
Identification of Tissue-Specific Gene Clusters Induced by DNA Demethylation in Lung Adenocarcinoma: More Than Germline Genes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. LUAD Cell Lines Datasets
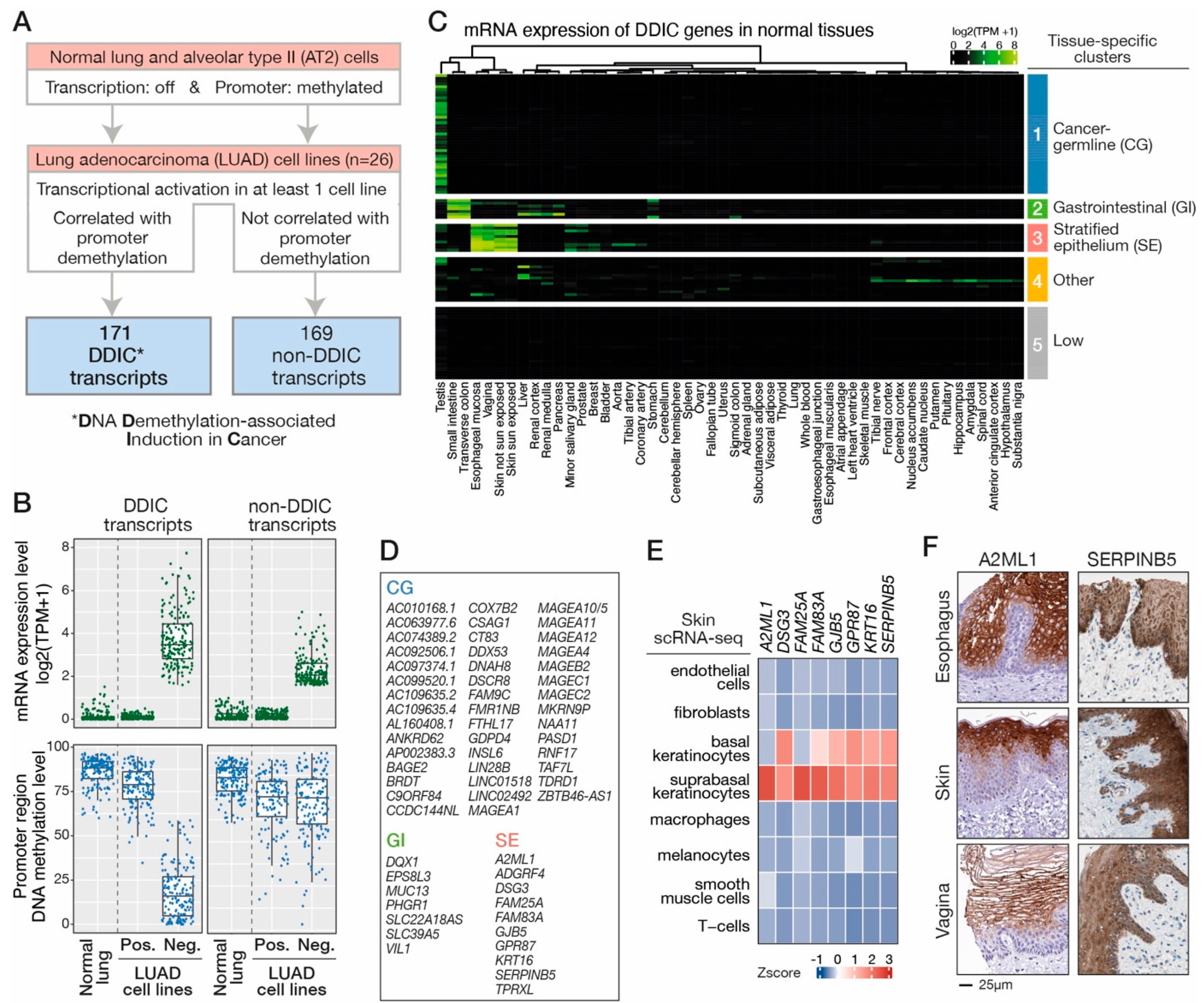
2.2. Procedure for the Identification of DDIC and Non-DDIC Transcripts in LUAD Cell Lines
- (1)
- Merging of transcripts originating from the same promoter region in LUAD cell lines. Transcripts arising from a transcriptional start site (TSS) located less than 50bp apart were considered to originate from the same promoter region. Their expression levels were therefore summed for transcript quantification. For each pooled transcript group, the TSS located at the closest 5′ end was chosen as reference.
- (2)
- Selection of repressed transcripts in normal lung. Transcripts that were initially silent in AT2 cells (RNA-seq data, [24]) and normal lung (RNA-seq, Roadmap Epigenomics [25]), and exhibited a methylation level of their promoter region ≥60% in normal lung (WGBS, Roadmap Epigenomics) and AT2 cells (WGBS, [26]) were retained. Transcript repression was defined as TPM <1 and/or lack of RNA-seq reads mapping to the genomic region of each transcript. The level of methylation of the promoter region was computed by averaging the methylation values of each CpG located at TSS −/+ 400 bp for each transcript. Only genomic regions harboring at least 3 CpGs were considered for this analysis, and when coverage information was available, CpGs covered by at least 2 reads were analyzed.
- (3)
- Selection of activated transcripts in LUAD cell lines. Among the previous transcript selection, we selected those that fulfilled the following criteria: first, transcripts were repressed in at least 2/26 cell lines (expression quantile 10 ≤ 0.1 TPM) and activated in at least 1/26 cell line with their maximum expression ≥2 TPM. Secondly, the minimum value of the level of methylation of their promoter region was below 40% and the maximum above 60%. These transcripts were considered as being repressed in normal lung tissue and ectopically expressed in LUAD cell lines.
