRef] [PubMed]
- Oscorbin, I.P.; Smertina, M.A.; Pronyaeva, K.A.; Voskoboev, M.E.; Boyarskikh, U.A.; Kechin, A.A.; Demidova, I.A.; Filipenko, M.L. Multiplex Droplet Digital PCR Assay for Detection of MET and HER2 Genes Amplification in Non-Small Cell Lung Cancer. Cancers 2022, 14, 1458. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, J.T.; Mollerup, J. Companion Diagnostics and Predictive Biomarkers for MET-Targeted Therapy in NSCLC. Cancers 2022, 14, 2150. [Google Scholar] [CrossRef] [PubMed]
- Lai, G.G.Y.; Lim, T.H.; Lim, J.; Liew, P.J.R.; Kwang, X.L.; Nahar, R.; Aung, Z.W.; Takano, A.; Lee, Y.Y.; Lau, D.P.X.; et al. Clonal MET Amplification as a Determinant of Tyrosine Kinase Inhibitor Resistance in Epidermal Growth Factor Receptor-Mutant Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2019, 37, 876–884. [Google Scholar] [CrossRef]
- Fan, Y.; Sun, R.; Wang, Z.; Zhang, Y.; Xiao, X.; Liu, Y.; Xin, B.; Xiong, H.; Lu, D.; Ma, J. Detection of MET Amplification by Droplet Digital PCR in Peripheral Blood Samples of Non-Small Cell Lung Cancer. J. Cancer Res. Clin. Oncol. 2022. [Google Scholar] [CrossRef]
- Friedlaender, A.; Subbiah, V.; Russo, A.; Banna, G.L.; Malapelle, U.; Rolfo, C.; Addeo, A. EGFR and HER2 Exon 20 Insertions in Solid Tumours: From Biology to Treatment. Nat. Rev. Clin. Oncol. 2022, 19, 51–69. [Google Scholar] [CrossRef]
- Culy, C.R.; Faulds, D. Gefitinib. Drugs 2002, 62, 2237–2248. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.K.; Enderle, D.; Noerholm, M.; Breakefield, X.O.; Skog, J.K. Exosome-Based Liquid Biopsies in Cancer: Opportunities and Challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Spaargaren, M.; Pals, S.T. Syndecan-1 and Stromal Heparan Sulfate Proteoglycans: Key Moderators of Plasma Cell Biology and Myeloma Pathogenesis. Blood 2021, 137, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Shinde, R.; Miralles, M.S.; Workman, P.; de Bono, J. Applications of Liquid Biopsy in the Pharmacological Audit Trail for Anticancer Drug Development. Nat. Rev. Clin. Oncol. 2021, 18, 454–467. [Google Scholar] [CrossRef]
- Hu, S.; Ren, S.; Cai, Y.; Liu, J.; Han, Y.; Zhao, Y.; Yang, J.; Zhou, X.; Wang, X. Glycoprotein PTGDS Promotes Tumorigenesis of Diffuse Large B-Cell Lymphoma by MYH9-Mediated Regulation of Wnt-Beta-Catenin-STAT3 Signaling. Cell Death Differ. 2022, 29, 642–656. [Google Scholar] [CrossRef]
- Lin, X.; Li, A.M.; Li, Y.H.; Luo, R.C.; Zou, Y.J.; Liu, Y.Y.; Liu, C.; Xie, Y.Y.; Zuo, S.; Liu, Z.; et al. Silencing MYH9 Blocks HBx-Induced GSK3beta Ubiquitination and Degradation to Inhibit Tumor Stemness in Hepatocellular Carcinoma. Signal Transduct. Target. Ther. 2020, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Zimmermannova, O.; Doktorova, E.; Stuchly, J.; Kanderova, V.; Kuzilkova, D.; Strnad, H.; Starkova, J.; Alberich-Jorda, M.; Falkenburg, J.H.F.; Trka, J.; et al. An Activating Mutation of GNB1 Is Associated with Resistance to Tyrosine Kinase Inhibitors in ETV6-ABL1-Positive Leukemia. Oncogene 2017, 36, 5985–5994. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, B.; Li, Y.M.; Yang, Q.Y.; Tu, K.J.; Li, L.Y. ALOX12B Promotes Carcinogenesis in Cervical Cancer by Regulating the PI3K/ERK1 Signaling Pathway. Oncol. Lett. 2020, 20, 1360–1368. [Google Scholar] [CrossRef]