- (4)
- Correlation between transcriptional activation and methylation of promoter region. We computed a Pearson correlation of the expression and promoter methylation status of each transcript in LUAD cell lines. At least 7/26 LUAD cell lines had to have both transcript expression and promoter methylation information available for a correlation to be computed. We selected transcripts that showed a significant inverse correlation between their transcriptional activation and promoter methylation (r ≤ −0.4, p-value < 0.05).
- (5)
- Manual curation. We visualized BAM files of the LUAD cell lines using IGV (Integrative Genomics Viewer, [27]) to determine the accuracy of the chosen TSS, confirm the transcriptional activation status, and determine transcript backbone for each of the selected transcripts. Finally, we also verified the correlation between expression and promoter methylation of each transcript by generating heatmaps (ComplexHeatmap R package v2.8.0) depicting the methylation state of each CpG located at TSS −/+ 400 bp. Transcripts that passed all the above filters were referred to as DNA-Demethylation-associated Induction in Cancer transcripts (DDIC transcripts). Non-DDIC transcripts were selected using the same procedure with different criteria for the following parameters: transcripts were originating from a gene giving rise only to that transcript (i.e., not alternative gene promoters), the corresponding gene is repressed in normal lung (lung tissue samples of GTEx show a median TPM < 0.5 and the one from Roadmap Epigenomics <1 TPM) and repressed in AT2 cells (TPM < 1). They showed no correlation between transcriptional activation and promoter demethylation (−0.1 ≤ r ≤ 0.1). No manual curation step was performed for non-DDIC transcripts.
2.3. RNA-Seq Public Datasets
- (1)
- Bulk RNA-seq. Normalized expression data of samples coming from 50 normal tissues were obtained from GTEx portal (v8, [28]). Both gene expression of individual samples per tissue and median expression of all samples per tissue were downloaded. FastQ files of the following cells and tissues were downloaded from Sequence Read Archive (SRA): AT2, lung, testis, sigmoid colon, small intestine, stomach, pancreas, liver, esophagus, skin, adipose, cerebral cortex, heart and thyroid. Files were processed as previously described [29]. All accession numbers are listed in Table S3.
- (2)
2.4. RNA-Seq of Cell Lines after DNA Demethylation Treatment
2.5. DNA Methylation Public Datasets
- (1)
- Normal tissues. For cerebral cortex samples, FastQ files of whole genome bisulfite-seq (WGBS) were downloaded from SRA. Trim Galore! software v0.5.0 was used to read quality control and trimming of low-quality reads. Read alignment and methylation calling were performed using Bismark v0.20.0. For liver sample, BAM file of WGBS was downloaded from ENCODE consortium database [39], and methylation calling was performed using Bismark v0.20.0. For the rest of the normal tissues, normalized hg38 WGBS data were obtained from the ENCODE consortium database.
- (2)
- Primary cells. For keratinocytes and sperm cells, WGBS hg19 normalized data were obtained from SRA [25]. Data were converted to hg38 coordinates using liftOver v1.10.0 R package. Normalized hg38 WGBS data of HUES64 were obtained from the ENCODE consortium database. For AT2 cells, FastQ files of WGBS were downloaded from SRA [26]. Trim Galore! software v0.5.0 was used to read quality control and trimming of low-quality reads. Read alignment onto hg38 genome and methylation calling were performed using Bismark v0.20.0. For all the above studies, when available, methylation information for the same CpG sequenced in forward and reverse strand were averaged. For duodenal crypt cell samples, normalized Infinium HumanMethylation 450 assays and EPICarrays data were obtained [40]. All accession numbers are listed in Table S3.
2.6. Immunohistochemical Data of Normal Tissues
2.7. Cell Culture
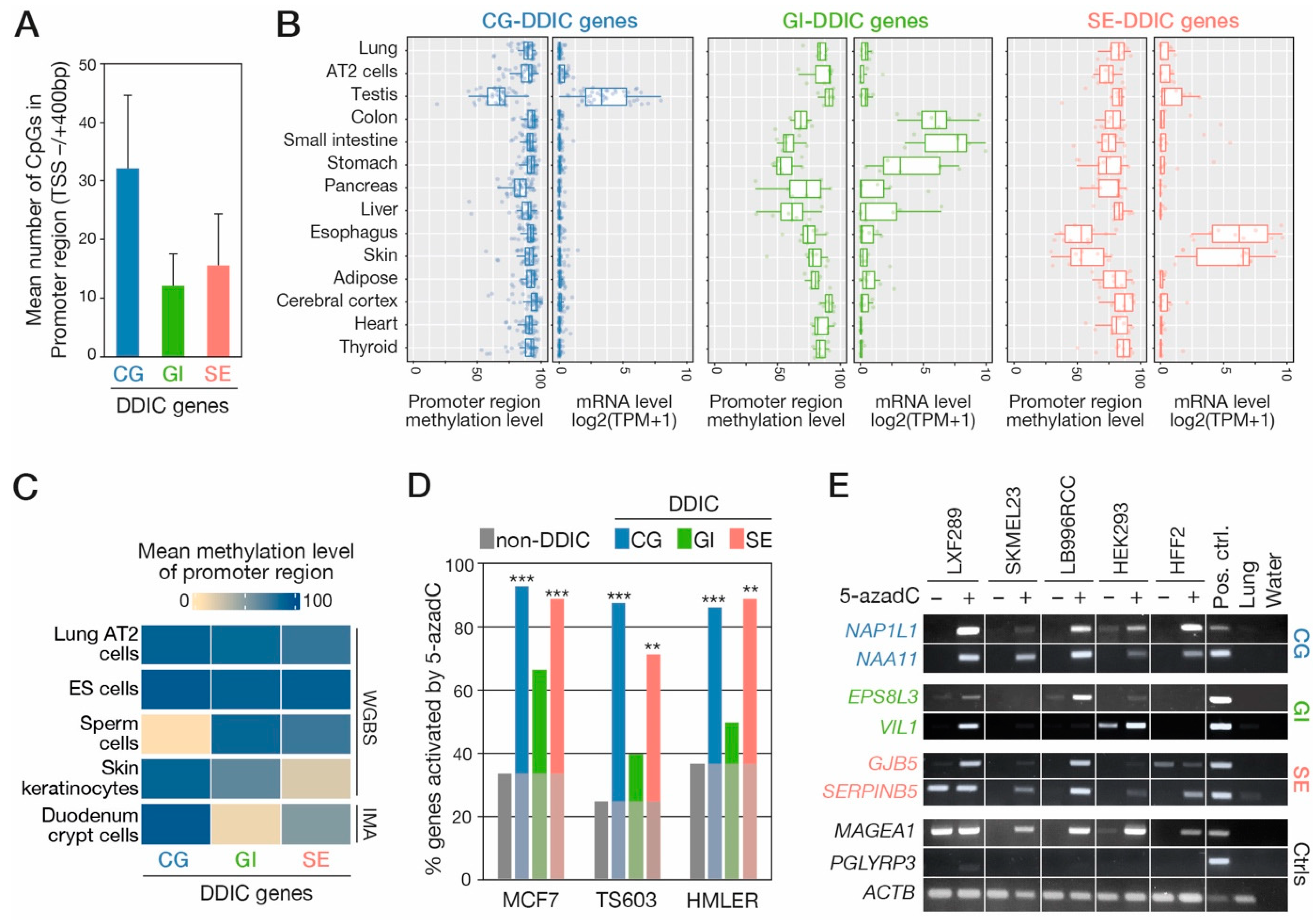
2.8. Cell Treatment with 5-azadC and siRNAs
2.9. RT-PCR and qPCR Analyses
2.10. Western Blot
2.11. The Cancer Genome Atlas Consortium Datasets and Analyses
- (1)
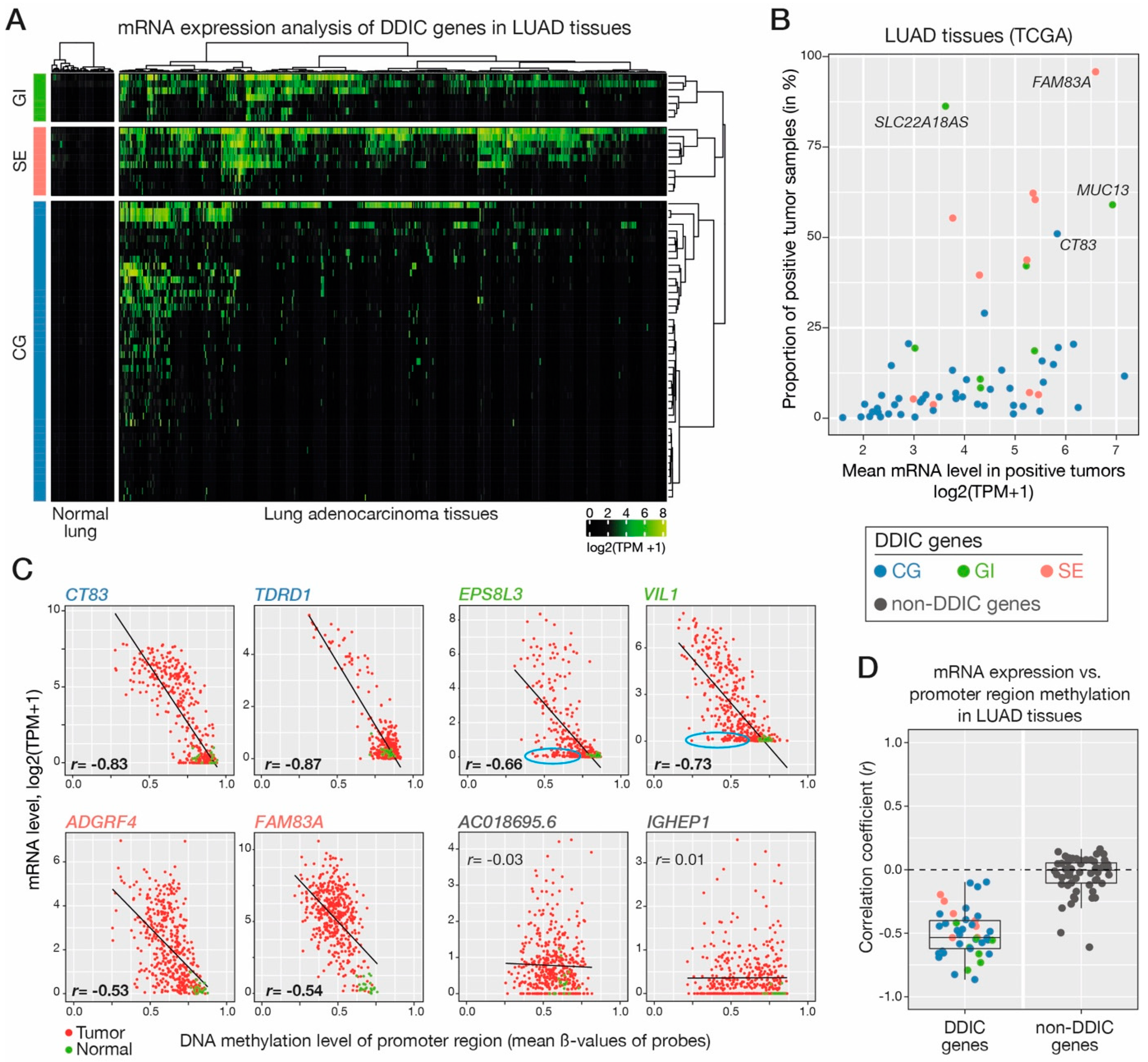
- Expression analysis. FPKM expression data were converted to TPM by dividing each FPKM gene expression in a sample by the sum of all gene expressions for that sample (×106). LUAD tumor samples were considered positive for the expression of a gene if they exhibited a TPM ≥ 2. They were considered negative when they showed a TPM <1. To define activating and non-activating LUAD tumor groups for each DDIC, expression quantile 20 and 80 of each gene were used as activation cut-offs (i.e., tumor samples that show a DDIC expression TPM <q20 were considered as repressing, and conversely, when TPM >q80 were considered as activating). When q80 value was <1 TPM, then all tumor samples showing a gene expression ≥1 TPM were considered positive.