- Liu, Z.; Li, L.; Li, X.; Hua, M.; Sun, H.; Zhang, S. Prediction and Prognostic Significance of ALOX12B and PACSIN1 Expression in Gastric Cancer by Genome-Wide RNA Expression and Methylation Analysis. J. Gastrointest. Oncol. 2021, 12, 2082–2092. [Google Scholar] [CrossRef]
- Schekman, R. Peroxisomes: Another Branch of the Secretory Pathway? Cell 2005, 122, 1–2. [Google Scholar] [CrossRef]
- Yamashita, S.; Hattori, N.; Fujii, S.; Yamaguchi, T.; Takahashi, M.; Hozumi, Y.; Kogawa, T.; El-Omar, O.; Liu, Y.Y.; Arai, N.; et al. Multi-Omics Analyses Identify HSD17B4 Methylation-Silencing as a Predictive and Response Marker of HER2-Positive Breast Cancer to HER2-Directed Therapy. Sci. Rep. 2020, 10, 15530. [Google Scholar] [CrossRef] [PubMed]
- Audet-Walsh, E.; Bellemare, J.; Lacombe, L.; Fradet, Y.; Fradet, V.; Douville, P.; Guillemette, C.; Levesque, E. The Impact of Germline Genetic Variations in Hydroxysteroid (17-Beta) Dehydrogenases on Prostate Cancer Outcomes after Prostatectomy. Eur. Urol. 2012, 62, 88–96. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical characteristics | Overall | |
|---|---|---|
| (n = 33) | ||
| Age | ||
| Median [Range] | 58.4 [29.3–73.5] | |
| Gender (%) | ||
| Female | 8 (24.2%) | |
| Male | 25 (75.8%) | |
| Smoking history (%) | ||
| No | 14 (42.4%) | |
| Yes | 19 (57.6%) | |
| Pathology (%) | ||
| Adenocarcinoma | 32 (97.0%) | |
| Pulmonary sarcomatoid carcinoma | 1 (3.0%) | |
| Stage (%) | ||
| III | 1 (3.0%) | |
| IV | 32 (97.0%) | |
| Performance status score (%) | ||
| 1 | 32 (97.0%) | |
| 2 | 1 (3.0%) | |
| Brain metastasis (%) | ||
| No | 23 (69.7%) | |
| Yes | 10 (30.3%) | |
| EGFR mutation (%) | ||
| 19DEL | 5 (15.2%) | |
| L858R | 8 (24.2%) | |
| Negative | 20 (60.6%) | |
| MET FISH (%) | ||
| Negative | 7 (21.2%) | |
| Positive | 16 (48.5%) | |
| NA | 10 (30.3%) | |
| MET IHC (%) | ||
| Negative | 11 (33.3%) | |
| Positive | 22 (66.7%) | |
| Treatment (%) | ||
| MET inhibitor + EGFR-TKI | 12 (36.4%) | |
| MET inhibitor | 21 (63.6%) | |
| Treatment line (%) | ||
| 1 | 7 (21.2%) | |
| ≥2 | 26 (78.2%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jie, G.-L.; Peng, L.-X.; Zheng, M.-M.; Sun, H.; Wang, S.-R.; Liu, S.-Y.M.; Yin, K.; Chen, Z.-H.; Tian, H.-X.; Yang, J.-J.; et al. Longitudinal Plasma Proteomics-Derived Biomarkers Predict Response to MET Inhibitors for MET-Dysregulated NSCLC. Cancers 2023, 15, 302. https://doi.org/10.3390/cancers15010302
Jie G-L, Peng L-X, Zheng M-M, Sun H, Wang S-R, Liu S-YM, Yin K, Chen Z-H, Tian H-X, Yang J-J, et al. Longitudinal Plasma Proteomics-Derived Biomarkers Predict Response to MET Inhibitors for MET-Dysregulated NSCLC. Cancers. 2023; 15(1):302. https://doi.org/10.3390/cancers15010302
Chicago/Turabian StyleJie, Guang-Ling, Lun-Xi Peng, Mei-Mei Zheng, Hao Sun, Song-Rong Wang, Si-Yang Maggie Liu, Kai Yin, Zhi-Hong Chen, Hong-Xia Tian, Jin-Ji Yang, and et al. 2023. "Longitudinal Plasma Proteomics-Derived Biomarkers Predict Response to MET Inhibitors for MET-Dysregulated NSCLC" Cancers 15, no. 1: 302. https://doi.org/10.3390/cancers15010302