- (2)
- Tumor grade analysis. Tumor grades were compared in the activating and non-activating tumor groups using a Chi-squared test. Comparison was computed if there were at least 15 tumors in each group. LUAD samples qualified as grades 1.5 and 2.5 were categorized as grade 1 and grade 2, respectively.
- (3)
- Survival analysis. Patient median survival time were compared between the activating and non-activating LUAD tumor groups using a Log rank test from the survival R package v3.2-11. At least 15 tumor samples had to constitute each group, to compare survival time between patients. When median survival time was not reached for a tumor group, the length of the TCGA-LUAD survival study was taken as the median survival time.
- (4)
- Methylation analysis. Infinium CG probes in hg19 coordinates were converted to hg38 coordinates using liftOver v1.10.0 R package. For correlation analyses between gene expression and promoter DNA methylation, LUAD tumor samples that exhibited information for both expression and promoter methylation were analyzed. CG probes located at −/+ 400 bp of TSS of each gene were averaged to define the level of methylation of the promoter region. Pearson correlation was computed for each gene.
- (5)
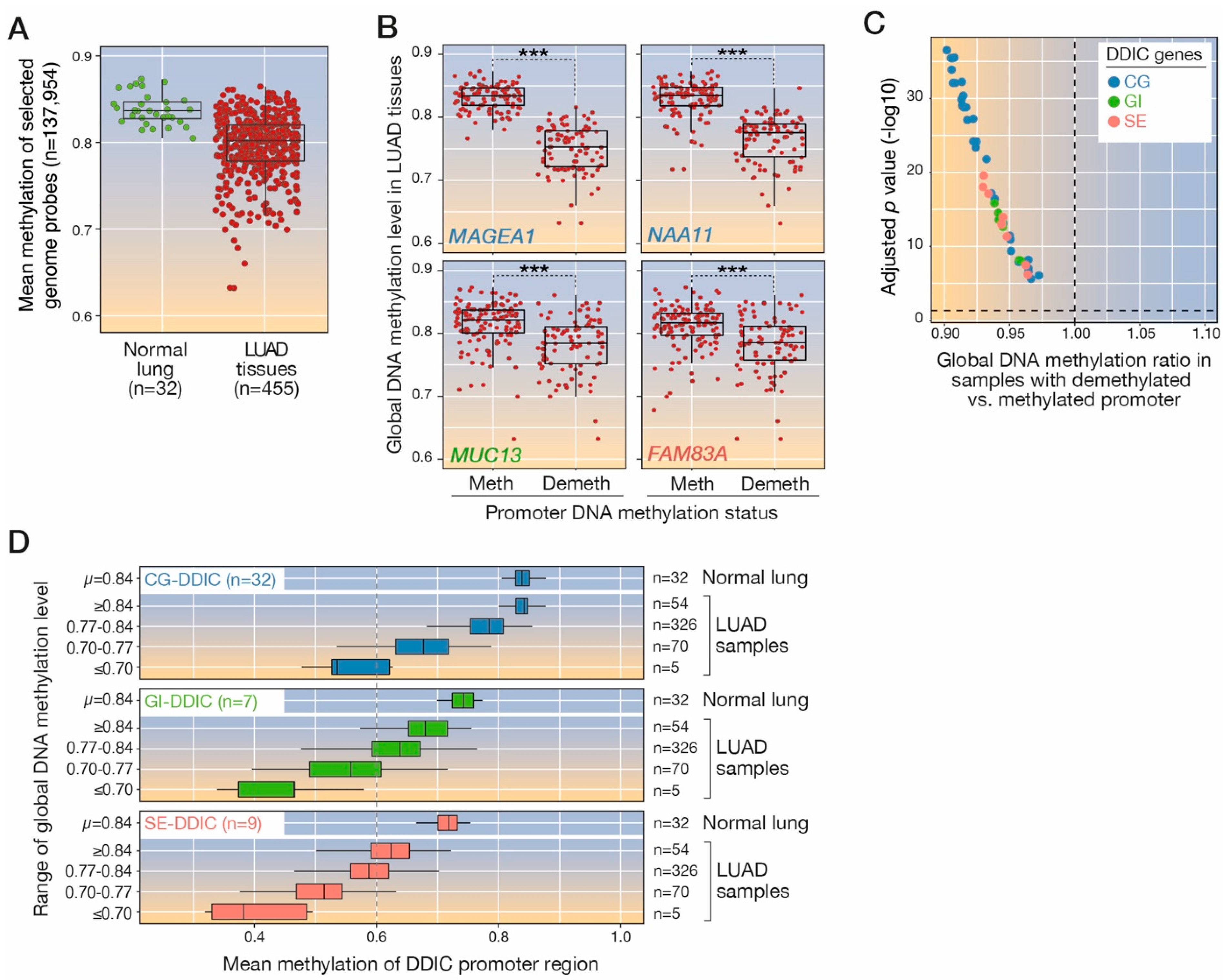
- Global DNA methylation analysis. Autosomal probes that showed an average of methylation higher than 0.7 in all normal lung samples (n = 32) were selected as a proxy for the assessment of global methylation levels in each tissue sample (n = 137,954 probes). To assess promoter methylation status in regard to global methylation levels, we defined two tumor subgroups based on the promoter methylation values of each DDIC in all LUAD tumor samples. We considered a first group of tumor samples that showed a methylated promoter region of each gene (i.e., the promoter methylation level was ≥quantile 80 of methylation values for that gene) and a second group that showed a demethylated promoter of each gene (promoter methylation ≤ quantile 20). Then, the global methylation level was compared between these two tumor groups using a Student’s t-test.
2.12. Statistical Analysis and Graphical Representations
3. Results
3.1. Search for Transcripts Showing DNA Demethylation-Associated Induction in LUAD Cell Lines
3.2. DDIC Genes Belonging to Tissue-Specific Expression Programs
3.3. DDIC Promoters Show Tissue- and Cell Type-Specific DNA Demethylation
3.4. Experimental Evaluation of the Role of DNA Methylation in Regulating DDIC Transcripts
3.5. Validating DNA Demethylation-Associated Induction of DDIC Genes In Vivo in LUAD Tumors
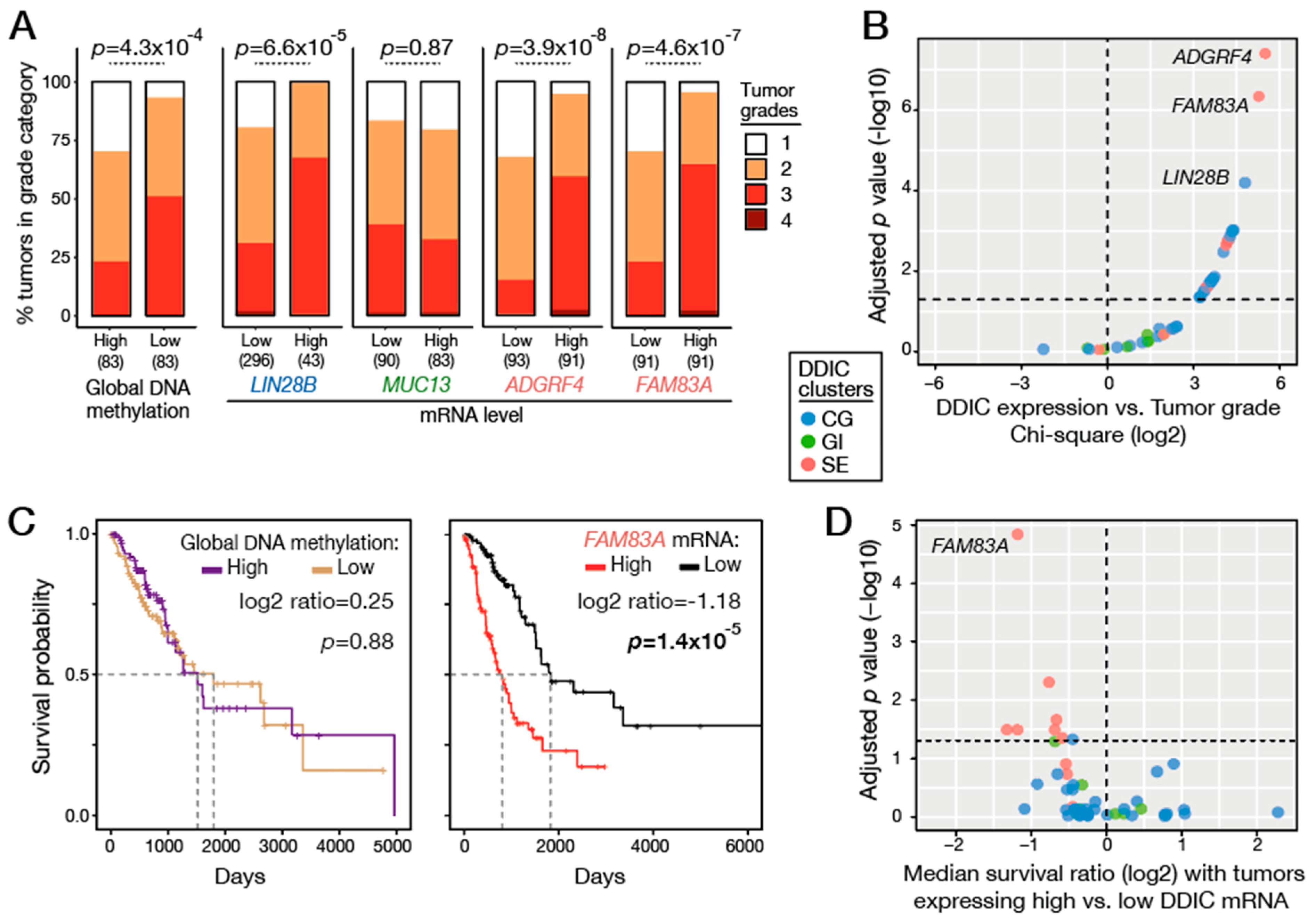
3.6. Global Genome Hypomethylation Is Associated with Local Demethylation of DDIC Genes
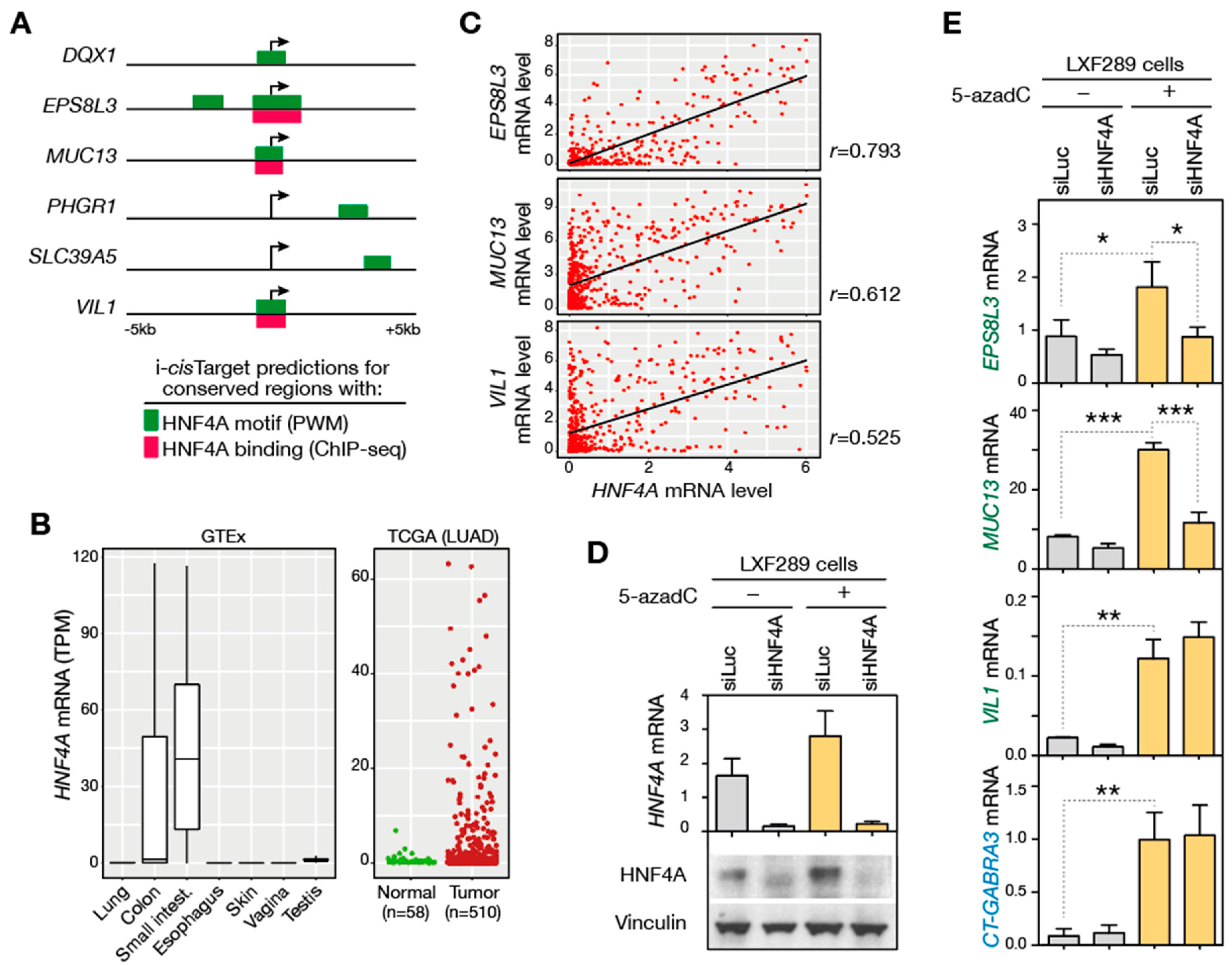
3.7. Assessing the Role of Transcription Factors in the Activation of GI-DDIC Genes
3.8. Associating Genome Hypomethylation and DDIC Gene Activation with Tumor Grade and Patient Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggs, A.D. DNA methylation and cell memory. Cell Biophys. 1989, 15, 1–13. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Feinberg, A.P. Epigenetic stochasticity, nuclear structure and cancer: The implications for medicine. J. Intern. Med. 2014, 276, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucl. Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef] [PubMed]
- De Smet, C.; Loriot, A. DNA hypomethylation and activation of germline-specific genes in cancer. Adv. Exp. Med. Biol. 2013, 754, 149–166. [Google Scholar]
- Cannuyer, J.; Loriot, A.; Parvizi, G.K.; De Smet, C. Epigenetic hierarchy within the MAGEA1 cancer-germline gene: Promoter DNA methylation dictates local histone modification. PLoS ONE 2013, 8, e58743. [Google Scholar] [CrossRef]
- De Smet, C.; Lurquin, C.; Lethé, B.; Martelange, V.; Boon, T. DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol. Cell. Biol. 1999, 19, 7327–7335. [Google Scholar] [CrossRef] [Green Version]
- Grunwald, C.; Koslowski, M.; Arsiray, T.; Dhaene, K.; Praet, M.; Victor, A.; Morresi-Hauf, A.; Lindner, M.; Passlick, B.; Lehr, H.A.; et al. Expression of multiple epigenetically regulated cancer/germline genes in nonsmall cell lung cancer. Int. J. Cancer 2006, 118, 2522–2528. [Google Scholar] [CrossRef] [PubMed]
- Sigalotti, L.; Coral, S.; Nardi, G.; Spessotto, A.; Cortini, E.; Cattarossi, I.; Colizzi, F.; Altomonte, M.; Maio, M. Promoter methylation controls the expression of MAGE2, 3 and 4 genes in human cutaneous melanoma. J. Immunother. 2002, 25, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Akers, S.N.; Odunsi, K.; Karpf, A.R. Regulation of cancer germline antigen gene expression: Implications for cancer immunotherapy. Future Oncol. 2010, 6, 717–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tongelen, A.; Loriot, A.; De Smet, C. Oncogenic roles of DNA hypomethylation through the activation of cancer-germline genes. Cancer Lett. 2017, 396, 130–137. [Google Scholar] [CrossRef]
- Whitehurst, A.W. Cause and consequence of cancer/testis antigen activation in cancer. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 251–272. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szyf, M.; Pakneshan, P.; Rabbani, S.A. DNA methylation and breast cancer. Biochem. Pharm. 2004, 68, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Dokun, O.Y.; Florl, A.R.; Seifert, H.H.; Wolff, I.; Schulz, W.A. Relationship of SNCG, S100A4, S100A9 and LCN2 gene expression and DNA methylation in bladder cancer. Int. J. Cancer 2008, 123, 2798–2807. [Google Scholar] [CrossRef]
- Bestor, T.H.; Edwards, J.R.; Boulard, M. Notes on the role of dynamic DNA methylation in mammalian development. Proc. Natl. Acad. Sci. USA 2015, 112, 6796–6799. [Google Scholar] [CrossRef] [Green Version]
- Blanpain, C. Tracing the cellular origin of cancer. Nat. Cell Biol. 2013, 15, 126–134. [Google Scholar] [CrossRef]
- Suzuki, A.; Makinoshima, H.; Wakaguri, H.; Esumi, H.; Sugano, S.; Kohno, T.; Tsuchihara, K.; Suzuki, Y. Aberrant transcriptional regulations in cancers: Genome, transcriptome and epigenome analysis of lung adenocarcinoma cell lines. Nucl. Acids Res. 2014, 42, 13557–13572. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.S.; Van Tongelen, A.; Loriot, A.; De Smet, C. Epigenetic Coactivation of MAGEA6 and CT-GABRA3 Defines Orientation of a Segmental Duplication in the Human X Chromosome. Cytogenet. Genome Res. 2019, 159, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Marconett, C.N.; Zhou, B.; Sunohara, M.; Pouldar, T.M.; Wang, H.; Liu, Y.; Rieger, M.E.; Tran, E.; Flodby, P.; Siegmund, K.D.; et al. Cross-Species Transcriptome Profiling Identifies New Alveolar Epithelial Type I Cell-Specific Genes. Am. J. Respir. Cell Mol. Biol. 2017, 56, 310–321. [Google Scholar] [CrossRef]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Zuber, V.; Marconett, C.N.; Shi, J.; Hua, X.; Wheeler, W.; Yang, C.; Song, L.; Dale, A.M.; Laplana, M.; Risch, A.; et al. Pleiotropic Analysis of Lung Cancer and Blood Triglycerides. J. Natl. Cancer Ins. 2016, 108, djw167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Fain, J.S.; Loriot, A.; Diacofotaki, A.; Van Tongelen, A.; De Smet, C. Transcriptional overlap links DNA hypomethylation with DNA hypermethylation at adjacent promoters in cancer. Sci. Rep. 2021, 11, 17346. [Google Scholar] [CrossRef]
- Sole-Boldo, L.; Raddatz, G.; Schutz, S.; Mallm, J.P.; Rippe, K.; Lonsdorf, A.S.; Rodriguez-Paredes, M.; Lyko, F. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 2020, 3, 188. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Bjorling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessio, A.C.; Weaver, I.C.; Szyf, M. Acetylation-induced transcription is required for active DNA demethylation in methylation-silenced genes. Mol. Cell Biol. 2007, 27, 7462–7474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandin, M.; Mathot, P.; Devailly, G.; Bidet, Y.; Ghantous, A.; Favrot, C.; Gibert, B.; Gadot, N.; Puisieux, I.; Herceg, Z.; et al. Inhibition of DNA methylation promotes breast tumor sensitivity to netrin-1 interference. EMBO Mol. Med. 2016, 8, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Sahm, F.; Steffl, B.; Arrillaga-Romany, I.; Cahill, D.; Monje, M.; Herold-Mende, C.; Wick, W.; Turcan, S. TERT and DNMT1 expression predict sensitivity to decitabine in gliomas. Neuro-Oncology 2021, 23, 76–87. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Lewis, S.K.; Nachun, D.; Martin, M.G.; Horvath, S.; Coppola, G.; Jones, D.L. DNA Methylation Analysis Validates Organoids as a Viable Model for Studying Human Intestinal Aging. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 527–541. [Google Scholar] [CrossRef] [Green Version]
- Tilman, G.; Arnoult, N.; Lenglez, S.; Van Beneden, A.; Loriot, A.; De Smet, C.; Decottignies, A. Cancer-linked satellite 2 DNA hypomethylation does not regulate Sat2 non-coding RNA expression and is initiated by heat shock pathway activation. Epigenetics 2012, 7, 903–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucl. Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Yu, K.H.; Zhang, C.; Berry, G.J.; Altman, R.B.; Re, C.; Rubin, D.L.; Snyder, M. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat. Commun. 2016, 7, 12474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billingham, R.E.; Silvers, W.K. The origin and conservation of epidermal specificities. N. Engl. J. Med. 1963, 268, 539–545. [Google Scholar] [CrossRef] [PubMed]
- D’Anna, F.; Van Dyck, L.; Xiong, J.; Zhao, H.; Berrens, R.V.; Qian, J.; Bieniasz-Krzywiec, P.; Chandra, V.; Schoonjans, L.; Matthews, J.; et al. DNA methylation repels binding of hypoxia-inducible transcription factors to maintain tumor immunotolerance. Genome Biol. 2020, 21, 182. [Google Scholar] [CrossRef]
- Devailly, G.; Grandin, M.; Perriaud, L.; Mathot, P.; Delcros, J.G.; Bidet, Y.; Morel, A.P.; Bignon, J.Y.; Puisieux, A.; Mehlen, P.; et al. Dynamics of MBD2 deposition across methylated DNA regions during malignant transformation of human mammary epithelial cells. Nucl. Acids Res. 2015, 43, 5838–5854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Cadieux, B.; Ching, T.T.; VandenBerg, S.R.; Costello, J.F. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006, 66, 8469–8476. [Google Scholar] [CrossRef] [Green Version]
- De Smet, C.; De Backer, O.; Faraoni, I.; Lurquin, C.; Brasseur, F.; Boon, T. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc. Natl. Acad. Sci. USA 1996, 93, 7149–7153. [Google Scholar] [CrossRef] [Green Version]
- Woloszynska-Read, A.; Mhawech-Fauceglia, P.; Yu, J.; Odunsi, K.; Karpf, A.R. Intertumor and intratumor NY-ESO-1 expression heterogeneity is associated with promoter-specific and global DNA methylation status in ovarian cancer. Clin. Cancer Res. 2008, 14, 3283–3290. [Google Scholar] [CrossRef] [Green Version]
- Imrichova, H.; Hulselmans, G.; Atak, Z.K.; Potier, D.; Aerts, S. i-cisTarget 2015 update: Generalized cis-regulatory enrichment analysis in human, mouse and fly. Nucl. Acids Res. 2015, 43, W57-64. [Google Scholar] [CrossRef] [PubMed]
- De Smet, C.; Courtois, S.J.; Faraoni, I.; Lurquin, C.; Szikora, J.P.; De Backer, O.; Boon, T. Involvement of two Ets binding sites in the transcriptional activation of the MAGE1 gene. Immunogenetics 1995, 42, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Futscher, B.W.; Oshiro, M.M.; Wozniak, R.J.; Holtan, N.; Hanigan, C.L.; Duan, H.; Domann, F.E. Role for DNA methylation in the control of cell type specific maspin expression. Nat. Genet. 2002, 31, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Sugano, M.; Nagasaka, T.; Sasaki, E.; Murakami, Y.; Hosoda, W.; Hida, T.; Mitsudomi, T.; Yatabe, Y. HNF4alpha as a marker for invasive mucinous adenocarcinoma of the lung. Am. J. Surg. Pathol. 2013, 37, 211–218. [Google Scholar] [CrossRef]
- Soares, J.; Pinto, A.E.; Cunha, C.V.; Andre, S.; Barao, I.; Sousa, J.M.; Cravo, M. Global DNA hypomethylation in breast carcinoma: Correlation with prognostic factors and tumor progression. Cancer 1999, 85, 112–118. [Google Scholar] [CrossRef]
- Zhang, W.; Klinkebiel, D.; Barger, C.J.; Pandey, S.; Guda, C.; Miller, A.; Akers, S.N.; Odunsi, K.; Karpf, A.R. Global DNA Hypomethylation in Epithelial Ovarian Cancer: Passive Demethylation and Association with Genomic Instability. Cancers 2020, 12, 764. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Luo, D.; Yao, B.; Yang, D.M.; Lin, S.; Girard, L.; DeBerardinis, R.J.; Minna, J.D.; Xie, Y.; Xiao, G. Systematic Analysis of Gene Expression in Lung Adenocarcinoma and Squamous Cell Carcinoma with a Case Study of FAM83A and FAM83B. Cancers 2019, 11, 886. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Meier, R.; Furuta, S.; Lenburg, M.E.; Kenny, P.A.; Xu, R.; Bissell, M.J. FAM83A confers EGFR-TKI resistance in breast cancer cells and in mice. J. Clin. Investig. 2012, 122, 3211–3220. [Google Scholar] [CrossRef] [Green Version]
- Snijders, A.M.; Lee, S.Y.; Hang, B.; Hao, W.; Bissell, M.J.; Mao, J.H. FAM83 family oncogenes are broadly involved in human cancers: An integrative multi-omics approach. Mol. Oncol. 2017, 11, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Locke, W.J.; Guanzon, D.; Ma, C.; Liew, Y.J.; Duesing, K.R.; Fung, K.Y.C.; Ross, J.P. DNA Methylation Cancer Biomarkers: Translation to the Clinic. Front. Genet. 2019, 10, 1150. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diacofotaki, A.; Loriot, A.; De Smet, C. Identification of Tissue-Specific Gene Clusters Induced by DNA Demethylation in Lung Adenocarcinoma: More Than Germline Genes. Cancers 2022, 14, 1007. https://doi.org/10.3390/cancers14041007
Diacofotaki A, Loriot A, De Smet C. Identification of Tissue-Specific Gene Clusters Induced by DNA Demethylation in Lung Adenocarcinoma: More Than Germline Genes. Cancers. 2022; 14(4):1007. https://doi.org/10.3390/cancers14041007
Chicago/Turabian StyleDiacofotaki, Anna, Axelle Loriot, and Charles De Smet. 2022. "Identification of Tissue-Specific Gene Clusters Induced by DNA Demethylation in Lung Adenocarcinoma: More Than Germline Genes" Cancers 14, no. 4: 1007. https://doi.org/10.3390/cancers14041007